

c-Abl inhibits replication of hepatitis B virus by promoting CRL4Cdt2-mediated destruction of the viral polymerase.
Abstract
About 257 million people with chronic infection of hepatitis B virus (HBV) worldwide are at high risk of developing terminal liver diseases. Reactivation of virus replication has been frequently reported in those patient populations receiving imatinib (an Abl kinase inhibitor) or bortezomib (a proteasome inhibitor) to treat concurrent diseases, but the underlying mechanism for this reactivation is unknown. We report that the HBV polymerase protein is recruited by Cdt2 to the cullin-RING ligase 4 (CRL4) for ubiquitination and proteasome degradation and that this process is stimulated by the c-Abl nonreceptor tyrosine kinase. Genetic ablation of the Abl-CRL4Cdt2 axis or pharmaceutical inhibition of this process stabilizes HBV polymerase protein and increases viral loads in HBV-infected liver cancer cell lines. Our study reveals a kinase-dependent activation of CRL4 ubiquitin ligase that can be targeted for blocking HBV replication.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a global health threat. It affects approximately 257 million individuals worldwide and exposes this population to increased risk of liver cirrhosis and cancer, which causes 887,000 deaths annually (1). HBV is a small enveloped DNA virus with a 3.2-kb genome of partially double-stranded relaxed-circular DNA and can be converted to an episomal covalently closed-circular DNA in the host nucleus, which serves as a template for transcription during virus replication (2). HBV replicates via an RNA intermediate (pgRNA) catalyzed by the reverse transcriptase activity of the viral polymerase (2, 3). HBV polymerase binds specifically to the stem-loop structure of the 5′ end of the pregenome and initiates encapsidation by core proteins and DNA synthesis (3). Several nucleoside/nucleotide analogs have been approved by the U.S. Food and Drug Administration to block HBV replication in patients by inhibiting the activity of viral polymerase (4).
Patients with chronic HBV infection are at high risk of reactivation of HBV replication after receiving immunosuppressive or targeted therapies for concurrent diseases (5, 6). HBV reactivation severely affects patients’ continuation with specific treatment, reduces their life quality and survival, and often results in liver inflammation and injury, acute liver failure, and even death. Immunosuppressive drugs, such as CD20 monoclonal antibodies ofatumumab and rituximab, can reactivate dormant HBV, likely by compromising the effectiveness of host immunity (7). Frequent HBV reactivation was also reported in patients with chronic HBV receiving imatinib for the treatment of chronic myeloid leukemia (CML) (8–10) and gastrointestinal stromal tumors (GISTs) (11) or reported in patients with multiple myeloma (MM) treated with bortezomib (12, 13). Imatinib (Gleevec, STI-571) is a small-molecule tyrosine kinase inhibitor (TKI) that inactivates Abl kinases, including the constitutively active BCR-ABL oncogene, and is approved to treat CML (14). Bortezomib is a proteasome inhibitor that blocks protein turnover and is approved as an MM therapy in combination with dexamethasone (15). However, the mechanism underlying HBV reactivation in imatinib- and bortezomib-treated patients is unknown.
The Abl family of nonreceptor tyrosine kinases, c-Abl (Abl-1) and Arg (Abl-2), are implicated in various cellular processes, including regulation of cell growth and survival, cellular stress responses, and cytoskeletal dynamics (16). The kinase activity of c-Abl is controlled by autoinhibition that involves intramolecular interactions between the N terminus, the SH3-SH2 domain, and the PTK (protein tyrosine kinase) domain (16). Upon activation, c-Abl can phosphorylate the damaged DNA-binding protein 1 (DDB1) (17) and promote the cullin-RING ligase 4 (CRL4) complex (CUL4-DDB1-Roc1/2) to target its substrates for ubiquitination (18). Phosphorylated Y316 of DDB1 recruits a small regulatory protein DDA1 to achieve enhanced CRL4 E3 ligase activity (19). In addition, CRL4 E3 activity is also modified by neddylation (20), CAND1 (cullin-associated and neddylation-dissociated 1) (21), and the CSN (COP9 signalosome) complex (22). A variety of host factors are targeted by CRL4 via about 90 DDB1- and CUL4-associated factor (DCAF) adaptor proteins to regulate DNA repair and chromatin remodeling, cell cycle progression, embryogenesis, hematopoiesis, spermatogenesis, and tumorigenesis (23).
Some substrates are diverted to CRL4 only by viral proteins or small-molecule chemicals. For instance, HBV protein HBx hijacks CRL4 to target the Smc5/6 complex for degradation to counteract the host restriction of extrachromosomal viral DNA transcription (24). Similarly, V proteins, expressed by simian virus 5 and type 2 human parainfluenza virus, function as exogenous DCAFs to degrade host antiviral STAT (signal transducers and activators of transcription) proteins (25, 26). Vpr and Vpx proteins encoded by human or simian immunodeficient viruses were also found to route CRL4DCAF1 to destroy various host restrictions (27, 28). Chemicals, such as immunomodulatory drugs (29, 30) and protein targeting chimera (31), can mimic these viral accessory proteins and recruit host proteins to CRL4CRBN E3 ligase to control their turnover and inhibit cancer progression.
In this study, we demonstrate that HBV polymerase protein is ubiquitinated by CRL4Cdt2 ligase, a process stimulated by c-Abl tyrosine kinase. Both c-Abl kinase inhibitor (imatinib) and proteasome inhibitor (bortezomib) stabilize the viral polymerase and increase the viral loads in HBV-infected cells, thus providing a mechanistic explanation for the mechanism by which these two drugs reactivate HBV replication in patients with chronic HBV in the clinic.
