

Dynamically reprogrammed neutrophils serve as critical initiators for the pathogenesis and treatment of atherosclerosis.
Abstract
Nonresolving inflammation perpetuated by innate leukocytes is involved in the pathogenesis of unstable atherosclerosis. However, the role and regulation of neutrophils related to nonresolving inflammation and atherosclerosis are poorly understood. We report herein that chronic subclinical endotoxemia, a risk factor for atherosclerosis, skewed neutrophils into a nonresolving inflammatory state with elevated levels of inflammatory mediators (Dectin-1, MMP9, and LTB4) and reduced levels of homeostatic mediators (LRRC32, TGFβ, and FPN). The polarization of neutrophils was due to ROS-mediated activation of oxCAMKII, caused by altered peroxisome homeostasis and reduced lysosome fusion. Application of 4-phenylbutyrate (4-PBA) enhanced peroxisome homeostasis of neutrophils, reduced oxCAMKII, and rebalanced the expression profiles of pro- and anti-inflammatory mediators. Adoptive transfer of neutrophils programmed by subclinical endotoxemia rendered exacerbated atherosclerosis. In contrast, transfer of ex vivo programmed neutrophils by 4-PBA reduced the pathogenesis of atherosclerosis. Our data define novel neutrophil dynamics associated with the progression and regression of atherosclerosis.
INTRODUCTION
Atherosclerosis and related cardiovascular complications are leading causes of morbidity and mortality worldwide with serious economic and health tolls. One of the key risk factors for atherosclerosis is the establishment of nonresolving inflammation. However, the limited understanding of underlying mechanisms presents a major road block for effective prevention and treatment (1, 2).
Emerging studies suggest that the lack of inflammation resolution during atherosclerosis may occur because of reprogramming and polarization of innate leukocytes under persistent low-grade inflammatory challenges (3, 4). Some of the well-studied aspects of polarized “memory” macrophages during atherosclerosis favor foam cell formation, with reduced efferocytosis and elevated necrosis, which collectively contribute to the pathogenesis of atherosclerosis (5, 6). In addition to the well-studied macrophages during atherosclerosis, neutrophils may also be involved through less-characterized mechanisms (7). Neutrophils constitute 50 to 70% of circulating white blood cells and have been shown to be elevated in circulating blood and atherosclerotic plaques from human patients and experimental animals with unstable plaques (8–11). Although the correlation of higher neutrophil populations with increased risks of unstable atherosclerotic plaques has been increasingly appreciated, the role of differentially polarized neutrophils in the pathogenesis of atherosclerosis under chronic inflammatory conditions is still poorly understood.
Using a previously developed murine model of low-grade inflammation and atherosclerosis that we developed through repetitive injections of subclinical-dose lipopolysaccharide (LPS), we tested the hypothesis that the differentially reprogrammed neutrophils under chronic inflammatory conditions may play a critical role during the pathogenesis of atherosclerosis. To test this hypothesis, we characterized the unique polarization of neutrophils by subclinical low-dose LPS both in vitro and in vivo, as well as the underlying molecular and cellular mechanisms. Through transfusing uniquely programmed neutrophils into recipient mice, we directly examined the role of reprogrammed neutrophils during the pathogenesis of atherosclerosis and plaque stability. We identified that disruption of peroxisome homeostasis by subclinical low-dose LPS is uniquely responsible for the inflammatory polarization of neutrophils. We further examined the efficacy of restoring neutrophil peroxisome homeostasis with 4-phenylbutyrate (4-PBA) and the therapeutic potential of 4-PBA–programmed neutrophil transfusion as a treatment for experimental atherosclerosis.
RESULTS
Reduced plaque stability in mice challenged with subclinical-dose endotoxin
We first confirmed our previous report that subclinical low-dose LPS exacerbates atherosclerosis progression through Oil-Red O and hematoxylin and eosin (H&E) staining (4). High-fat diet (HFD)–fed ApoE−/− mice injected weekly with subclinical low-dose LPS (5 ng/kg body weight) for 4 weeks developed significantly larger atherosclerotic plaques as compared to mice injected with phosphate-buffered saline (PBS) (Fig. 1, A and B). It is most noteworthy that chronic injection of subclinical low-dose LPS significantly reduced plaque stability as reflected in significantly reduced collagen content within the plaque (Fig. 1C). Matrix metallopeptidases such as matrix metalloproteinase 9 (MMP9), responsible for degrading collagen, and inflammatory lipid mediators such as leukotriene B4 (LTB4) have been closely associated with reduced plaque stability (12, 13). We then tested the levels of MMP9 and observed a significant elevation of plasma MMP9 and LTB4 levels in mice injected with LPS as compared to mice injected with PBS (Fig. 1D). LPS administration also induced remarkable elevation of circulating myeloperoxidase (MPO) (fig. S1), another proinflammatory mediator promoting plaque instability (14). On the other hand, the circulating levels of the anti-inflammatory mediator transforming growth factor–β (TGFβ) were significantly reduced in mice injected with low-dose LPS (Fig. 1D). Similar effects were observed in regular chow diet (RD)–fed mice injected with low-dose LPS (fig. S2).
Fig. 1. Subclinical endotoxemia exacerbates atherosclerotic pathogenesis.
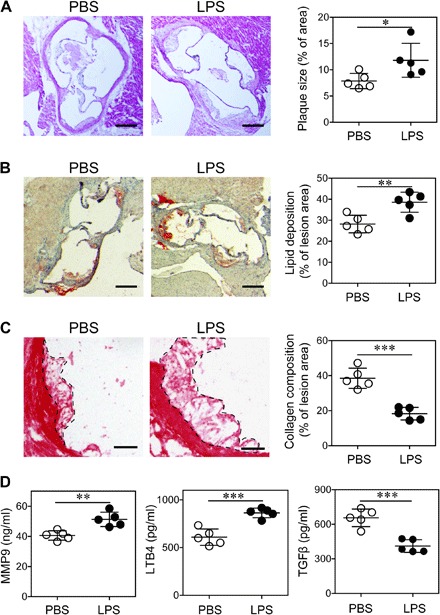
ApoE−/− mice were administrated with PBS or superlow-dose LPS, together with HFD for 4 weeks. (A) Representative images of H&E-stained atherosclerotic lesions and quantification of plaque size demonstrated as the percentage of lesion area within aortic root area. Scale bars, 300 μm. (B) Representative images of Oil Red O–stained atherosclerotic plaques and quantification of lipid deposition within lesion area. Scale bars, 300 μm. (C) Representative images of Picrosirius red–stained atherosclerotic plaques and quantification of collagen content within lesion area. Scale bars, 100 μm. (D) Determination of circulating MMP9, LTB4, and TGFβ levels by enzyme-linked immunosorbent assay (ELISA). Data are representative of two independent experiments, and error bars represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001; Student’s t test (n = 5 for each group).
