

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of cardiometabolic syndrome, which often also includes obesity, diabetes, and dyslipidemia. It is rapidly becoming the most prevalent liver disease worldwide. A sizable minority of NAFLD patients develop nonalcoholic steatohepatitis (NASH), which is characterized by inflammatory changes that can lead to progressive liver damage, cirrhosis, and hepatocellular carcinoma. Recent studies have shown that in addition to genetic predisposition and diet, the gut microbiota affects hepatic carbohydrate and lipid metabolism as well as influences the balance between pro‐inflammatory and anti‐inflammatory effectors in the liver, thereby impacting NAFLD and its progression to NASH. In this review, we will explore the impact of gut microbiota and microbiota‐derived compounds on the development and progression of NAFLD and NASH, and the unexplored factors related to potential microbiome contributions to this common liver disease.
Keywords: microbiome/microbiota, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis
Subject Categories: Digestive System; Microbiology, Virology & Host Pathogen Interaction
Glossary
- Bacteremia
Presence of bacteria in the blood.
- Bile acids
A group of molecules synthesized from cholesterol in hepatocytes that are major component of the bile. They function in facilitation of nutrient absorption in the gut and metabolic signaling in several tissues in the body.
- Dysbiosis
Imbalance in composition of microbiota that has a negative effect on the physiology of the host.
- Fecal microbiota transplantation (FMT)
Medical procedure that involves transfer of fecal matter from a donor or donors into the intestinal tract of recipient.
- Gut epithelial barrier
A layer of epithelial cells of the gut that forms a physical barrier between contents of the gut lumen and the host while allowing transport of desired molecules such as nutrients.
- Gut–liver axis
The interaction between physiology of the gut and the liver that is based on the physical connection between these organs via portal vein.
- Histone deacetylases (HDACs)
Family of enzymes that cleave acetyl group from histones resulting in more positive charge of histone tails and tighter binding of histones to negatively charged DNA.
- Inflammasome
A term describing multiprotein complexes formed in response to variety of signals such as pathogens as well as sterile stress signals and result in secretion of pro‐inflammatory signals such as IL‐1β or IL‐18 and eventually induction of pyroptosis.
- Intestinal bacterial outgrowth
Abnormally high amount of bacteria present in the intestine.
- Kupffer cells
Macrophages of the liver.
- NAFLD
Presence of at least 5% hepatic steatosis in patients without excessive alcohol consumption, including the entire spectrum of fatty liver disease ranging from simple fatty liver to nonalcoholic steatohepatitis, fibrosis, and cirrhosis.
- NASH
Presence of at least 5% hepatic steatosis with histological characterization of liver inflammation and hepatocyte injury with or without fibrosis.
- Oxidative stress
A situation when there are more free radicals produced than the cell can neutralize with antioxidants. In this situation, free radicals interact and damage cellular components such as proteins, lipids, or nucleic acids.
- Reactive oxygen species (ROS)
Unstable, very reactive molecules containing oxygen, such as peroxide, superoxide, hydroxyl radical.
- S‐adenosyl methionine (SAM) cycle
Reactions that produce, consume, and regenerate S‐adenosyl methionine. Regeneration of SAM allows it to function as a key donor of methyl group in the cell.
- Short‐chain fatty acids
Fatty acids with less than six carbon atoms.
- Steatosis
A process defined by accumulation of excessive amount of lipids in the liver cells, mainly inside the hepatocytes.
Introduction
NAFLD and NASH
Nonalcoholic fatty liver disease (NAFLD) is defined as the presence of at least 5% hepatic steatosis but lacking common causes of secondary hepatic fat accumulation, such as excessive alcohol consumption, chronic viral hepatitis, autoimmune hepatitis, congenital hepatic disorders, or long‐term use of steatosis‐inducing medications (Chalasani et al, 2018). NAFLD often, but not always, occurs together with type 2 diabetes, obesity, dyslipidemia, and hypertension, which constitute cardiometabolic disease (Younossi et al, 2016).
The global prevalence of NAFLD is estimated to be 25.24%, with the highest rates in the Middle East (31.79%) and South America (30.45%), followed by Asia (27.37%), North America (24.13%), Europe (23.71%), and Africa (13.48%; Younossi et al, 2016). The incidence of NAFLD is reported to range from 20 to 50 cases per 1,000 person‐years in different countries (Chalasani et al, 2018). These alarming numbers make NAFLD a major clinical and economic burden and the new epidemic in global chronic liver disease (Younossi et al, 2018). A substantial portion of NAFLD patients develop nonalcoholic steatohepatitis (NASH), which is histologically characterized by the presence of hepatic inflammation and liver injury. NAFLD, and particularly NASH, can potentially progress to fibrosis, cirrhosis, and eventually hepatocellular carcinoma (Pais et al, 2013; Calzadilla Bertot & Adams, 2016). In this context, NAFLD is currently the leading cause of chronic liver disease in the USA and Europe. Indeed, NASH‐associated cirrhosis has become the second leading indication for adult liver transplantation in the USA and continues to grow (Wong et al, 2015). Current treatment of NAFLD focuses on lifestyle modification. Liver‐targeted pharmacotherapy is reserved for high‐risk patients and is mostly experimental (Tilg, 2017; Chalasani et al, 2018).
The pathogenesis of NAFLD is poorly understood. It is thought to involve complex interactions among genetic susceptibility variants, environmental factors, insulin resistance, and changes in the gut microbiota (Arab et al, 2017). The interplay between these factors results in altered lipid metabolism and excessive lipid accumulation in hepatocytes, culminating in the development of NAFLD. Furthermore, the microbiota is involved in alternating the balance between pro‐inflammatory or anti‐inflammatory signals, thereby contributing to inflammation that may result in progression to NASH. There is thus an urgent need to fully understand the pathogenesis of NAFLD, and the contribution of the microbiome to NAFLD and its progression to NASH, which may enable to improve diagnostics, patient stratification, and identification of new therapeutic targets.
The gut microbiome
Recent years brought enhancing understanding that the microbiota, an ecosystem consisting of bacteria, archaea, protists, fungi, and viruses living in the human gut, is not an idle bystander but an interconnected, active player in human physiology. The microbiota composition and function are shaped by a variety of host and environmental factors including diet, physical activity, medication, circadian activity, and geographical localization. This complex community of microorganisms bears several folds more genetic information than the human genome, for example, enzymes capable of biochemical functions lacking in the human host, such as deconjugation of primary bile acids or breakdown of indigestible carbohydrates. This person‐specific microbiota configuration cooperates with the unique host genetics in contributing to individualized traits and phenotypes.
The gut microbiota actively participates in the digestion of food and facilitates the absorption of the dietary molecules into the portal and systemic circulation (Jumpertz et al, 2011; Ridaura et al, 2013), interacts with the mucosal immune system, to shape development of its antigen recognition, recruitment, proliferation, and effector function, and may impact the host even at far sites from where it actually resides, via modulation of migrating immune cells and through metabolites influxing from the gut into the systemic circulation, where they can affect other distant tissues. The bacterial components and metabolites that cross the epithelial barrier enter the circulation and may feature a systemic bioactive effect. For example, l‐carnitine found in red meat is metabolized by gut bacteria and the host liver to trimethylamine‐N‐oxide (TMAO), which promotes atherosclerosis. Consortia of bacteria of vegans and vegetarians produce less TMAO than those of omnivorous humans when given the same amount of l‐carnitine (Koeth et al, 2013). As in the case of TMAO, changes in the number and composition of microbes in the gut may lead to an altered signaling to the host via microbiome‐derived metabolites, mediated through (i) changes in concentration and composition of bacterially derived molecules recognized by pattern recognition receptors, (ii) changes in abundance and composition of the antigen pool available for immune system sampling, and (iii) altered concentration and composition of bacterial metabolites that influx systemically into the host (Kau et al, 2011; Sharon et al, 2014; Neis et al, 2015; Levy et al, 2017). The effect mediated by the molecules that are absorbed into the portal blood is particularly important in the liver, as it is the first organ exposed to portal blood.
In the following sections, we aim to shed light on mechanisms by which gut microbiota both positively and negatively affects development and progression of NAFLD and NASH.