RESULTS
Inhibition of c-Abl kinase stimulates HBV replication
To confirm the implications of the clinical observations, we treated HepG2.2.15, an HBV-replicating cell line, with imatinib or dasatinib and found that the HBV DNA load was elevated after treatment with either drug (Fig. 1, A and B). In contrast, treatment with several other kinase inhibitors including crizotinib [ALK (anaplastic lymphoma kinase) inhibitor], erlotinib [EGFR (epidermal growth factor receptor) inhibitor], or ruxolitinib [Janus kinase (JAK) inhibitor] did not exhibit a significant effect on capsid-associated viral DNA level (fig. S1, A to C).
Fig. 1. c-Abl kinase reduces HBV replication and HBV polymerase protein level.
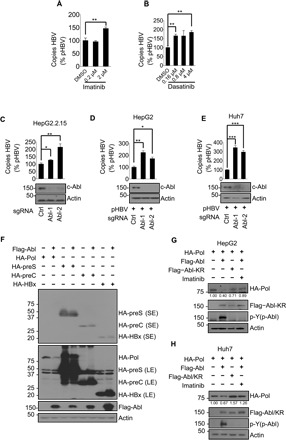
(A and B) Quantitation of capsid-associated viral DNA by real-time polymerase chain reaction (PCR). HepG2.2.15 cells were treated with dimethyl sulfoxide (DMSO) or imatinib (A) or dasatinib (B) in indicated concentrations for 24 hours before harvest. Mean copy number from cells treated with DMSO was set to 100% and compared with others. Statistical significance compared with DMSO is noted by asterisks (n = 3 to 4 per group). (C) Quantitation of capsid-associated viral DNA by real-time PCR in HepG2.2.15 cells knocking out control (sgCtrl) or Abl (sgAbl-1/2). Mean copy number from sgCtrl cells was set to 100% and compared with others (n = 3 per group). (D and E) Quantitation of capsid-associated viral DNA by real-time PCR in HepG2 cells (D) or Huh7 cells (E) knocking out control or Abl. Cells were transfected with pHBV for 48 hours before harvest. Mean copy number from sgCtrl cells was set to 100% and compared with others (n = 3 per group). (F) Human embryonic kidney (HEK) 293T cells were cotransfected with constructs expressing hemagglutinin (HA)–tagged polymerase (HA-Pol), preS (HA-preS), preC (HA-preC), and HBx (HA-HBx), and Flag-tagged Abl (Flag-Abl) or empty vector controls. SE, short exposure; LE, long exposure. Western blot was performed 48 hours after transfection. HepG2 cells (G) or Huh7 cells (H) were transfected as shown. Cells were treated with DMSO or 2 μM imatinib for 24 hours before harvest. Total cell lysates were then analyzed for the indicated proteins. *P < 0.05, **P < 0.01, and ***P < 0.001.
Both imatinib and dasatinib inhibit the constitutively active BCR-ABL kinase that causes CML in patients (32) and endogenous Abl family kinases (33). Deletion of Abl1, encoding c-Abl, in HepG2.2.15 (Fig. 1C), or HepG2 and Huh7 cell lines transfected with HBV expression construct (pHBV) (Fig. 1, D and E) by CRISPR-Cas9 led to increased viral loads, indicating that c-Abl kinase inhibits HBV replication.
c-Abl kinase reduces the level of HBV polymerase
To determine how c-Abl inhibits HBV replication, we examined the levels of the four proteins encoded by the HBV genome (namely, polymerase, S protein, C protein, and HBx) by transient transfection assays in human embryonic kidney (HEK) 293T cells. Only the level of viral polymerase was significantly decreased by overexpression of c-Abl (Fig. 1F). Down-regulation of polymerase by c-Abl was also confirmed in the HepG2 and Huh7 liver cancer cell lines (Fig. 1, G and H). In all three cell lines, imatinib treatment blocked a decrease in c-Abl–induced polymerase, and expression of the kinase-deficient mutant (Abl-KR) (34) failed to reduce the polymerase level, suggesting that c-Abl kinase activity is essential for its effect on polymerase (Fig. 1, G and H, and fig. S2A). Conversely, loss of endogenous c-Abl increased the polymerase protein level (fig. S2, B and C). Expression of Arg, a c-Abl homolog, similarly decreased the polymerase level in a kinase-dependent manner (fig. S2D). Together, these results demonstrate that c-Abl kinase negatively regulates HBV polymerase expression. We next set out to explore the underlying mechanism of this kinase and its implications for the clinically reported instances of HBV reactivation.
c-Abl kinase promotes HBV polymerase ubiquitination and proteasome-dependent degradation
To determine how c-Abl down-regulates polymerase, we first examined the mRNA levels of polymerase by quantitative polymerase chain reaction (PCR) and found no difference after c-Abl expression (fig. S3A). In contrast, cycloheximide (CHX) chasing assays revealed that c-Abl decreased the protein stability of polymerase in HEK293T cells (Fig. 2A), shifting its half-life from 1.04 to 0.35 hours (Fig. 2B). Treatment with MG132 (a proteasome inhibitor) or MLN4924 (a neddylation-activing enzyme inhibitor) abrogated the effect of c-Abl on polymerase protein levels (Fig. 2C). As MLN4924 interferes with all cullin-RING E3 ligase activities (20), these data suggest that the HBV polymerase was likely a c-Abl–dependent target of a cullin-RING ligase (CRL) for proteasome degradation.
Fig. 2. c-Abl kinase promotes HBV polymerase ubiquitination and proteasome-dependent degradation.
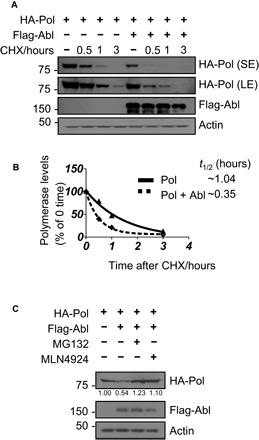
(A) CHX chasing analysis for polymerase stability in HEK293T cells expressing Flag-Abl. Cells were treated with CHX (100 μg/ml) for the indicated time. (B) Time course of polymerase protein decay from (A). Each band of the Western blot for polymerase was quantified with National Institutes of Health (NIH) ImageJ software. Half-life (t1/2) was estimated as the time for degradation of 50% of the protein. (C) HEK293T cells were transfected with HA polymerase, Flag-Abl, or empty vector control and were treated with MG132 (20 μM) or MLN4924 (0.2 μM) for 8 hours before harvest. Total cell lysates were subjected to Western blotting.
