

Summary
Considerable progress has been made in identifying microenvironmental signals that effect the reversible phenotypic transitions underpinning the early steps in the metastatic cascade. However, although the general principles underlying metastatic dissemination have been broadly outlined, a common theme that unifies many of the triggers of invasive behavior in tumors has yet to emerge. Here we discuss how many diverse signals that induce invasion converge on reprogramming of protein translation via phosphorylation of eIF2α, a hallmark of the starvation response. These include starvation as a consequence of nutrient or oxygen limitation, or pseudo-starvation imposed by cell extrinsic microenvironmental signals or by cell intrinsic events including oncogene activation. Since in response to resource limitation single cell organisms undergo phenotypic transitions remarkably similar to those observed within tumors, we propose that a starvation/pseudo-starvation model to explain cancer progression provides an integrated and evolutionarily conserved conceptual framework to understand progression of this complex disease.
Keywords: Phenotypic plasticity, Cancer heterogeneity, Nutrient supply and demand, Translation reprogramming, Pseudo-starvation, Invasion, eIF2α
eTOC blurb
García-Jiménez and Goding discuss how many of the triggers of invasion in cancer converge on translation reprogramming, a hallmark of the starvation response. They propose that cancer invasion reflects an evolutionarily conserved adaptation to nutrient limitation in which the starvation response has been hijacked by microenvironmental signals including inflammation.
Introduction
Cancer initiation is underpinned by epigenetic and genetic events that rewire the signaling networks that respond to environmental cues. As a consequence, cancer cells exhibit deregulated proliferation and metabolism (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Although cancer can originate from a single cell, within tumors cancer cells are frequently both genetically and phenotypically heterogeneous. Genetic lesions are largely maintained and may be used to study Darwinian tumor evolution occurring in response to stresses encountered within the intra-tumor microenvironment (Brooks et al., 2015; Burrell et al., 2013; McGranahan and Swanton, 2015). By contrast, mounting evidence indicates that phenotypic heterogeneity generated as a consequence of bidirectional interactions between (epi)genetically heterogeneous tumor cells and the complex tumor microenvironment may be dynamic and reversible (Beerling et al., 2016; Brabletz et al., 2005; Chaffer et al., 2011; Chaffer et al., 2016; Gupta et al., 2009a; Gupta et al., 2011; Hoek and Goding, 2010; Hoek et al., 2004; Huang et al., 2013; Mlecnik et al., 2016; Nieto et al., 2016; Ocana et al., 2012; Pinner et al., 2009; Quail and Joyce, 2013; Tsai et al., 2012; Zhao et al., 2016). Although genetic lesions may affect the response of a cell to its environment, even genetically uniform cancer cells can adopt functionally distinct cell states (Kreso et al., 2013) that may exhibit profoundly different biological properties including proliferation, invasion and dormancy. Some phenotypes are also associated with stem cell-like properties (Clarke et al., 2006; Gupta et al., 2009a; Rosen and Jordan, 2009; Shackleton, 2010; Visvader and Lindeman, 2008) or resistance to targeted therapies, including immune checkpoint inhibitors (Emmons et al., 2016; Holzel et al., 2013; Hugo et al., 2016; Rambow et al., 2018; Smith and Bhowmick, 2016; Tsoi et al., 2018). Genetic (Kandoth et al., 2013; McGranahan and Swanton, 2015) and phenotypic (Marusyk and Polyak, 2010) heterogeneity shape cancer progression and represent a considerable barrier to effective therapy (Brooks et al., 2015). While genetic diversity within and between tumors in the same patient poses a major challenge to targeted ‘pharmacogenetic’ therapies, the presence of plastic phenotypic states (eg. proliferative, invasive) common to most tumors offers interesting therapeutic opportunities. For example, using small molecules to effect transitions from drug-resistant to drug-sensitive phenotypes (Saez-Ayala et al., 2013) or targetting specific phenotypic sub-populations of cancer cells (Beug, 2009; Gupta et al., 2009b; Huang et al., 2013). However, to have an enduring impact on patient survival, such ‘pharmacophenomic’ therapies require an in depth and integrated understanding of how and why different phenotypic states are established, and the molecular mechanisms that govern and maintain phenotypic transitions.
In this article we highlight how the state transitions that fuel cancer progression reflect a proliferative to invasive phenotype switch by single cell organisms confronted by resource limitation. In cancer, a restricted nutrient/oxygen supply within tumor can promote invasion, but even in regions of tumors where nutrients may be abundant, non-nutritional signals can hijack the response to nutrient limitation to impose a pseudo-starvation state that drives invasiveness.
The transition to an invasive phenotype
Invasion is a prerequisite for metastatic dissemination in which cells from the primary tumor invade surrounding tissue, subsequently enter and then exit the blood or lymphatic vessels, and finally colonize and proliferate in new locations (Coghlin and Murray, 2010; Klein, 2009; Lambert et al., 2017). Genetic and microenvironmental events may work in concert to effect the phenotypic transitions that support metastatic spread, and it has been suggested that oncogene activation and loss of tumor suppressor genes early in tumorigenesis predispose to metastatic dissemination (Bernards and Weinberg, 2002). However the transition from benign (non-metastasising) to malignant (potentially metastasising) tumors is usually characterized by the acquisition of additional genetic lesions that may increase the probability of a cell adopting an invasive phenotype (Klein, 2013). Thus in mouse models of melanoma the BRAFV600E mutation alone gives rise to benign non-metastatic lesions (Dankort et al., 2009), but a transition from a benign to a malignant phenotype in which melanomas metastasize to lung and lymph nodes is observed if PTEN loss is combined with BRAFV600E (Dankort et al., 2009). It is important to note however, that the majority of cells in the primary tumor do not migrate. PTEN loss therefore increases the probability of metastatic dissemination and/or colonization by reducing the threshold for metastasis, rather than imposing an inevitable transition to an invasive phenotype. As proposed previously (Vogelstein et al., 2013), metastasis may therefore be a stochastic process in which genetic lesions confer sensitivity to changes in the microenvironment leading to phenotypic instability (Hoek and Goding, 2010). This phenomenon may not be exclusive to cancer cells and fate transitions may occur in a stochastic fashion in normal (non-cancer) cells, with the probability of a phenotype switch occurring being modulated by signals (Moris et al., 2016). One example would be the switch to migration of the neural crest during development (Theveneau and Mayor, 2012).
The evolutionary origin of phenotype transitions in cancer
If the probability of a cancer cell adopting an invasive phenotype is a stochastic process that ultimately depends on its sensitivity to microenvironmental cues, then what are the signals that drive the transition to invasiveness? Or more simply put, why does any cell move? Understanding the reason why proliferating cancer cells become invasive is important as metastases cause the great majority of cancer-related deaths (Siegel et al., 2016). Since evolution preserves effective survival strategies, a clue as to why cancer cells adopt an invasive phenotype may be provided by the proliferative to invasive phenotype switch that occurs in single cell organisms.
One of the most primitive drivers of behavior is the need to assimilate nutrients to fuel growth and cell division. Among single cell organisms the development of motility would have conferred a major selective advantage by allowing cells to seek nutrient-rich environments actively rather than waiting passively for nutrients to arrive. Thus, post-exponential phase E. coli undergoing nutrient limitation respond by increasing the levels of nutrient transporters and subsequently becoming highly motile if the limitation in nutrient supply is not resolved (Serra and Hengge, 2014). If this response is insufficient to restore the supply-demand balance then bacteria will undergo an additional phenotypic transition leading ultimately to sporulation or a persister phenotype (depending on species), a dormant and highly stress and drug-resistant state (Serra and Hengge, 2014). Similarly, in response to hypoxia protein levels within E. coli are stochastically altered to generate regulated phenotypic diversity that sustains survival of the population as a whole by generating different levels of fitness between individual cells (Carey et al., 2018). Both temporal and spatial phenotypic transitions have also been well-characterized in Bacillus subtilis biofilms; as resources become limiting cells switch from proliferation to invasion, followed by matrix secretion and ultimately dormancy (Vlamakis et al., 2008; Vlamakis et al., 2013). Even non-motile species such as the yeast S. cerevisiae, can forage via a switch to hyphal growth (Gancedo, 2001; Gimeno et al., 1992), enabling cells at the tip of the hyphae to escape a nutrient-depleted environment and seek out new resources.
At first sight it seems unlikely that the phenotypic transitions observed in cancer represent an evolutionarily conserved survival strategy related to a starvation response. In contrast to single cell organisms, multicellular organisms have developed an efficient nutrient transport and storage system that ensures a constant and limited supply of nutrients to cells, sufficient to satisfy the needs of development or adult tissue homeostasis despite discontinuous eating behavior. Moreover, unlike bacteria or yeast, the presence of an extracellular nutrient supply does not normally trigger proliferation of mammalian cells (Lloyd, 2013; Rathmell et al., 2000). Growth and proliferation of normal adult mammalian cells is carefully coordinated with neighboring cells such that their proliferation rate is adjusted to match the needs of tissue renewal and nutrient availability. Instructions to divide are provided by mitogens and growth factors that act through their receptors and downstream signaling pathways to drive cell division, reprogram the Rb-E2F1 axis that maintains quiescence (Yao, 2014), and increase uptake of nutrients necessary to sustain growth and proliferation (Ward and Thompson, 2012). The availability of key nutrients can also regulate the activity of drivers of proliferation. For example, the activity of the pro-proliferative Wnt/β-catenin pathway, that can trigger stem cell activation and proliferation in a range of cancer types (Clevers, 2006), is dependent on glucose levels in colon cancer cells (Chocarro-Calvo et al., 2013).