Associations between NAFLD and NASH and alterations in the gut microbiome
Several studies implicate the involvement of the gut microbiome with NASH or NAFLD in mice and humans. High‐fat diet‐fed germ‐free mice exhibit lower levels of lipids in the liver in comparison with the conventionally housed mice (Rabot et al, 2010). Microbiome transferred into germ free mice, from mice that developed fasting hyperglycemia and insulinemia, but not from healthy mice, led to the development of NAFLD in recipient mice (Le Roy et al, 2013). Using 16S rDNA profiling of mice, two bacterial species, Lachnospiraceae bacterium 609 and Barnesiella intestinihominis, were found to be significantly overrepresented in the stool with a potency to induce NAFLD, while Bacteroides vulgatus was underrepresented in comparison with the control (Le Roy et al, 2013). Furthermore, a correlation analysis of NAFLD‐associated parameters and the abundance of bacterial species in mice fed with low‐fat and high‐fat diet showed association between Lactobacillus gasseri and Lactobacillus taiwanensis and the area of lipidic droplets in the liver (Zeng et al, 2013).
Nonalcoholic fatty liver disease and NASH in humans often co‐occur with obesity and poor dietary habits making it difficult to disentangle effects of diet and accompanying metabolic changes in liver disease from the effects mediated by the altered microbiome under these same conditions. Nevertheless, some species in humans were associated with NAFLD, with the abundance of bacterial species, such as Proteobacteria, Enterobacteria, and Escherichia (Zhu et al, 2013), or Bacteroides (Boursier et al, 2016), being higher in patients with NASH as compared to matched healthy individuals. Profiling stool microbiome of children with NAFLD showed more abundant Gammaproteobacteria and Prevotella in comparison with the microbiota of obese children without NAFLD (Michail et al, 2015). Of note, an increase in Proteobacteria and decrease in Firmicutes were observed during progression of NAFLD, suggesting that the gut microbiome may not be stably dysbiotic during disease progression (Loomba et al, 2017). Interestingly, in a dietary intervention study, NAFLD patients were subjected to low‐choline diet for 6 weeks resulting in microbiome alterations, which included Gammaproteobacteria correlating positively, while Erysipelotrichia correlating negatively with the hepatic lipid content (Spencer et al, 2011). The impact of nonabsorbable antibiotic treatment in modulating disease features in patients with NAFLD and NASH also supports the potential role of microbiome in its pathogenesis. Short‐term treatment of patients with steatosis and NASH with the nonabsorbable antibiotic rifaximin led to improvement in liver function (Gangarapu et al, 2015). Similarly, long‐term antibiotic treatment led to decrease in small intestinal bacterial outgrowth, correlating with an improvement in liver function (Madrid et al, 2001).
Collectively, these association studies show that correlations may exist between bacterial composition and distinct taxa and NAFLD or NASH. However, these observations are limited by the lack of reproducibility between cohorts, the absence of a mechanistic explanation for dysbiosis or of its effects on NAFLD and NASH, and lack of evidence pointing toward causality. Furthermore, in most studies microbiome is sampled from stool bacterial composition of which is distinct from the communities present in the more proximal sites of the intestine (Zmora et al, 2018).
Mechanisms contributing to microbiome modulation of NAFLD and NASH
Several potential mechanisms by which gut microbiota regulates NAFLD and NASH have been studied in recent years. Suggested mechanisms include dysbiotic bacteria and their derived products translocating to the liver through a disrupted gut barrier, where they evoke a hepatic inflammatory reaction and commensal or metabolite‐induced interplays with dietary factors in inducing steatosis.
The microbiome–gut–liver anatomical axis
The concept of gut bacteria having influence on liver homeostasis stems from the intimate anatomical interaction between the gastrointestinal tract and the liver, which is often termed “gut–liver axis”. Indeed, such connection is derived from the embryonic development, when the liver buds directly from the foregut. The liver plays a crucial role at the nexus of host–microbe interactions because it is the first organ to drain the gut through the portal vein. The portal blood, in addition to nutrients, contains other molecules that either actively or passively cross from the gut to the blood (Fig 1). This makes the liver one of the most exposed organs to intestinal bacteria and bacterial‐derived products (Macpherson et al, 2016). Disturbance of the gut–liver axis was shown to play a pivotal role in the pathogenesis of NAFLD. These include gut barrier disruption, bacterial translocation and inflammatory response in the liver, such as Toll‐like receptor (TLR) signaling and inflammasome activation, and changes in composition of bacterial products.
Figure 1. Gut microbiota‐derived compounds affecting liver metabolism.
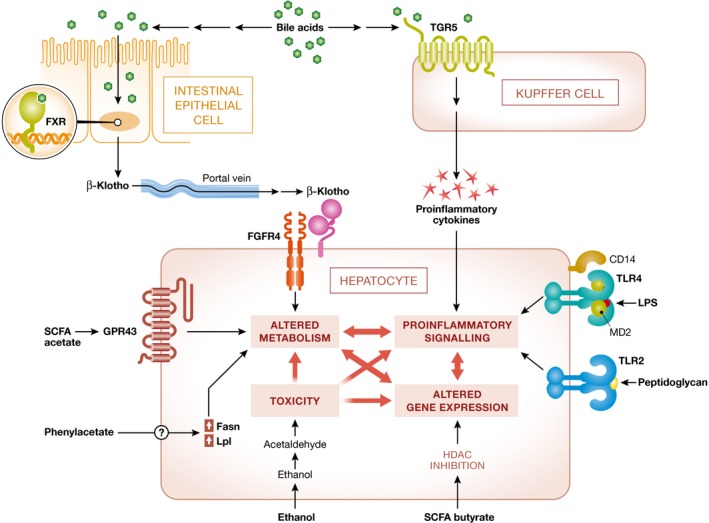
Microbiome‐derived compounds affect the hepatocytes via small molecules leading to highly interconnected effects including pro‐inflammatory signaling, changes in gene expression, and alteration in metabolism and toxicity. Bile acids bind their receptor in the intestinal epithelial cells leading to release of βKlotho that travels via portal vein to the liver where it binds FGFR4 on the surface of hepatocytes and leads to changes in metabolism. Bile acids also activate the TGR5 receptor on the Kupffer cells leading to secretion of pro‐inflammatory cytokines that in turn signal to hepatocytes. Bacterial pattern molecules, i.e., peptidoglycan and LPS, signal via TRL2 and TRL4. Short‐chain fatty acids act via binding to their receptors (acetate binds GPR43) and alter metabolism; additionally, butyrate is histone deacetylase (HDAC) inhibitor and regulates gene expression through modulation of chromatin state. Phenylacetate by unknown mechanism affects expression of metabolic genes such as Fasn and Lpl leading to metabolic changes. Finally, acetaldehyde, toxic derivative of ethanol, exacerbates high oxidative stress on the hepatocytes.
Microbiome impact on gut barrier function
The healthy intestinal epithelium forms a tightly sealed physical barrier that separates the host from the contents of the gut. Tight junctions are one of the most important physiological and pathological regulators of intestinal permeability. Under physiological conditions, tight junction proteins, such as zonula occludens, seal adjacent epithelial cells at the apical sites and prevent bacteria from entering the intestinal mucosa and the bloodstream (Turner, 2009). However, such regulation becomes pathological with disruption of tight junctions and excessive paracellular leakage of non‐self antigens to the lamina propria, which subsequently leads to the development of NASH (Fasano, 2008). The gut barrier consists of not only tight junctional complexes but also mucus layer produced by goblet cells, antimicrobial defense mediated by Paneth cells, and the complicated network of innate and adaptive immune cell populations. A delicate crosstalk and balance among gut microbiota, intestinal epithelial cells, and gut mucosal system are important to maintain intestinal permeability and tissue homeostasis (Peterson & Artis, 2014). There is emerging evidence for a dysfunctional gut barrier or altered gut permeability in NAFLD patients (Miele et al, 2009; Volynets et al, 2012; Luther et al, 2015). A meta‐analysis based on five clinical studies shows that NAFLD and NASH patients are more likely to have altered gut permeability in comparison with healthy controls (Luther et al, 2015). The association of increased intestinal permeability is stronger particularly in NASH patients, demonstrating that the inflammatory changes observed in NASH might be caused by the increased intestinal permeability (Luther et al, 2015). On the molecular level, NAFLD patients exhibit decreased expression of zonula occludens‐1 (ZO‐1) and junctional adhesion molecule A (JAM‐A; Miele et al, 2009; Rahman et al, 2016).
However, the co‐occurrence of NAFLD/NASH and disruption of the gut epithelial barrier do not prove causation. Whether the alteration of gut permeability is a cause or a consequence of NAFLD is an intriguing question but remains unclear to date. The study by Luther et al (2015) suggested that initial liver damage might precede the development of disrupted gut permeability. Using methionine–choline‐deficient (MCD) diet‐induced NASH model in vivo, the authors found that the disruption of tight junction proteins and altered intestinal permeability only occurred after initial hepatic injury (Luther et al, 2015). However, whether this holds true in other animal models or in humans remains to be investigated.
