

SUMMARY
Expression quantitative trait locus (eQTL) analyses identify genetic markers associated with the expression of a gene. Most up-to-date eQTL studies consider the connection between genetic variation and expression in a single tissue. Multi-tissue analyses have the potential to improve findings in a single tissue, and elucidate the genotypic basis of differences between tissues. In this article, we develop a hierarchical Bayesian model (MT-eQTL) for multi-tissue eQTL analysis. MT-eQTL explicitly captures patterns of variation in the presence or absence of eQTL, as well as the heterogeneity of effect sizes across tissues. We devise an efficient Expectation–Maximization (EM) algorithm for model fitting. Inferences concerning eQTL detection and the configuration of eQTL across tissues are derived from the adaptive thresholding of local false discovery rates, and maximum a posteriori estimation, respectively. We also provide theoretical justification of the adaptive procedure. We investigate the MT-eQTL model through an extensive analysis of a 9-tissue data set from the GTEx initiative.
Keywords: GTEx, Hierarchical Bayesian model, Local false discovery rate, MT-eQTL, Tissue specificity
1. Introduction
Genetic variation in a population is commonly studied through the analysis of single nucleotide polymorphisms (SNPs), which are genetic variants occurring at specific sites in the genome. Expression quantitative trait locus (eQTL) analysis seeks to identify genetic variants affecting the expression of one or more genes: a gene–SNP pair for which the expression of the gene is associated with the value of the SNP is referred to as an eQTL. Identification of eQTL has proven to be a useful tool in the study of pathways and networks that underlie disease in human and other populations (cf. Kendziorski and Wang, 2006; Wright and others, 2012).
To date, most eQTL studies have considered the effects of genetic variation on expression within a single tissue. A natural next step in understanding the genomic variation of expression is the simultaneous analysis of eQTL in multiple tissues. Multi-tissue eQTL analysis has the potential to improve the findings of single tissue analyses by borrowing strength across tissues, and to address fundamental biological questions about the nature and source of variation between tissues. An important feature of multiple tissue studies is that a SNP may be associated with the expression of a gene in some tissues, but not in others. Thus a full multi-tissue analysis must identify complex patterns of association across multiple tissues.
Until recently, understanding of multi-tissue eQTL relationships was limited by a shortage of true multi-tissue data sets, requiring the assimilation of data or results from different studies involving distinct populations, measurement platforms, and analysis protocols. In contrast, the GTEx initiative (The GTEx Consortium, 2015) and related projects are currently generating genetic data from dozens of tissues in several hundred individuals, greatly expanding our potential understanding of eQTLs across multiple tissues. The size and complexity of these emerging multi-tissue data sets have created the need to expand existing statistical tools for eQTL analysis.
In this article, we introduce and study a hierarchical Bayesian model for the simultaneous
analysis of eQTL in multiple tissues. We particularly focus on cis-eQTL, where a SNP is
located near the transcription start site of a gene. We call the method
MT-eQTL (MT stands for multi-tissue). The dimension of
the MT-eQTL model is equal to the number of tissues. In this article, we primarily consider
a moderate dimension, typically between 1 and 10. Importantly, we do not seek to model the
full expression and genotype data, but focus instead on the vector 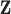 of
Fisher transformed correlations between expression and genotype across tissues. Figure 1b (upper panel) shows a density scatter plot of the
z-statistics for the lung and thyroid data from GTEx pilot data freeze as reported by The GTEx Consortium (2015). The lower panel illustrates
the results of the MT-eQTL model: z pairs close to the origin for which no eQTL are detected
have been removed, resulting in the central white region; detected eQTL are colored
according to whether an eQTL is detected in both tissues (light gray points) or a single
tissue (dark gray and black points). Our model explicitly captures patterns of variation in
the presence or absence of eQTL, as well as the heterogeneity of effect sizes across
tissues.
of
Fisher transformed correlations between expression and genotype across tissues. Figure 1b (upper panel) shows a density scatter plot of the
z-statistics for the lung and thyroid data from GTEx pilot data freeze as reported by The GTEx Consortium (2015). The lower panel illustrates
the results of the MT-eQTL model: z pairs close to the origin for which no eQTL are detected
have been removed, resulting in the central white region; detected eQTL are colored
according to whether an eQTL is detected in both tissues (light gray points) or a single
tissue (dark gray and black points). Our model explicitly captures patterns of variation in
the presence or absence of eQTL, as well as the heterogeneity of effect sizes across
tissues.
Fig. 1.
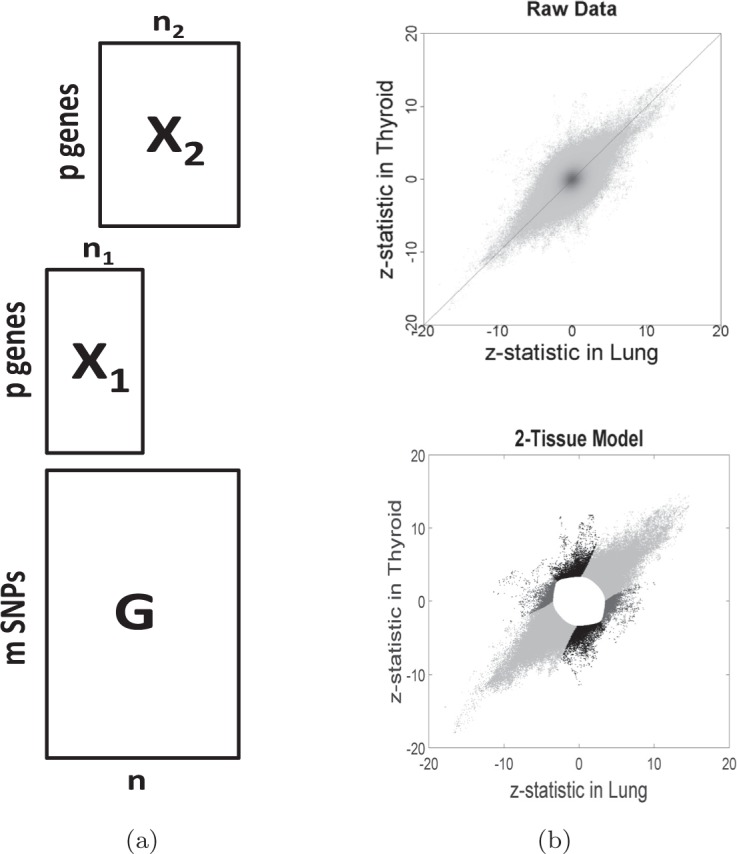
(a) Illustration of the typical data format with two tissues. Genotype data
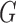 is available for
is available for
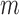 SNPs and each of
SNPs and each of
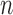 samples. Expression measurements are
available for
samples. Expression measurements are
available for 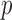 genes; sample sets for different
tissues may not be the same. (b) z-statistics for lung and thyroid: density plot for all
local gene–SNP pairs (top), and scatter plot for significant local gene–SNP pairs with
tissue specificity by gray scale (bottom). The gene–SNP pairs deemed insignificant are
omitted, leading to the white space at the center of the plot. The remaining points are
colored according to their assessed tissue specificity: dark gray points correspond to
the Lung-specific eQTL; black points correspond to the Thyroid-specific eQTL; light gray
points correspond to the cross-tissue eQTL.