In principle therefore, mammalian cells should not be subject to the nutrient limitation that drives phenotypic transitions in single cell organisms. However in cancer, deregulated pro-proliferative signaling increases nutrient demand (Keibler et al., 2016) that may or may not be satisfied, and disrupts signaling networks that coordinate demand with supply (Pavlova and Thompson, 2016; Ward and Thompson, 2012). As tumors grow, the risk of both nutritional and hypoxic stress is enhanced by their frequently chaotic neovasculature (Nagy et al., 2009). For example, poor blood flow in functionally and structurally abnormal vessels can lead to perfusion-limited hypoxia such that even cells close to vessels may be hypoxic while cells at a distance from vessels will be exposed to more severe diffusion-limited hypoxia (Vaupel et al., 2004). Reduced flow combined with high demand from tumor cells may therefore lead to limitation of oxygen, as well as amino acids and glucose, even in close proximity to vessels, and mounting evidence suggests that nutrient limitation may be a feature of some cancers.
Glutamine, the most abundant amino acid in the blood (Brosnan, 2003) can be depleted in tumors (Kamphorst et al., 2015; Pan et al., 2016; Roberts et al., 1956), most likely as a result of it being used to fuel anabolic metabolism (Altman et al., 2016; DeBerardinis et al., 2008; Wise and Thompson, 2010). Aspartate limitation has also been recognized recently as restricting tumor growth in vivo, especially under conditions of hypoxia (Garcia-Bermudez et al., 2018; Sullivan et al., 2018). Depletion of essential amino acids can also occur via expression of specific amino acid degrading enzymes such as indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) and greatly impacts cancer biology by contributing to an immune-suppressive microenvironment (Platten et al., 2012; Timosenko et al., 2016; Zhai et al., 2014). Similarly, the high glycolytic activity of many tumors can trigger glucose limitation within the tumor microenvironment leading to metabolic restriction of tumor-associated T-cells (Chang et al., 2015). In such an environment competition between cells for resources may play a key role both in generating phenotypic heterogeneity and in selecting for specific genetic or phenotypic subpopulations. For example, oral carcinoma cells expressing high levels of the fatty acid importer CD36 are better able to initiate tumors than their low-expressing counterparts (Pascual et al., 2016), presumably because they have a greater potential capacity for taking up lipids required to fuel metastatic outgrowth. A second example is provided by ovarian cancer, where metastasis relies on the ability of ovarian cancer cells to stimulate lipid release from adipocytes (Nieman et al., 2011). Similar observations have also been made in melanoma where adipocyte-derived lipids facilitate melanoma progression (Zhang et al., 2018)
Adaptations to nutrient and oxygen limitation
Although resource limitation occurs within tumors, an excess demand over supply of any essential nutrient is not sustainable in the long-term. Consequently cancer cells, like bacteria, employ strategies directed towards increasing supply and decreasing demand.
Strategies that increase supply include: metabolic adaptation to use alternative fuels, including glucose, glutamine, alanine, pyruvate, lactate and lipids (Allen et al., 2016; Jimenez-Valerio and Casanovas, 2016; Nieman et al., 2011; Pavlova and Thompson, 2016; Pisarsky et al., 2016); increased macropinocytosis to take up and degrade proteins and lipids from their environment, including extracellular matrix and necrotic cell debris (Commisso et al., 2013; Davidson et al., 2016; Kamphorst et al., 2015; Kim et al., 2018; Muranen et al., 2017; Palm et al., 2015); secretion of ‘feed me’ signals that trigger release of nutrients including amino and fatty acids from other cells in the vicinity, a process termed metabolic symbiosis (Martinez-Outschoorn et al., 2014; Mikkilineni et al., 2017; Nakajima and Van Houten, 2013; Sonveaux et al., 2008; Sousa et al., 2016); and enhancing autophagy, a lysosome-dependent process by which non-essential intracellular components are recycled (Rzymski et al., 2009), that is a key survival strategy in several cancers (Goulielmaki et al., 2016; Guo et al., 2011; Perera et al., 2015; White, 2013; Yang et al., 2011). Tumor cells may also ensure a longer-term increase in nutrient supply by stimulating neo-angiogenesis through the expression of Vascular Endothelial Growth Factor (VEGF) that promotes formation of new blood vessels (Simons et al., 2016).
A key route to decreasing nutrient demand is to reduce protein synthesis, a highly nutrient and energy dependent process (Ma and Blenis, 2009). Global protein synthesis is controlled by the mTORC1 complex that promotes translation initiation by phosphorylating ribosomal protein S6 kinase (S6K) and the eukaryotic translation initiation factor 4E (elF4E)-binding protein (4E-BP1). Nutrient limitation can be coupled to reduced translation since signaling via the mTORC1 complex is restricted either by reduced amino acid supply, or under energy limiting conditions by activation of AMP-activated protein kinase (AMPK) (Ng et al., 2012; Sancak et al., 2008; Saxton and Sabatini, 2017).
Translational output is also modulated by regulation of the eIF2 translation initiation complex. Phosphorylation on Ser51 of eIF2α, the smallest subunit of the eIF2 translation initiation complex, by GCN2, PRKR-like ER kinase (PERK) or PKR (Figure 1) in response to a range of stresses and signals (Koritzinsky et al., 2013; Muaddi et al., 2010; Wang and Kaufman, 2014) converts eIF2α from a substrate of the eIF2B guanine exchange factor to a competitive inhibitor. Consequently, drivers of eIF2α phosphorylation suppress initiation of global protein synthesis (Koromilas, 2015). Notably, p-eIF2α diminishes translation initiation to reduce nutrient demand, but also promotes selective translation of a specific subset of proteins that operate to resolve the supply-demand imbalance. These include Activating Transcription Factor 4 (ATF4) (Pakos-Zebrucka et al., 2016), a key mediator of the integrated stress response (ISR) (Cubillos-Ruiz et al., 2017; Rzymski et al., 2009)
Figure 1.
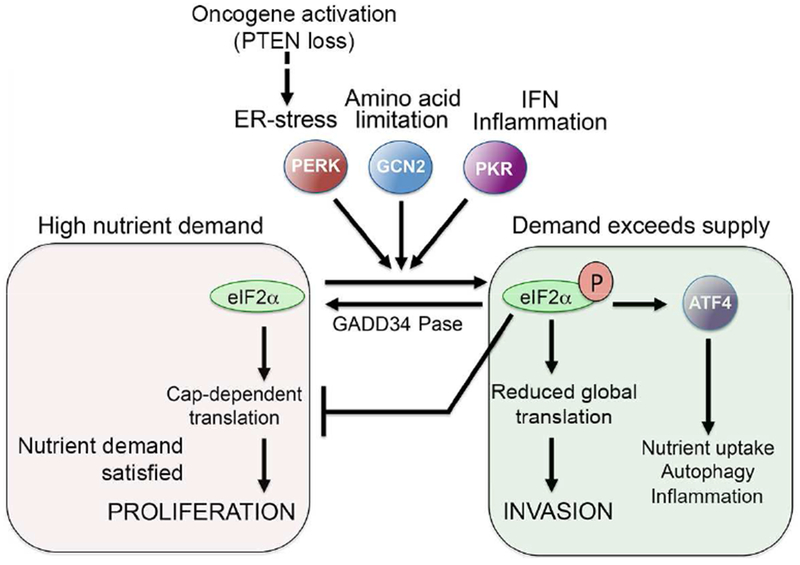
Control of Translation reprogramming by eIF2α phosphorylation
Translation initiation factor eIF2α can be phosphorylated by multiple upstream kinases, including GCN2 that senses amino acid limitation, PERK that responds to ER-stress, and PKR that lies downstream from interferon and inflammation. Phosphorylation can be reversed by the action of phosphatases such as GADD34. Phosphorylated eIF2α triggers inhibition of global translation to suppress nutrient demand, but also increases translation of a restricted set of mRNAs that includes the transcription factor ATF4 that promotes nutrient uptake and autophagy to increase nutrient supply. Phosphorylated eIF2α can also drive invasion, allowing cells to escape resource-poor environments and seek new sources of nutrient supply. A pseudo-starvation state can be imposed in nutrient–rich conditions through cell extrinsic signals such as inflammation that trigger eIF2α phosphorylation, or cell intrinsic signals that lead to heightened protein synthesis and ER-stress such as activation of oncogenes or in some circumstances loss of PTEN.
ATF4 promotes expression of the CHOP/DDIT3 transcription factor that increases nutrient supply by cooperating with ATF4 to activate genes implicated in amino acid transport and autophagy (B’Chir et al., 2013; Han et al., 2013; Harding et al., 2000). Moreover, although transcription up-regulation of VEGF expression by hypoxia-induced transcription factor (HIF) is well known (Semenza, 2015), its transcription is also stimulated by ATF4 as a consequence of the unfolded protein response (Pereira et al., 2010), a limited supply of amino acids (Longchamp et al., 2018) or low glucose (Wang et al., 2012). Significantly, eIF2α phosphorylation is also required for translation of VEGF (Stein et al., 1998), as well as genes implicated in autophagy (Yaman et al., 2003). The phosphorylation of eIF2α therefore increases nutrient supply by stimulating angiogenesis and recycling organelles, but reduces demand by restricting global translation. The response to nutrient limitation is therefore tightly coupled with translation reprogramming.