Inflammatory polarization of neutrophils during atherosclerosis
Both MMP9 and LTB4 are strongly associated with unstable atherosclerotic plaques, and among innate leukocytes, neutrophils are the primary producers of MMP9 and LTB4 (15, 16). However, the potential role of neutrophils in this process has not been studied. Therefore, we decided to test the activation status of neutrophils in mice injected with subclinical low-dose LPS by examining key surface activation markers using flow cytometry. Select inflammatory markers such as CD11b and Dectin-1 were significantly elevated in LPS-injected mice as compared to PBS-injected mice (Fig. 2). In addition, these neutrophils had a significant reduction of surface-attached CD62L (Fig. 2), another classical indicator of neutrophil activation (17). We observed a similar trend in neutrophils harvested from circulating blood (Fig. 2A), spleen (Fig. 2B), and bone marrow (BM) (Fig. 2C). In addition to increased activation, we also observed higher percentages of neutrophils in the blood, spleens, and atherosclerotic plaques of mice chronically injected with LPS (fig. S3). Along with markers of neutrophil activation, we also examined the expression of key homeostatic molecules such as leucine-rich repeat containing 32 (LRRC32) and ferroportin (FPN) in neutrophils from LPS-treated mice. LRRC32 is a cell surface–conjugated molecule involved in the processing of latent TGFβ to its soluble and active form (18), while FPN is involved in the polarization of innate leukocytes into an anti-inflammatory state through the modulation of intracellular iron content (19). We observed that neutrophils from mice chronically injected with LPS had significantly reduced surface levels of both LLRC32 and FPN (Fig. 2, A to C). Instead of mean fluorescence intensity (MFI), we also used geometric MFI as a parameter to analyze the expressions of tested molecules, demonstrating a similar modulation of neutrophil phenotype after LPS treatment (fig. S4). Together, our data reveal an in vivo proinflammatory polarization of neutrophils in mice chronically injected with low-dose LPS.
Fig. 2. Subclinical endotoxin primes neutrophils into a proinflammatory state in atherosclerotic mice.
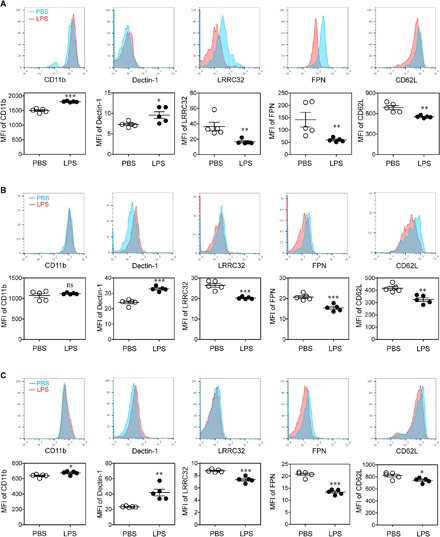
ApoE−/− mice were administrated with PBS or superlow-dose LPS, together with HFD for 4 weeks. The surface phenotypes of Ly6G+ neutrophils in the peripheral blood (A), spleen (B), and BM (C) were analyzed with flow cytometry. Data are representative of two independent experiments, and error bars represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t test (n = 5 for each group). ns, not significant.
Mechanisms underlying the inflammatory polarization of neutrophils
After characterizing the polarization of neutrophils in vivo by subclinical-dose LPS challenge, we further examined whether low-dose LPS could directly polarize neutrophils in vitro. BM-derived neutrophils were cultured with granulocyte colony-stimulating factor (G-CSF), together with or without LPS overnight. The activation status of the neutrophils was determined through measurement of secreted inflammatory mediators by enzyme-linked immunosorbent assay (ELISA) and examination of key cell surface markers by flow cytometry. We observed significantly higher levels of MMP9, LTB4, and MPO in the supernatant of neutrophils cultured with low-dose LPS as compared to those cultured with G-CSF alone (Fig. 3A). We also observed significantly elevated cell surface levels of CD11b and Dectin-1 and significantly reduced levels of CD62L, LRRC32, and FPN on neutrophils cultured with low-dose LPS (Fig. 3B). Coculture of neutrophils with low-dose LPS and oxidized low-density lipoprotein (oxLDL) further synergized the induction of MMP9, LTB4, and MPO (Fig. 3A). In addition to inflammatory lipid mediators, we also examined the expression of selected microRNAs (miRNAs) expressed by neutrophils. Our previous study revealed that subclinical-dose LPS could potently induce the expression of miR-24 in monocytes, a critical miRNA involved in the propagation of nonresolving inflammation (4). We observed that neutrophils challenged with subclinical-dose LPS also express miR-24 (Fig. 3C). Subclinical dose LPS not only induced the inflammatory miR-24 but also potently suppressed the expression of miR-126 (Fig. 3C), a key miRNA involved in tissue homeostasis and vascular integrity (20).
Fig. 3. Superlow-dose LPS induces inflammatory polarization of neutrophils in vitro.
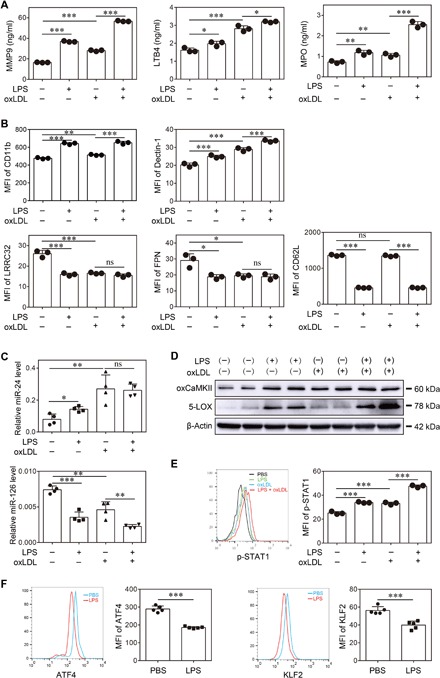
Neutrophils were purified from the BM of wild-type C57BL/6 mice and treated with PBS, superlow-dose LPS (100 pg/ml), and/or oxLDL (10 μg/ml) for 2 days. (A) Levels of MMP9, LTB4, and MPO were determined by ELISA (n = 3 for each group). (B) The surface phenotype of neutrophils was analyzed by flow cytometry (n = 3 for each group). (C) Levels of miR-24 and miR-126 were determined by real-time reverse transcription polymerase chain reaction (RT-PCR) (n = 4 for each group). (D) The expressions of oxCaMKII and 5-LOX were determined by Western blot. (E) Representative histogram and quantification of p-STAT1 level as determined by flow cytometry (n = 3 for each group). (F) Representative histograms and quantification of ATF4 and KLF2 levels as determined by flow cytometry (n = 5 for each group). Data are representative of three independent experiments, and error bars represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, (A to C and E) one-way analysis of variance (ANOVA) and (F) Student’s t test.
Given our finding of polarized inflammatory neutrophils both in vitro and in vivo by subclinical-dose LPS, we next examined the potential underlying mechanism. Since the activation of 5-lipoxygenase (5-LOX) mediated by oxidized calmodulin-dependent protein kinase II (oxCAMKII) has been shown to be important for the expression of inflammatory mediators such as LTB4, we tested the activation status of oxCAMKII and 5-LOX in neutrophils challenged with subclinical-dose LPS. As shown in Fig. 3D, we observed that neutrophils cultured with subclinical-dose LPS had increased levels of oxCAMKII and 5-LOX. Through in situ immunohistochemical staining, we further confirmed that the levels of oxCAMKII were significantly elevated in either RD-fed or HFD-fed ApoE−/− mice chronically injected with low-dose LPS as compared to corresponding control mice with PBS injection (fig. S5).