Regardless of the triggering event, impaired gut permeability aggravates NASH. As an example, induction of a leaky and inflamed gut by dextran sulfate sodium (DSS) leads to translocation of lipopolysaccharide (LPS) to the systemic circulation and consequently worsens liver inflammation and fibrosis in mice fed with high‐fat diet (Gäbele et al, 2011). Similarly, JAM‐A‐deficient mice, a genetic model of gut barrier dysfunction, when fed with high‐fat, fructose, and cholesterol diet, develop more severe steatohepatitis than control mice (Rahman et al, 2016).
Microbiota impact on liver steatosis
Fat absorption is impacted by the gut microbiota (Bäckhed et al, 2004), including the duodenal microbiota (Martinez‐Guryn et al, 2018), and is considered a contributing factor to liver steatosis (Le Roy et al, 2013). Several effector molecules have been linked to these effects.
Short‐chain fatty acids
Complex carbohydrates, such as dietary fiber and resistant starch, are digested by many different gut bacterial species leading to release of short‐chain fatty acids (SCFAs): acetate, propionate, butyrate, pentanoic (valeric) acid, and hexanoic (caproic) acid (Høverstad & Midtvedt, 1986). Although most SCFAs are utilized in the gut, some amount is transported to the bloodstream through the monocarboxylate transporter 1 (MCT‐1) and the sodium‐coupled monocarboxylate transporter 1 (SMCT‐1) receptors and via the portal vein reach the liver, where they can be channeled into the tricarboxylic acid (TCA) cycle and become source of energy. In addition, SCFAs are also potent signaling molecules through binding to G‐protein‐coupled receptors GPR41, GPR43, and GPR109A (Samuel et al, 2008; Maslowski et al, 2009). While GPR41 binds propionate and GPR43 primarily recognizes acetate, both can also bind all other SCFAs with lower affinity (Brown et al, 2003). Moreover, other GPCRs, such as OLFR78 and GPR109A, can also bind SCFA with varying affinity adding complexity to the system (Pluznick et al, 2013; Kasubuchi et al, 2015). Importantly, butyrate and to some level propionate and acetate can act as histone deacetylase (HDAC) inhibitors. Acetylation of histone tails leads to introduction of negative charge and to less efficient binding of histones to the DNA, resulting in opening of the chromatin and gene expression. Hence, SCFAs acting as inhibitors of histone deacetylases positively regulate gene expression. The repertoire of genes affected depends on the cellular context. Moreover, in the liver short‐chain fatty acids regulate synthesis of cholesterol, a precursor of bile acids, which are potent mediators of communication between gut microbiome and the host (Hara et al, 1999).
One of the mechanisms by which short‐chain fatty acids affect fat accumulation both in the liver and in the adipose tissue is regulation of insulin sensitivity via GPR43. Short‐chain fatty acid activation of GPR43 signaling in adipose tissue promotes energy expenditure and inhibits fat accumulation in adipose tissue as well as in the liver (Kimura et al, 2013). Unlike colonized mice, germ‐free mice in which short‐chain fatty acids are not produced by the microbiome maintain normal weight regardless of GPR43 being knocked out (Kimura et al, 2013). Similarly, mice treated with antibiotics developed more severe NAFLD and had higher insulin resistance and adiposity (Mahana et al, 2016).
Another possible mechanism of action of short‐chain fatty acids to limit NASH is by reducing inflammatory signals. In murine models of colitis, arthritis, and asthma, deletion of GPR43 leads to increased pro‐inflammatory cytokine production and immune cell recruitment (Maslowski et al, 2009). Similar phenotypes are observed in germ‐free mice that lack short‐chain fatty acid‐producing bacteria (Maslowski et al, 2009). In humans, increases in the amount of consumed fiber lead to higher expression of GPR41 and GPR43 and lower inflammatory markers and improved lung function in patients with asthma (Halnes et al, 2017).
While many studies investigated the roles of SCFAs on gut physiology (Macia et al, 2015), their function in liver disease remains to be fully elucidated. An initial study indicated an association between NASH and low‐fiber diet (Cortez‐Pinto et al, 2006), while another study showed some improvement in blood alanine transaminase (ALT) and aspartate transaminase (AST) activity (measure of liver damage) and cholesterol in NAFLD patients fed on high‐fiber diet (Rocha et al, 2007). Interestingly, overweight and obese patients have higher level of propionate in their stool, suggesting that either it is overproduced or its absorption is disrupted (Schwiertz et al, 2010). Larger studies are needed to validate these observations and determine if and how SCFAs may play a role in the pathogenesis of NAFLD and NASH.
Bile acids
Primary bile acids (BAs) are synthesized by the liver, are secreted to the gallbladder, and released into the duodenum following food ingestion. In the gut, they are metabolized to secondary bile acids by the gut bacteria. Then, they are reabsorbed into the portal vein and most of the molecules are captured by the liver and are recirculated, but some remain in the blood and act as signaling molecules. Bile acids function in the host in a dual manner: (i) they facilitate lipid solubilization, digestion, and absorption and (ii) they act as signaling molecules via, among others, TGR5 and FXR receptors (Sinal et al, 2000; Pols et al, 2011). Chemical identity and composition of the bile acid pool affect lipid absorption and potency to activate receptors.
The enzymes for dehydroxylation of the hydroxyl group at the C‐7α position of deconjugated BAs to form the secondary BAs are expressed throughout the entire bacterial kingdom, but Clostridium XIVa was identified to have the most potent enzymes for this conversion (Ridlon et al, 2013). The other BA transformation that is mediated by bacteria is the dehydrogenation and epimerization of 3‐, 7‐, and 12‐hydroxyl groups. Ridlon et al (2006) summarized the complex metabolism of bile acids and the reactions carried out by specific bacteria.
Patients with NAFLD feature elevated levels of bile acids in liver tissue, serum, and urine with a significantly higher ratio of more hydrophobic and cytotoxic bile acid species (Aranha et al, 2008; Ferslew et al, 2015). Perturbations in gut microbiota levels and community composition are very likely to affect the bile acid pool, but this causal relationship has not been explicitly showed yet in vivo. It is hypothesized that commensal microbes modulate host cellular metabolism through bile acid conversions, including lipid and glucose homeostasis, but this merits further studies. Furthermore, it is hypothesized that changes in microbiome resulting from bariatric surgeries may have beneficial effect on NASH, but the contribution of microbiome and other factors was not disentangled yet (Chavez‐Tapia et al, 2010).
Choline
The effect of choline deficiency on the development of NAFLD and NASH is well established, corroborated by the fact that choline‐deficient diet is a commonly used murine model of NASH (Blumberg & McCollum, 1941). In the liver, choline is mainly used in biogenesis of phosphatidylcholine or is directed to maintain the S‐adenosyl methionine (SAM) cycle. Moreover, the liver is an important storage compartment for choline in the body. Deletion of genes involved in choline metabolism and its availability to the SAM cycle or deletion of genes of the SAM cycle leads to NAFLD. Furthermore, in the absence of choline, very‐low‐density lipoprotein (VLDL) levels are down‐regulated, due to insufficient phosphatidylcholine. The hepatic ratio of phosphatidylcholine to its unmethylated precursor phosphatidylethanolamine in NAFLD is lower than in healthy subjects, suggesting deficiency in methyl donor SAM (Arendt et al, 2013). Choline has two origins: exogenous and endogenous. Diet provides about 70% of choline, while the remainder is synthesized in the body. The amount of available choline depends on its amount in the diet and on its metabolism in the gut by microbiota, which is also regulated by choline levels (Spencer et al, 2011).
Commensal bacteria, e.g., E. coli, Desulfovibrio desulfuricans, can convert choline to methylamines such as trimethylamine (TMA), dimethylamine (DMA), and monomethylamine (MMA; Ackermann & Schutze, 1910; Zeisel et al, 1983; Craciun & Balskus, 2012). The choline utilization cluster, present in many different bacterial species, carries the function of metabolism of choline to TMA (Al‐Waiz et al, 1992; Zhang et al, 1999; Craciun & Balskus, 2012; Martínez‐del Campo et al, 2015). Germ‐free mice inoculated with bacterial isolates predicted to metabolize choline to TMA in comparison with mice inoculated with bacteria that do not metabolize choline, featured choline depletion both in blood serum and in feces (Romano et al, 2015). Species shown to metabolize choline to TMA in vitro include Anaerococcus hydrogenalis, Clostridium asparagiforme, Clostridium hathewayi, Clostridium sporogenes, Escherichia fergusonii, Proteus penneri, Providencia rettgeri and Edwardsiella tarda (Romano et al, 2015). Most studies focus on the role of TMA derivative TMAO, as it was shown to play role in atherosclerosis (Koeth et al, 2013). Additionally, dysbiotic microbiota featuring enhanced conversion of choline to methylamines can potentially lead to deficiency of choline and contribute to NASH. Indeed, it was shown that NAFLD is associated with lower levels of phosphatidylcholine and higher levels of TMA in the blood, implicating the role of microbiota in the imbalance of choline metabolic flux (Dumas et al, 2006).