genes; sample sets for different
tissues may not be the same. (b) z-statistics for lung and thyroid: density plot for all
local gene–SNP pairs (top), and scatter plot for significant local gene–SNP pairs with
tissue specificity by gray scale (bottom). The gene–SNP pairs deemed insignificant are
omitted, leading to the white space at the center of the plot. The remaining points are
colored according to their assessed tissue specificity: dark gray points correspond to
the Lung-specific eQTL; black points correspond to the Thyroid-specific eQTL; light gray
points correspond to the cross-tissue eQTL.
The contribution of the article is 5-fold: (i) introduction of a novel hierarchical Bayesian model for multi-tissue eQTL analysis; (ii) development of an efficient EM algorithm for estimating the parameters of the model; (iii) analysis of the properties of the model; (iv) rigorous theoretical arguments showing that model-based testing procedures control FDR under realistic assumptions; (v) applications to the GTEx data.
1.1. Related work
Most existing multi-tissue analyses extract eQTL individually from each tissue and then apply post hoc procedures to assess commonality and specificity (Dimas and others, 2009; Fu and others, 2012; Nica and others, 2011; Brown and others, 2013). Recently, several joint analysis approaches were proposed. Gerrits and others (2009) used an ANOVA model to study the genotype effect on a transcript across several cell types. Petretto and others (2010) used a sparse Bayesian multivariate regression model to identify eQTL at multiple loci for same transcripts in many tissues. More recently, Flutre and others (2013) developed a Bayesian model and a permutation-based approach to identify eQTL in multiple tissues. The computation is prohibitive for a moderate number of tissues and a large number of gene–SNP pairs. Sul and others (2013) proposed a “Meta-Tissue” method that combines linear mixed models with meta-analysis. It focuses on one gene–SNP pair at a time. However, the method cannot borrow strength across gene–SNP pairs for eQTL detection, or provide global parameter estimates to characterize eQTL patterns.
In the literature, eQTL analyses are generally divided into two categories: gene-level analysis and SNP-level analysis. The former focuses on the identification of eQTL genes, typically by averaging evidence over all candidate SNPs. The latter treats all gene–SNP pairs equally and aims at identifying significantly associated pairs. Both Gerrits and others (2009) and Sul and others (2013) studied eQTL at the SNP level while Petretto and others (2010) and Flutre and others (2013) are gene-level studies. Gene-level analysis tries to address linkage disequilibrium by assuming there is at most one causal SNP for each gene. However, it cannot provide a list of candidate SNP loci which are potential eQTL for a gene. In this article, we shall focus on the SNP level study, providing a complementary view of the problem. We will also address the computational issue and the lack-of-power concern by exploiting an empirical Bayes approach.
2. The MT-eQTL model
2.1. Format of multi-tissue eQTL data
The general data format for the multi-tissue eQTL problem is as follows. For each of
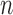 donors we have full genotype information,
and measurements of gene expression in at least one of
donors we have full genotype information,
and measurements of gene expression in at least one of 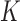 tissues.
Let
tissues.
Let 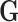 be an
be an 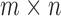 matrix containing the measured genotype of each donor in the study at
matrix containing the measured genotype of each donor in the study at
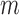 SNPs. The entries take values
SNPs. The entries take values
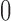 ,
, 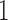 , and
, and
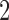 , typically coded as the number of minor
allele variants. Each column of
, typically coded as the number of minor
allele variants. Each column of 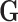 corresponds to a
donor, and each row corresponds to a SNP. The measured transcript levels for tissue
corresponds to a
donor, and each row corresponds to a SNP. The measured transcript levels for tissue
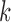 are contained in a
are contained in a
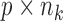 matrix
matrix
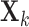 , where
, where 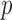 is the
number of genes, and
is the
number of genes, and 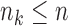 is the number of donors for
tissue
is the number of donors for
tissue 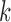 . The number of donors
. The number of donors
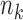 can vary widely among tissues, and even
if two tissues have similar numbers of samples, they may have few common donors. The data
available for the purposes of multi-tissue eQTL analysis has the form
can vary widely among tissues, and even
if two tissues have similar numbers of samples, they may have few common donors. The data
available for the purposes of multi-tissue eQTL analysis has the form
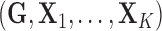 .
Figure 1a gives an illustration of the typical data
format with two tissues.
.
Figure 1a gives an illustration of the typical data
format with two tissues.
In most cases, eQTL analysis is preceded by several preprocessing steps and covariate adjustment. Covariate adjustment is necessary because genotype and expression data usually contain confounding factors. Some confounders, such as gender, are observed, while others are of unknown technical or biological origin. To identify the unknown confounding factors, most studies use principal components, surrogate variables (Leek and Storey, 2007), or PEER factors (Stegle and others, 2012) as covariates. In Section 4.1, we shall discuss the preprocessing procedure of the GTEx data. For now, we just assume that the expression data and genotype data have been appropriately residualized for confounders, so the comparison of these residualized quantities are partial correlations adjusted for covariates.
2.2. Multivariate z-statistic from single tissue correlations
Denote a gene by 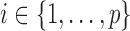 and a SNP by
and a SNP by
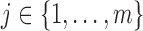 . We focus on a
subset
. We focus on a
subset 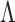 of the full index set
of the full index set
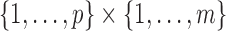 that consists of pairs
that consists of pairs 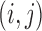 such that SNP
such that SNP
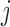 is located within a fixed distance
(usually 100 Kilobases or 1 Megabase) of the transcription start site of gene
is located within a fixed distance
(usually 100 Kilobases or 1 Megabase) of the transcription start site of gene
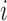 .
.