While p-eIF2α plays a key role in resolving any supply-demand imbalance, if the upstream stresses persist it can also promote apoptosis by activating a negative feedback loop (Figure 2). eIF2α phosphorylation leads to translation of ATF4 which in turn increases expression of CHOP (Tabas and Ron, 2011) that promotes expression of the eIF2α phosphatase subunit GADD34 leading to translation recovery (Marciniak et al., 2004). CHOP also suppresses expression of BCL2 (McCullough et al., 2001), a key pro-survival and anti-apoptotic protein. Consequently under prolonged stress induced by severe hypoxia, nutrient limitation, ROS or chemotherapy, the p-eIF2α/ATF4/CHOP axis reduces BCL2 activity to sensitize cells to death, and restores protein synthesis, thereby increasing nutrient demand and proteotoxic stress to drive apoptosis (Han et al., 2013; Hetz, 2012; Marciniak et al., 2004; Rozpedek et al., 2016). If death is to be avoided, therefore, the stresses driving eIF2α phosphorylation must be resolved in a timely manner.
Figure 2.
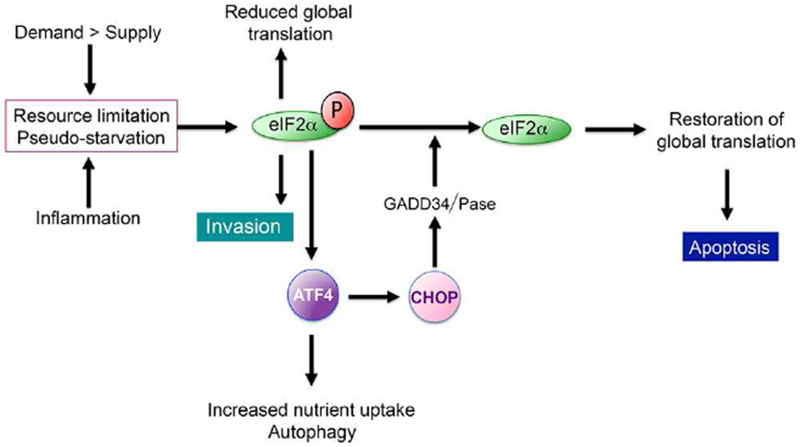
The translational feedback loop downstream from eIF2α phosphorylation. Starvation or Pseudo-starvation promote eIF2α phosphorylation and consequently invasion and a reduction of global translation. As part of the translation response, ATF4 is induced and transcriptionally activates CHOP/DDIT3. In turn, CHOP activates an eIF2α phosphatase to restore protein translation and induce apoptosis.
Notably, oxygen may also be considered as a key ‘nutrient’. Hypoxia, commonly encountered within tumours, leads to reduced activity of the oxygen-dependent prolyl hydroxylases FIH (Factor Inhibiting Hypoxia-induced factor) (Mahon et al., 2001) and VHL (Von Hippel-Lindau) (Ivan et al., 2001; Jaakkola et al., 2001) and stabilization of the hypoxia-inducible factors HIF1α, HIF1β and HIF2. Increased HIF activity mediates an adaptive response to hypoxia directed towards promoting survival including metabolic reprogramming, regulation of microenvironmental pH and up-regulation of VEGF expression to promote new blood vessel formation (Marchiq and Pouyssegur, 2016; Semenza, 2013,2015. While oxygen is critically required for oxidative phosphorylation in the electron transport chain, it is also required for formation of disulphide bonds in the ER (Koritzinsky et al., 2013) and for fatty acid desaturation via the iron-dependent enzyme stearoyl CoA desaturase (Koeberle et al., 2016). Since fatty acid composition and correct protein folding are critical to the health of the ER, hypoxia, like nutrient limitation, triggers ER-stress, phosphorylation of eIF2α and translation reprogramming.
Translation reprogramming and invasion
In addition to adjusting metabolism and protein translation, single cell organisms also undergo a transition to invasiveness under nutrient-depleted conditions. For example, in response to nutrient limitation the yeast S. cerevisiae, switches from budding to invasive pseudohyphal growth (Gancedo, 2001; Gimeno et al., 1992), a phenotypic transition critically dependent on eIF2α phosphorylation (Falletta et al., 2017) that may be considered an evolutionary conserved hallmark of the starvation response. Invasiveness complements adjustments to supply and demand by enabling cells to escape a nutrient depleted environment and to forage for nutrients elsewhere. But is invasion in cancer also linked to translation reprogramming, and in particular to eIF2α phosphorylation? Increasing evidence suggests that it is. In melanoma, glutamine limitation triggers a G1 arrest and invasiveness that is critically dependent on eIF2α phosphorylation (Falletta et al., 2017). Similarly, hypoxia, that drives several steps in metastasis including intra- and extravasation (Rankin and Giaccia, 2016; Semenza ,2016) , triggers invasion in breast cancer that is associated with activation of the eIF2α kinase PERK (Nagelkerke et al., 2013). Increased PERK activity in breast cancer is considered a key feature of cells with an epithelial-to-mesenchymal (EMT)-related phenotype (Feng et al., 2014). Significantly, suppressing the PERK/eIF2α/ATF4 axis in pancreatic cancer cells using acriflavine led to inhibition of morphological EMT and invasion as well as reversal of acquired drug-resistance (Dekervel et al., 2017). eIF2α phosphorylation is also a driver of increased invasiveness in chronic myeloid leukemia (Podszywalow-Bartnicka et al., 2016). Note however, that although ATF4 can be induced by mTORC1 signaling downstream from serum stimulation in a p-eIF2α-independent fashion (Ben-Sahra et al., 2016), doxycycline-mediated induction of ATF4 without accompanying eIF2α phosphorylation does not drive invasion in melanoma (Falletta et al., 2017); while ATF4 may contribute to the generation of an invasive phenotype it is not sufficient. Thus, whereas translation reprogramming is clearly associated with cancer progression (Sendoel et al., 2017), how changes in translation of specific mRNAs lead cells to undergo a proliferative to invasive phenotypic transition is only beginning to be understood. For example, PERK’s pro-metastatic function in breast cancer has been attributed in part to activation of the transcription factor CREB3L1/OASIS (Feng et al., 2017). Interestingly, translation of the EMT-associated transcription factors SNAIL1, TWIST and ZEB2 is promoted by YB-1, an RNA-binding protein and transcription factor that plays a pleiotropic role in cancer progression (Lasham et al., 2013), that like eIF2α phosphorylation also suppresses global cap-dependent translation (Evdokimova et al., 2009). YB-1 plays a similar role in sarcoma where it promotes invasion and translation of the hypoxia response factor HIF1α (El-Naggar et al., 2015). However the relationship between YB-1 and the triggers of eIF2α phosphorylation remains to be explored. Nevertheless, current evidence suggests that selective translation of specific mRNAs (translation reprogramming) is a key contributing factor in disease progression in several cancer types.
Pseudo-starvation
Since invasion in response to starvation is an evolutionarily conserved survival strategy designed to resolve a nutrient supply-demand imbalance, a model to explain why cancer cells adopt an invasive phenotype based on exposure to a resource-limited environment is potentially attractive. Note that limitation of just one essential nutrient will be sufficient to induce a starvation response, meaning that the energy required for cell migration may be provided by using alternative fuels to generate ATP. However, a model based on nutrient restriction as the sole driver of invasion might be insufficient. Although nutrient limitation may play a key role in invasion within poorly vascularized regions of tumors, invasion is often observed at the invasive front of carcinomas located at the tumor edge (Christofori, 2006) where it is unlikely that nutrient limitation will be a driving force for migration. In this location cancer cells are in close juxtaposition with the tumor-associated stroma comprised of a combination of fibroblasts, epithelial cells, adipocytes and a variety of immune cell types. Bi-directional interactions between the stromal components and cancer cells are instrumental in promoting invasion and an EMT, notably through the secretion of signaling molecules associated with an inflammatory response (Shalapour and Karin, 2015).
Inflammation is an evolutionarily conserved process implicated in normal tissue regeneration and is frequently deregulated in chronic and degenerative disease (Karin and Clevers, 2016). Inflammation can have a positive role in attenuating tumorigenesis, but it has also been linked with stemness, drug-resistance, metastatic dissemination and EMT (Chaffer et al., 2016; Lambert et al., 2017; Nieto et al., 2016; Quail and Joyce, 2013; Shalapour and Karin, 2015). Indeed, in a mouse model of pancreatic cancer, for example, inflammation was closely associated with cancer cell dissemination that occurred even prior to primary tumor formation (Rhim et al., 2012).
Although historically described as a binary state transition, EMT has more recently been proposed to be associated with more intermediate phenotypes (Bierie et al., 2017; Hong et al., 2015; Huang et al., 2013; Jolly et al., 2016; Nieto et al., 2016; Zhang et al., 2014). Support for a model in which regional inflammatory signaling contributes to the generation of intermediate EMT states in vivo has been provided by Pastushenko et al (2018) using mouse models of squamous cell carcinoma and mammary tumors as well as patient-derived xenografts. The results obtained using a combination of flow cytometry, immunofluorescence and single cell RNA-sequencing, provided clear evidence for six distinct mesenchymal populations that had undergone EMT to different extents and which exhibited different degrees of plasticity defined as an ability to generate phenotypic heterogeneity in derived tumors. Notably, different phenotypic subpopulations were shown to be spatially organized within tumors, with the location of different phenotypes related to the distribution of both adaptive and innate immune cells. For example, regions with a more epithelial phenotype were characterized by reduced endothelial CD31+ cells and low numbers of infiltrating CD45+ T cells and cancer-associated fibroblasts. By contrast, regions with tumor cells that had undergone EMT were associated with high levels of infiltrating CD45+ immune cells as well as macrophages and monocytes and elevated expression by the tumor cells of chemokines and pro-inflammatory and pro-angiogenic molecules. Since antibodies targeting macrophages increased the proportion of epithelial-like tumor cells in the population at the expense of those undergoing EMT, the results suggest that, consistent with previous models, bi-directional signaling between the cancer cells and immune cells plays a key role in generating phenotypic heterogeneity including the switch to different EMT-associated states.