CRL4 E3 ubiquitin ligase targets HBV polymerase for ubiquitination
We recently reported that c-Abl phosphorylates DDB1 to recruit the regulatory protein DDA1 to stimulate ubiquitination of CRL4 substrates (19). Since MG132 and MLN4924 blocked the down-regulation of HBV polymerase by c-Abl (Fig. 2C), we performed experiments to determine whether the polymerase is a substrate of CRL4. First, the HBV polymerase exhibited DDB1-dependent interaction with CUL4A or CUL4B (CUL4A/4B) after being transiently expressed in HEK293T cells by coimmunoprecipitation (co-IP) analysis (Fig. 3A). The polymerase also formed a complex with endogenous DDB1 and CUL4A/4B (Fig. 3B). Knocking down the expression of DDB1 or CUL4A/4B by RNA interference decreased ubiquitination of polymerase, assayed by precipitating ubiquitin (Fig. 3C) or polymerase (fig. S3B), with a reciprocal increase in polymerase protein abundance (Fig. 3D). We therefore conclude that CRL4 ubiquitin ligase can target HBV polymerase for proteasome destruction.
Fig. 3. CRL4cdt2 E3 ubiquitin ligase targets HBV polymerase for ubiquitination.
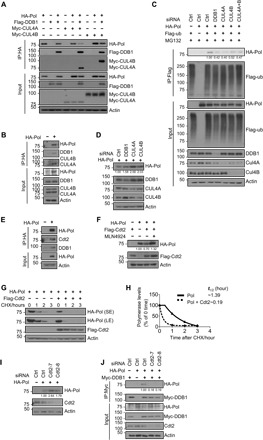
(A) Co-IP of HA polymerase with CRL4 components in HEK293T cells by overexpression HA polymerase, Myc-CUL4A, Myc-CUL4B, and Flag-DDB1. (B) Co-IP of HA polymerase with endogenous CRL4 in HEK293T cells. (C) Ubiquitination of polymerase by Flag IP in Huh7 cells expressing HA polymerase, Flag-ub, and siDDB1, siCUL4A, siCUL4B, or a combination of siCUL4A and siCUL4B and treated with MG132 for 8 hours before harvest. Flag immunoprecipitates (top) or total cell lysates (bottom) were then analyzed by Western blot. (D) Immunoblot for polymerase protein level in Huh7 cells knocking down CRL4 components. (E) Co-IP of HA polymerase with endogenous Cdt2 and DDB1 in Huh7 cells. (F) The transfected HEK293T cells were treated with DMSO or MLN4924 (0.2 μM) for 8 hours before harvest. (G) CHX chasing analysis for polymerase stability in HEK293T cells expressing Flag-Cdt2. Cells were treated with CHX (100 μg/ml) for the indicated time. (H) Time course of polymerase protein decay from Fig. 3G. Each band of the Western blot for polymerase was quantified with NIH ImageJ software. Half-life (t1/2) was estimated as the time for degradation of 50% of the protein. (I) HEK293T cells were transfected with control siRNA or siCdt2 and HA polymerase. Total cell lysates were subjected to Western blotting. (J) Co-IP of DDB1 with polymerase in Huh7 cells by overexpressing HA polymerase, Myc-DDB1, and siCdt2
Cdt2 is a CRL4 substrate receptor for HBV polymerase
Caenorhabditis elegans polymerase η, which replicates damaged DNA, is recruited to CRL4 by Cdt2, a DCAF protein (35). Similar to polymerase η, HBV polymerase also interacted with endogenous Cdt2 in Huh7 cells (Fig. 3E). Overexpression of Cdt2 reduced HBV polymerase protein abundance, which can be abrogated by MLN4924 treatment in HEK293T cells (Fig. 3F), without altering its mRNA level (fig. S3C). CHX chasing assays revealed that Cdt2 decreased the protein stability of polymerase (Fig. 3G), shifting its half-life from 1.39 to 0.19 hours (Fig. 3H). Knocking down Cdt2 expression consistently increased polymerase protein abundance (Fig. 3I) and decreased the interaction of polymerase with DDB1 (Fig. 3J). These data support the hypothesis that the viral polymerase is a substrate of CRL4Cdt2 ubiquitin ligase.
c-Abl promotes CRL4Cdt2 to target HBV polymerase
We next sought to determine the role of c-Abl in CRL4Cdt2-mediated destruction of HBV polymerase. We found that the c-Abl–induced decrease in HBV polymerase protein level was partially abrogated by knocking down the expression of DDB1, CUL4A/4B, or Cdt2 in HEK293T cells (Fig. 4, A and B), with reciprocal alterations observed in polymerase ubiquitination (Fig. 4C). c-Abl was reported to phosphorylate DDB1 for DDA1 docking to activate CRL4 (19). We found that c-Abl–dependent decrease of polymerase protein abundance was enhanced by DDA1 overexpression (Fig. 4D) but blocked by DDA1 knockdown (Fig. 4E). A DDB1 triple mutant (Y182F, Y718F, and Y316F), which abolishes its phosphorylation by c-Abl and its interaction with DDA1 (19), was found to block c-Abl–induced polymerase degradation when overexpressed in HEK293T cells (Fig. 4F). Loss of endogenous c-Abl does not affect the assembly of the DDB1-Cdt2-Pol complex (fig. S3D), indicating that c-Abl does not enhance the CRL4 E3 activity by promoting the complex assembly (19). On the basis of these results, we conclude that c-Abl kinase promotes degradation of HBV polymerase via stimulation of CRL4Cdt2-DDA1 E3 ligase.