Let 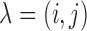 be a gene-SNP pair of
interest. Let
be a gene-SNP pair of
interest. Let 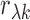 and
and 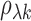 denote, respectively, the
sample and population correlation of transcript
denote, respectively, the
sample and population correlation of transcript 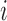 and SNP
and SNP
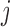 in tissue
in tissue 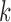 . We use
the Pearson product-moment correlation for several reasons: (i) with proper transformation
of transcript data, the sample correlation has a known, normal distribution (Winterbottom, 1979), which is the basis of the proposed
multi-tissue model; (ii) the Pearson correlation has close connection with the regression
coefficient in a simple linear model relating transcript abundance and genotype (the
foundation of most single-tissue eQTL studies). Note that the sample correlation
. We use
the Pearson product-moment correlation for several reasons: (i) with proper transformation
of transcript data, the sample correlation has a known, normal distribution (Winterbottom, 1979), which is the basis of the proposed
multi-tissue model; (ii) the Pearson correlation has close connection with the regression
coefficient in a simple linear model relating transcript abundance and genotype (the
foundation of most single-tissue eQTL studies). Note that the sample correlation
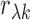 depends only on the
depends only on the
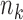 measurements from donors of tissue
measurements from donors of tissue
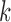 . The vector of correlations
. The vector of correlations
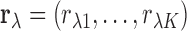 captures the association between the expression of transcript
captures the association between the expression of transcript 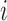 and the
value of genotype
and the
value of genotype 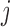 in
in 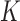 tissues. Relationships between different tissues will be reflected in correlations between
the entries of
tissues. Relationships between different tissues will be reflected in correlations between
the entries of 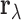 . These features make
. These features make
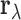 a natural starting
point for a multi-tissue eQTL model.
a natural starting
point for a multi-tissue eQTL model.
We build a multivariate model for the correlation vector 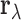 . Let
. Let
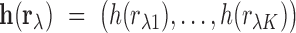 be the vector obtained by applying the Fisher transformation
be the vector obtained by applying the Fisher transformation 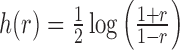 to each component of
to each component of 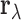 . Let
. Let
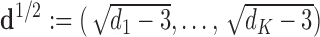 be a scaling vector, where
be a scaling vector, where 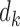 is the degrees of freedom for
is the degrees of freedom for
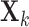 and
and 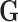 , equal to
, equal to
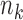 minus the number of covariates used to
correct genotype and expression for samples in tissue
minus the number of covariates used to
correct genotype and expression for samples in tissue 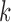 .
Finally, define the vector
.
Finally, define the vector 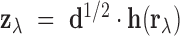 where
where 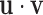 denotes the
Hadamard (entry-wise) product of vectors
denotes the
Hadamard (entry-wise) product of vectors 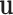 and
and
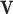 . Let
. Let 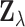 denote the random vector for
denote the random vector for
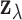 . If we assume that the
expression measurements
. If we assume that the
expression measurements 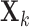 are approximately normal,
standard arguments for the Fisher transformation (Winterbottom, 1979) imply that
are approximately normal,
standard arguments for the Fisher transformation (Winterbottom, 1979) imply that 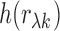 is
approximately normal with mean
is
approximately normal with mean 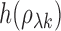 and
variance
and
variance 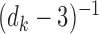 . By a routine multivariate
extension of this fact,
. By a routine multivariate
extension of this fact, 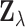 is approximately normally
distributed with mean
is approximately normally
distributed with mean 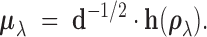 The variance stabilizing property of the Fisher transformation and our choice of scaling
ensures that the variance of each entry
The variance stabilizing property of the Fisher transformation and our choice of scaling
ensures that the variance of each entry 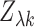 of
of
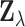 is close to one,
regardless of
is close to one,
regardless of 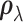 . In
particular, if the true correlation
. In
particular, if the true correlation 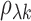 between
transcript
between
transcript 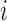 and SNP
and SNP 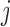 for
tissue
for
tissue 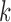 is zero, then
is zero, then 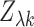 is approximately standard
normal. Thus the
is approximately standard
normal. Thus the 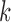 -th entry of the observed vector
-th entry of the observed vector
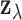 is a z-statistic for
testing
is a z-statistic for
testing 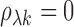 versus
versus
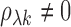 .
.
The use of z-statistics greatly reduces the data complexity and magnitude, without losing
much information regarding gene-SNP associations. It facilitates statistical modeling and
computation. Importantly, the components of 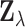 are not
independent due to the correlation of effect sizes and sample overlaps in different
tissues. Capturing this dependence is one of the key features of the MT-eQTL model, which
is described in detail below.
are not
independent due to the correlation of effect sizes and sample overlaps in different
tissues. Capturing this dependence is one of the key features of the MT-eQTL model, which
is described in detail below.
2.3. Hierarchical model
Let 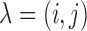 be a gene–SNP pair in
be a gene–SNP pair in
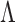 . MT-eQTL is a multivariate,
hierarchical Bayesian model for the random vector
. MT-eQTL is a multivariate,
hierarchical Bayesian model for the random vector 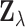 . In detail, we assume that
. In detail, we assume that
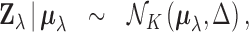 |
(2.1) |
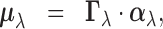 |
(2.2) |
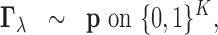 |
(2.3) |
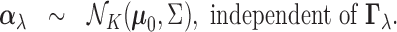 |
(2.4) |
We briefly explain the rationale behind the model setup. The first relation is a
consequence of the Fisher transformation, where 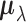 denotes the true
effect sizes of the gene–SNP pair
denotes the true
effect sizes of the gene–SNP pair 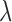 across the
across the
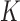 tissues. The
tissues. The 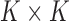 covariance matrix
covariance matrix
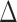 has diagonal values 1; its
off-diagonal values capture the correlations between any two tissues arising from the
underlying sampling process. In practice, the off-diagonal values are typically weakly
positive due to overlapping donors for different tissues. Since the true effect sizes are
unknown in practice, in (2.2), we
build a hierarchical Bayesian model for
has diagonal values 1; its
off-diagonal values capture the correlations between any two tissues arising from the
underlying sampling process. In practice, the off-diagonal values are typically weakly
positive due to overlapping donors for different tissues. Since the true effect sizes are
unknown in practice, in (2.2), we
build a hierarchical Bayesian model for 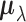 based on two
assumptions: when the SNP has no effect on the gene in a tissue, the true effect size is
0; when the SNP regulates the gene in a tissue, the true effect size follows a random
distribution. Thus
based on two
assumptions: when the SNP has no effect on the gene in a tissue, the true effect size is
0; when the SNP regulates the gene in a tissue, the true effect size follows a random
distribution. Thus 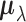 is represented
as a Hadamard product of two random vectors,
is represented
as a Hadamard product of two random vectors, 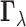 and
and
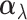 .
.