Bacteria can also utilize choline to synthesize phosphatidylcholine via the phosphatidylcholine synthase (Pcs) pathway. In eukaryotes, phosphatidylcholine is a membrane component, but it is estimated to be present in the membranes of 10–15% of bacterial species including L. pneumophila and P. aeruginosa (Sohlenkamp et al, 2003; Geiger et al, 2013). In the context of NAFLD‐associated small intestinal bacterial bloom and overgrowth, bacterial demand for choline, aimed at synthesizing phosphatidyl choline for growth and division, may increase and contribute to choline deficiency in the host and resultant NAFLD and NASH.
Microbial fermentation to alcohol
Microbiota harbors genes that can ferment dietary sugars into ethanol that can then enter the bloodstream. The amount of alcohol produced depends on the availability of carbohydrates from the diet and is increased in obese mice (Cope et al, 2000). In children with NASH as well as in adults with NAFLD, there is significantly more bacteria associated with increased alcohol levels in the blood in comparison with obese children without NASH (Volynets et al, 2012; Zhu et al, 2013; Michail et al, 2015).
Alcohol exacerbates oxidative stress and inflammation in the liver. Moreover, in the liver alcohol dehydrogenase metabolizes ethanol to toxic acetaldehyde, which due to its electrophilic nature forms adducts with proteins and other molecules in cells leading to loss of structure and function. Acetaldehyde is converted to acetate by CYP2E1, but this pathway can be saturated leading to accumulation of this toxic intermediate. Expression of CYP2E1 is up‐regulated in the presence of ethanol. CYP2E1 does metabolize not only acetaldehyde, but also other molecules, and its function is linked to the level of oxidative stress in the liver, which is a potent pro‐fibrotic signal. Interestingly, patients suffering from NASH exhibit increased CYP2E1 levels (Zong et al, 2012).
Phenylacetate
It was recently suggested that the microbially derived metabolite of phenylalanine, phenylacetate, is up‐regulated in blood of females with NASH (Hoyles et al, 2018). This molecule's blood levels have higher predictive power of the disease than conventional diagnostics. Microbiota of patients with NASH is enriched with genes for phenylalanine metabolism to phenylacetate, potentially suggesting an involvement in generation of this NASH‐associated molecule. Interestingly, mice chronically treated with phenylacetate feature elevated expression of genes related to fat metabolism (Lpl, Fasn), increased levels of hepatic triglycerides, and eventually developed liver steatosis (Hoyles et al, 2018). In NAFLD and NASH patients in addition to phenylalanine, phenylacetate precursor, abundances of other amino acids, notably branched chain amino acids (BCAAs), are altered and associate with disease severity, suggesting that there may be other metabolites produced by microbiome that play role in the disease (Gaggini et al, 2018).
Microbiome‐induced induction of liver inflammation
Gut barrier disruption leads to the translocation of overgrowing bacteria and their products to mucosa and circulation, and hence initiates or enhances liver inflammation (Fig 2). There is already an increased bacterial translocation from the gut toward blood and remote tissues at the early onset of HFD‐induced type 2 diabetes, which leads to a continuous metabolic bacteremia (Amar et al, 2011). In this context, the most extensively studied microbial molecule is LPS, a cell wall component of gram‐negative bacteria, also known as endotoxin (Soares et al, 2010). Systemic LPS concentration is significantly elevated in NAFLD in both human and animal studies (Yang et al, 1997; Bergheim et al, 2008; Harte et al, 2010; Sharifnia et al, 2015). High‐fat diet in mouse models induces LPS elevation to a level, which sufficiently initiates obesity and insulin resistance, and similar metabolic responses are triggered in normal diet‐fed mice with chronic infusion of LPS (Cani et al, 2007). The effect of LPS on the development of NASH has also been shown in genetically obese fatty/fatty rats and obese/obese mice, which are more susceptible to LPS‐mediated liver injury (Yang et al, 1997).
Figure 2. Pattern diagram featuring gut permeability, bacterial translocation, and TLR signaling in NAFLD/NASH .
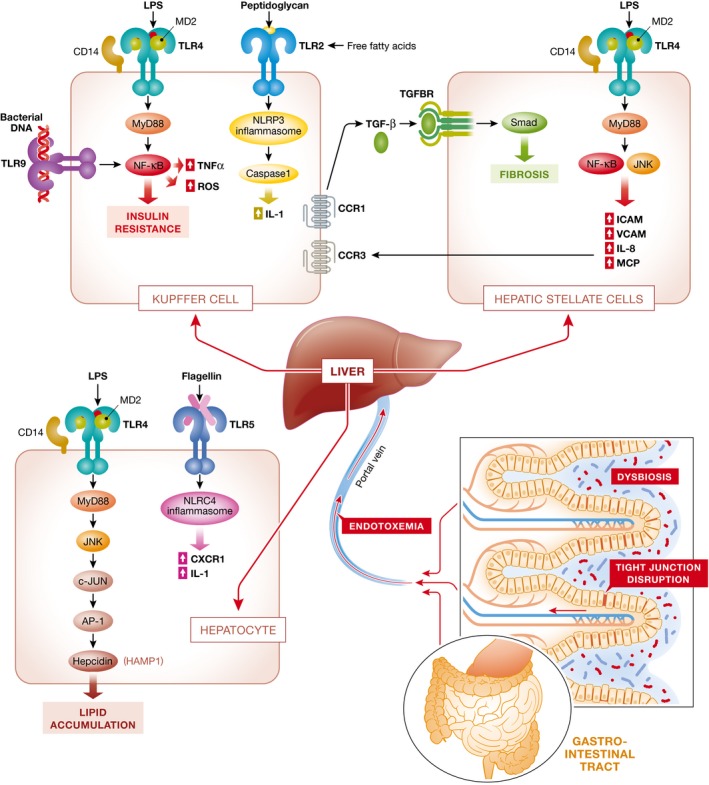
Gut microbiota alteration might contribute to increased gut permeability, translocation of bacteria, and microbiota products from the gut to liver through the portal vein. Different TLRs (TLR4, TLR9, TLR2, TLR5) in different cell types of the liver (Kupffer cells, hepatic stellate cells, hepatocytes) sense bacterial products and trigger downstream inflammatory responses as well as cytokine production. Different cell types, such as Kupffer cells and hepatic stellate cells, might interact with each other in inducing inflammation and fibrosis.
On the molecular level, LPS, a pathogen‐associated molecular pattern (PAMP), is recognized by the specific pattern recognition receptor, called Toll‐like receptor 4 (TLR4) and its co‐receptors, LPS binding protein (LBP) and CD14. Activation of TLR4 triggers downstream inflammatory cascade, involving, depending on the context, NF‐ĸB, AP‐1, and IRF3 activation (Zhai et al, 2004; Abu‐Shanab & Quigley, 2010). TLR4‐deficient mice display decreased liver injury, inflammation, and lipid accumulation in comparison with wild‐type mice in NAFLD models induced by high fructose or MCD diet (Rivera et al, 2007; Spruss et al, 2009), which confirms the role of TLR4 in inducing inflammation in NAFLD. Consistently, CD14 mutant mice are resistant to LPS‐initiated metabolic features including obesity, diabetes, and liver fat accumulation and inflammation, indicating that metabolic endotoxemia triggers NAFLD in a CD14‐dependent approach (Cani et al, 2007).