OxCAMKII-mediated signal transducer and activator of transcription 1 (STAT1) activation is responsible for the expression of Dectin-1 (21, 22). We therefore measured the activation status of STAT1 through detecting the levels of phosphorylated STAT1 (p-STAT1) by flow cytometry and observed significantly elevated levels of p-STAT1 in neutrophils challenged with subclinical-dose LPS (Fig. 3E). On the other hand, homeostatic transcription factors such as Krüppel-like factor-2 (KLF2) and activating transcription factor 4 (ATF4) are involved in transcribing homeostatic molecules such as FPN and LRRC32 and reducing reactive oxygen species (ROS)–mediated activation of oxCAMKII (23–26). We observed that subclinical-dose LPS significantly reduced the cellular levels of KLF2 and ATF4 in neutrophils (Fig. 3F). Collectively, the activation of oxCAMKII and the reduction of homeostatic KLF2 and ATF4 may be responsible for polarizing neutrophils into a nonresolving inflammatory state conducive to atherosclerosis.
In vitro polarized nonresolving inflammatory neutrophils are sufficient to confer plaque instability
To test whether polarized neutrophils by subclinical-dose LPS are directly responsible for exacerbated atherosclerosis and plaque instability, we performed experiments by transfusing in vitro polarized neutrophils into recipient animals. BM neutrophils were polarized through an in vitro culture with G-CSF and either low-dose LPS or PBS overnight. HFD-fed ApoE−/− mice were injected intravenously with neutrophils cultured in vitro with either PBS or LPS on a weekly basis for 1 month. The viability of transferred neutrophils was determined by annexin V/PI (propidium iodide) staining, and the results demonstrated that >95% of cultured neutrophils remained viable after in vitro polarization for 24 hours (fig. S6, A and B) and ~90% viable for 48 hours (fig. S6, C and D), consistent with the literature reports showing that murine BM neutrophils have a longer life span than circulating neutrophils and G-CSF delays neutrophil apoptosis (27, 28). Moreover, we tested the surface phenotype of the neutrophils before transfer. The neutrophils polarized by low-dose LPS for 24 hours exhibited elevated expressions of CD11b and Dectin-1 and reduced expressions of LRRC32, FPN, and CD62L (fig. S6E). One week after the final injection, mice were sacrificed and examined. We observed that mice injected with LPS-polarized neutrophils had significantly larger plaque sizes and higher lipid deposition within the plaques (Fig. 4, A and B). The collagen content within the plaques from mice injected with LPS-polarized neutrophils was significantly lower compared to that of mice injected with PBS-treated neutrophils (Fig. 4C). The mice receiving LPS-polarized neutrophils demonstrated significantly higher levels of plasma cholesterol and triglycerides than their counterparts receiving PBS-treated neutrophils (fig. S7A). We further tested the status of key enzymes such as oxCAMKII responsible for generating inflammatory mediators within the plaques through immunohistochemical staining and observed a significantly elevated signal of oxCAMKII within the plaques of mice transfused with LPS-polarized neutrophils as compared to mice transfused with PBS-treated control neutrophils (Fig. 4D). Adoptive transfer of LPS-primed neutrophils also resulted in the elevation of total macrophage load and the frequency of SR-A+ macrophages in atherosclerotic plaques, suggesting that primed neutrophils may communicate with monocyte/macrophage population and induce macrophage activation, further amplifying the detrimental effect (fig. S7, B to D). Consequently, we observed significantly higher levels of plasma MPO, LTB4, and MMP9 in mice transfused with LPS-polarized neutrophils as compared to mice transfused with PBS-treated control neutrophils (Fig. 4E). Thus, our data indicate that polarized neutrophils by subclinical-dose LPS are directly responsible for exacerbated atherosclerosis in vivo.
Fig. 4. Neutrophils polarized by superlow-dose LPS aggravate atherosclerosis.
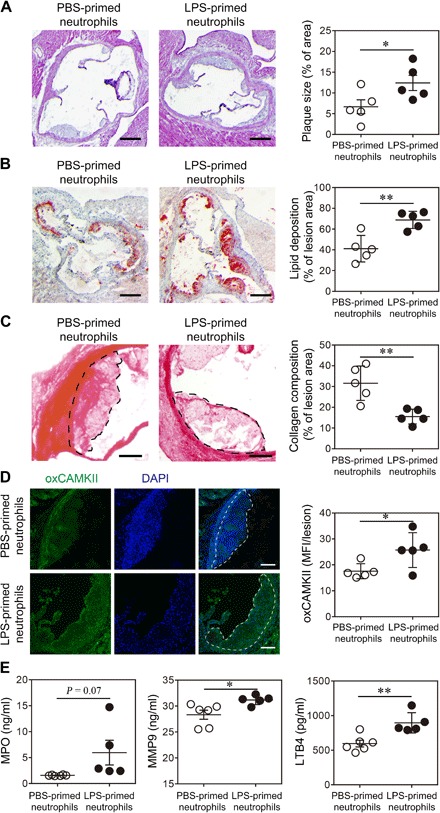
Neutrophils purified from ApoE−/− mice were treated with PBS or superlow-dose LPS (100 pg/ml) for 24 hours. PBS- or LPS-polarized neutrophils (2 × 106 cells per mouse) were then adoptively transferred by intravenous injection to HFD-fed ApoE−/− mice once a week for 4 weeks. Samples were collected 1 week after the last neutrophil transfer. (A) Representative images of H&E-stained atherosclerotic lesions and quantification of plaque size exhibited as the percentage of lesion area within aortic root area. Scale bars, 300 μm. (B) Representative images of Oil Red O–stained atherosclerotic plaques and quantification of lipid deposition within lesion area. Scale bars, 300 μm. (C) Representative images of Picrosirius red–stained atherosclerotic plaques and quantification of collagen content within lesion area. Scale bars, 100 μm. (D) Representative images and quantification of lesional oxCaMKII levels by confocal microscopy. Scale bars, 100 μm. DAPI, 4′,6-diamidino-2-phenylindole. (E) Determination of circulating MPO, MMP9, and LTB4 levels by ELISA. Data are representative of two independent experiments, and error bars represent means ± SEM. *P < 0.05 and **P < 0.01, Student’s t test (n = 5 to 6 for each group).
Polarized inflammatory neutrophils exhibit disrupted peroxisome homeostasis and elevated ROS
Our data reveal a novel aspect of neutrophil polarization that is conducive for exacerbated atherosclerosis. Our findings complement emerging studies that suggest that neutrophils may be differentially programmed into distinct functional states with significant pathophysiological implications (29). However, the cellular and molecular mechanisms underlying neutrophil polarization remain less studied. Given our above observation that neutrophils programmed by subclinical-dose LPS exhibit elevated oxCAMKII, a critical signaling molecule involved in the expression of inflammatory mediators (30), we further tested whether ROS were involved. We measured the levels of intracellular ROS in neutrophils cultured with PBS and LPS through staining with a ROS-specific fluorescent probe followed by flow cytometry. We observed that neutrophils programmed with subclinical-dose LPS or oxLDL exhibited significantly higher levels of ROS as compared to control neutrophils cultured with PBS (fig. S8 and Fig. 5A). Because altered peroxisome homeostasis is critically important for ROS generation, we examined peroxisome homeostasis with particular focus on its ability to communicate with the lysosome in neutrophils treated with subclinical-dose LPS. As shown in Fig. 5C, neutrophils cultured with subclinical-dose LPS exhibited a disruption of proper fusion between the peroxisome and the lysosome. In contrast, PBS-cultured control neutrophils exhibited efficient fusion of the peroxisome with the lysosome.
Fig. 5. 4-PBA enhances peroxisome homeostasis in neutrophils.