The random vector 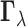 is a
configuration vector for the gene–SNP pair
is a
configuration vector for the gene–SNP pair 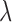 , indicating
whether there is an eQTL in each of the K tissues. As in (2.3), the prior distribution of
, indicating
whether there is an eQTL in each of the K tissues. As in (2.3), the prior distribution of 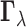 is a
multinomial distribution with
is a
multinomial distribution with 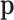 being the probability mass
function. The multinomial distribution has
being the probability mass
function. The multinomial distribution has 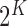 components, each being
a length-
components, each being
a length-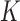 vector of
vector of 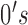 and
and
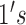 . In particular,
. In particular,
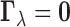 indicates there is no eQTL in any tissue for the gene–SNP pair
indicates there is no eQTL in any tissue for the gene–SNP pair 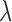 ,
and
,
and 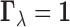 indicates there are eQTL in all tissues for this particular gene–SNP pair. The random
vector
indicates there are eQTL in all tissues for this particular gene–SNP pair. The random
vector 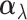 is an eQTL
effect size vector for the gene–SNP pair
is an eQTL
effect size vector for the gene–SNP pair 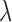 , capturing the
true effect size in each tissue if there is an eQTL. In (2.4), we give
, capturing the
true effect size in each tissue if there is an eQTL. In (2.4), we give 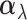 a Gaussian
prior, with mean
a Gaussian
prior, with mean 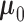 and covariance
and covariance
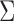 . The mean parameter
. The mean parameter
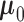 is a
length-
is a
length-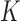 vector capturing the average eQTL effect
sizes in all tissues, and the
vector capturing the average eQTL effect
sizes in all tissues, and the 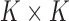 matrix
matrix
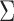 represents the covariance structure
of eQTL effect sizes across multiple tissues. The diagonal values indicate the variation
of effect sizes in different tissues; and the off-diagonal values, typically strongly
positive, reflect the relations of effect sizes between tissues.
represents the covariance structure
of eQTL effect sizes across multiple tissues. The diagonal values indicate the variation
of effect sizes in different tissues; and the off-diagonal values, typically strongly
positive, reflect the relations of effect sizes between tissues.
In the model, there are four major parameters, 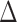 ,
,
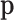 ,
, 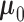 and
and
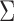 . The parameters characterize
multi-tissue effect sizes for all gene–SNP pairs, and carry important biological
interpretations. We will exploit an empirical Bayes approach to estimate all parameters
from data.
. The parameters characterize
multi-tissue effect sizes for all gene–SNP pairs, and carry important biological
interpretations. We will exploit an empirical Bayes approach to estimate all parameters
from data.
2.4. Mixture model and estimation
The hierarchical model (2.1)–(2.4) describing the distribution of
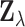 is fully specified by
is fully specified by
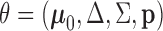 ,
which consists of
,
which consists of 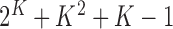 real-valued parameters.
Estimation of, and inference from, the hierarchical model is based on an equivalent
mixture representation.
real-valued parameters.
Estimation of, and inference from, the hierarchical model is based on an equivalent
mixture representation.
If  is distributed as
is distributed as
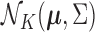 and
and 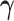 is a fixed vector in
is a fixed vector in
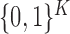 , then one may readily verify that
the entrywise product
, then one may readily verify that
the entrywise product 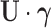 is
distributed as
is
distributed as 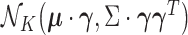 .
A straightforward argument then shows that the hierarchical model (2.1)–(2.4) is equivalent to a mixture model
.
A straightforward argument then shows that the hierarchical model (2.1)–(2.4) is equivalent to a mixture model
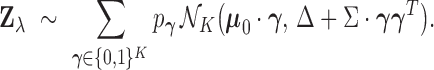 |
(2.5) |
The mixture model is readily interpretable. Each component of the model corresponds to a
unique configuration 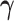 , or equivalently, a
unique pattern of tissue specificity. The model component corresponding to
, or equivalently, a
unique pattern of tissue specificity. The model component corresponding to
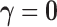 represents
the case in which there are no eQTL in any tissue, and has associated (null) distribution
represents
the case in which there are no eQTL in any tissue, and has associated (null) distribution
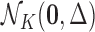 . The model
component corresponding to
. The model
component corresponding to 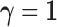 represents
the case in which there are eQTL in every tissue, and has associated distribution
represents
the case in which there are eQTL in every tissue, and has associated distribution
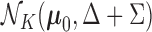 .
Other values of
.
Other values of 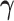 represent
intermediate cases in which there are eQTL in some tissues (those with
represent
intermediate cases in which there are eQTL in some tissues (those with
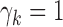 ) and not in others (those with
) and not in others (those with
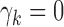 ).
).
We adopt an empirical Bayes approach, estimating the model parameters
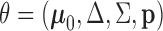 from the observed z-statistics
from the observed z-statistics 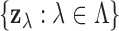 by maximizing the likelihood derived from (2.5). Beginning with the work of Newton
and others (2001) and Efron
and others (2001), empirical Bayes approaches have been
applied to hierarchical models in a number of genetic applications, most notably the study
of differential expression and co-expression in gene expression microarrays (Newton and others, 2004; Smyth and others, 2004; Efron, 2008; Dawson and
Kendziorski, 2012).
by maximizing the likelihood derived from (2.5). Beginning with the work of Newton
and others (2001) and Efron
and others (2001), empirical Bayes approaches have been
applied to hierarchical models in a number of genetic applications, most notably the study
of differential expression and co-expression in gene expression microarrays (Newton and others, 2004; Smyth and others, 2004; Efron, 2008; Dawson and
Kendziorski, 2012).
Directly maximizing the joint log likelihood of the model (2.5) across gene–SNP pairs is computationally intractable. On
the one hand, observations for different gene–SNP pairs may be correlated, as each gene
may contain multiple SNPs and neighboring SNPs may have relatively strong linkage
disequilibrium. On the other hand, the likelihood function for each gene–SNP pair has
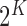 components, each corresponding to a
weighted multivariate Gaussian likelihood function with overlapping model parameters. Note
that the parameters in the model (2.5) determine, and are determined by, the marginal
distribution of the vectors
components, each corresponding to a
weighted multivariate Gaussian likelihood function with overlapping model parameters. Note
that the parameters in the model (2.5) determine, and are determined by, the marginal
distribution of the vectors 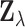 , and do not depend on their
joint distribution. We address the issue of correlated observations by maximizing a
marginal composite likelihood, which is defined as the product of the marginal likelihoods
of all considered gene–SNP pairs. As such, it does not attempt to capture correlation
between different gene–SNP pairs. For typical eQTL analyses, in which the number of
gene–SNP pairs is large and average pairwise correlations are low, we expect the use of
marginal composite likelihood estimation has little effect on statistical efficiency.
, and do not depend on their
joint distribution. We address the issue of correlated observations by maximizing a
marginal composite likelihood, which is defined as the product of the marginal likelihoods
of all considered gene–SNP pairs. As such, it does not attempt to capture correlation
between different gene–SNP pairs. For typical eQTL analyses, in which the number of
gene–SNP pairs is large and average pairwise correlations are low, we expect the use of
marginal composite likelihood estimation has little effect on statistical efficiency.
To address the difficulty of parameter estimation, we exploit an EM algorithm by treating
the underlying configuration vector for each gene–SNP pair as a latent variable. As a
result, the estimation of the probability mass function 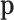 can be separated from the estimation of
can be separated from the estimation of 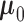 ,
,
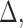 and
and 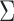 .