Fig. 4. c-Abl kinase activates CRL4-DDA1 to reduce the level of HBV polymerase.

(A) Immunoblot for polymerase protein level in HEK293T cells overexpressing Flag-Abl and knocking down DDB1 or CUL4A/4B. (B) Immunoblot for polymerase protein level in HEK293T cells overexpressing Flag-Abl and knocking down Cdt2. (C) Ubiquitination of polymerase by IP in Huh7 cells expressing HA polymerase, Flag-Abl, Myc-ub, and siDDB1, siCUL4A, siCUL4B, or a combination of siCUL4A and siCUL4B and treated with MG132 for 8 hours. IP-Myc (top) or total cell lysates (bottom) were then analyzed for the indicated proteins. (D) Immunoblot for polymerase protein level in HEK293T cells overexpressing Flag-Abl and Flag-DDA1. (E) Immunoblot for polymerase protein level in HEK293T cells overexpressing Flag-Abl and knocking down DDA1. (F) Immunoblot for polymerase protein level in HEK293T cells overexpressing Flag-Abl, Myc-DDB1 wild type, and triple mutant (TM).
Inhibition of c-Abl kinase enhances HBV replication by stabilizing viral polymerase
We have demonstrated that inhibition or deletion of c-Abl increases viral loads in cell culture (Fig. 1) and that c-Abl stimulates CRL4Cdt2 ligase to target HBV polymerase for proteasome degradation (Fig. 4). We next addressed whether the regulation of polymerase abundance by c-Abl accounted for the alteration of viral replication. To monitor the viral polymerase levels in the context of HBV replication, we engineered a hemagglutinin (HA) tag to the C terminus of HBV polymerase in a replication-competent construct (pHBV) and, at the same time, introduced a translation stop codon after the initial seven codons of the HBx gene without altering the protein coding of the overlapping polymerase gene (36)(Fig. 5A). This new construct, designated as pHBV(Pol-HA)ΔX, allowed for HBV replication when transfected to HepG2 and Huh7 cells, which can be enhanced by cotransfection with an HBx-expressing construct (fig. S4, A and B), consistent with the role of HBx in HBV replication (37). With this new composite HBV construct (compHBV; Fig. 5A), we found that both c-Abl–induced decrease of HBV polymerase abundance and suppression of HBV replication were abrogated by c-Abl kinase inhibition (Fig. 5, B and C), proteasome or CRL inhibition (Fig. 5, D and E), or CRL4 expression knockdown (Fig. 5, F and G) in HepG2 and Huh7 cells. Expression of exogenous HBV polymerase dose-dependently rescued the inhibitory effect of c-Abl on HBV replication in Huh7 (Fig. 6A) and HepG2 cells (Fig. 6B). Knocking down the expression of CRL4Cdt components in these two cell lines also enhanced HBV viral loads, with concomitant increase of HBV polymerase protein abundance (fig. S4, C to H), as observed in cells treated with MG132 or MLN4924 (fig. S4, I and J), implicating a role of the endogenous c-Abl kinase in regulating HBV replication.
Fig. 5. Inhibition of c-Abl kinase activates HBV replication by stabilizing the level of HBV polymerase.

(A) Strategy (see text for description). (B) HepG2 cells or (C) Huh7 cells were cotransfected with indicated plasmids and were treated with or without imatinib for 24 hours before harvest, whole-cell lysates were prepared for Western blotting (bottom), and capsid-associated viral DNAs were quantitated by real-time PCR (top). Mean copy number from cells only transfected with compHBV was set to 100% and compared with others (n = 3 per group). (D) HepG2 cells or (E) Huh7 cells were cotransfected with indicated plasmids and were treated with DMSO, MG132, or MLN4924 for 8 hours before harvest; whole-cell lysates were prepared for Western blotting (bottom); and capsid-associated viral DNAs were quantitated by real-time PCR (top). Mean copy number from cells treated with DMSO was set to 100% and compared with others (n = 3 to 4 per group). (F) HepG2 cells or (G) Huh7 cells were transfected with indicated siRNAs and plasmids, whole-cell lysates were prepared for Western blotting (bottom), and capsid-associated viral DNAs were quantitated by real-time PCR (top). Mean copy number from cells transfected with control siRNA was set to 100% and compared with others (n = 3 to 4 per group). *P < 0.05, **P < 0.01, and ***P < 0.001.
Fig. 6. c-Abl inhibits HBV replication in vitro and in vivo.
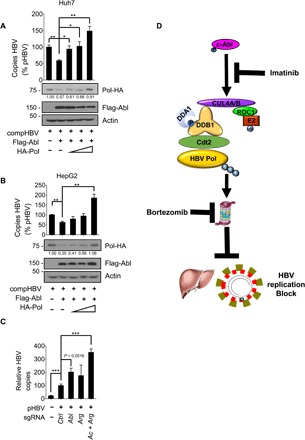
(A) Huh7 cells and (B) HepG2 cells were cotransfected with indicated plasmids, whole-cell lysates were prepared for Western blotting (bottom), and capsid-associated viral DNAs were quantitated by real-time PCR (top). Mean copy number from cells only transfected with compHBV was set to 100% and compared with others (n = 3 per group). (C) ICR mice were hydrodynamically injected with plasmid DNA, and capsid-associated HBV DNAs were purified from liver tissue. Mean copy number from liver of ICR mice hydrodynamic injected into sgCtrl was set to 100% and compared with others. Statistical significance compared with sgCtrl is noted by asterisks (lanes 1, 4, and 5: n = 4 per group; lane 2: n = 3; lane 3: n = 6). (D) Schematic model. c-Abl promotes CRL4Cdt2-mediated ubiquitination of HBV polymerase and further suppresses HBV replication. Imatinib promotes HBV reactivation through c-Abl kinase abrogation to down-regulate CRL4 activity, and bortezomib inhibits proteasome activity to protest the ubiquitination of HBV polymerase. *P < 0.05, **P < 0.01, and ***P < 0.001.