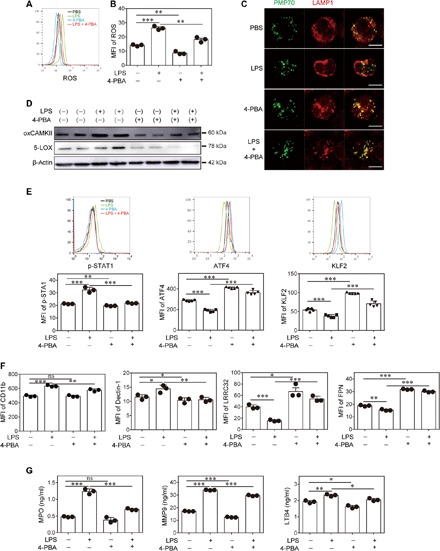
Neutrophils were purified from the BM of wild-type C57BL/6 mice and treated with PBS, superlow-dose LPS (100 pg/ml), and/or 4-PBA (1 mM) for 2 days. (A) Representative histogram of ROS level determined by CellROX labeling. (B) Quantification of ROS levels in neutrophils (n = 3 for each group). (C) Representative confocal microscopy images of the neutrophils stained with anti-PMP70 (peroxisomal membrane protein 70) and anti-LAMP1 (lysosomal-associated membrane protein 1) antibodies to demonstrate the localization and fusion of peroxisomes and lysosomes. Scale bars, 5 μm. (D) Western blot data of oxCaMKII and 5-LOX expression. (E) Representative histograms and quantification of p-STAT1 (n = 3 for each group), ATF4, and KLF2 (n = 5 for each group) levels as determined by flow cytometry. (F) Surface phenotype of neutrophils was analyzed by flow cytometry (n = 3 for each group). (G) The levels of MPO, MMP9, and LTB4 were determined by ELISA (n = 3 for each group). Data are representative of three independent experiments, and error bars represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA.
To test whether the disruption of peroxisome homeostasis is responsible for LPS-mediated neutrophil polarization, we used a selective compound, 4-PBA, which is known to induce effective peroxisome homeostasis (31). As shown in Fig. 5C, the application of 4-PBA effectively restored the fusion of the peroxisome and lysosome in cells treated with LPS. Furthermore, 4-PBA treatment effectively ameliorated the induction of ROS by LPS in neutrophils (Fig. 5, A and B). The activation of oxCAMKII and the induction of 5-LOX by LPS were also effectively ameliorated by the addition of 4-PBA (Fig. 5D). Incubation with 4-PBA also led to a reduction of p-STAT1 and restoration of KLF2/ATF4 in neutrophils challenged with LPS (Fig. 5E). Our data indicate that altered peroxisome homeostasis in neutrophils programmed by subclinical-dose LPS is responsible for elevated ROS and inflammatory polarization and that restoration of peroxisome homeostasis may hold promise for maintaining proper neutrophil function and alleviating atherosclerosis progression.
4-PBA enhances neutrophil homeostasis
Given our finding that 4-PBA can effectively ameliorate LPS-induced induction of ROS and activation of oxCAMKKII, we next performed functional tests to examine whether 4-PBA can restore functional homeostasis of neutrophils. To test this, we examined the levels of cell surface markers, expressed inflammatory mediators, and key miRNAs involved in vascular inflammatory and homeostasis. As shown in Fig. 5F, 4-PBA effectively reduced the expression of inflammatory neutrophil surface markers such as CD11b and Dectin-1 induced by LPS. The addition of 4-PBA was also sufficient for the induction of the homeostatic surface markers FPN and LRRC32 (Fig. 5F). Through ELISA, we observed that 4-PBA can potently reduce the induction of MPO, LTB4, and MMP9 in neutrophils by LPS (Fig. 5G). In terms of the miRNAs, 4-PBA treatment also reduced the induction of proinflammatory miR-24 by LPS and restored the induction of homeostatic miR-126 (fig. S9). Therefore, our data reveal that 4-PBA can effectively enhance neutrophil homeostasis.
Enhanced neutrophil homeostasis alleviates atherosclerosis
On the basis of our finding that 4-PBA could potently enhance neutrophil homeostasis, we next tested whether these neutrophils with enhanced homeostasis could alleviate atherosclerosis. To test this, ApoE−/− mice fed with HFD were transfused weekly for 4 weeks with neutrophils cultured with either PBS or 4-PBA. The neutrophils polarized with 4-PBA for 24 hours demonstrated reduced CD11b and Dectin-1 expression and elevated LRRC32, FPN, and CD62L expression (fig. S6E), exhibiting a similar phenotype as the neutrophils primed with 4-PBA for 48 hours. One week after the final transfusion, mice were sacrificed for analysis.
We observed that mice transfused with 4-PBA–programmed neutrophils had a threefold reduction in plaque sizes and a twofold reduction of the plaque lipid content (Fig. 6, A and B). Collagen staining also revealed that mice transfused with 4-PBA–programmed neutrophils had significantly higher levels of plaque collagen as compared to mice transfused with control neutrophils (Fig. 6C). Mice transferred with 4-PBA–polarized neutrophils exhibited a significant reduction of plasma cholesterol and triglycerides as compared to mice transferred with PBS-treated neutrophils (fig. S10A). Furthermore, we observed a decline in activated SR-A+ macrophages within the plaques of mice transferred with 4-PBA–polarized neutrophils (fig. S10, B to D).
Fig. 6. Neutrophils reprogrammed by 4-PBA alleviate atherosclerosis.
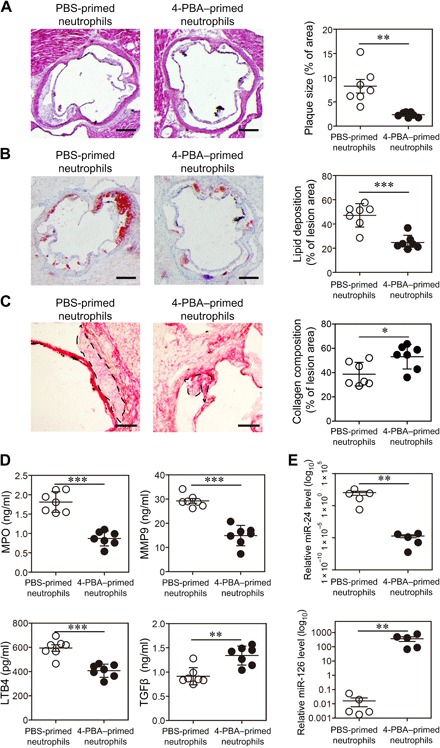
Neutrophils purified from ApoE−/− mice were treated with PBS or 4-PBA (1 mM) for 24 hours. PBS- or 4-PBA–polarized neutrophils (2 × 106 cells per mouse) were then adoptively transferred by intravenous injection to HFD-fed ApoE−/− mice once a week for 4 weeks. Samples were collected 1 week after the last neutrophil transfer. (A) Representative images of H&E-stained atherosclerotic lesions and quantification of plaque size exhibited as the percentage of lesion area within aortic root area. Scale bars, 300 μm. (B) Representative images of Oil Red O–stained atherosclerotic plaques and quantification of lipid deposition within lesion area. Scale bars, 300 μm. (C) Representative images of Picrosirius red–stained atherosclerotic plaques and quantification of collagen content within lesion area. Scale bars, 100 μm. (D) Determination of circulating MPO, MMP9, LTB4, and TGFβ levels by ELISA. (E) Determination of circulating miR-24 and miR-126 levels by real-time RT-PCR. Data are representative of two independent experiments, and error bars represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t test (n = 5 to 7 for each group).