The optimization with respect to
.
The optimization with respect to 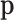 has a closed-form
solution in each iteration. Furthermore, in cis-eQTL analysis, the null configuration
has a closed-form
solution in each iteration. Furthermore, in cis-eQTL analysis, the null configuration
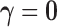 and the full
alternative configuration
and the full
alternative configuration 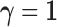 together
usually account for the majority of the prior weight. When estimating
together
usually account for the majority of the prior weight. When estimating
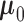 ,
,
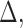 and
and 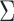 ,
if we only focus on the log likelihood terms corresponding to these two configurations,
each parameter has an explicit estimate. As such, we use a modified EM algorithm with the
two-term approximation, which greatly reduces the computational cost. Simulation studies
show that such approximation has little affect on the accuracy of the estimation. More
details of the model fitting algorithm can be found in Section 1 of Supplementary
material available at Biostatistics online.
,
if we only focus on the log likelihood terms corresponding to these two configurations,
each parameter has an explicit estimate. As such, we use a modified EM algorithm with the
two-term approximation, which greatly reduces the computational cost. Simulation studies
show that such approximation has little affect on the accuracy of the estimation. More
details of the model fitting algorithm can be found in Section 1 of Supplementary
material available at Biostatistics online.
2.5. Marginal compatibility
In eQTL studies with multiple tissues, it is desirable if the model for a subset of tissues is compatible with the model for a superset of tissues in the sense that the former can be obtained from the latter via marginalization. We refer to this property as marginal compatibility. From the model interpretation point of view, the property guarantees that parameters (e.g. prior probabilities of different eQTL configurations, covariance of effect sizes in different tissues) corresponding to a set of tissues do not depend on whether we observe just those tissues or a superset of the tissues. It is crucial in multi-tissue eQTL studies as we essentially always analyze a set of some hypothetical superset of tissues that we do not observe. From the model fitting point of view, with the property, we only need to fit the full model with all available tissues once. The model for any subset of tissues can be obtained directly through marginalization without refitting.
To elaborate, let 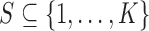 be a subset of
be a subset of
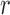 tissues, with
tissues, with 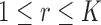 . The mixture model (2.5) has two important compatibility
properties: (i) the marginalization of the full model to
. The mixture model (2.5) has two important compatibility
properties: (i) the marginalization of the full model to 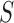 has
the same general form as the model derived from
has
the same general form as the model derived from 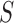 alone; and (ii) the
parameters of the marginal model are obtained by restricting the parameters of the full
model to
alone; and (ii) the
parameters of the marginal model are obtained by restricting the parameters of the full
model to 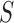 . The following definition and lemma makes
these statements precise. See Section 2 of Supplementary material available at
Biostatistics online for a proof of the lemma.
. The following definition and lemma makes
these statements precise. See Section 2 of Supplementary material available at
Biostatistics online for a proof of the lemma.
Definition: Let 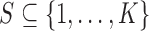 with
cardinality
with
cardinality 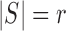 . For each vector
. For each vector
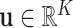 let
let
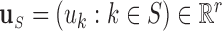 be the vector obtained by restricting
be the vector obtained by restricting 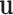 to the entries in
to the entries in
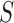 . Similarly, for each matrix
. Similarly, for each matrix
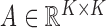 let
let
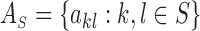 be the
be the 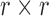 matrix obtained by retaining
only the rows and columns with indices in
matrix obtained by retaining
only the rows and columns with indices in 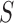 . Note that if
. Note that if
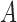 is non-negative (positive) definite, then
is non-negative (positive) definite, then
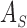 is non-negative
(positive) definite as well.
is non-negative
(positive) definite as well.
Lemma 2.1
If
be a random vector having the mixture distribution (2.5), then
where
is the probability mass function on
obtained by marginalizing
to
, i.e.
.
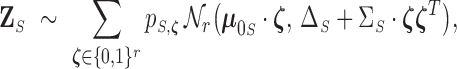
3. Multi-tissue eQTL inference
Once fit, the mixture model (2.5)
provides the basis for inference about eQTL across tissues. When the number of gene–SNP
pairs is large, as in the GTEx example in Section 4,
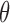 can be accurately estimated from data.
At the level of posterior inference for gene–SNP pairs, we therefore regard
can be accurately estimated from data.
At the level of posterior inference for gene–SNP pairs, we therefore regard
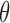 as fixed and known. For data sets with
small sample sizes, approximate standard errors for the components of
as fixed and known. For data sets with
small sample sizes, approximate standard errors for the components of
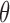 can be obtained from the likelihood
via the observed information matrix.
can be obtained from the likelihood
via the observed information matrix.
Denote the density of the distribution 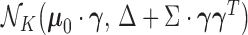 associated with the configuration
associated with the configuration 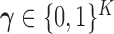 by
by
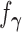 . Thus under the
mixture model (2.5) the random vector
. Thus under the
mixture model (2.5) the random vector
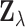 has density
has density
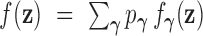 ,
,
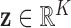 . In view of this
expression and the hierarchical model (2.1)–(2.4), one may regard
. In view of this
expression and the hierarchical model (2.1)–(2.4), one may regard
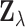 as one element of a jointly
distributed pair
as one element of a jointly
distributed pair 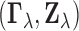 ,
where
,
where
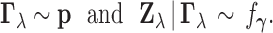 |
(3.1) |
We carry out multi-tissue eQTL analysis based on the posterior distribution of the
configuration 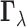 given the
observed vector of z-statistics
given the
observed vector of z-statistics 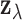 . Two
inference problems are of central interest: one is eQTL detection, in all tissues and in a
subset of tissues; the other is the assessment of eQTL tissue specificity given eQTL is
present in at least one tissue.
. Two
inference problems are of central interest: one is eQTL detection, in all tissues and in a
subset of tissues; the other is the assessment of eQTL tissue specificity given eQTL is
present in at least one tissue.
3.1. Detection of eQTL using the local false discovery rate
A primary goal of multi-tissue analysis is testing each transcript–SNP pair for the presence of an eQTL in at least one tissue. This can be formulated as a multiple testing problem:
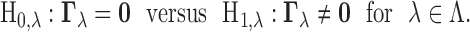 |
(3.2) |
For 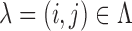 the null
hypothesis
the null
hypothesis 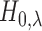 asserts that SNP
asserts that SNP
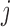 is not an eQTL for transcript
is not an eQTL for transcript
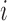 in any tissue, while the alternative
in any tissue, while the alternative
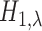 asserts that there is an eQTL
between
asserts that there is an eQTL
between 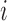 and
and 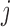 in at
least one tissue.
in at
least one tissue.