As a mechanism to promote invasion, the interaction of cancer cells with a reactive stroma would at first sight appear unrelated to invasion driven by starvation. However, is it possible that the diverse inputs driving cancer cell invasion in response to stromal signaling do so by hijacking the evolutionarily conserved starvation response mediated by eIF2α phosphorylation? The convergence of nutritional and non-nutritional triggers of invasion on translation reprogramming would provide a foundation for an integrated model of cancer cell invasion underpinned by the lessons of evolution.
As highlighted above, in addition to GCN2 that responds to amino acid limitation, eIF2α phosphorylation can be mediated by PERK which is activated by ER-stress, and by PKR in response to interferon and double-stranded RNA (Leprivier et al., 2015). Consequently, microenvironmental signals unrelated to nutrient limitation that activate these kinases will trigger eIF2α phosphorylation and translation reprogramming. This implies that invasive cells may exist in two states. First, starvation defined by nutrient limitation where cells will be unable to divide, but will be invasive; and second a state we term ‘psuedo-starvation’ where features of the starvation response, including phosphorylation of eIF2α and translation reprogramming, are imposed by cell extrinsic signals arising from the microenvironment or by cell intrinsic events such as deregulated signaling arising from genetic lesions linked to cancer progression. Thus, in a pseudo-starvation state migration/invasion may occur even under conditions of nutrient abundance and individual cells may consequently be both invasive and proliferative. Under pseudo-starvation we envisage that eIF2α phosphorylation may promote invasion and reduced global translation, but that translation of mRNAs associated with proliferation would need to be maintained. It may be relevant that TORC1 activity, that is usually reduced under amino acid limiting conditions, can be maintained by signaling through SRC (Pal et al.,2018) , a non-receptor tyrosine kinase implicated in cell invasion (Patel et al., 2016).
Triggers of pseudo-starvation
Within the tumor microenvironment several inflammatory signaling molecules have a potential to impose a pseudo-starvation state.
Tumor necrosis factor alpha (TNFα), a primary inflammatory cytokine, is frequently found at high levels in tumors (Balkwill, 2009) and in wound healing (Koh and DiPietro, 2011) primarily as a result of secretion by infiltrating immune cells. The initial short-term (within 4 h to 24 h) response to TNFα is up-regulation of NFκB signaling that drives expression of a secretome including inflammatory cytokines (Balkwill, 2009; Greten et al., 2004). TNFα can also induce an unfolded protein response (UPR) and consequent eIF2α phosphorylation in fibroblasts via activation of PKR (Srivastava et al., 1998). Similarly, long-term exposure (between 24 h and 96 h) of melanoma cells to TNFα invokes a response remarkably similar to that driven by nutrient deprivation. In vitro, these include eIF2α phosphorylation, ATF4 protein expression, and increased invasiveness and de-differentiation (Falletta et al., 2017). In vivo, secretion of TNFα by tumor-infiltrating T-cells and macrophages can de-differentiate melanoma cells and is also associated with induction of ATF4 target genes (Falletta et al., 2017; Landsberg et al., 2012), a hallmark of the starvation response downstream from eIF2α phosphorylation. Although it has not been investigated directly, the time taken for cultured cells exposed to TNFα to re-program translation might indicate that it is an indirect outcome mediated by the cytokines released by the earlier NFκB activation. The role of NFκB in triggering invasion is underscored by the observation that NFκB can be activated in response to eIF2α phosphorylation (Deng et al., 2004; Tam et al., 2012) and that NFκB blockade inhibits invasion of breast cancer cells in 3D culture (Becker-Weimann et al., 2013).
TGFβ is a key cytokine that plays a critical role in development, and is implicated in fibrosis during tissue repair (Nieto et al., 2016). It also plays a major role in tumor biology and is a potent inducer of EMT in a range of cancer cell types (Bierie and Moses, 2006; Nieto et al., 2016; Pastushenko et al., 2018; Pickup et al., 2013; Pinner et al., 2009). TGFβ regulates transcription primarily through the activation of SMAD transcription factors. Although activation of SMADs may lead to context-specific patterns of gene expression resulting from cooperative binding with lineage specific transcription factors (David and Massague, 2018), TGFβ signaling can lead to up-regulation of a repertoire of chemokines and signaling molecules, including VEGF (Feng et al., 2014), a ‘feed-me’ signal that promotes angiogenesis. Considerable emphasis has been placed on the transcriptional outputs downstream from TGFβ signaling, but importantly, in breast cancer cells TGFβ-induced EMT is associated with activation of PERK (Feng et al., 2014). Consequently inhibition of PERK blocks TGFβ-stimulated invasiveness. This implies that the biological consequences of TGFβ signaling requires translation reprograming if the full impact of its transcriptional output is to be manifest. Thus, both TNFα and TGFβ reprogram translation by inducing eIF2α phosphorylation to impose a pseudostarvation state associated with invasion. Although the contribution of each of the factors secreted in response to TNFα and TGFβ have yet to be determined, in effect these cytokines act as ‘get-out-of-here’ signals that promote invasiveness by hijacking the evolutionarily conserved link between starvation and migration.
Additional non-nutritional signals known to drive eIF2α phosphorylation and invasion include Leukemia inhibitory factor (LIF), bone morphogenic protein 4 (BMP4), and interferon. LIF triggers invasiveness in stromal fibroblasts (Albrengues et al., 2014) and in several cancer types (Gulluoglu et al., 2017; Lee et al., 2010; Liu et al., 2015) through inhibition of an eIF2α phosphatase (Friend et al., 2015). BMP4 activates PERK (Friend et al., 2015) and also triggers invasiveness in a range of cancers (Guo et al., 2012; Maegdefrau and Bosserhoff, 2012; Rothhammer et al., 2005; Yang et al., 2014). Lastly, interferon drives eIF2α phosphorylation via activation of PKR and is required for invasion in breast cancer cells (Bennett et al., 2012). Although many inflammatory signals trigger eIF2α phosphorylation and promote invasion, the contribution of translation reprogramming to inflammation-driven cancer progression needs to be explored further. Nevertheless it is evident that translation reprogramming plays a key role in mediating phenotypic transitions that drive cancer progression.
Beyond natural triggers, phenotypic transitions related to pseudo-starvation may also be induced by at least some targeted therapies. In this respect, the importance of a ‘starvation’ phenotype in generating phenotypic heterogeneity in vivo was recently highlighted by using single cell RNA-sequencing to generate expression profiles of individual melanoma cells derived from drug naïve patient-derived xenografts (Rambow et al., 2018). Gene expression profiles were derived from tumors before, during and after development of resistance to treatment with a BRAF inhibitor to determine the degree of tumor phenotypic heterogeneity underlying resistance and relapse. The results revealed a striking degree of phenotypic heterogeneity that can co-exist within tumors and especially within the minimal residual disease state. While a small minority of cells exhibited a hyper-differentiated drug-resistant state, the remaining de-differentiated cells could be grouped into three distinct phenotypes: A neural crest stem cell (NCSC)-like state; a subpopulation exhibiting an invasive gene expression signature; and cells exhibiting hallmarks of a starvation response including high expression of the CD36 fatty acid importer. Significantly, bioinformatic analysis of the phenotypes detected suggested that proliferating melanoma cells in vivo would transition through the starved phenotype in order to generate the other subpopulations observed. The RNA-seq profiles were used to identify specific markers of each subpopulation which were then applied in multiplexed immunohistochemistry to reveal that populations of cells with similar phenotypes were spatially organized into clusters, an observation reminiscent of the regional distribution of EMT subpopulations (Pastushenko et al., 2018). Notably cells exhibiting a CD36-high starvation gene expression profile were located within regions of tumors distant from blood vessels, consistent with the possibility that a restricted nutrient or oxygen supply would contribute to the generation of the starvation phenotype in vivo. These observations are in line with previous work demonstrating that BRAF inhibitor-induced eIF2α phosphorylation and ER-stress is a key determinant of phenotypic resistance to BRAF targeted therapy (Ma et al., 2014).
While pseudo-starvation can be triggered by a range of cell extrinsic signals including starvation, inflammation and therapies, it can also originate by cell intrinsic mechanisms including activation of oncogenes or inactivation of tumor suppressors. For example, induction of MYC expression can drive ER-stress and eIF2α phosphorylation (Hart et al., 2012) as can activated Ha-Ras (Battcock et al., 2006). Moreover, activation of BRAF in melanocytes can lead to eIF2α phosphorylation (Corazzari et al., 2015; Ferretta et al., 2016) and an E- to N-cadherin switch in adhesion molecules that may prime for invasive behavior (Boyd et al., 2013). These observations are consistent with the results from a study of activated RAS-driven squamous cell carcinoma (Sendoel et al., 2017) in which the authors concluded that eIF2α phosphorylation occurred early in tumor formation. Similarly, genetic or epigenetic inactivation of PTEN, a common occurrence in cancer linked to increased metastatic potential, is associated with increased translation initiation as a consequence of elevated PI3K signaling to mTORC1 (Figure 1) (Milella et al., 2015). Consequently cells may phosphorylate eIF2α to dial down global protein synthesis so as to rebalance protein synthesis capacity with the elevated translation initiation arising as a consequence of PTEN loss (Nguyen et al., 2018; Zeng et al., 2011). However in other systems it has been reported that PTEN inactivation can suppress eIF2α phosphorylation (Mounir et al., 2009), indicating that the relationship between eIF2α and PTEN may be context dependent. Nevertheless, pseudo-starvation driven by cell intrinsic mechanisms may provide some mechanistic underpinning for the hypothesis proposed by Bernards and Weinberg (Bernards and Weinberg, 2002) that oncogene activation and senescence bypass, events early in tumorigenesis, predispose to metastatic dissemination.