In addition to significantly reduced plaque sizes, we further measured inflammatory markers in the experimental mice. We observed that mice transfused with 4-PBA–programmed neutrophils had significantly lower plasma levels of MPO, LTB4, and MMP9 and significantly elevated levels of TGFβ (Fig. 6D). As compared to mice transfused with control neutrophils, mice transfused with 4-PBA–programmed neutrophils had significantly lower proinflammatory miR-24 and higher homeostatic miR-126 in circulation (Fig. 6E). Our data indicate that 4-PBA–programmed neutrophils can be effectively used to enhance tissue homeostasis in vivo and hold promising potential to treat atherosclerosis.
DISCUSSION
Our study reveals a novel programming dynamic of neutrophils involved in the generation of unstable atherosclerotic plaques. We showed that pathologically relevant superlow-dose endotoxin can program neutrophils into a distinct state, with altered balance of nonresolving inflammation that is conducive to the development of unstable atherosclerotic plaques. The nature of nonresolving inflammatory neutrophils is reflected in elevated ROS due to disrupted peroxisome homeostasis, resulting in the skewed activation of oxCAMKII and downstream expression of LTB4, MMP9, and miR24 and reduced ATF4/KLF2-mediated expression of resolving mediators such as LRRC32 and miR-126. We further demonstrated that the restoration of neutrophil peroxisome homeostasis through the application of 4-PBA can effectively resolve neutrophil inflammation and that neutrophils reprogrammed by 4-PBA can potently attenuate atherosclerosis progression.
Our findings build on the emerging correlation of elevated circulating neutrophil counts with the development of unstable atherosclerotic plaques previously documented in both experimental animals and human patients with atherosclerosis (8–11). In human atherosclerosis, high-circulating neutrophil ratios have been associated with characteristics of rupture-prone atherosclerotic lesions (10). In experimental animals, neutrophilia promoted by hyperlipidemia particularly accelerates early atherosclerosis, which is apparent within 4 weeks of HFD feeding (8). The sizes of atherosclerotic lesions were shown to be positively correlated with circulating neutrophils counts, and the depletion of neutrophils was shown to reduce the initial phase of atherosclerotic lesion burden (8). However, the fundamental nature and underlying mechanisms of proatherosclerotic neutrophils are poorly appreciated or understood.
Our data fill this critical void and reveal that, in addition to the elevated counts of neutrophils, the quality of polarized neutrophils may bear more significant relevance to atherosclerosis. We found that mice subjected to chronic injection of pathologically relevant subclinical low-dose LPS developed atherosclerotic plaques with elevated lipid deposition and reduced collagen content, characteristics of unstable plaques. We observed that neutrophils underwent unique polarization in mice injected with low-dose LPS and adopted a skewed nonresolving inflammatory state represented by increased expression of proinflammatory mediators such as LTB4, MPO, and MMP9, known risk factors for unstable plaques (12–14). This was in addition to the reduced expression of resolving mediators such as LRRC32 and miR-126. LRRC32 is responsible for the generation of mature TGFβ, while miR-126 is critically involved in facilitating vascular integrity (18, 20). Through adoptive transfer studies, we demonstrated that the transfer of neutrophils polarized by superlow-dose LPS was sufficient to significantly aggravate atherosclerosis and reduce plaque stability as compared to the transfer of equal numbers of PBS-treated control neutrophils. Our study complements a recent report that the reduction of LRRC32 levels on human immune cells is associated with reduced human atherosclerotic plaque stability (32).
Our data reveal a novel programming dynamic in neutrophils under chronic low-grade inflammatory conditions and are consistent with a recent independent study regarding the effects of endotoxemia on plaque destabilization (13). Mawhin et al. (13) recently reported that mice repeatedly injected with LPS (1.5 mg/kg) (~30 to 60 μg per mouse) exhibited elevated numbers of circulating neutrophils potentially mediated by elevated LTB4 and severe atherosclerotic vulnerability with reduced collagen content and enlarged necrotic cores. However, that study did not define the fundamental nature of neutrophils affected by LPS and did not provide a causal connection between neutrophils and plaque instability. Our study not only complements the phenotypic observation of unstable plaques elicited by LPS injection but also provides compelling mechanistic principles for neutrophil polarization directly responsible for the development of unstable plaques. Here, we provide both in vitro and in vivo data that define the unique skewing of neutrophils by very low dose LPS into a nonresolving inflammatory state directly responsible for unstable atherosclerosis. Our study also better resembles the pathological conditions in humans and experimental animals with endotoxemia (33–36). Circulating endotoxin levels in humans and experimental animals are extremely low and within the picogram-nanogram per kilogram ranges (37–41), which are thousands-fold less than the microgram-milligram per kilogram range that most studies used. The unique adaptations of innate leukocytes to varying dosages of bacteria endotoxin are highly complex and bear profound pathological relevance (42–45). However, to our knowledge, this study provides the first characterization of unique neutrophil polarization by pathologically relevant concentrations of endotoxin. We observed that neutrophils are skewed to a polarized inflammatory state with reduced ability for homeostatic resolution. The identified features of polarized neutrophils such as elevated surface Dectin-1 and CD11b and reduced surface LRRC32 and FPN, as well as increased secretion of MPO, LTB4, and miR-24 and reduced secretion of miR-126, may serve as potential markers for characterizing neutrophils involved in various chronic inflammatory diseases such as atherosclerosis.
The qualitative natures, rather than the simple elevation of neutrophil counts, have been increasingly implicated in the pathogenesis of other chronic diseases such as cancer (46). Although neutrophils are known to be pleiotropic in nature, the exact characterization of neutrophil polarization and the underlying mechanisms are poorly defined. To fill this critical gap, our study reveals that the novel disruption of peroxisome homeostasis within neutrophils may be one of the critical culprits. We observed that polarized neutrophils by superlow-dose endotoxin exhibit a disruption of proper fusion among peroxisomes and lysosomes, which contributes to the accumulation of ROS and the development of nonresolving inflammatory neutrophils. The accumulation of ROS is associated with elevated oxCAMKII responsible for the expression of LTB4, MPO, and Dectin-1. On the other hand, polarized neutrophils have reduced ATF4 and KLF2, key transcription factors beneficial for the resolution of inflammation through the expression of homeostatic molecules such as LRRC32 and miR-126. ATF4 and KLF2 are also known to closely cooperate with NRF2 [nuclear factor (erythroid-derived 2)–like 2] in reducing ROS and restoring cellular homeostasis. Collectively, our study defines the disrupted molecular balance in neutrophils leading to the accumulation of ROS and the development of a nonresolving inflammatory state conducive for atherosclerosis.
This study further defines a novel and effective strategy in rebalancing the polarized neutrophils back to the homeostatic state through the application of 4-PBA. Our work builds on previous biochemical characterizations of 4-PBA in other cells, which demonstrated that 4-PBA can restore peroxisome homeostasis (31, 47–49). We observed that 4-PBA can restore peroxisome-lysosome fusion in neutrophils and reduce LPS-mediated elevation of neutrophil ROS. 4-PBA can effectively reduce the induction of oxCAMKII, LTB4, MPO, Dectin-1, CD11b, and miR-24 in neutrophils by superlow-dose LPS and also restore the expression of ATF4, FPN, LRRC32, and miR-126 in neutrophils suppressed by LPS. Functionally, we demonstrated that rejuvenated homeostatic neutrophils in vitro by 4-PBA treatment can potently reduce the pathogenesis of atherosclerosis, as reflected in drastically reduced lesion sizes and elevated collagen contents.