The null hypotheses can also be expressed in the form 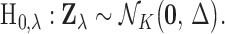 One may derive a p-value for each
One may derive a p-value for each 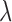 directly from the
null distribution, and convert it to control the overall false discovery rate (FDR) (cf.
Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003). However, this procedure
ignores relevant information about the distribution of
directly from the
null distribution, and convert it to control the overall false discovery rate (FDR) (cf.
Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003). However, this procedure
ignores relevant information about the distribution of 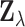 under the alternative that
is contained in the mixture model.
under the alternative that
is contained in the mixture model.
We address the multiple testing problem (3.2) using the local FDR introduced by Efron
and others (2001) in the context of an empirical Bayes
analysis of differential expression in microarrays. Other applications of the local FDR to
genomic problems can be found in Newton and
others (2004), Efron (2007), and
Efron (2008). To simplify notation, let
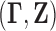 denote a
generic pair distributed as
denote a
generic pair distributed as 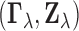 .
.
Definition: The local FDR of an observed z-statistic vector
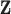 under the model (2.5) is defined by
under the model (2.5) is defined by
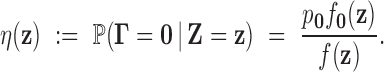 |
(3.3) |
Let 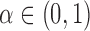 be a target FDR for the
multiple testing problem (3.2).
Vectors
be a target FDR for the
multiple testing problem (3.2).
Vectors 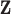 for which the local false discovery
rate
for which the local false discovery
rate 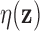 is small provide evidence for
the alternative
is small provide evidence for
the alternative 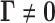 . We
carry out testing of gene–SNP pairs using a step-up procedure applied to the running
average of the ordered local false discover rates (Newton
and others, 2004; Cai and Sun,
2009).
. We
carry out testing of gene–SNP pairs using a step-up procedure applied to the running
average of the ordered local false discover rates (Newton
and others, 2004; Cai and Sun,
2009).
Local FDR Step-Up Procedure: Target FDR 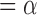
1. Given: Observed
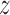 -statistic vectors
-statistic vectors
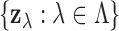 .
.2. Enumerate the elements of
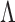 as
as
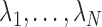 so that
so that
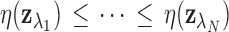 .
.3. Reject hypotheses
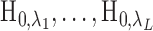 ,
where
,
where 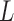 is the largest integer such that
is the largest integer such that
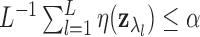 .
.
3.2. Theoretical justification of the local FDR approach
In order to better understand the local FDR step-up procedure, and to assess its
performance, it is useful to express the procedure in an equivalent form. As noted by
Efron and others (2001), the
false discovery rate associated with a rejection region 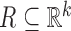 for the multiple
testing problem (3.2) is given by
for the multiple
testing problem (3.2) is given by
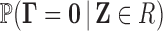 .
They establish the following elementary fact, which exhibits a connection between FDR and
local FDR.
.
They establish the following elementary fact, which exhibits a connection between FDR and
local FDR.
Proposition 3.1
If
is such that
, then
.
As noted above, vectors 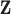 for which
for which 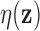 is small provide evidence
against
is small provide evidence
against  , so it is
natural to reject
, so it is
natural to reject 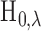 when
when
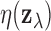 falls below an
appropriate threshold. Consider rejection regions of the form
falls below an
appropriate threshold. Consider rejection regions of the form 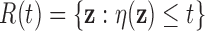 for
for 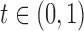 . Given a target FDR
. Given a target FDR
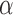 , we wish to find
, we wish to find
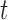 such that
such that 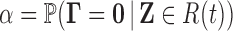 .
By Proposition 3.1 this is equivalent to finding
.
By Proposition 3.1 this is equivalent to finding 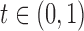 such that
such that
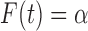 , where
, where
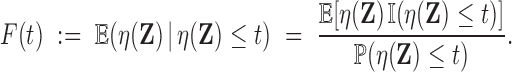 |
The empirical analog of 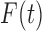 is the ratio
is the ratio
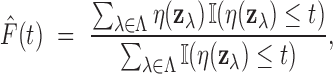 |
which depends only on 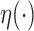 and the
observed vectors
and the
observed vectors 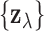 . The function
. The function
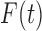 is strictly increasing and continuous
(see Section 3.1 of Supplementary material available at
Biostatistics online for proof). Thus if
is strictly increasing and continuous
(see Section 3.1 of Supplementary material available at
Biostatistics online for proof). Thus if 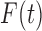 and
and
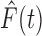 were equal, the local FDR
step-up procedure and the idealized threshold procedure would coincide. In general,
were equal, the local FDR
step-up procedure and the idealized threshold procedure would coincide. In general,
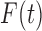 and
and 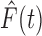 will be different, but
multiplying the numerator and denominator of
will be different, but
multiplying the numerator and denominator of 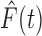 by
by
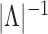 it is evident that the two
functions will be close if
it is evident that the two
functions will be close if 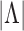 is large and the dependence among
the observed
is large and the dependence among
the observed 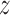 -vectors is not extreme. Asymptotic
control of the FDR by the step-up procedure is established in Theorem 3.2 below. The proof
can be found in Section 3 of Supplementary material available at
Biostatistics online.
-vectors is not extreme. Asymptotic
control of the FDR by the step-up procedure is established in Theorem 3.2 below. The proof
can be found in Section 3 of Supplementary material available at
Biostatistics online.
Let 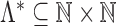 be an infinite index set, and let
be an infinite index set, and let 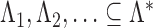 be a sequence of finite subsets of
be a sequence of finite subsets of 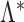 . Let
. Let
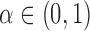 be a target FDR that is
less than the maximum value of
be a target FDR that is
less than the maximum value of 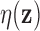 . For each
. For each
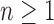 let
let 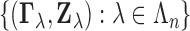 be jointly distributed pairs having the same distribution as
be jointly distributed pairs having the same distribution as 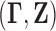 . We wish
to assess the performance of the local FDR step-up procedure, which rejects
. We wish
to assess the performance of the local FDR step-up procedure, which rejects
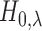 when
when
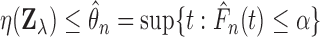 where
where
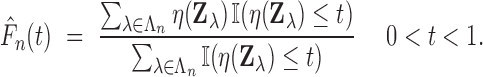 |
The number of false discoveries and total discoveries for the procedure are equal to
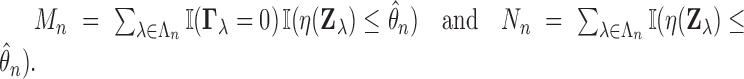
Theorem 3.2
Let
have joint distribution given by Model (3.1) with parameters
. Assume that
is positive definite and that the diagonal entries of
are positive. If
in probability for each
then
as
.