Collectively these observations suggest a model (Figure 3) in which activation of oncogenes and loss of tumor suppressors would lead to low level eIF2α phosphorylation that would facilitate tumor outgrowth by balancing nutrient supply and demand: the elevated nutrient demand associated with initial tumor expansion could outstrip supply, but by increasing eIF2α phosphorylation to limit protein synthesis, demand would be restrained. Additional stresses driven by nutrient limitation would trigger increased eIF2α phosphorylation, invasion, ATF4-dependent VEGF mRNA expression and its translation and secretion to promote angiogenesis. ATF4 can also drive transcription of several genes encoding cytokines such as IL-8, IL-6 or CCl2 (Gargalovic et al., 2006; Huang et al., 2015; Zhang et al., 2013). Additional inflammatory signaling would arise through activation of NFκB downstream of p-eIF2α (Deng et al., 2004; Lawrence, 2009; Tam et al., 2012). Importantly, phosphorylation of eIF2α not only leads to transcriptional up-regulation of pro-inflammatory cytokines, but also their translation (Gameiro and Struhl, 2018). The consequent pro-inflammatory environment attracts immune cells that will further increase inflammation within and around the tumor. Depending on the repertoire of inflammatory molecules expressed, the microenvironment may impose a pseudo-starvation phenotype that reinforces eIF2α phosphorylation. Downstream of eIF2α phosphorylation a phenotypic transition to invasion will take place that may be accompanied by proliferation if nutrients are in sufficient abundance.
Figure 3.
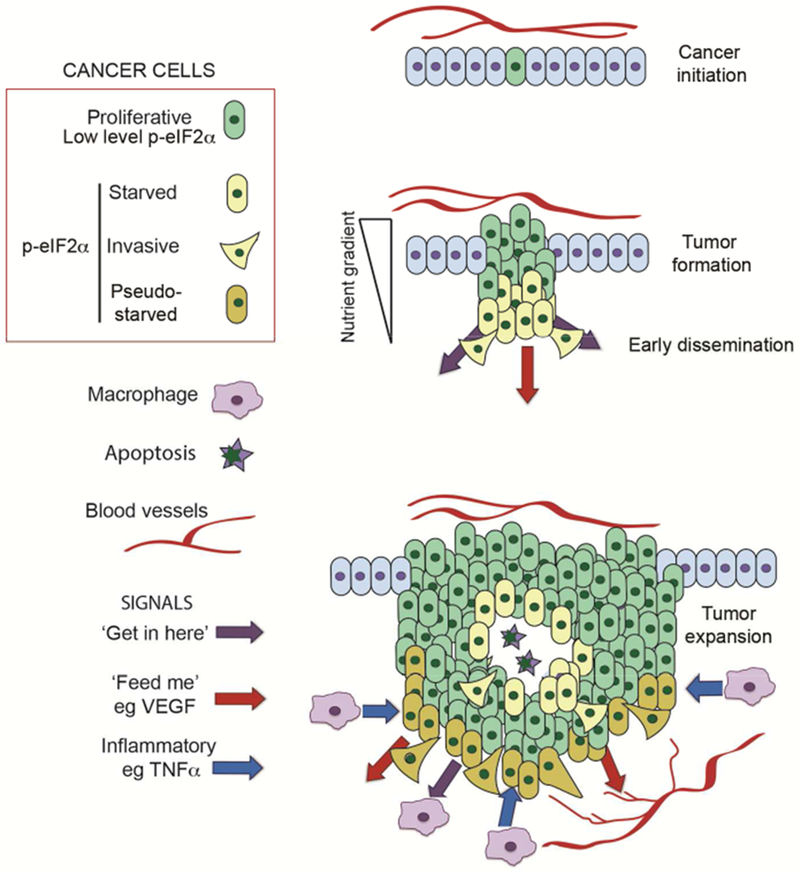
Starvation and pseudo-starvation generate invasive cancer cells.
Tumor initiation (top) driven by activation of oncogenes and loss of tumor suppressors will generate cells that exhibit deregulated proliferation and which may be primed for invasion via cell intrinsic pathways that generate a low level eIF2α phosphorylation and phenotypic instability. As tumors form (middle) gradients of nutrients and oxygen will arise as a consequence of poor vasculature. Cells will respond to low nutrient/oxygen levels by increasing eIF2α phosphorylation and will as a consequence adopt an invasive phenotype, activate inflammatory ‘get-in-here’ signaling downstream from NFκB and ATF4, translate the VEGF ‘feed-me’ pro-angiogenesis signal, and participate in metabolic symbiosis. As tumors expand (bottom) the arrival of immune cells will lead to additional inflammatory signaling that will trigger further eIF2α phosphorylation to impose a pseudo-starved state that reduces the threshold for cells to adopt an invasive phenotype even in regions of the tumor where cells may proliferate if nutrients are abundant. The depiction is simplified for the sake of clarity an does not include multiple other tumor-associated cell types including tumor-associated lymphocytes, cancer-associated fibroblasts or adipocytes.
Stabilization of the invasive state
It might be reasoned that once invasive cells enter the relatively nutrient rich bloodstream, they would escape the microenvironment responsible for triggering starvation or pseudo-starvation triggered by cell extrinsic stimuli. In this scenario rapid reversal of a phospho-eIF2α-driven invasive phenotype could create a barrier to extravasation. Although environmentally-induced cellular phenotypes may potentially be reversed when conditions change, reversal may take time. Estimates of the half-life of circulating tumor cells in the blood range from a few minutes in mice (Sasportas and Gambhir, 2014) to up to 2.4 hours in humans (Meng et al., 2004). It is plausible therefore that such a short time in the bloodstream may not be sufficient to reverse an invasive phenotype, especially if phenotypic states become stabilized, for example through feed-back transcriptional regulatory loops or through metabolically-dependent epigenetic modifications.
Feedback loops that act as binary switches are common in transcription regulation and can establish multiple stable stationary states within a population that may not readily be reversed (Macarthur et al., 2009). Alternatively a transient state may be stabilized via epigenetic reprogramming. For example, recent characterization of single melanoma cells within tumors revealed a subset of rare, phenotypically distinct, cells that exhibited a transient transcriptional state that conferred resistance to BRAF inhibitors (Shaffer et al., 2017). Exposure to a BRAF inhibitor led to epigenetic reprogramming that fixed the transient transcriptional state to generate a stable drug-resistant phenotype. A similar epigenetic reprogramming event may explain why cells that undergo a transition to invasion in response to nutrient limitation or a pseudo-starvation event do not readily reverse their phenotype within vessels or as they migrate within tumors or tissue. Phenotype fixation is consistent with the fact that the majority of the cells that survive dissemination and extravasate to invade surrounding tissues do not readily take up proliferation (Giancotti, 2013; Naumov et al., 2002; Sosa et al., 2014). Thus invasive behavior is maintained for a period in the foreign location, presumably in the absence of the initial trigger for invasion, before most cells become dormant and a few resume proliferation. Nevertheless further studies are required to characterize better at the single cell level the epigenetic changes induced and their persistence after exposure to short-term or prolonged pro-invasive signals in vivo.
Translation reprogramming and survival during metastatic dissemination
Phenotypic plasticity in cancer enables cells to adapt to their environment and consequently adopt a spectrum of survival strategies that range between maximized proliferation at the cost of increased vulnerability, versus reduced proliferation to maximize tolerance to stressful conditions (Aktipis et al., 2013; Chen et al., 2011). If translation reprogramming by p-eIF2α mediates the transition to invasiveness, for example in response to starvation or pseudo-starvation signals, does reprogramming of translation also increase tolerance to stresses encountered during metastatic dissemination?
The process of metastasis is highly inefficient. Many cells may enter the blood or lymphatic system daily (Chang et al., 2000; Kang and Pantel, 2013), but few survive and fewer are competent to form metastases (Cameron et al., 2000; Lambert et al., 2017; Luzzi et al., 1998; Massague and Obenauf, 2016; Vanharanta and Massague, 2013). Understanding why some cells are endowed with a greater capacity to initiate metastases than others is of significant clinical relevance. Increasing evidence suggests that translation reprogramming may play a key role in promoting survival in the bloodstream, for example by suppressing oxidative stress or anoikis. Circulating cancer cells (CCCs) from a number of tumor types exhibiting an EMT–related phenotype also possess enhanced mitochondrial respiration, increased oxygen consumption and high ATP generation supported by high levels of PGC1α, a transcription cofactor implicated in mitochondrial biogenesis (Dupuy et al., 2015). In melanoma, ROS are higher in CCCs than melanoma cells in a subcutaneous environment (Piskounova et al., 2015). In this model, suppressing oxidative stress increased the survival of CCCs and promoted successful metastatic colonization of visceral organs, an observation in agreement with antioxidants such as glutathione enhancing cancer progression (Harris et al., 2015). Significantly, ROS promote eIF2α phosphorylation (Rajesh et al., 2015). In turn p-eIF2α promotes translation of mRNAs from genes that suppress oxidative stress (Harding et al., 2003) to increase survival in a feedback loop that enhances metastatic colonization.