TLR4 is expressed by the immune and parenchymal cells of the liver; hence, the effect of LPS on the liver is compounded, and disentangling it requires understanding of all the components and interactions between them. Depletion of Kupffer cells with clodronate liposomes in MCD diet‐fed mice decreases the overall TLR4 expression in the liver and blunts the histological evidence of NASH (Rivera et al, 2007). Kupffer cells activated by LPS produce pro‐inflammatory cytokines IL‐18, IL‐1β, and IL‐12, leading to induction of NK cells and cytotoxic T cells (Takahashi et al, 1996; Seki et al, 2001). In fructose‐induced NAFLD in mice, the activation of Kupffer cells through TLR4‐dependent mechanism is mediated via a MyD88‐dependent signaling pathway, leading to induction of hepatic TNF‐α mRNA, formation of reactive oxygen species (ROS), and insulin resistance (Rivera et al, 2007; Spruss et al, 2009). TLR4 expressed by hepatocytes responds to LPS and regulates the expression of hepcidin, a key iron‐regulatory protein implicated in obesity and NAFLD, via the MyD88‐IRAK‐TRAF6‐JNK‐AP‐1 axis (Lu et al, 2016; Lee et al, 2017). In liver sinusoidal endothelial cells (LSEC), TLR4 activation leads to induced production of TNF‐α and ROS, but in vivo they are mostly exhibiting tolerance to LPS and its effect is probably minimal (Sarphie et al, 1996; Uhrig et al, 2005). TLR4 signaling also plays an important role in activation of fibrogenic phenotype in hepatic stellate cells (HSCs). Activated HSCs produce various chemokines and adhesion molecules, which in turn induce chemotaxis of liver macrophages—Kupffer cells. Recruited Kupffer cells enhance TGF‐β production, which binds to TGF‐β receptor on HSCs, further activating fibrogenesis (Paik et al, 2003; Seki et al, 2007).
In addition to TLR4, other TLRs are also found to play role in the development of NASH, including TLR9, TLR5, and TLR2. The role of TLR2, a receptor for peptidoglycan, is contradictory in current studies. In some studies, TLR2 knockout enhances NASH in murine NASH models induced by methionine–choline‐deficient diet, accompanied by elevated expression of inflammatory and fibrosis markers, suggesting a protective role of TLR2‐mediated signals (Szabo et al, 2006; Rivera et al, 2010). Inversely, TLR2‐deficient mice fed with choline‐deficient amino acid (CDAA) defined diet‐suppressed progression of NASH (Miura et al, 2013), which is consistent with findings in high‐fat‐induced metabolic syndrome in murine models (Ehses et al, 2010; Himes & Smith, 2010). Indeed, TLR2 signaling and palmitic acid signaling in Kupffer cells and macrophages are both required to activate NLRP3 inflammasome and secrete IL‐1 that results in progression to NASH (Miura et al, 2013). The contradictory role of TLR2 might be due to different NASH models used in different studies. It is proposed that CDAA diet model is more relevant to human NASH, which is characterized by obesity, insulin resistance, and liver fibrosis (Miura et al, 2013). Flagellin receptor (TLR5)‐deficient mice develop metabolic syndrome including hepatic steatosis (Vijay‐Kumar et al, 2010). Furthermore, hepatocytes lacking TLR5 show impairment in elimination of flagellated bacteria and are more susceptible to the liver injury induced by MCD diet or high‐fat diet (Etienne‐Mesmin et al, 2016). In a murine model of NASH, TLR9 bacterial DNA sensing and downstream signaling up‐regulates IL‐1β production by Kupffer cells, induces chemotaxis of macrophages and neutrophils, and leads to steatosis, inflammation, and fibrosis (Miura et al, 2010; Mridha et al, 2017). In mice, administration of a TLR9 antagonist protected from NASH, but it was also suggested that it was host mitochondrial DNA rather than bacterial DNA that activated this receptor in disease (Garcia‐Martinez et al, 2016). The roles of other TLRs in regulating NASH remain largely unknown and needs to be further investigated.
Signals from TLRs converge at the inflammasome that orchestrates effector functions, including IL‐1β and IL‐18 secretion. So far, NLRP3 and NLRP6 were implicated to have a role in microbiome induction of NASH (Henao‐Mejia et al, 2012). Kupffer cells activated by palmitic acid, in a NLRP3 inflammasome‐dependent manner, secrete pro‐inflammatory IL‐1β and IL‐18, thus contributing to NASH development (Cai et al, 2017). Another study showed that besides Kupffer cells, inflammasome components are also present in HSCs and are required for the development of liver fibrosis (Watanabe et al, 2009).
Causal evidence to microbiome involvement in the pathogenesis of NAFLD and NASH
Although only a minority of cause‐and‐effect studies linking the microbiome to NAFLD and NASH pathogenesis have been conducted to date, evidence regarding the causal role of gut microbiota in NAFLD is increasing. In contrast to conventionally raised mice, germ‐free mice fed with hypercaloric diet exhibit an impaired weight gain and lipid metabolism as well as lack of liver steatosis (Kaden‐Volynets et al, 2018; Martinez‐Guryn et al, 2018). However, conventionalization of germ‐free mice with normal mouse microbiota induces de novo hepatic lipogenesis, in addition to increased body fat content and insulin resistance (Bäckhed et al, 2004). Moreover, germ‐free mice colonized with high‐fat diet‐induced jejunal microbiota showed increased lipid absorption even on low‐fat diet (Martinez‐Guryn et al, 2018), suggesting the crucial role of intestinal microbiota in triggering metabolic response. Inflammasome‐deficient mice aggravate NAFLD/NASH progression via modulation of gut microbiota, and more interesting, the increased severity of NASH in these mice was transmissible through animal cohousing leading to microbiome transfer between co‐housed mice, highlighting that modulated microbiota contribute to the pathogenesis of NAFLD (Henao‐Mejia et al, 2012). The decisive role of microbiota in NAFLD development is further confirmed by a study where differences in microbiota composition between the NAFLD‐resistant and NAFLD‐susceptible phenotypes were identified (Le Roy et al, 2013). In this study, only germ‐free mice receiving NAFLD‐prone microbiota developed hyperglycemia, hyperinsulinemia, and hepatic steatosis, indicating that the tendency to develop NAFLD is transmissible through gut microbiota transplantation. Further exciting evidence comes from recent studies using human donors. Gut microbiota from obese humans induced the onset of hepatic steatosis through modulation of lipid metabolism transcriptional profiles in germ‐free mice. Furthermore, mice receiving microbiota from the same donor but after weight loss exhibit normal liver physiology (Wang et al, 2018). In another study, high‐fat diet‐fed germ‐free mice inoculated with microbiota of NASH patients, rather than healthy donors, showed an exacerbated NASH phenotype, as manifested by increased liver steatosis and inflammation (Chiu et al, 2017). Based on these promising animal models, future studies are needed to establish a definite cause–effect link between dysbiosis and human NAFLD.
The microbiome as a potential therapeutic target in NAFLD and NASH
The targeting of gut microbiota as a noninvasive diagnostic tool that correlates with the severity of NAFLD/NASH holds promise as a future therapeutic modality in these currently cureless disorders. Initial studies suggest that the progression of NAFLD and NASH may be monitored by blood serum metabolites such as phenylacetate (Hoyles et al, 2018) and microbiome composition (Loomba et al, 2017). For more specific and sensitive diagnostic tools, multiple measurements can be integrated. Indeed, in a recent study by Hoyles et al (2018) data from metagenomics, liver transcriptomics, and metabolomics are integrated to shed light on the importance of microbial biomarkers as a potential diagnostic tool for metabolic and fibrotic liver diseases, which can be used in conjunction with current invasive approaches to determine stages of NAFLD. Among patients with NAFLD, the early diagnosis of hepatic fibrosis is an important but challenging clinical question. In a recent study with both discovery and validation cohorts, blood microbiota alterations are associated with liver fibrosis in obese patients, featuring increased 16S rRNA concentration and decreased bacterial diversity in the blood, pointing out that blood dysbiosis might be used as a potential noninvasive biomarker (Lelouvier et al, 2016). Certainly, more future studies will pave the way to microbiota diagnostics as reliable predictive markers of disease onset and progression.
A number of studies investigated the feasibility of using the intestinal microbiota as a potential therapeutic target for NAFLD, including modulation by antibiotic treatment, prebiotics, probiotics, and synbiotics. These treatment strategies have been systematically reviewed elsewhere (Ma et al, 2017). Such therapies are expected to attenuate the disease by altering the contribution of gut microbiota to its pathogenesis. Although many animal studies presented exciting results, reproducible human clinical trial results are in great need.
The therapeutic effect of fecal microbiota transplantation (FMT) in NASH has been suggested by a few animal studies. Zhou et al (2017a) reported that FMT attenuated high‐fat diet‐induced NASH in mice via beneficial regulation of gut microbiota. García‐Lezana et al (2018) showed that FMT restored a healthy intestinal microbiota and normalized portal hypertension in a rat model of NASH. The effect of FMT on NAFLD/NASH is just beginning to be investigated and requires more animal and human clinical studies. Notably, a few clinical trials assessing treatment of NASH patients with FMT are currently underway (NCT02469272, NTR4339).