The ratio of expectations 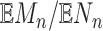 is
sometimes referred to as the marginal false discovery rate (m-FDR). Cai and Sun (2009) established optimality properties and m-FDR control
of several local FDR based testing procedures, including the step-up procedure used here,
under independence and monotonicity assumptions. However, these assumptions are typically
violated in the setting of interest to us here. The monotonicity assumption, which in the
present case involves the relationship between the distributions of the local FDR
is
sometimes referred to as the marginal false discovery rate (m-FDR). Cai and Sun (2009) established optimality properties and m-FDR control
of several local FDR based testing procedures, including the step-up procedure used here,
under independence and monotonicity assumptions. However, these assumptions are typically
violated in the setting of interest to us here. The monotonicity assumption, which in the
present case involves the relationship between the distributions of the local FDR
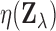 under
under
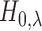 and
and 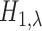 , does not appear to hold.
Moreover, in eQTL data there are typically significant correlations between nearby SNPs
(linkage disequilibrium), leading to to complex, non-stationary correlations between the
gene-SNP based vectors
, does not appear to hold.
Moreover, in eQTL data there are typically significant correlations between nearby SNPs
(linkage disequilibrium), leading to to complex, non-stationary correlations between the
gene-SNP based vectors 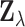 .
.
Theorem 3.2 makes no explicit assumptions on the joint distribution of the vectors
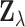 ; instead it relies on the
relatively weak condition that
; instead it relies on the
relatively weak condition that 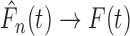 in
probability. This condition holds, for example, under the (very mild) assumption that the
variance of the numerator and the denominator of
in
probability. This condition holds, for example, under the (very mild) assumption that the
variance of the numerator and the denominator of 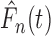 are of order
are of order
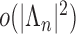 . The variance decay
assumption concerns the family of all gene–SNP pairs, across all measured genes instead of
a single gene. Although the SNPs co-located with a particular gene may be highly
correlated, correlations are generally weak, or zero, across distant genes. These distal
pairs dominate the index set
. The variance decay
assumption concerns the family of all gene–SNP pairs, across all measured genes instead of
a single gene. Although the SNPs co-located with a particular gene may be highly
correlated, correlations are generally weak, or zero, across distant genes. These distal
pairs dominate the index set 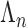 , and so the variance decay
assumption should be satisfied in any cis-eQTL analysis involving a large number of genes.
When the assumption holds, the conclusion of the theorem may be strengthened to
, and so the variance decay
assumption should be satisfied in any cis-eQTL analysis involving a large number of genes.
When the assumption holds, the conclusion of the theorem may be strengthened to
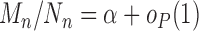 .
.
3.3. Analysis for subsets of tissues
In some problems, a subset 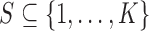 of the available
tissues may be of primary interest. The multiple testing framework described above can be
adapted to the tissues in
of the available
tissues may be of primary interest. The multiple testing framework described above can be
adapted to the tissues in 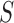 in two primary ways. The first is to
construct a model based only on the tissues in
in two primary ways. The first is to
construct a model based only on the tissues in 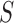 and use the resulting
local FDR to identify multi-tissue eQTL. However, this approach does not make use of the
available data from tissues outside
and use the resulting
local FDR to identify multi-tissue eQTL. However, this approach does not make use of the
available data from tissues outside 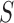 and as such it does not
borrow strength from commonalities among tissues. As an alternative, one may use the
marginal local FDR for
and as such it does not
borrow strength from commonalities among tissues. As an alternative, one may use the
marginal local FDR for 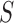 , defined by
, defined by
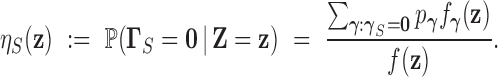 |
(3.4) |
Here 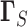 and
and
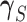 denote,
respectively, the restriction of the vectors
denote,
respectively, the restriction of the vectors 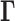 and
and
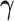 to the tissues in
to the tissues in
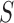 , while
, while 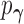 ,
,
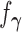 and
and
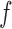 correspond to the full model (2.5). We emphasize that the marginal
local FDR
correspond to the full model (2.5). We emphasize that the marginal
local FDR 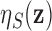 is a function of the
complete vector of z-statistics, and therefore depends on the fitted model for the full
set of tissues. In Section 4.3, we have shown
that the marginal local FDR derived from the full data set is uniformly more powerful than
the local FDR derived from a subset of the data in detecting eQTLs in a subset of tissues.
More numerical results can be found in Section
4.3 of Supplementary
material available at Biostatistics online.
is a function of the
complete vector of z-statistics, and therefore depends on the fitted model for the full
set of tissues. In Section 4.3, we have shown
that the marginal local FDR derived from the full data set is uniformly more powerful than
the local FDR derived from a subset of the data in detecting eQTLs in a subset of tissues.
More numerical results can be found in Section
4.3 of Supplementary
material available at Biostatistics online.
3.4. Assessments of tissue specificity
Testing gene–SNP pairs is typically the first step in multi-tissue eQTL analysis.
Rejection of 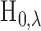 is based on evidence
that
is based on evidence
that 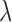 is an eQTL in at least one of the
available tissues. More detailed statements about the pattern of eQTL across tissues can
be made using information about the full configuration vector
is an eQTL in at least one of the
available tissues. More detailed statements about the pattern of eQTL across tissues can
be made using information about the full configuration vector 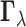 . If the
hypothesis
. If the
hypothesis 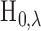 is rejected, a natural
estimate of
is rejected, a natural
estimate of 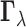 is the
maximum a posteriori (MAP) configuration defined by
is the
maximum a posteriori (MAP) configuration defined by
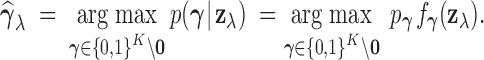 |
As an alternative, one may compute the marginal posterior probability of an eQTL in each
tissue 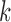 , namely
, namely 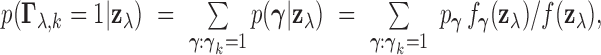 and declare an eQTL in tissue
and declare an eQTL in tissue 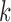 if this marginal
probability exceeds a predefined threshold. Both MAP and thresholding of the marginal
posterior extend to subsets of tissues.
if this marginal
probability exceeds a predefined threshold. Both MAP and thresholding of the marginal
posterior extend to subsets of tissues.
3.5. Testing a family of configurations
The goal of the multiple testing problem (3.2) is to determine whether the configuration 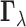 of a gene–SNP
pair is equal to
of a gene–SNP
pair is equal to 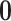 or belongs to the complementary set
or belongs to the complementary set
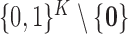 . More
generally, one may test membership of
. More
generally, one may test membership of 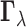 in any fixed
subset
in any fixed
subset 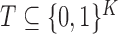 of configurations.