A second major cause of death in cells entering the bloodstream is likely to be anoikis (Cao et al., 2016), an apoptotic death program associated with inappropriate or inadequate interactions with extracellular matrix. Significantly, in breast cancer cells activation of PERK, upstream from eIF2α-phosphorylation can promote anoikis-resistance (Avivar-Valderas et al., 2011). By promoting resistance to both anoikis and oxidative stress, eIF2α phosphorylation may enhance CCC survival and increase the probability of invasive cells initiating a successful metastasis. Collectively, the evidence suggests that while many other factors may contribute, the altered translational landscape and transcription program downstream from eIF2α phosphorylation enhances the probability of successful metastatic colonization.
Conclusions
Although regulation of transcription underlying phenotypic transitions in cancer has historically been a major focus of attention, there is an increasing appreciation that protein translation represents a critical nexus for the control of phenotypic identity. In this article we highlight how eIF2α phosphorylation and downstream reprogramming of protein translation can underpin invasiveness. In part this reflects a response to nutrient (and oxygen) limitation that echoes the adaptation to resource limitation in single cell organisms where the imperative to balance nutrient supply and demand is instrumental in inducing phenotypic transitions remarkably similar to those that occur within the tumor microenvironment. As organisms evolved and became more complex, the invasive response to starvation was maintained, but was also hijacked by signals that can promote invasion by imposing a pseudo-starvation state on cells. These include a range of inflammatory signaling molecules and perhaps many others, including components of the senescence-associated secretome. Such signaling would potentially promote survival in a stressful microenvironment by enabling recipient cells to pre-adapt before being exposed directly to the stress that induced the donor cell to send the signal. Moreover, while translation reprogramming is recognized as a key event in promoting invasion in an increasing number of cancers, whether it represents a universal driver of metastatic dissemination is not yet clear. However, since phosphorylation of eIF2α is necessary for neural crest migration as well as invasion in yeast (Falletta et al., 2017), it seems likely that translation reprogramming is an evolutionarily conserved mechanism that may play a widespread role in cancer progression.
Finally, to understand fully the origins of the ‘Hallmarks of Cancer’ (Hanahan and Weinberg, 2011) the contribution of the altered translation and transcription that occurs as a consequence of eIF2α phosphorylation should be taken into account. Beyond the initial genetic/epigenetic events that underpin tumor initiation to give replicative immortality - and consequently deregulated cellular energetics -eIF2α phosphorylation is necessary for angiogenesis by facilitating translation of VEGF (a ‘feed-me’ signal). eIF2α phosphorylation lies both upstream and downstream of inflammatory signaling that provides both ‘get-in-here’ signals to attract immune cells to the tumor and ‘get-out-of-here’ signals that induce neighboring cancer cells to reprogram translation. eIF2α phosphorylation also facilitates resistance to cell death, for example by counteracting oxidative stress and anoikis. Moreover although little is known of how translation reprogramming might affect genome instability in cancer, in bacteria exposure to nutritional or other stresses leads to increased expression of error-prone DNA polymerases that increase the genetic diversity within the population (Foster, 2007; MacLean et al., 2013). We therefore anticipate that similar mechanisms may operate in cancer cells to increase genetic diversity that, together with epigenetic/phenotypic heterogeneity, would fuel the emergence of therapy resistance.
In summary, eIF2α phosphorylation can suppress global translation and facilitate survival under conditions of protein synthesis overload caused by oncogene activation and loss of tumor suppressors such as PTEN, and can promote invasion in response to starvation or pseudostarvation. However, it seems likely that altered translation of specific subsets of mRNAs will be key to cells adopting an invasive phenotype. We do not know whether it is increased translation of some genes or decreased translation of others, or more likely a combination of both, that drives cells to invade. The identification of the critical mRNAs in this process will represent a major advance in our understanding of how ‘starvation’ in response to the microenvironment can impose specific phenotypic transitions in cancer, and more importantly how a moderate starvation phenotype can act as an intermediate state between proliferation and stem cells and dormancy. As such, therapies targeting the downstream events associated with eIF2α phosphorylation are likely to have significant clinical benefit as has already been seen in pre-clinical studies in prostate cancer and melanoma (Nguyen et al., 2018; Falletta el al., 2017). However, since many key signaling events provoke different outcomes dependent on cell context, it is likely that the impact of eIF2α phosphorylation on cell behavior may be influenced by cell type, genetic background or the associated microenvironment. Nevertheless, while the role of starvation and pseudo-starvation-induced invasion in many cancers remains to be fully explored, a model for cancer cell invasion that invokes translation reprogramming provides an evolutionarily conserved framework to decipher the complexities of microenvironment-driven phenotype-switching and the biology of cancer progression.
Acknowledgements
CRG is funded by the Ludwig Institute for Cancer Research and NIH grant PO1 CA128814-06A1, and CG-J by Instituto de Salud Carlos III, Grant PI13/01150 and Ministerio de Economia y Competitividad Grant SAF2016-79837-R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aktipis CA, Boddy AM, Gatenby RA, Brown JS, and Maley CC (2013). Life history trade-offs in cancer evolution. Nat. Rev. Cancer 13, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrengues J, Bourget I, Pons C, Butet V, Hofman P, Tartare-Deckert S, Feral CC, Meneguzzi G, and Gaggioli C (2014). LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Reports 7, 1664–1678. [DOI] [PubMed] [Google Scholar]
- Allen E, Mieville P, Warren CM, Saghafinia S, Li L, Peng MW, and Hanahan D (2016). Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Reports 15, 1144–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, and Dang CV (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, and Aguirre-Ghiso JA (2011). PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol 31, 3616–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, and Bruhat A (2013). The eIF2αlpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F (2009). Tumour necrosis factor and cancer. Nat. Rev. Cancer 9, 361–371. [DOI] [PubMed] [Google Scholar]
- Battcock SM, Collier TW, Zu D, and Hirasawa K (2006). Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J. Virol 80, 4422–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker-Weimann S, Xiong G, Furuta S, Han J, Kuhn I, Akavia UD, Pe’er D, Bissell MJ, and Xu R (2013). NFkB disrupts tissue polarity in 3D by preventing integration of microenvironmental signals. Oncotarget 4, 2010–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, Maynard C, Schafer R, van Diest P, Voest E, van Oudenaarden A, et al. (2016). Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Reports 14, 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, and Manning BD (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RL, Carruthers AL, Hui T, Kerney KR, Liu X, and May WS Jr. (2012). Increased expression of the dsRNA-activated protein kinase PKR in breast cancer promotes sensitivity to doxorubicin. PloS One 7, e46040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernards R, and Weinberg RA (2002). A progression puzzle. Nature 418, 823. [DOI] [PubMed] [Google Scholar]
- Beug H (2009). Breast cancer stem cells: eradication by differentiation therapy? Cell 138, 623–625. [DOI] [PubMed] [Google Scholar]
- Bierie B, and Moses HL (2006). Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520. [DOI] [PubMed] [Google Scholar]
- Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, Liu Donaher J, Reinhardt F, Chaffer CL, Keckesova Z, et al. (2017). Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci USA 114, E2337–E2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SC, Mijatov B, Pupo GM, Tran SL, Gowrishankar K, Shaw HM, Goding CR, Scolyer RA, Mann GJ, Kefford RF, et al. (2013). Oncogenic B-RAF(V600E) signaling induces the T-Box3 transcriptional repressor to repress E-cadherin and enhance melanoma cell invasion. J. Invest. Dermatol 133, 1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Spaderna S, Hlubek F, and Kirchner T (2005). Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 5, 744–749. [DOI] [PubMed] [Google Scholar]
- Brooks MD, Burness ML, and Wicha MS (2015). Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 17, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnan JT (2003). Interorgan amino acid transport and its regulation. J. Nutr 133, 2068S–2072S. [DOI] [PubMed] [Google Scholar]
- Burrell RA, McGranahan N, Bartek J, and Swanton C (2013). The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345. [DOI] [PubMed] [Google Scholar]
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, and MacDonald IC (2000). Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60, 2541–2546. [PubMed] [Google Scholar]
- Cao Z, Livas T, and Kyprianou N (2016). Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit. Rev. Oncol 21, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey JN, Mettert EL, Roggiani M, Myers KS, Kiley PJ, and Goulian M (2018). Regulated Stochasticity in a Bacterial Signaling Network Permits Tolerance to a Rapid Environmental Change. Cell 173, 196–207 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. (2011). Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 108, 7950–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, San Juan BP, Lim E, and Weinberg RA (2016). EMT, cell plasticity and metastasis. Cancer Metastasis Rev 35, 645–654. [DOI] [PubMed] [Google Scholar]
- Chang C-HH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YS, di Tomaso E, McDonald DM, Jones RG, Jain RK, and Munn LL (2000). Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc.Natl. Acad. Sci. USA 97, 14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sprouffske K, Huang Q, and Maley CC (2011). Solving the puzzle of metastasis: the evolution of cell migration in neoplasms. PloS One 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chocarro-Calvo A, Garcia-Martinez JM, Ardila-Gonzalez S, De la Vieja A, and Garcia-Jimenez, (2013). Glucose-induced beta-catenin acetylation enhances Wnt signaling in cancer. Mol. Cell 49, 474–486. [DOI] [PubMed] [Google Scholar]
- Christofori G (2006). New signals from the invasive front. Nature 441, 444–450. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, and Wahl GM (2006). Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66, 9339–9344. [DOI] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480. [DOI] [PubMed] [Google Scholar]
- Coghlin C, and Murray GI (2010). Current and emerging concepts in tumour metastasis. J. Path 222, 1–15. [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S,Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, and Piacentini M (2015). Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 22, 946–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, and Bosenberg M (2009). Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet 41, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, and Massague J (2018). Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol 19, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. (2016). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med 23, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20. [DOI] [PubMed] [Google Scholar]