Together, our study not only provides compelling mechanistic data that address a unique aspect of neutrophil polarization relevant to the pathogenesis of unstable atherosclerotic plaques but also demonstrates the novel feasibility of using reprogrammed neutrophils to directly treat experimental atherosclerosis.
MATERIALS AND METHODS
Experimental animals and LPS injection
Male ApoE−/− mice (6 to 8 weeks old) on the C57BL/6 background were purchased from the Jackson laboratory and fed with RD or HFD. Either PBS or subclinical-dose LPS (5 ng/kg body weight) was intraperitoneally injected every 3 days for 1 month. Then, the mice were sacrificed, and tissues were harvested for further analyses. All animal procedures were in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Intuitional Animal Care and Use Committee of Virginia Tech.
Analyses of neutrophil phenotype in vivo
Peripheral blood cells, BM cells, and splenocytes were harvested from the mice treated as described above. Cells were stained with anti-Ly6G (1:200 dilution; BioLegend, no. 127618), anti-CD11b (1:200 dilution; BioLegend, no. 101206), anti-CD62L (1:200 dilution; BioLegend, no. 104407), anti–Dectin-1 (1:200 dilution; BioLegend, no. 144305), anti-LLRC32 (1:200 dilution; BioLegend, no. 142904), and anti-FPN (1:200 dilution; Novus, no. NBP1’21502) antibodies. The surface phenotype of Ly6G+ neutrophils was analyzed using FACSCanto II (BD Biosciences). The data were processed by FlowJo (TreeStar).
Adoptive transfer of in vitro primed neutrophils
BM cells were isolated from ApoE−/− mice, and BM neutrophils (Ly6G+ population) were purified by FACS (fluorescence-activated cell sorter) sorting with >99.5% purity. The cells were cultured in complete RPMI with G-CSF (100 ng/ml) in the presence of subclinical-dose LPS (100 pg/ml) or 4-PBA (1 mM) for 24 hours. Cells were washed three times with PBS and suspended in PBS for injection. HFD-fed ApoE−/− mice were transfused with 2 × 106 neutrophils in 200 μl of PBS once weekly through intravenous injection for 4 weeks. One week after the last cell transfer, mice were sacrificed and tissues were harvested for subsequent analyses.
Analyses of atherosclerotic lesions
Histological analyses were performed on fresh-frozen and optimal cutting temperature (OCT) compound–embedded proximal aortic sections (8 μm). Slides were fixed in 4% neutral buffered formalin for 5 min, followed by H&E or Oil Red O staining. Collagen staining was performed using the Picrosirius Red Stain Kit (PolySciences) according to the manufacturer’s instructions. The samples were observed under a light microscope. The percentages of total lesion area, lipid deposition, and collagen composition were calculated. For immunofluorescence staining, proximal aortic sections were fixed with 4% neutral buffered formalin for 5 min, permeabilized with 0.1% saponins, and blocked with 10% normal goat serum (Jackson ImmunoResearch) for 1 hour. To detect lesional neutrophils, the samples were stained with Alexa Fluor 647–conjugated anti-Ly6G antibody (1:50 dilution; BioLegend, no. 127610). To detect lesional macrophages, the samples were costained with eFluor 660–conjugated anti-CD68 (1:100 dilution; Thermo Fisher Scientific, no. 50-0681-82) and rabbit anti–SR-A (1:100 dilution; Thermo Fisher Scientific, no. PA5-22956) antibodies, followed by staining with DyLight 488–conjugated goat anti-rabbit immunoglobulin G (IgG) (1:100 dilution; Thermo Fisher Scientific, no. 35552) in the dark at room temperature for 1 hour. To detect oxCAMKII levels, the samples were stained with rabbit anti-oxCaMKII antibody (1:100 dilution; Millipore, no. 07-1387) at 4°C overnight and then with DyLight 488–conjugated goat anti-rabbit IgG (1:100 dilution; Thermo Fisher Scientific, no. 35552) in the dark at room temperature for 1 hour. 4′,6-Diamidino-2-phenylindole was used to stain nucleus. The samples were observed under a confocal microscope.
In vitro neutrophil priming and FACS analyses
BM neutrophils were isolated from C57BL/6 mice and cultured in complete RPMI medium containing 10% fetal bovine serum, 2 mM l-glutamine, and 1% penicillin/streptomycin in the presence of G-CSF (100 ng/ml). PBS, subclinical-dose LPS (100 pg/ml), oxLDL (10 μg/ml), or 4-PBA (1 mM) was added to cell cultures. After 2 days, the cells were harvested and stained with anti-Ly6G (1:200 dilution; BioLegend, nos. 127606, 127610, or 127618), anti-CD11b (1:200 dilution; BioLegend, no. 101206), anti-CD62L (1:200 dilution; BioLegend, no. 104407), anti–Dectin-1 (1:200 dilution; BioLegend, no. 144305), anti-LLRC32 (1:200 dilution; BioLegend, no. 142904), and anti-FPN (1:200 dilution; Novus, no. NBP1-21502) antibodies. In some experiments, the cells were fixed and permeabilized using a transcription factor phospho buffer set (BD Biosciences) and then stained with primary anti–p-STAT1 (1:50 dilution; Cell Signaling Technology, no. 8009S), anti-ATF4 (1:50 dilution; Proteintech, no. 10835-1-AP), and anti-KLF2 (1:100 dilution; Novus, no. NBP2-61812) antibodies, followed by staining with Alexa Fluor 488–conjugated goat anti-rabbit IgG (1:2000 dilution; Abcam, no. ab150077). Surface phenotype and transcription factor phosphorylation of Ly6G+ neutrophils were analyzed using FACSCanto II (BD Biosciences). The viability of the neutrophils primed for 24 and 48 hours was determined with an annexin V/PI staining kit (Thermo Fisher Scientific), followed by flow cytometry. For intracellular ROS detection, 5 μM CellROX Green Reagent (Thermo Fisher Scientific) was added to neutrophil cultures 30 min before harvesting, and cells were analyzed using FACSCanto II (BD Biosciences). The data were processed by FlowJo (TreeStar).
ELISA and determination of plasma lipids
For in vivo analyses, plasma was collected from the mice at time of sacrificing. For in vitro analyses, purified BM neutrophils were cultured in complete RPMI medium with G-CSF (100 ng/ml) in the presence of PBS, subclinical-dose LPS (100 pg/ml), oxLDL (10 μg/ml), or 4-PBA (1 mM), and supernatant was collected after 2 days. ELISA kit of MPO was purchased from Thermo Fisher Scientific, and ELISA kits of MMP9, LTB4, and TGFβ1 were purchased from R&D Systems. Cholesterol quantitation kit was purchased from Sigma-Aldrich, and triglyceride quantification kit was purchased from BioVision.
Immunoblotting
Purified BM neutrophils were cultured in complete RPMI medium with G-CSF (100 ng/ml) in the presence of PBS, subclinical-dose LPS (100 pg/ml), oxLDL (10 μg/ml), or 4-PBA (1 mM), and total cell lysate was extracted on day 2. Protein samples were separated with SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes, which were probed with anti-oxCaMKII (1:500 dilution; Millipore, no. 07-1387), anti–5-LOX (1:500 dilution; Cell Signaling Technology, no. 3289S), and anti–β-actin (1:1000 dilution; Santa Cruz Biotechnology, no. sc-47778) primary antibodies, followed by horseradish peroxidase–conjugated anti-rabbit IgG (1:1000 dilution; Cell Signaling Technology, no. 7074S) or anti-mouse IgG (1:1000 dilution; Cell Signaling Technology, no. 7076S) secondary antibodies. Images were developed with an enhanced chemiluminesence (ECL) detection kit (Thermo Fisher Scientific).