Other potential therapeutic targets, such as anti‐LPS immunoglobulin and microbiota‐associated metabolite treatment, have been suggested by several studies but merit independent validation and mechanistic evaluation. For example, oral administration of IMM‐124E, an anti‐LPS‐enriched bovine colostrum, was suggested to alleviate chronic inflammation, liver damage, and insulin resistance associated with NASH in mice models (Adar et al, 2012) and a small cohort of patients with biopsy‐proven NASH (Mizrahi et al, 2012). Pharmaceuticals targeting bile acid dysregulation in NAFLD, including FXR agonists, PPARα agonists, ursodeoxycholic acid (UDCA), and its derivatives, have entered different phases of clinical trials, and some of them have shown promising therapeutic effects (Yu et al, 2018). The administration of butyrate, a SCFA, effectively ameliorates lipid accumulation and liver inflammation in animal NAFLD models, through modulation of gut microbiota and gut barrier function (Zhou et al, 2017b), attenuation of inducible nitric oxide synthase (iNOS) induction (Jin et al, 2016), and suppression of inflammatory pathways (Sun et al, 2018).
Perspectives and future directions
Given the high prevalence and increasing incidence of NAFLD worldwide and its close association with gut microbiota, research focusing on microbiome provides great opportunities as well as challenges in both the pathogenesis and treatment options of NAFLD/NASH. Since most previous studies adopted 16S rRNA sequencing which provided low resolution limited to genus level, it is imperative to identify NAFLD‐related microbes at strain level using highly accurate metagenomics sequencing, which also offers functional information of intestinal microbiome. Beyond association studies, future research work will aim to elucidate the direct causative relationship between gut dysbiosis and NAFLD in both animal models and human studies, aiming at microbiota‐targeted therapeutics. Despite great advances in correlating microbiota changes with NAFLD, specific molecular mechanism underlying the host–environment–microbiome interplay involved in the development and progression of NAFLD/NASH remains largely elusive. Therefore, a deeper and further understanding of gut microbiota‐associated metabolic profile in NAFLD/NASH subjects should be discovered. This will uncover novel treatment strategies to inhibit bacterial‐derived pathways that in turn lead to disease progression.
The heterogeneous clinical features of NAFLD can be explained by incorporation of dietary factors, genetics, and interpersonal gut microbiome variability. Precision medicine as a novel clinical approach is imperative in developing therapeutic approaches in such complex diseases. In fact, recent study has shown that individual glycemic response can be accurately predicted by combining personal, dietary, and microbiome features, successfully targeting personalized nutrition (Zeevi et al, 2015). Therefore, a precision medicine approach based on host, diet, and microbiome profiles could facilitate risk stratification and predict the variable clinical phenotypes, diagnostic accuracy, and therapeutic response of NAFLD. The implication of gut microbiome‐based precision medicine, including personalized probiotics (Suez et al, 2018; Zmora et al, 2018) and postbiotic interventions (Thaiss et al, 2016), might also be an important consideration for NAFLD treatment.
Altogether, opportunities and challenges provided by microbiome research open a new window for future studies, which will hopefully elucidate the more specific role of gut microbiome in NAFLD and establish microbiota‐targeted personalized treatment approaches.
Pending issues.
-
(i)
Identification of alterations in microbiota composition and microbial function in NAFLD/NASH patients.
-
(ii)
Elucidation of specific gut microbiota‐associated molecular mechanism underlying the pathogenesis of NAFLD/NASH.
-
(iii)
Proving the directly causal role of gut microbiota alteration in NAFLD/NASH development.
-
(iv)
Establishment of the gut microbiota‐targeted precision medicine in the treatment of NAFLD/NASH.
Conflict of interest
Eran Elinav is a payed consultant to BiomX and DayTwo.
Acknowledgements
We thank the Elinav lab for fruitful discussions and apologize to those authors whose works could not be cited due to space limitations. AAK received funding from EMBO Long Term Fellowship 2016‐1088 and the European Union's Horizon 2020 research and innovation programme under the Marie Sk?odowska‐Curie Grant Agreement No. 747114. DPZ is the recipient of the European Crohn's and Colitis Organization (ECCO) Fellowship, and is supported by the Ke Lin Program of the First Affiliated Hospital, Sun Yat‐sen University. O.S is supported by a research grant from the Alexander Grass foundation. E.E. is a senior fellow, Canadian Institute of Advanced Research (CIFAR) and an international scholar, The Bill & Melinda Gates Foundation & Howard Hughes Medical Institute (HHMI).
EMBO Mol Med (2019) 11: e9302
See the Glossary for abbreviations used in this article.
References
- Abu‐Shanab A, Quigley EMM (2010) The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7: 691–701 [DOI] [PubMed] [Google Scholar]
- Ackermann D, Schutze H (1910) The formation of trimethylamine by bacterium prodigiosum. Zentralb Physiol 24: 210–211 [Google Scholar]
- Adar T, Ben Ya'acov A, Lalazar G, Lichtenstein Y, Nahman D, Mizrahi M, Wong V, Muller B, Rawlin G, Ilan Y (2012) Oral administration of immunoglobulin G‐enhanced colostrum alleviates insulin resistance and liver injury and is associated with alterations in natural killer T cells. Clin Exp Immunol 167: 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Waiz M, Mikov M, Mitchell SC, Smith RL (1992) The exogenous origin of trimethylamine in the mouse. Metabolism 41: 135–136 [DOI] [PubMed] [Google Scholar]
- Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez‐Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S et al (2011) Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3: 559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M (2017) Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 65: 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranha MM, Cortez‐Pinto H, Costa A, da Silva IBM, Camilo ME, de Moura MC, Rodrigues CMP (2008) Bile acid levels are increased in the liver of patients with steatohepatitis. Eur J Gastroenterol Hepatol 20: 519–525 [DOI] [PubMed] [Google Scholar]
- Arendt BM, Ma DWL, Simons B, Noureldin SA, Therapondos G, Guindi M, Sherman M, Allard JP (2013) Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl Physiol Nutr Metab 38: 334–340 [DOI] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, McClain CJ, Bischoff SC (2008) Antibiotics protect against fructose‐induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol 48: 983–992 [DOI] [PubMed] [Google Scholar]
- Blumberg H, McCollum EV (1941) The prevention by choline of liver cirrhosis in rats on high fat, low protein diets. Science 93: 598–599 [DOI] [PubMed] [Google Scholar]
- Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo‐Perez F, Guy CD, Seed PC, Rawls JF, David LA et al (2016) The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63: 764–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ et al (2003) The orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278: 11312–11319 [DOI] [PubMed] [Google Scholar]
- Cai C, Zhu X, Li P, Li J, Gong J, Shen W, He K (2017) NLRP3 deletion inhibits the non‐alcoholic steatohepatitis development and inflammation in Kupffer cells induced by palmitic acid. Inflammation 40: 1875–1883 [DOI] [PubMed] [Google Scholar]
- Calzadilla Bertot L, Adams LA (2016) The natural course of non‐alcoholic fatty liver disease. Int J Mol Sci 17: E774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772 [DOI] [PubMed] [Google Scholar]
- Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ (2018) The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67: 328–357 [DOI] [PubMed] [Google Scholar]
- Chavez‐Tapia NC, Tellez‐Avila FI, Barrientos‐Gutierrez T, Mendez‐Sanchez N, Lizardi‐Cervera J, Uribe M (2010) Bariatric surgery for non‐alcoholic steatohepatitis in obese patients. Cochrane Database Syst Rev 20: CD007340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C‐C, Ching Y‐H, Li Y‐P, Liu J‐Y, Huang Y‐T, Huang Y‐W, Yang S‐S, Huang W‐C, Chuang H‐L (2017) Nonalcoholic fatty liver disease is exacerbated in high‐fat diet‐fed gnotobiotic mice by colonization with the gut microbiota from patients with nonalcoholic steatohepatitis. Nutrients 9: E1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope K, Risby T, Diehl AM (2000) Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology 119: 1340–1347 [DOI] [PubMed] [Google Scholar]
- Cortez‐Pinto H, Jesus L, Barros H, Lopes C, Moura MC, Camilo ME (2006) How different is the dietary pattern in non‐alcoholic steatohepatitis patients? Clin Nutr 25: 816–823 [DOI] [PubMed] [Google Scholar]
- Craciun S, Balskus EP (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci USA 109: 21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas M‐E, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC et al (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin‐resistant mice. Proc Natl Acad Sci USA 103: 12511–12516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, Schraenen A, Lemaire K, Debray S, Van Lommel L et al (2010) Toll‐like receptor 2‐deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high‐fat diet. Diabetologia 53: 1795–1806 [DOI] [PubMed] [Google Scholar]