- Dekervel J, Bulle A, Windmolders P, Lambrechts D, Van Cutsem E, Verslype C, and van Pelt J (2017). Acriflavine Inhibits Acquired Drug Resistance by Blocking the Epithelial-to-Mesenchymal Transition and the Unfolded Protein Response. Transl. Oncol 10, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, and Ron D (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. cell. Biol 24, 10161–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al. (2015). PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 22, 577–589. [DOI] [PubMed] [Google Scholar]
- El-Naggar AM, Veinotte CJ, Cheng H, Grunewald TG, Negri GL, Somasekharan SP, Corkery DP, Tirode F, Mathers J, Khan D, et al. (2015). Translational Activation of HIF1alpha by YB-1 Promotes Sarcoma Metastasis. Cancer Cell 27, 682–697. [DOI] [PubMed] [Google Scholar]
- Emmons MF, Faiao-Flores F, and Smalley KS (2016). The role of phenotypic plasticity in the escape of cancer cells from targeted therapy. Biochem. Pharmacol 122, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, et al. (2009). Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 15, 402–415. [DOI] [PubMed] [Google Scholar]
- Falletta P, Sanchez-del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, Siddaway R, Lisle R, Freter R, Daniels M, et al. (2017). Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 31, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YX, Jin DX, Sokol ES, Reinhardt F, Miller DH, and Gupta PB (2017). Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun 8, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q, et al. (2014). Epithelial-to-mesenchymal transition activates PERK-eIF2αlpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 4, 702–715. [DOI] [PubMed] [Google Scholar]
- Ferretta A, Maida I, Guida S, Azzariti A, Porcelli L, Tommasi S, Zanna P, Cocco T, Guida M, and Guida G (2016). New insight into the role of metabolic reprogramming in melanoma cells harboring BRAF mutations. Biochimi. Biophys. Acta 1863, 2710–2718. [DOI] [PubMed] [Google Scholar]
- Foster PL (2007). Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 42, 373–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend K, Brooks HA, Propson NE, Thomson JA, and Kimble J (2015). Embryonic Stem Cell Growth Factors Regulate eIF2αlpha Phosphorylation. PloS One 10, e0139076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gameiro PA, and Struhl K (2018). Nutrient Deprivation Elicits a Transcriptional and Translational Inflammatory Response Coupled to Decreased Protein Synthesis. Cell Reports 24, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gancedo JM (2001). Control of pseudohyphae formation in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 25, 107–123. [DOI] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA, et al. (2018). Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell. Biol 20, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, et al. (2006). Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc. Natl. Acad. Sci. USA 103, 12741–12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG (2013). Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno CJ, Ljungdahl PO, Styles CA, and Fink GR (1992). Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell 68, 1077–1090. [DOI] [PubMed] [Google Scholar]
- Goulielmaki M, Koustas E, Moysidou E, Vlassi M, Sasazuki T, Shirasawa S, Zografos G, Oikonomou E, and Pintzas A (2016). BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 7, 9188–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, and Karin M (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296. [DOI] [PubMed] [Google Scholar]
- Gulluoglu S, Sahin M, Tuysuz EC, Yaltirik CK, Kuskucu A, Ozkan F, Sahin F, Ture U, and Bayrak OF (2017). Leukemia Inhibitory Factor Promotes Aggressiveness of Chordoma. Oncol. Res 25, 1177–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Huang J, and Gong J (2012). Bone morphogenetic protein 4 (BMP4) is required for migration and invasion of breast cancer. Mol. Cell. Biochem 363, 179–190. [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Chaffer CL, and Weinberg RA (2009a). Cancer stem cells: mirage or reality? Nat. Med 15, 1010–1012. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, and Lander ES (2011). Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146, 633–644. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, and Lander ES (2009b). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. [DOI] [PubMed] [Google Scholar]
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. (2012). ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 122, 4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews Mol. Cell Biol. 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Hoek K, and Goding CR (2010). Cancer stem cells versus phenotype switching in melanoma. Pigment Cell Melanoma Res 23, 746–759. [DOI] [PubMed] [Google Scholar]
- Hoek K, Rimm DL, Williams KR, Zhao H, Ariyan S, Lin A, Kluger HM, Berger AJ, Cheng E, Trombetta ES, et al. (2004). Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 64, 5270–5282. [DOI] [PubMed] [Google Scholar]
- Holzel M, Bovier A, and Tuting T (2013). Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Hong T, Watanabe K, Ta CH, Villarreal-Ponce A, Nie Q, and Dai X (2015). An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 11, e1004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Jing G, Wang JJ, Sheibani N, and Zhang SX (2015). ATF4 is a novel regulator of MCP-1 in microvascular endothelial cells. J. Inflamm 12, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY, Wong MK, Tan TZ, Kuay KT, Ng AH, Chung VY, Chu YS, Matsumura N, Lai HC, Lee YF, et al. (2013). An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death & Disease 4, e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, and Kaelin WG Jr. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- Jimenez-Valerio G, and Casanovas O (2016). Antiangiogenic Resistance: Novel Angiogenesis Axes Uncovered by Antiangiogenic Therapies Research. Curr. Drug Targets 17, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E, and Levine H (2016). Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 7, 27067–27084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, and Lu C (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, and Pantel K (2013). Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, and Clevers H (2016). Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG, and Stephanopoulos G (2016). Metabolic requirements for cancer cell proliferation. Cancer & Metab. 4,16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. (2018). PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov. 8, 866–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CA (2009). Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 9, 302–312. [DOI] [PubMed] [Google Scholar]
- Klein CA (2013). Selection and adaptation during metastatic cancer progression. Nature 501, 365–372. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Loser K, and Thurmer M (2016). Stearoyl-CoA desaturase-1 and adaptive stresssignaling. Biochim. Biophys. Acta 1861, 1719–1726. [DOI] [PubMed] [Google Scholar]
- Koh TJ, and DiPietro LA (2011). Inflammation and wound healing: the role of the macrophage. Expert Rev. Mol. Med 13, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, Boutros PC, Braakman I, and Wouters BG (2013). Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 203, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koromilas AE (2015). Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 1849, 871–880. [DOI] [PubMed] [Google Scholar]
- Kreso A, O’Brien CA, van Galen P, and Gan OI (2013). Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 339, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, et al. (2012). Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416. [DOI] [PubMed] [Google Scholar]
- Lasham A, Print CG, Woolley AG, Dunn SE, and Braithwaite AW (2013). YB-1: oncoprotein, prognostic marker and therapeutic target? Biochemical J. 449, 11–23. [DOI] [PubMed] [Google Scholar]
- Lawrence T (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspectives in Biology 1, a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Jan HJ, Lai JH, Ma HI, Hueng DY, Lee YC, Cheng YY, Liu LW, Wei HW, and Lee HM (2010). Nodal promotes growth and invasion in human gliomas. Oncogene 29, 3110–3123. [DOI] [PubMed] [Google Scholar]
- Leprivier G, Rotblat B, Khan D, Jan E, and Sorensen PH (2015). Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta 1849, 845–860. [DOI] [PubMed] [Google Scholar]
- Liu B, Lu Y, Li J, Liu Y, Liu J, and Wang W (2015). Leukemia inhibitory factor promotes tumor growth and metastasis in human osteosarcoma via activating STAT3. APMIS 123, 837–846. [DOI] [PubMed] [Google Scholar]
- Lloyd AC (2013). The Regulation of cell size. Cell 154, 1194–1205. [DOI] [PubMed] [Google Scholar]
- Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, Hine C, Ben-Sahra I, Knudsen NH, Brace LE, et al. (2018). Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 173, 117–129 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, and Groom AC (1998). Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Path 153, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. (2014). Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest 124, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, and Blenis J (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318. [DOI] [PubMed] [Google Scholar]
- Macarthur BD, Ma’ayan A, and Lemischka IR (2009). Systems biology of stem cell fate and cellular reprogramming. Nat. Rev. Mol. Cell. Biol 10, 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean RC, Torres-Barcelo C, and Moxon R (2013). Evaluating evolutionary models of stress-induced mutagenesis in bacteria. Nat. Rev. Genet 14, 221–227. [DOI] [PubMed] [Google Scholar]
- Maegdefrau U, and Bosserhoff AK (2012). BMP activated Smad signaling strongly promotes migration and invasion of hepatocellular carcinoma cells. Exp. Mol. Pathol 92, 74–81. [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, and Semenza GL (2001). FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiq I, and Pouyssegur J (2016). Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med 94, 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, and Ron D (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Lisanti MP, and Sotgia F (2014). Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 25, 47–60. [DOI] [PubMed] [Google Scholar]
- Marusyk A, and Polyak K (2010). Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, and Obenauf AC (2016). Metastatic colonization by circulating tumour cells. Nature 529, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, and Holbrook NJ (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol 21, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, and Swanton C (2015). Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26. [DOI] [PubMed] [Google Scholar]
- Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, et al. (2004). Circulating tumor cells in patients with breast cancer dormancy. Clinical cancer research : an official journal of the American Association for Cancer Res. 10, 8152–8162. [DOI] [PubMed] [Google Scholar]
- Mikkilineni L, Whitaker-Menezes D, Domingo-Vidal M, Sprandio J, Avena P, Cotzia P, Dulau-Florea A, Gong J, Uppal G, Zhan T, et al. (2017). Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol 44, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, et al. (2015). PTEN: Multiple Functions in Human Malignant Tumors. Frontiers Oncology 5, 24. doi: 10.3389/fonc.2015.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M, et al. (2016). The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med 8, 327ra326. [DOI] [PubMed] [Google Scholar]
- Moris N, Pina C, and Arias AM (2016). Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev.Genet 17, 693–703. [DOI] [PubMed] [Google Scholar]