Real-time reverse transcription polymerase chain reaction for detection of miRNAs
For in vivo analyses, plasma was collected from the mice at time of sacrificing. Circulating miRNAs were isolated with miRNeasy Serum/Plasma kit (Qiagen). For in vitro analyses, purified BM neutrophils were cultured in complete RPMI medium with G-CSF (100 ng/ml) in the presence of PBS, subclinical-dose LPS (100 pg/ml), or 4-PBA (1 mM), and miRNAs were isolatedusing the miRNeasy Mini Kit (Qiagen) after 2 days. TaqMan miRNA assay kits for detection of U6, miR-16, miR-24, and miR-126 were purchased from Thermo Fisher Scientific. Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed following the manual. U6 expression was used as the internal control for miRNA expressions in neutrophil cultures, and miR-16 expression was used as the internal control for plasma miRNA expressions.
Confocal microscopy
Purified BM neutrophils were cultured in complete RPMI medium with G-CSF (100 ng/ml) in the presence of PBS, subclinical-dose LPS (100 pg/ml), or 4-PBA (1 mM) for 2 days. To determine lysosome-peroxisome fusion, the cells were fixed with 4% paraformaldehyde, deposited on slides through cytospin, and permeabilized with 0.2% Triton X-100. The cells were blocked and stained with primary rabbit anti-mouse PMP70 antibody (1:1000) supplied in the SelectFX Alexa Fluor 488 Peroxisome Labeling Kit (Thermo Fisher Scientific, #S34201), followed by staining with Alexa Fluor 488 goat anti-rabbit secondary antibody (1:1000) supplied in the kit. After extensive washing with PBS, the cells were then stained with Cy3 anti-LAMP1 antibody (1:1000 dilution; Abcam, no. Ab67283) and observed under a confocal microscope.
Statistical analyses
Statistical analysis was performed using Prism 6 software (GraphPad Software, La Jolla, CA), and data were expressed as means ± SEM. The significance of the differences was assessed by Student’s t test (for two groups) or one-way analysis of variance (ANOVA) (for multiple groups). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the members of the Li laboratory for technical assistance and animal husbandry. We also thank A. Rahtes for careful and critical proofreading of the manuscript. Funding: This work was supported by grants from the National Institutes of Health (grant no. R01 HL115835 to L.L). Author contributions: S.G. performed most of the in vitro and in vivo studies. Y.Z. and C.L. performed the in vivo adoptive transfer studies. L.L. designed the study and wrote the manuscript. Competing interests: Y.Z., S.G., and L.L. are inventors on a patent disclosure related to this work filed by the Virginia Tech intellectual property office (no. VTIP 19-021; filed on 22 August 2018). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav2309/DC1
Fig. S1. Subclinical endotoxin up-regulates MPO level in HFD-fed mice.
Fig. S2. Subclinical endotoxemia exacerbates atherosclerotic pathogenesis in RD-fed mice.
Fig. S3. Subclinical endotoxin causes neutrophil expansion in atherosclerotic mice.
Fig. S4. Subclinical endotoxin primes neutrophils into a proinflammatory state in atherosclerotic mice.
Fig. S5. Subclinical endotoxin induces oxCAMKII elevation in vivo.
Fig. S6. Neutrophils maintain viability after in vitro polarization.
Fig. S7. Transfusion of superlow-dose LPS–polarized neutrophils elevates plasma lipid levels and modulates lesional macrophages.
Fig. S8. Superlow-dose LPS and oxLDL treatment elevates ROS accumulation in neutrophils.
Fig. S9. 4-PBA reverses superlow-dose LPS–induced differential regulation of miR-24 and miR-126 in neutrophils.
Fig. S10. Transfusion of 4-PBA–polarized neutrophils down-regulates plasma lipid levels and reduces lesional macrophage activation.
REFERENCES AND NOTES
- 1.Kasikara C., Doran A. C., Cai B., Tabas I., The role of non-resolving inflammation in atherosclerosis. J. Clin. Invest. 128, 2713–2723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabas I., Garcia-Cardeña G., Owens G. K., Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 209, 13–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christ A., Bekkering S., Latz E., Riksen N. P., Long-term activation of the innate immune system in atherosclerosis. Semin. Immunol. 28, 384–393 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Geng S., Chen K., Yuan R., Peng L., Maitra U., Diao N., Chen C., Zhang Y., Hu Y., Qi C.-F., Pierce S., Ling W., Xiong H., Li L., The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat. Commun. 7, 13436 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekkering S., Joosten L. A. B., van der Meer J. W. M., Netea M. G., Riksen N. P., The epigenetic memory of monocytes and macrophages as a novel drug target in atherosclerosis. Clin. Ther. 37, 914–923 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Leentjens J., Bekkering S., Joosten L. A. B., Netea M. G., Burgner D. P., Riksen N. P., Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ. Res. 122, 664–669 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Soehnlein O., Multiple roles for neutrophils in atherosclerosis. Circ. Res. 110, 875–888 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Drechsler M., Megens R. T. A., van Zandvoort M., Weber C., Soehnlein O., Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Horne B. D., Anderson J. L., John J. M., Weaver A., Bair T. L., Jensen K. R., Renlund D. G., Muhlestein J. B., Intermountain Heart Collaborative Study Group , Which white blood cell subtypes predict increased cardiovascular risk? J. Am. Coll. Cardiol. 45, 1638–1643 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Ionita M. G., van den Borne P., Catanzariti L. M., Moll F. L., de Vries J.-P. P. M., Pasterkamp G., Vink A., de Kleijn D. P. V., High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 30, 1842–1848 (2010). [DOI] [PubMed] [Google Scholar]
- 11.van Leeuwen M., Gijbels M. J. J., Duijvestijn A., Smook M., van de Gaar M. J., Heeringa P., de Winther M. P. J., Tervaert J. W. C., Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler. Thromb. Vasc. Biol. 28, 84–89 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Luttun A., Lutgens E., Manderveld A., Maris K., Collen D., Carmeliet P., Moons L., Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E–deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation 109, 1408–1414 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Mawhin M.-A., Tilly P., Zirka G., Charles A.-L., Slimani F., Vonesch J.-L., Michel J.-B., Bäck M., Norel X., Fabre J.-E., Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxaemia. Cardiovasc. Res. 114, 1656–1666 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Nicholls S. J., Hazen S. L., Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 1102–1111 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Ford-Hutchinson A. W., Bray M. A., Doig M. V., Shipley M. E., Smith M. J. H., Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 286, 264–265 (1980). [DOI] [PubMed] [Google Scholar]
- 16.Yabluchanskiy A., Ma Y., Iyer R. P., Hall M. E., Lindsey M. L., Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology 28, 391–403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishimoto T. K., Jutila M. A., Berg E. L., Butcher E. C., Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science 245, 1238–1241 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Wang R., Kozhaya L., Mercer F., Khaitan A., Fujii H., Unutmaz D., Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13439–13444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganz T., Nemeth E., Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 15, 500–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S., Aurora A. B., Johnson B. A., Qi X., McAnally J., Hill J. A., Richardson J. A., Bassel-Duby R., Olson E. N., The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biswas S. K., Gangi L., Paul S., Schioppa T., Saccani A., Sironi M., Bottazzi B., Doni A., Vincenzo B., Pasqualini F., Vago L., Nebuloni M., Mantovani A., Sica A., A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 107, 2112–2122 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Nair J. S., DaFonseca C. J., Tjernberg A., Sun W., Darnell J. E. Jr., Chait B. T., Zhang J. J., Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-γ. Proc. Natl. Acad. Sci. U.S.A. 99, 5971–5976 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson J. R., Joiner M.-l. A., Guan X., Kutschke W., Yang J., Oddis C. V., Bartlett R. K., Lowe J. S., O'Donnell S. E., Aykin-Burns N., Zimmerman M. C., Zimmerman K., Ham A.-J. L., Weiss R. M., Spitz D. R., Shea M. A., Colbran R. J., Mohler P. J., Anderson M. E., A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fledderus J. O., Boon R. A., Volger O. L., Hurttila H., Ylä-Herttuala S., Pannekoek H., Levonen A.-L., Horrevoets A. J. G., KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 1339–1346 (2008). [DOI] [PubMed] [Google Scholar]