- Etienne‐Mesmin L, Vijay‐Kumar M, Gewirtz AT, Chassaing B (2016) Hepatocyte toll‐like receptor 5 promotes bacterial clearance and protects mice against high‐fat diet‐induced liver disease. Cell Mol Gastroenterol Hepatol 2: 584–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano A (2008) Physiological, pathological, and therapeutic implications of zonulin‐mediated intestinal barrier modulation: living life on the edge of the wall. Am J Pathol 173: 1243–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, Brouwer KLR, Barritt AS IV (2015) Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci 60: 3318–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C, Obermeier F (2011) DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol 55: 1391–1399 [DOI] [PubMed] [Google Scholar]
- Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M et al (2018) Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology 67: 145–158 [DOI] [PubMed] [Google Scholar]
- Gangarapu V, Ince AT, Baysal B, Kayar Y, Kılıç U, Gök Ö, Uysal Ö, Şenturk H (2015) Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 27: 840–845 [DOI] [PubMed] [Google Scholar]
- García‐Lezana T, Raurell I, Bravo M, Torres‐Arauz M, Salcedo MT, Santiago A, Schoenenberger A, Manichanh C, Genescà J, Martell M et al (2018) Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 67: 1485–1498 [DOI] [PubMed] [Google Scholar]
- Garcia‐Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ (2016) Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 126: 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger O, López‐Lara IM, Sohlenkamp C (2013) Phosphatidylcholine biosynthesis and function in bacteria. Biochim Biophys Acta 1831: 503–513 [DOI] [PubMed] [Google Scholar]
- Halnes I, Baines KJ, Berthon BS, MacDonald‐Wicks LK, Gibson PG, Wood LG (2017) Soluble fibre meal challenge reduces airway inflammation and expression of GPR43 and GPR41 in asthma. Nutrients 9: E57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Haga S, Aoyama Y, Kiriyama S (1999) Short‐chain fatty acids suppress cholesterol synthesis in rat liver and intestine. J Nutr 129: 942–948 [DOI] [PubMed] [Google Scholar]
- Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef‐Elabd EM, Tripathi G, Ashour E, Abdalla MS, Sharada HM et al (2010) Elevated endotoxin levels in non‐alcoholic fatty liver disease. J Inflamm 7: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao‐Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ et al (2012) Inflammasome‐mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himes RW, Smith CW (2010) Tlr2 is critical for diet‐induced metabolic syndrome in a murine model. FASEB J 24: 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høverstad T, Midtvedt T (1986) Short‐chain fatty acids in germfree mice and rats. J Nutr 116: 1772–1776 [DOI] [PubMed] [Google Scholar]
- Hoyles L, Fernández‐Real J‐M, Federici M, Serino M, Abbott J, Charpentier J, Heymes C, Luque JL, Anthony E, Barton RH et al (2018) Molecular phenomics and metagenomics of hepatic steatosis in non‐diabetic obese women. Nat Med 24: 1070–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin CJ, Engstler AJ, Sellmann C, Ziegenhardt D, Landmann M, Kanuri G, Lounis H, Schröder M, Vetter W, Bergheim I (2016) Sodium butyrate protects mice from the development of the early signs of non‐alcoholic fatty liver disease: role of melatonin and lipid peroxidation. Br J Nutr 116: 1682–1693 [DOI] [PubMed] [Google Scholar]
- Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, Krakoff J (2011) Energy‐balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr 94: 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden‐Volynets V, Basic M, Neumann U, Pretz D, Rings A, Bleich A, Bischoff SC (2018) Lack of liver steatosis in germ‐free mice following hypercaloric diets. Eur J Nutr 10.1007/s00394-018-1748-4 [DOI] [PubMed] [Google Scholar]
- Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I (2015) Dietary gut microbial metabolites, short‐chain fatty acids, and host metabolic regulation. Nutrients 7: 2839–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI (2011) Human nutrition, the gut microbiome and the immune system. Nature 474: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T et al (2013) The gut microbiota suppresses insulin‐mediated fat accumulation via the short‐chain fatty acid receptor GPR43. Nat Commun 4: 1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L et al (2013) Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19: 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A et al (2013) Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut 62: 1787–1794 [DOI] [PubMed] [Google Scholar]
- Lee Y‐S, Kim Y‐H, Jung YS, Kim K‐S, Kim D‐K, Na S‐Y, Lee J‐M, Lee C‐H, Choi H‐S (2017) Hepatocyte toll‐like receptor 4 mediates lipopolysaccharide‐induced hepcidin expression. Exp Mol Med 49: e408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelouvier B, Servant F, Païssé S, Brunet A‐C, Benyahya S, Serino M, Valle C, Ortiz MR, Puig J, Courtney M et al (2016) Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology 64: 2015–2027 [DOI] [PubMed] [Google Scholar]
- Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E (2017) Dysbiosis and the immune system. Nat Rev Immunol 17: 219–232 [DOI] [PubMed] [Google Scholar]
- Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK et al (2017) Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab 25: 1054–1062.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Bennett RG, Kharbanda KK, Harrison‐Findik DD (2016) Lack of hepcidin expression attenuates steatosis and causes fibrosis in the liver. World J Hepatol 8: 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, Motola DL, Luther S, Bohr S, Jeoung SW et al (2015) Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol 1: 222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Zhou Q, Li H (2017) Gut microbiota and nonalcoholic fatty liver disease: insights on mechanisms and therapy. Nutrients 9: E1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, Ian McKenzie C, Hijikata A, Wong C et al (2015) Metabolite‐sensing receptors GPR43 and GPR109A facilitate dietary fibre‐induced gut homeostasis through regulation of the inflammasome. Nat Commun 6: 6734 [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Heikenwalder M, Ganal‐Vonarburg SC (2016) The liver at the nexus of host‐microbial interactions. Cell Host Microbe 20: 561–571 [DOI] [PubMed] [Google Scholar]
- Madrid AM, Hurtado C, Venegas M, Cumsille F, Defilippi C (2001) Long‐term treatment with cisapride and antibiotics in liver cirrhosis: effect on small intestinal motility, bacterial overgrowth, and liver function. Am J Gastroenterol 96: 1251–1255 [DOI] [PubMed] [Google Scholar]
- Mahana D, Trent CM, Kurtz ZD, Bokulich NA, Battaglia T, Chung J, Müller CL, Li H, Bonneau RA, Blaser MJ (2016) Antibiotic perturbation of the murine gut microbiome enhances the adiposity, insulin resistance, and liver disease associated with high‐fat diet. Genome Med 8: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP (2015) Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio 6: e00042‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM et al (2018) Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe 23: 458–469.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D et al (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, Reo NV (2015) Altered gut microbial energy and metabolism in children with non‐alcoholic fatty liver disease. FEMS Microbiol Ecol 91: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G et al (2009) Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49: 1877–1887 [DOI] [PubMed] [Google Scholar]
- Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E (2010) Toll‐like receptor 9 promotes steatohepatitis by induction of interleukin‐1beta in mice. Gastroenterology 139: 323–334.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E (2013) Toll‐like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 57: 577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi M, Shabat Y, Ben Ya'acov A, Lalazar G, Adar T, Wong V, Muller B, Rawlin G, Ilan Y (2012) Alleviation of insulin resistance and liver damage by oral administration of Imm124‐E is mediated by increased Tregs and associated with increased serum GLP‐1 and adiponectin: results of a phase I/II clinical trial in NASH. J Inflamm Res 5: 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mridha AR, Haczeyni F, Yeh MM, Haigh WG (2017) TLR9 is upregulated in human and murine NASH: pivotal role for inflammatory recruitment and cell survival. Clin Sci 131: 2145–2159 [DOI] [PubMed] [Google Scholar]
- Neis EPJG, Dejong CHC, Rensen SS (2015) The role of microbial amino acid metabolism in host metabolism. Nutrients 7: 2930–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik Y‐H, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA (2003) Toll‐like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 37: 1043–1055 [DOI] [PubMed] [Google Scholar]
- Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, Ratziu V, LIDO Study Group (2013) A systematic review of follow‐up biopsies reveals disease progression in patients with non‐alcoholic fatty liver. J Hepatol 59: 550–556 [DOI] [PubMed] [Google Scholar]
- Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153 [DOI] [PubMed] [Google Scholar]
- Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan L‐X, Rey F, Wang T et al (2013) Olfactory receptor responding to gut microbiota‐derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA 110: 4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols TWH, Noriega LG, Nomura M, Auwerx J, Schoonjans K (2011) The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol 54: 1263–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabot S, Membrez M, Bruneau A, Gérard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ (2010) Germ‐free C57BL/6J mice are resistant to high‐fat‐diet‐induced insulin resistance and have altered cholesterol metabolism. FASEB J 24: 4948–4959 [DOI] [PubMed] [Google Scholar]
- Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, Smith T, Neish AS, Li H, Tan S et al (2016) Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 151: 733–746.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR et al (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang D‐J, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47: 241–259 [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Alves JM, Hylemon PB, Bajaj JS (2013) Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 4: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M (2007) Toll‐like receptor‐4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non‐alcoholic steatohepatitis. J Hepatol 47: 571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, Pruitt K (2010) Toll‐like receptor‐2 deficiency enhances non‐alcoholic steatohepatitis. BMC Gastroenterol 10: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha R, Cotrim HP, Siqueira AC, Floriano S (2007) Non alcoholic fatty liver disease: treatment with soluble fibres. Arq Gastroenterol 44: 350–352 [DOI] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador‐Noguez D, Rey FE (2015) Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine‐N‐oxide. MBio 6: e02481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M et al (2008) Effects of the gut microbiota on host adiposity are modulated by the short‐chain fatty‐acid binding G protein‐coupled receptor, Gpr41. Proc Natl Acad Sci USA 105: 16767–16772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarphie TG, D'Souza NB, Deaciuc IV (1996) Kupffer cell inactivation prevents lipopolysaccharide‐induced structural changes in the rat liver sinusoid: an electron‐microscopic study. Hepatology 23: 788–796 [DOI] [PubMed] [Google Scholar]
- Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, Hardt PD (2010) Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18: 190–195 [DOI] [PubMed] [Google Scholar]
- Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O et al (2001) Lipopolysaccharide‐induced IL‐18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL‐12 and IL‐1beta. J Immunol 166: 2651–2657 [DOI] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF (2007) TLR4 enhances TGF‐beta signaling and hepatic fibrosis. Nat Med 13: 1324–1332 [DOI] [PubMed] [Google Scholar]
- Sharifnia T, Antoun J, Verriere TGC, Suarez G, Wattacheril J, Wilson KT, Peek RM Jr, Abumrad NN, Flynn CR (2015) Hepatic TLR4 signaling in obese NAFLD. Am J Physiol Gastrointest Liver Physiol 309: G270–G278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK (2014) Specialized metabolites from the microbiome in health and disease. Cell Metab 20: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102: 731–744 [DOI] [PubMed] [Google Scholar]
- Soares J‐B, Pimentel‐Nunes P, Roncon‐Albuquerque R, Leite‐Moreira A (2010) The role of lipopolysaccharide/toll‐like receptor 4 signaling in chronic liver diseases. Hepatol Int 4: 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohlenkamp C, López‐Lara IM, Geiger O (2003) Biosynthesis of phosphatidylcholine in bacteria. Prog Lipid Res 42: 115–162 [DOI] [PubMed] [Google Scholar]
- Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA (2011) Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140: 976–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I (2009) Toll‐like receptor 4 is involved in the development of fructose‐induced hepatic steatosis in mice. Hepatology 50: 1094–1104 [DOI] [PubMed] [Google Scholar]
- Suez J, Zmora N, Zilberman‐Schapira G, Mor U, Dori‐Bachash M, Bashiardes S, Zur M, Regev‐Lehavi D, Ben‐Zeev Brik R, Federici S et al (2018) Post‐antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 174: 1406–1423.e16 [DOI] [PubMed] [Google Scholar]
- Sun B, Jia Y, Hong J, Sun Q, Gao S, Hu Y, Zhao N, Zhao R (2018) Sodium butyrate ameliorates high‐fat‐diet‐induced non‐alcoholic fatty liver disease through peroxisome proliferator‐activated receptor α‐mediated activation of β oxidation and suppression of inflammation. J Agric Food Chem 66: 7633–7642 [DOI] [PubMed] [Google Scholar]
- Szabo G, Dolganiuc A, Mandrekar P (2006) Pattern recognition receptors: a contemporary view on liver diseases. Hepatology 44: 287–298 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ogasawara K, Takeda K, Hashimoto W, Sakihara H, Kumagai K, Anzai R, Satoh M, Seki S (1996) LPS induces NK1.1+ alpha beta T cells with potent cytotoxicity in the liver of mice via production of IL‐12 from Kupffer cells. J Immunol 156: 2436–2442 [PubMed] [Google Scholar]
- Thaiss CA, Itav S, Rothschild D, Meijer M, Levy M, Moresi C, Dohnalová L, Braverman S, Rozin S, Malitsky S et al (2016) Persistent microbiome alterations modulate the rate of post‐dieting weight regain. Nature 540: 544–551 [DOI] [PubMed] [Google Scholar]
- Tilg H (2017) How to approach a patient with nonalcoholic fatty liver disease. Gastroenterology 153: 345–349 [DOI] [PubMed] [Google Scholar]
- Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9: 799–809 [DOI] [PubMed] [Google Scholar]
- Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, Gerken G, Limmer A, Knolle PA (2005) Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol 77: 626–633 [DOI] [PubMed] [Google Scholar]
- Vijay‐Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll‐like receptor 5. Science 328: 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volynets V, Küper MA, Strahl S, Maier IB, Spruss A, Wagnerberger S, Königsrainer A, Bischoff SC, Bergheim I (2012) Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci 57: 1932–1941 [DOI] [PubMed] [Google Scholar]
- Wang R, Li H, Yang X, Xue X, Deng L, Shen J, Zhang M, Zhao L, Zhang C (2018) Genetically obese human gut microbiota induces liver steatosis in germ‐free mice fed on normal diet. Front Microbiol 9: 1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA et al (2009) Inflammasome‐mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 296: G1248–G1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A (2015) Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148: 547–555 [DOI] [PubMed] [Google Scholar]
- Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM (1997) Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 94: 2557–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 64: 73–84 [DOI] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15: 11–20 [DOI] [PubMed] [Google Scholar]
- Yu Q, Jiang Z, Zhang L (2018) Bile acid regulation: a novel therapeutic strategy in non‐alcoholic fatty liver disease. Pharmacol Ther 190: 81–90 [DOI] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben‐Yacov O, Lador D, Avnit‐Sagi T, Lotan‐Pompan M et al (2015) Personalized nutrition by prediction of glycemic responses. Cell 163: 1079–1094 [DOI] [PubMed] [Google Scholar]
- Zeisel SH, Wishnok JS, Blusztajn JK (1983) Formation of methylamines from ingested choline and lecithin. J Pharmacol Exp Ther 225: 320–324 [PubMed] [Google Scholar]
- Zeng H, Liu J, Jackson MI, Zhao F‐Q, Yan L, Combs GF Jr (2013) Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high‐fat diet–3. J Nutr 143: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Shen X, O'Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec‐Weglinski JW (2004) Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3‐dependent MyD88‐independent pathway. J Immunol 173: 7115–7119 [DOI] [PubMed] [Google Scholar]
- Zhang AQ, Mitchell SC, Smith RL (1999) Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol 37: 515–520 [DOI] [PubMed] [Google Scholar]
- Zhou D, Pan Q, Shen F, Cao H‐X, Ding W‐J, Chen Y‐W, Fan J‐G (2017a) Total fecal microbiota transplantation alleviates high‐fat diet‐induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep 7: 1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Pan Q, Xin F‐Z, Zhang R‐N, He C‐X, Chen G‐Y, Liu C, Chen Y‐W, Fan J‐G (2017b) Sodium butyrate attenuates high‐fat diet‐induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J Gastroenterol 23: 60–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57: 601–609 [DOI] [PubMed] [Google Scholar]
- Zmora N, Zilberman‐Schapira G, Suez J, Mor U, Dori‐Bachash M, Bashiardes S, Kotler E, Zur M, Regev‐Lehavi D, Brik RB‐Z et al (2018) Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 174: 1388–1405.e21 [DOI] [PubMed] [Google Scholar]
- Zong H, Armoni M, Harel C, Karnieli E, Pessin JE (2012) Cytochrome P‐450 CYP2E1 knockout mice are protected against high‐fat diet‐induced obesity and insulin resistance. Am J Physiol Endocrinol Metab 302: E532–E539 [DOI] [PMC free article] [PubMed] [Google Scholar]