To test the inhibitory role of endogenous Abl kinases, we introduced plasmid constructs expressing spCas9, small-guide RNA (sgRNA) sequences against the Abl1 and Arg genes, together with HBV genomic DNA to mouse liver by hydrodynamic tail vein injection, as we performed previously (38). Mice were sacrificed on day 4 after injection, and viral capsids were purified from equivalent amounts of liver tissues. The success of plasmid expression was demonstrated by the fluorescence of co-injected green fluorescent protein construct (fig. S4K). Viral DNA load was elevated significantly in the ICR mouse liver receiving both sgAbl and sgArg (Fig. 6C), indicating a physiological role of Abl kinases in HBV suppression.
DISCUSSION
c-Abl kinase regulates the life cycle of various pathogenic viruses by targeting host factors or viral proteins. For example, c-Abl activates the Wave2 complex and promotes Arp2/3-dependent actin polymerization to facilitate HIV-1 Env–mediated virus fusion and entry (39). The kinase also enhances the cell susceptibility to polyomavirus infection by increasing the levels of ganglioside receptors in the plasma membrane (40). In addition, c-Abl directly phosphorylates Ebola VP40 protein or HCV NS5A protein to promote release of highly pathogenic Ebola virus (41) or assembly of HCV viral particles (42), respectively. In this study, we showed that c-Abl kinase targets CRL4Cdt2 E3 ligase to enhance its activity to ubiquitinate the HBV polymerase protein for proteasome destruction (Fig. 6D). Inhibiting c-Abl kinase activity with imatinib or proteasome with bortezomib would result in accumulation of HBV polymerase protein and release of HBV from replication block (Fig. 6D). This study thus provides a mechanistic explanation for the many clinical reports that imatinib/dasatinib and bortezomib reactivate dormant HBV in chronic carrier patients when treated for concurrent CML, GISTs, or MM (8–13). Accordingly, prescription of preemptive HBV surveillance and anti-HBV therapy is advised for all patients with HBV receiving these target therapeutic agents to avoid the risk of severe, sometimes fatal, reactivation of HBV.
HBV encodes another protein, HBx, that also interacts with DDB1 to stimulate HBV replication (36, 38, 43). Unlike HBV polymerase, HBx is not a substrate of CRL4 ligase but hijacks CRL4 to target the host structural maintenance of chromosomes complex 5/6 (Smc5/6) for ubiquitination and degradation, resulting in derepression of episomal HBV genome expression (24). Therefore, CRL4 ligase appears to be a double-edged sword for HBV replication. On the one hand, it specifically targets the viral polymerase for degradation to inhibit HBV replication upon certain environmental cues. On the other hand, it is repurposed by the viral HBx protein to destroy host restriction factors Smc5/6 to allow for efficient HBV replication. It is intriguing that two of the four viral proteins encoded by HBV interact with the same host ubiquitin ligase, exemplifying a host strategy to silence virus infection and a viral strategy to escape host restriction of its replication. The role of HBx in CRL4Cdt2-mediated targeting of HBV polymerase is currently being investigated, with a particular focus on the polymerase turnover during host stress and virus evolution.
CRL4Cdt2 E3 ligase maintains genomic stability by targeted destruction of critical chromatin regulators such as Cdt1, p21, and Set8 in the G1 and G1/S phases of cell cycle in a proliferating cell nuclear antigen (PCNA)–dependent manner (44). Among these substrates, Cdt1 (chromatin licensing and DNA replication factor 1) has been most intensively investigated. Cdt1 is ubiquitinated by CRL4Cdt2 in the S phase and after DNA damage, and its destruction is strictly coupled to the process of DNA replication (45). Cdt1 docks onto chromatin-bound PCNA with its PCNA-interacting protein motif (PIP box) (46). CRL4Cdt2 is subsequently recruited to the PIP degron on Cdt1 and destroys Cdt1 on chromatin (44). Although we have demonstrated that viral polymerase is a substrate of CRL4Cdt2, it is unclear how polymerase destruction is coupled to cell proliferation and cell cycle and whether a PIP degron from viral polymerase is involved. HBV replication was reported to be cell cycle dependent and is inversely correlated with cellular DNA synthesis (47). Future experiments should be designed to investigate HBV polymerase turnover by CRL4Cdt2 and HBV replication in various stages of cell cycle and to validate the significance in animal model systems.
MATERIALS AND METHODS
Plasmids and cloning
A plasmid carrying a greater-than-unit-length (1.1) HBV genome (pcDNA3.1) and the same plasmid carrying a stop codon at position 8 of HBx open reading frame that prevents expression of HBx and adding an HA epitope tag (YPYDVPDYA) to the polymerase C terminus are referred to here as pHBV and pHBV(Pol-HA)ΔX, respectively. DNA fragments corresponding to HBV polymerase, HBV preC, HBV preS, and HBx proteins of HBV were amplified separately from HBV genome (pcDNA3.1), subcloned into pXF4H expression vector from X. H. Feng’s laboratory in Zhejiang University between the Bam HI and Eco RI restriction sites, and named as HA-tagged polymerase (HA-Pol), preC (HA-preC), preS (HA-preS), and HBx (HA-HBx). In addition, the HBx protein of HBV was amplified and subcloned into pXF6F expression vector from X. H. Feng between the Bam HI and Eco RI restriction sites and was named as Flag-HBx.