- 25.He C. H., Gong P., Hu B., Stewart D., Choi M. E., Choi A. M. K., Alam J., Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein - Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 276, 20858–20865 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Suragani R. N. V. S., Zachariah R. S., Velazquez J. G., Liu S., Sun C.-W., Townes T. M., Chen J.-J., Heme-regulated eIF2α kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 119, 5276–5284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boxio R., Bossenmeyer-Pourié C., Steinckwich N., Dournon C., Nüße O., Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 75, 604–611 (2004). [DOI] [PubMed] [Google Scholar]
- 28.van Raam B. J., Drewniak A., Groenewold V., van den Berg T. K., Kuijpers T. W., Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood 112, 2046–2054 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silvestre-Roig C., Hidalgo A., Soehnlein O., Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 127, 2173–2181 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Singh M. V., Anderson M. E., Is CaMKII a link between inflammation and hypertrophy in heart? J. Mol. Med. 89, 537–543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gondcaille C., Depreter M., Fourcade S., Lecca M. R., Leclercq S., Martin P. G. P., Pineau T., Cadepond F., ElEtr M., Bertrand N., Beley A., Duclos S., de Craemer D., Roels F., Savary S., Bugaut M., Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. J. Cell Biol. 169, 93–104 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joly A.-L., Seitz C., Liu S., Kuznetsov N. V., Gertow K., Westerberg L. S., Paulsson-Berne G., Hansson G. K., Andersson J., Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ. Res. 122, 1385–1394 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Cani P. D., Bibiloni R., Knauf C., Waget A., Neyrinck A. M., Delzenne N. M., Burcelin R., Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Erridge C., Attina T., Spickett C. M., Webb D. J., A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 86, 1286–1292 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Raparelli V., Basili S., Carnevale R., Napoleone L., Del Ben M., Nocella C., Bartimoccia S., Lucidi C., Talerico G., Riggio O., Violi F., Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 65, 571–581 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Wiedermann C. J., Kiechl S., Dunzendorfer S., Schratzberger P., Egger G., Oberhollenzer F., Willeit J., Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 34, 1975–1981 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Feroze U., Kalantar-Zadeh K., Sterling K. A., Molnar M. Z., Noori N., Benner D., Shah V., Dwivedi R., Becker K., Kovesdy C. P., Raj D. S., Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J. Ren. Nutr. 22, 317–326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goto T., Edén S., Nordenstam G., Sundh V., Svanborg-Eden C., Mattsby-Baltzer I., Endotoxin levels in sera of elderly individuals. Clin. Diagn. Lab. Immunol. 1, 684–688 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harte A. L., Varma M. C., Tripathi G., McGee K. C., Al-Daghri N. M., Al-Attas O. S., Sabico S., O’Hare J. P., Ceriello A., Saravanan P., Kumar S., McTernan P. G., High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care 35, 375–382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maury E., Blanchard H. S., Chauvin P., Guglielminotti J., Alzieu M., Guidet B., Offenstadt G., Circulating endotoxin and antiendotoxin antibodies during severe sepsis and septic shock. J. Crit. Care 18, 115–120 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Zhang R., Miller R. G., Gascon R., Champion S., Katz J., Lancero M., Narvaez A., Honrada R., Ruvalcaba D., McGrath M. S., Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 206, 121–124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chae B. S., Pretreatment of low-dose and super-low-dose LPS on the production of in vitro LPS-induced inflammatory mediators. Toxicol. Res. 34, 65–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirohashi N., Morrison D. C., Low-dose lipopolysaccharide (LPS) pretreatment of mouse macrophages modulates LPS-dependent interleukin-6 production in vitro. Infect. Immun. 64, 1011–1015 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopanakis K., Tzepi I.-M., Pistiki A., Carrer D.-P., Netea M. G., Georgitsi M., Lymperi M., Droggiti D.-I., Liakakos T., Machairas A., Giamarellos-Bourboulis E. J., Pre-treatment with low-dose endotoxin prolongs survival from experimental lethal endotoxic shock: Benefit for lethal peritonitis by Escherichia coli. Cytokine 62, 382–388 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Yuan R., Geng S., Li L., Molecular mechanisms that underlie the dynamic adaptation of innate monocyte memory to varying stimulant strength of TLR ligands. Front. Immunol. 7, 497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicolás-Ávila J. Á., Adrover J. M., Hidalgo A., Neutrophils in homeostasis, immunity, and cancer. Immunity 46, 15–28 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Li X., Baumgart E., Dong G.-X., Morrell J. C., Jimenez-Sanchez G., Valle D., Smith K. D., Gould S. J., PEX11α is required for peroxisome proliferation in response to 4-phenylbutyrate but is dispensable for peroxisome proliferator-activated receptor alpha-mediated peroxisome proliferation. Mol. Cell. Biol. 22, 8226–8240 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu N., Qiang W., Kuang X., Thuillier P., Lynn W. S., Wong P. K. Y., The peroxisome proliferator phenylbutyric acid (PBA) protects astrocytes from ts1 MoMuLV-induced oxidative cell death. J. Neurovirol. 8, 318–325 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Walter K. M., Schönenberger M. J., Trötzmüller M., Horn M., Elsässer H. P., Moser A. B., Lucas M. S., Schwarz T., Gerber P. A., Faust P. L., Moch H., Köfeler H. C., Krek W., Kovacs W. J., Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 20, 882–897 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav2309/DC1
Fig. S1. Subclinical endotoxin up-regulates MPO level in HFD-fed mice.
Fig. S2. Subclinical endotoxemia exacerbates atherosclerotic pathogenesis in RD-fed mice.
Fig. S3. Subclinical endotoxin causes neutrophil expansion in atherosclerotic mice.
Fig. S4. Subclinical endotoxin primes neutrophils into a proinflammatory state in atherosclerotic mice.
Fig. S5. Subclinical endotoxin induces oxCAMKII elevation in vivo.
Fig. S6. Neutrophils maintain viability after in vitro polarization.
Fig. S7. Transfusion of superlow-dose LPS–polarized neutrophils elevates plasma lipid levels and modulates lesional macrophages.
Fig. S8. Superlow-dose LPS and oxLDL treatment elevates ROS accumulation in neutrophils.
Fig. S9. 4-PBA reverses superlow-dose LPS–induced differential regulation of miR-24 and miR-126 in neutrophils.
Fig. S10. Transfusion of 4-PBA–polarized neutrophils down-regulates plasma lipid levels and reduces lesional macrophage activation.