- Mounir Z, Krishnamoorthy JL, Robertson GP, Scheuner D, Kaufman RJ, Georgescu MM, and Koromilas AE (2009). Tumor suppression by PTEN requires the activation of the PKR-eIF2αlpha phosphorylation pathway. Science Signaling 2, ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M, and Koromilas AE (2010). Phosphorylation of eIF2αlpha at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol. Biol. Cell 21, 3220–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Iwanicki MP, Curry NL, Hwang J, DuBois CD, Coloff JL, Hitchcock DS, Clish CB, Brugge JS, and Kalaany NY (2017). Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun 8, 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, and Span PN (2013). Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast cancer research : BCR 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, Chang SH, Dvorak AM, and Dvorak HF (2009). Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 100, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima EC, and Van Houten B (2013). Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol. Carcin 52, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, and Chambers AF (2002). Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 62, 2162–2168. [PubMed] [Google Scholar]
- Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, Davicioni E, Triche TJ, and Sorensen PH (2012). The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 19, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F, et al. (2018). Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med 10 DOI 10.1126/scitranslmed.aar2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, and Nieto MA (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724. [DOI] [PubMed] [Google Scholar]
- Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Reports 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Palmieri M, Chaudhury A, Klisch TJ, di Ronza A, Neilson JR, Rodney GG, and Sardiello M (2018). Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nat. Commun 9, 4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 18, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, Berenguer A, Prats N, Toll A, Hueto JA, et al. (2016). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45. [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556 463–468. [DOI] [PubMed] [Google Scholar]
- Patel A, Sabbineni H, Clarke A, and Somanath PR (2016). Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci 157, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Liao N, Neale GA, and Hendershot LM (2010). Transcriptional and posttranscriptional regulation of proangiogenic factors by the unfolded protein response. PloS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, and Moses HL (2013). The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, Bonvin E, Goding C, and Sahai E (2009). Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 69, 7969–7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann J, Hess C, and Christofori G (2016). Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Reports 15, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten M, Wick W, and Van den Eynde BJ (2012). Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 72, 5435–5440. [DOI] [PubMed] [Google Scholar]
- Podszywalow-Bartnicka P, Cmoch A, Wolczyk M, Bugajski L, Tkaczyk M, Dadlez M, Nieborowska-Skorska M, Koromilas AE, Skorski T, and Piwocka K (2016). Increased phosphorylation of eIF2αlpha in chronic myeloid leukemia cells stimulates secretion of matrix modifying enzymes. Oncotarget 7, 79706–79721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh K, Krishnamoorthy J, Kazimierczak U, Tenkerian C, Papadakis AI, Wang S, Huang S, and Koromilas AE (2015). Phosphorylation of the translation initiation factor eIF2αlpha at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 6, e1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaela M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Towards minimal residual disease-directed therapy in melanoma. Cell 174, 843–855. [DOI] [PubMed] [Google Scholar]
- Rankin EB, and Giaccia AJ (2016). Hypoxic control of metastasis. Science 352, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, and Thompson CB (2000). In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 6, 683–692. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. (2012). EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Tanaka KK, Tanaka T, and Simonsen DG (1956). Free amino acids in growing and regressing acites cell tumors: Host resistance and chemical agents. Cancer Res. 16, 970–978. [PubMed] [Google Scholar]
- Rosen JM, and Jordan CT (2009). The increasing complexity of the cancer stem cell paradigm. Science 324, 1670–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer T, Poser I, Soncin F, Bataille F, Moser M, and Bosserhoff AK (2005). Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 65, 448–456. [PubMed] [Google Scholar]
- Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, and Majsterek I (2016). The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med 16, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Singleton DC, and Harris AL (2009). Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8, 3838–3847. [DOI] [PubMed] [Google Scholar]
- Saez-Ayala M, Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, Chazarra S, Freter R, Middleton M, Pinero-Madrona A, Cabezas-Herrera J, Goding CR, et al. (2013). Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell 24, 105–119. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasportas LS, and Gambhir SS (2014). Imaging circulating tumor cells in freely moving awake small animals using a miniaturized intravital microscope. PloS One 9, e86759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2013). HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest 123, 3664–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2015). Targeting Hypoxia-Inducible Factor 1 to Stimulate Tissue Vascularization. J. Invest. Med 64, 361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2016). The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta 1863, 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, et al. (2017). Translation from unconventional 5’ start sites drives tumour initiation. Nature 541, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, and Hengge R (2014). Stress responses go three dimensional - the spatial order of physiological differentiation in bacterial macrocolony biofilms. Environ Microbiol 16, 1455–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M (2010). Normal stem cells and cancer stem cells: similar and different. Semin. Cancer Biol. 20, 85–92. [DOI] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S, and Karin M (2015). Immunity, inflammation, and cancer: an eternal fight between good and evil. J. Clin. Invest 125, 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2016). Cancer statistics, 2016. CA Cancer J Clin 66, 7–30. [DOI] [PubMed] [Google Scholar]
- Simons M, Gordon E, and Claesson-Welsh L (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nature reviews Mol. Cell. Biol 17, 611–625. [DOI] [PubMed] [Google Scholar]
- Smith BN, and Bhowmick NA (2016). Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med 5, 17; doi: 10.3390/jcm5020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. (2008). Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest 118, 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MSS, Bragado P, and Aguirre-Ghiso JA (2014). Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava SP, Kumar KU, and Kaufman RJ (1998). Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem 273, 2416–2423. [DOI] [PubMed] [Google Scholar]
- Stein I, Itin A, Einat P, Skaliter R, Grossman Z, and Keshet E (1998). Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol. Cell. Biol 18, 3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA, et al. (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 20, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, and Ron D (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam AB, Mercado EL, Hoffmann A, and Niwa M (2012). ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PloS One 7, e45078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, and Mayor R (2012). Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol 366, 34–54. [DOI] [PubMed] [Google Scholar]
- Timosenko E, Ghadbane H, Silk JD, Shepherd D, Gileadi U, Howson LJ, Laynes R, Zhao Q, Strausberg RL, Olsen LR, et al. (2016). Nutritional Stress Induced by Tryptophan-Degrading Enzymes Results in ATF4-Dependent Reprogramming of the Amino Acid Transporter Profile in Tumor Cells. Cancer Res. 76, 6193–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, and Yang J (2012). Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, and Massague J (2013). Origins of metastatic traits. Cancer Cell 24, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Mayer A, and Höckel M (2004). Tumor hypoxia and malignant progression. Methods in Enzymol. 381, 335–354. [DOI] [PubMed] [Google Scholar]
- Visvader JE, and Lindeman GJ (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768. [DOI] [PubMed] [Google Scholar]
- Vlamakis H, Aguilar C, Losick R, and Kolter R (2008). Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 22, 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlamakis H, Chai Y, Beauregard P, Losick R, and Kolter R (2013). Sticking together: building a biofilm the Bacillus subtilis way. Nat. Rev. Microbiol 11, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, and Kaufman RJ (2014). The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597. [DOI] [PubMed] [Google Scholar]
- Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, and Polverini PJ (2012). The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 72, 5396–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E (2013). Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27, 2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, and Thompson CB (2010). Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaman I, Fernandez J, Liu H, Caprara M, Komar AA, Koromilas AE, Zhou L, Snider MD, Scheuner D, Kaufman RJ, et al. (2003). The zipper model of translational control: a small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 113, 519–531. [DOI] [PubMed] [Google Scholar]
- Yang J, Wang J, Pan L, Li H, Rao C, Zhang X, Niu G, Qu J, and Hou L (2014). BMP4 is required for the initial expression of MITF in melanocyte precursor differentiation from embryonic stem cells. Exp. Cell Res. 320, 54–61. [DOI] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G (2014). Modelling mammalian cellular quiescence. Interface Focus 4, 20130074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng N, Li Y, He L, Xu X, Galicia V, Deng C, and Stiles BL (2011). Adaptive basal phosphorylation of eIF2αlpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Molecular cancer research : MCR 9, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Lauing KL, Chang AL, Dey M, Qian J, Cheng Y, Lesniak MS, and Wainwright DA (2014). The role of IDO in brain tumor immunotherapy. J. Neurooncol 123, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bai N, Chang A, Zhang Z, Yin J, Shen W, Tian Y, Xiang R, and Liu C (2013). ATF4 is directly recruited by TLR4 signaling and positively regulates TLR4-trigged cytokine production in human monocytes. Cell Mol. Immunol. 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, Elankumaran S, and Xing J (2014). TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Science Signal. 7, ra91. [DOI] [PubMed] [Google Scholar]
- Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, et al. (2018). Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Disc. 8, 1006–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhu X, Cui K, Mancuso J, Federley R, Fischer K, Teng GJ, Mittal V, Gao D, Zhao H, et al. (2016). In Vivo Visualization and Characterization of Epithelial-Mesenchymal Transition in Breast Tumors. Cancer Res. 76, 2094–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]