Plasmids encoding Flag-Abl/KR, Flag-ub, and Flag-DDA1 were described (19). The full-length ub was a gift from Z. P. Xia’s laboratory in Zhejiang University and were amplified with a Myc (MEQKLISEEDL) tag in the N terminus, subcloned into pXF4H expression vector between the Cla I and Eco RI sites, and named as Myc-ub. The full-length DDB1, DDB1-TM(Y182F/Y718F/Y316F), CUL4A, and CUL4B were amplified with a Myc tag in the N terminus, subcloned into pXF4H expression vector between the Cla I and Eco RI sites, and named as Myc-DDB1, Myc-DDB1-TM, Myc-CUL4A, and Myc-CUL4B.
Cell culture and transfections
HEK293T cells, Huh7 cells, HepG2 cells, and HepG2.2.15 cells obtained from the American Type Culture Collection were maintained at 37°C in 5% CO2 condition. Transfections were performed using PEI (polyethylenimine) for HEK293T cells or Liposomal Transfection Reagent (Hieff Trans) for Huh7 and HepG2 according to the manufacturer’s instructions.
Lentiviral vectors
The sgRNA sequences targeting c-Abl and sgcontrol were annealed and subcloned into lentiCRISPRv2 vectors as previously described (19). The lentivirus packaging and collection were described (19). c-Abl or control knocking out in Huh7, HepG2, and HepG2.2.15 cell lines was generated by corresponding lentiviruses infection with polybrene (6 μg/ml; Sigma-Aldrich) for 24 hours, followed by puromycin (2 μg/ml) selection.
pX459 vector was digested with Bbs I and ligated with annealed oligonucleotides. The sgRNA sequences were as follows: GTCTCAGCGAAGCAGCTCGA (sgAbl) and GCGTCTGCGGTACGCCCCGC (sgArg).
Hydrodynamic injection
Animal care and experimental procedures were in accordance with the Animal Research Committee guidelines of Zhejiang University. Mice were maintained under specific pathogen–free conditions in a controlled environment of 20° to 22°C, with a 12-hour/12-hour light/dark cycle, 50 to 70% humidity, and food and water provided ad libitum.
For hydrodynamic injection, plasmid DNA suspended in 2 ml of PBS was injected into 8-week-old male ICR mice via the tail vein in 5 to 7 s. The amount of injected DNA was 60 μg sgCtrl +60 μg pHBV, 60 μg sgAbl +60 μg pHBV, 60 μg sgArg + 60 μg pHBV, and 60 μg sgAbl + 60 μg sgArg +60 μg pHBV. All the mice were sacrificed on day 4 after hydrodynamic tail vein injection (38, 48).
Compounds and antibodies
Imatinib (S2475), MG132 (S2619), bortezomib (S1013), and MLN4924 (S7109) were purchased from Selleck Chemicals. CHX (C7698) was purchased from Sigma-Aldrich Technology. Dasatinib, crizotinib, erlotinib, and ruxolitinib were gifts from WuXi AppTec. All compounds mentioned were dissolved in dimethyl sulfoxide.
Anti-Flag (F3165, Sigma-Aldrich), anti-HA (H6908, Sigma-Aldrich), anti-Myc (sc-40, Santa Cruz Biotechnology), anti-CUL4A (A300-739A, Bethyl Laboratories), anti-CUL4A/B (ab76470, Abcam), anti-p-Y (9411S, Cell Signaling Technology), anti-c-Abl (sc-131, Santa Cruz), anti-DDB1 (37-6200, Invitrogen), anti-Cdt2 (ab72264, Abcam), anti–β-actin (4967S, Cell Signaling Technology), and anti-DDA1 (AP14094C, Abgent) were purchased and used according to the manufacturers’ recommendations. Secondary antibodies were goat anti-mouse immunoglobulin–horseradish peroxidase (IgG-HRP; LK-GAM007, Lianke Bio) and goat anti-rabbit IgG-HRP (LK-GAR007, Lianke Bio).
RNA extraction and real-time PCR detection of cDNA
Cells were transiently transfected with indicated plasmids for 48 hours, and the RNA was extracted by TRIzol Reagent (15596-018, Invitrogen). The concentration of RNA was measured by NanoDrop 2000. The complementary DNA (cDNA) was reverse transcripted by PrimeScript RT Master Mix (RR036A, Takara) according to the manufacturer’s protocol.
The quantification of cDNA was performed using SYBR Green Master Mix (11201ES08-5, Yeasen Biotechnology) on Applied Biosystems 7500 Real-Time PCR System. The PCR primer sequences were as follows: Pol, 5′-AGAAACAACACATAGCGCCTCAT-3′ (forward) and 5′-TGCCCCATGCTGTAGATCTTG-3′ (reverse); glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-CGACCACTTTGTCAAGCTCA-3′ (forward) and 5′-TTACTCCTTGGAGGCCATGT-3′ (reverse); Cdt2, 5′-TGGTCTTCACAATACCCTCTTCA-3′ (forward) and 5′-CTTCATTGGCAACTGCTAG-TACA-3′ (reverse).
siRNA sequences and transfection
Small interfering RNAs (siRNAs) were transfected into HEK293T cells, Huh7 cells, and HepG2 cells using Liposomal Transfection Reagent (Hieff Trans) according to the manufacturer’s instructions. The siRNA sequences for CUL4A, CUL4B, DDB1, DDA1, and Cdt2 were as follows: siCUL4A, GAACUUCCGAGACAGACCUdTdT; siCUL4B, AAGCCUAAAUUACCAGAAAdTdT; siDDB1, ACUAGAUCGCGAUAAUAAAdTdT; siDDA1, CCUCAUAGGAGCCGAUGUAdTdT; siCdt2-7, GAAUUAUACUGCUUAUCGAdTdT; and siCdt2-8, GUCAAGACCUGGCCUAGUAdTdT.
Co-IP assays
HEK293T cells, Huh7 cells, and HepG2 cells were transiently transfected with indicated plasmids for 48 hours or with siRNAs for 72 hours and then lysed in NETN lysis buffer [150 mM NaCl, 1% NP-40, 50 mM tris-HCl, pH8.0, 1 mM Na3VO4, supplemented with complete protease inhibitors (MCE)]. After centrifugation at 13,000 rpm for 15 min, supernatants were subjected to IP by incubation with anti-HA Affinity Gel (E6779, Sigma-Aldrich), anti-Flag Affinity Gel (A2220, Sigma-Aldrich), and anti-Myc Affinity Gel (B23401, BioTool). The co-IP was separated with SDS–polyacrylamide gel electrophoresis and analyzed by Western blot.
Purification of capsid-associated viral DNA and real-time PCR detection of HBV DNA
Extraction of capsid-associated DNA was described previously (38, 49). Equivalent amounts of cells or liver tissues were used to isolate capsid-associated viral DNA using the QIAamp MinElute Virus Spin Kit (Qiagen).
The quantification of capsid-associated DNA was performed using SYBR Green Master Mix (11201ES08-5, Yeasen Biotechnology) on Applied Biosystems 7500 Real-Time PCR System (49). The percentage of the copy number was measured as described previously (38).
Quantitation and statistical analysis
Western blots were exposed on a Kodak BioMax MS film and quantitated by ImageJ software. All results were confirmed in at least three independent experiments. Statistical significance was determined using the Student’s t test (GraphPad Software). Error bars shown in the figures indicate the SEM, unless otherwise indicated. Asterisks in the figures were used to indicate statistically significant differences (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
We thank Z. P. Xia and X. H. Feng for the plasmids; Z. G. Peng for technical help; and W. J. Wang, X. X. Zou, and P. T. Lin for administrative support. Funding: This work was supported in part by funds from the National 973 Plan for Basic Research (2015CB553803), National Natural Science Foundation (91429302), and Fundamental Research Funds for the Central Universities and Key Construction Program of the National “985” Project. Authors contributions: L.H., J.Z., and Y.C. designed the study. L.H., J.Z., and Y.L. performed all the experiments except the mouse studies, which were contributed by T.J., J.W., and L.H. T.S., C.G., and M.L. packaged the lentiviruses. S.G., J.C., and H.L. provided technical assistance. L.H., J.Z., X.C., and Y.C. analyzed the data. L.H. and Y.C. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau7130/DC1
Fig. S1. Capsid-associated viral DNA level in the treatment of TKIs.
Fig. S2. c-Abl kinase reduces the level of HBV polymerase.
Fig. S3. c-Abl–CRL4Cdt2 reduces HBV polymerase by promoting its ubiqutination but not transcription level.
Fig. S4. Inhibition of CRL4 E3 ubiquitin ligase enhances HBV replication by stabilizing viral polymerase.
Fig. S5. Western blot–scanned films.
Fig. S6. Western blot–scanned films.
REFERENCES AND NOTES
- 1.WHO, Global Hepatitis Report, 2017 (2017).
- 2.Gerlich W. H., Medical virology of hepatitis B: How it began and where we are now. Virol. J. 10, 239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nassal M., Hepatitis B viruses: Reverse transcription a different way. Virus Res. 134, 235–249 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Tawada A., Kanda T., Imazeki F., Yokosuka O., Prevention of hepatitis B virus-associated liver diseases by antiviral therapy. Hepatol. Int. 10, 574–593 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Hwang J. P., Lok A. S.-F., Management of patients with hepatitis B who require immunosuppressive therapy. Nat. Rev. Gastroenterol. Hepatol. 11, 209–219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H. Y., Kim W., Chemotherapy-related reactivation of hepatitis B infection: Updates in 2013. World J. Gastroenterol. 20, 14581–14588 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Bisceglie A. M., Lok A. S., Martin P., Terrault N., Perrillo R. P., Hoofnagle J. H., Recent US Food and Drug Administration warnings on hepatitis B reactivation with immune-suppressing and anticancer drugs: Just the tip of the iceberg? Hepatology 61, 703–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda K., Shiga Y., Takahashi A., Kai T., Kimura H., Takeyama K., Noji H., Ogawa K., Nakamura A., Ohira H., Sato Y., Maruyama Y., Fatal hepatitis B virus reactivation in a chronic myeloid leukemia patient during imatinib mesylate treatment. Leuk. Lymphoma 47, 155–157 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Wang Y. D., Cui G. H., Li M., Gowrea B., Xia J., Hu Y., Hepatitis B virus reactivation in a chronic myeloid leukemia patient treated with imatinib mesylate. Chin. Med. J. 125, 2636–2637 (2012). [PubMed] [Google Scholar]
- 10.Lai G. M., Yan S. L., Chang C. S., Tsai C. Y., Hepatitis B reactivation in chronic myeloid leukemia patients receiving tyrosine kinase inhibitor. World J. Gastroenterol. 19, 1318–1321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker E. J., Simko J. P., Ko A. H., Hepatitis B viral reactivation secondary to imatinib treatment in a patient with gastrointestinal stromal tumor. Anticancer Res. 34, 3629–3634 (2014). [PubMed] [Google Scholar]
- 12.Tanaka H., Sakuma I., Hashimoto S., Takeda Y., Sakai S., Takagi T., Shimura T., Nakaseko C., Hepatitis B reactivation in a multiple myeloma patient with resolved hepatitis B infection during bortezomib therapy: Case report. J. Clin. Exp. Hematop. 52, 67–69 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Hussain S., Jhaj R., Ahsan S., Ahsan M., Bloom R. E., Jafri S.-M. R., Bortezomib induced hepatitis B reactivation. Case Rep. Med. 2014, 964082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roskoski R., Jr., A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 100, 1–23 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Manasanch E. E., Orlowski R. Z., Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 14, 417–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hantschel O., Superti-Furga G., Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5, 33–44 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Cong F., Tang J., Hwang B. J., Vuong B. Q., Chu G., Goff S. P., Interaction between UV-damaged DNA binding activity proteins and the c-Abl tyrosine kinase. J. Biol. Chem. 277, 34870–34878 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X., Zhang J., Lee J., Lin P. S., Ford J. M., Zheng N., Zhou P., A kinase-independent function of c-Abl in promoting proteolytic destruction of damaged DNA binding proteins. Mol. Cell 22, 489–499 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Gao S., Geng C., Song T., Lin X., Liu J., Cai Z., Cang Y., Activation of c-Abl kinase potentiates the anti-myeloma drug lenalidomide by promoting DDA1 protein recruitment to the CRL4 ubiquitin ligase. J. Biol. Chem. 292, 3683–3691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S., Cullis C. A., Doucette A., Garnsey J. J., Gaulin J. L., Gershman R. E., Lublinsky A. R., McDonald A., Mizutani H., Narayanan U., Olhava E. J., Peluso S., Rezaei M., Sintchak M. D., Talreja T., Thomas M. P., Traore T., Vyskocil S., Weatherhead G. S., Yu J., Zhang J., Dick L. R., Claiborne C. F., Rolfe M., Bolen J. B., Langston S. P., An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Petroski M. D., Deshaies R. J., Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Cavadini S., Fischer E. S., Bunker R. D., Potenza A., Lingaraju G. M., Goldie K. N., Mohamed W. I., Faty M., Petzold G., Beckwith R. E. J., Tichkule R. B., Hassiepen U., Abdulrahman W., Pantelic R. S., Matsumoto S., Sugasawa K., Stahlberg H., Thomä N. H., Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531, 598–603 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Jackson S., Xiong Y., CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Decorsière A., Mueller H., van Breugel P. C., Abdul F., Gerossier L., Beran R. K., Livingston C. M., Niu C., Fletcher S. P., Hantz O., Strubin M., Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Precious B., Childs K., Fitzpatrick-Swallow V., Goodbourn S., Randall R. E., Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J. Virol. 79, 13434–13441 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulane C. M., Kentsis A., Cruz C. D., Parisien J. P., Schneider K. L., Horvath C. M., Composition and assembly of STAT-targeting ubiquitin ligase complexes: Paramyxovirus V protein carboxyl terminus is an oligomerization domain. J. Virol. 79, 10180–10189 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrofelbauer B., Hakata Y., Landau N. R., HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. U.S.A. 104, 4130–4135 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hrecka K., Hao C., Gierszewska M., Swanson S. K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M. P., Skowronski J., Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu G., Middleton R. E., Sun H., Naniong M., Ott C. J., Mitsiades C. S., Wong K. K., Bradner J. E., Kaelin W. G., The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kronke J., Udeshi N. D., Narla A., Grauman P., Hurst S. N., McConkey M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S. L., Carr S. A., Ebert B. L., Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., Crews C. M., Modular protac design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55, 807–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apperley J. F., Chronic myeloid leukaemia. Lancet 385, 1447–1459 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Nagar B., Bornmann W. G., Pellicena P., Schindler T., Veach D. R., Miller W. T., Clarkson B., Kuriyan J., Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243 (2002). [PubMed] [Google Scholar]
- 34.Sawyers C. L., McLaughlin J., Goga A., Havlik M., Witte O., The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell 77, 121–131 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Kim S. H., Michael W. M., Regulated proteolysis of DNA polymerase eta during the DNA-damage response in C. elegans. Mol. Cell 32, 757–766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leupin O., Bontron S., Schaeffer C., Strubin M., Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 79, 4238–4245 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouchard M. J., Schneider R. J., The enigmatic X gene of hepatitis B virus. J. Virol. 78, 12725–12734 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodgson A. J., Hyser J. M., Keasler V. V., Cang Y., Slagle B. L., Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 426, 73–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harmon B., Campbell N., Ratner L., Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLOS Pathog. 6, e1000956 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swimm A. I., Bornmann W., Jiang M., Imperiale M. J., Lukacher A. E., Kalman D., Abl family tyrosine kinases regulate sialylated ganglioside receptors for polyomavirus. J. Virol. 84, 4243–4251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.García M., Cooper A., Shi W., Bornmann W., Carrion R., Kalman D., Nabel G. J., Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci. Transl. Med. 4, 123ra124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamauchi S., Takeuchi K., Chihara K., Sun X., Honjoh C., Yoshiki H., Hotta H., Sada K., Hepatitis C virus particle assembly involves phosphorylation of NS5A by the c-Abl tyrosine kinase. J. Biol. Chem. 290, 21857–21864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sitterlin D., Bergametti F., Tiollais P., Tennant B. C., Transy C., Correct binding of viral X protein to UVDDB-p127 cellular protein is critical for efficient infection by hepatitis B viruses. Oncogene 19, 4427–4431 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Havens C. G., Walter J. C., Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 25, 1568–1582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin J., Arias E. E., Chen J., Harper J. W., Walter J. C., A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Arias E. E., Walter J. C., PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Ozer A., Khaoustov V. I., Mearns M., Lewis D. E., Genta R. M., Darlington G. J., Yoffe B., Effect of hepatocyte proliferation and cellular DNA synthesis on hepatitis B virus replication. Gastroenterology 110, 1519–1528 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N. S., Cai W., Yang G., Bronson R., Crowley D. G., Zhang F., Anderson D. G., Sharp P. A., Jacks T., CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keasler V. V., Hodgson A. J., Madden C. R., Slagle B. L., Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 81, 2656–2662 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau7130/DC1
Fig. S1. Capsid-associated viral DNA level in the treatment of TKIs.
Fig. S2. c-Abl kinase reduces the level of HBV polymerase.
Fig. S3. c-Abl–CRL4Cdt2 reduces HBV polymerase by promoting its ubiqutination but not transcription level.
Fig. S4. Inhibition of CRL4 E3 ubiquitin ligase enhances HBV replication by stabilizing viral polymerase.
Fig. S5. Western blot–scanned films.
Fig. S6. Western blot–scanned films.