The associated testing problem can be written as
of configurations.
The associated testing problem can be written as
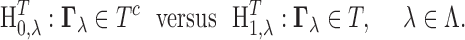 |
(3.5) |
A test statistic for (3.5) can be obtained by marginalizing the full local FDR (3.3) as
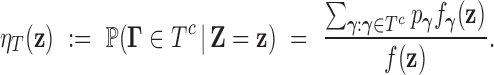 |
The local FDR step-up procedure can then be applied to the values
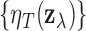 in order to
control the overall FDR in (3.5).
in order to
control the overall FDR in (3.5).
4. GTEx data analysis
In this section, we apply the MT-eQTL model and inference procedures to the GTEx pilot data freeze (The GTEx Consortium, 2015). A pointer to the publicly available data is at http://www.broadinstitute.org/gtex/.
4.1. Data preprocessing
We focus on nine primary tissues having between 80 and 160 samples: adipose, artery, blood, heart, lung, muscle, nerve, skin, and thyroid. In what follows, tissues will be ordered alphabetically. In total, there are 175 genotyped individuals with expression data in at least one of these tissues (the sample information can be found in Figure S1 of Supplementary material available at Biostatistics online).
Each entry of the genotype data matrix 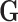 records the
number of minor allele variants of one donor at one SNP locus. Any missing value at a
locus was imputed by the corresponding row average. Loci with minor allele frequency less
than 5% in all genotyped individuals were
discarded, resulting in slightly less than 7 million SNPs. The expression level for each
gene in each tissue and sample is measured by the number of mapped reads per kilobase per
million reads (RPKM). Genes having fewer than
records the
number of minor allele variants of one donor at one SNP locus. Any missing value at a
locus was imputed by the corresponding row average. Loci with minor allele frequency less
than 5% in all genotyped individuals were
discarded, resulting in slightly less than 7 million SNPs. The expression level for each
gene in each tissue and sample is measured by the number of mapped reads per kilobase per
million reads (RPKM). Genes having fewer than 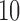 samples with RPKM
greater than
samples with RPKM
greater than 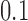 in some tissue were discarded, leaving
slightly more than 20,000 genes. To improve robustness, the gene expression values across
samples in a tissue were inverse quantile normalized.
in some tissue were discarded, leaving
slightly more than 20,000 genes. To improve robustness, the gene expression values across
samples in a tissue were inverse quantile normalized.
Fifteen PEER factors were identified from the expression data from each tissue, and three principal components were identified from the genotype data. With an additional covariate for gender, we obtained nineteen covariates in total. For each tissue, the confounding effects were adjusted by residualizing the expression data and the corresponding genotype data on nineteen covariates respectively. Consequently, the degree of freedom for each tissue is equal to the sample size in that tissue minus 19.
4.2. Model fit
We focus on testing of cis-eQTL, restricting our attention to SNPs that lie within 100
kilobases of the transcription start site of a gene, yielding roughly 10 million gene–SNP
pairs of interest. Subsequently, the full 9D MT-eQTL model was fit using the modified EM
algorithm described in Section 2.4 with the
parameter 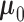 set to zero. Fitting
the full model took less than 24 hours, and required less than 8 gigabytes of RAM, on a
desktop computer with 2.93GHz Intel Xeon CPU. A comparison of timing results for fitting
sub-models of different dimensions between our method and the Meta-Tissue method (Sul and others, 2013) can be found in
Section 5 of Supplementary material available at
Biostatistics online.
set to zero. Fitting
the full model took less than 24 hours, and required less than 8 gigabytes of RAM, on a
desktop computer with 2.93GHz Intel Xeon CPU. A comparison of timing results for fitting
sub-models of different dimensions between our method and the Meta-Tissue method (Sul and others, 2013) can be found in
Section 5 of Supplementary material available at
Biostatistics online.
In what follows we denote the estimated model parameters by 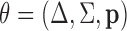 . Values
of the estimated parameters can be found in Section
5 of Supplementary
material available at Biostatistics online. The off-diagonal
values of
. Values
of the estimated parameters can be found in Section
5 of Supplementary
material available at Biostatistics online. The off-diagonal
values of 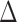 are all positive but small in scale
(between
are all positive but small in scale
(between 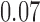 and
and 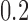 ),
suggesting that donor overlap among tissues and other features of the experimental design
have a weak but positive effect on the correlations of effect sizes across tissues. The
diagonal values of
),
suggesting that donor overlap among tissues and other features of the experimental design
have a weak but positive effect on the correlations of effect sizes across tissues. The
diagonal values of 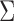 indicate modest heterogeneity of
effect size variation across tissues. The off-diagonal values of
indicate modest heterogeneity of
effect size variation across tissues. The off-diagonal values of 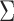 reflect positive, often large, correlation of effect sizes arising from commonalities
among tissues.
reflect positive, often large, correlation of effect sizes arising from commonalities
among tissues.
The fitted probability mass function 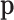 assigns
probabilities to each of the
assigns
probabilities to each of the 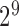 possible eQTL configurations. The most
likely configuration is
possible eQTL configurations. The most
likely configuration is 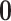 with
with 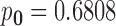 , indicating that about
68% of the gene–SNP pairs do not have an
eQTL in any tissue. This is consistent with previous studies (Wright and others, 2014). To summarize
, indicating that about
68% of the gene–SNP pairs do not have an
eQTL in any tissue. This is consistent with previous studies (Wright and others, 2014). To summarize  ,
we sum up the prior probabilities of configurations with the same Hamming weight (defined
as the number of 1s in a length-9 binary configuration sequence). This provides an
overview of the overall probability of seeing an cis-eQTL in k
tissues, where
,
we sum up the prior probabilities of configurations with the same Hamming weight (defined
as the number of 1s in a length-9 binary configuration sequence). This provides an
overview of the overall probability of seeing an cis-eQTL in k
tissues, where 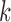 ranges from
ranges from 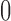 to
to
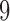 . We note, however, that the probabilities
for configurations with the same Hamming weight may be quite different. The total prior
probabilities are shown in Figure 2 in the log scale.
The U-shape curve indicates that for cis-eQTL analysis, the most likely configurations are
eQTL in no tissue, in a single tissue, or in all tissues, and the least likely
configurations are those with eQTL in roughly half the tissues. We remark that the pattern
may only apply to cis-eQTL but not to trans-eQTL.
. We note, however, that the probabilities
for configurations with the same Hamming weight may be quite different. The total prior
probabilities are shown in Figure 2 in the log scale.
The U-shape curve indicates that for cis-eQTL analysis, the most likely configurations are
eQTL in no tissue, in a single tissue, or in all tissues, and the least likely
configurations are those with eQTL in roughly half the tissues. We remark that the pattern
may only apply to cis-eQTL but not to trans-eQTL.
Fig. 2.
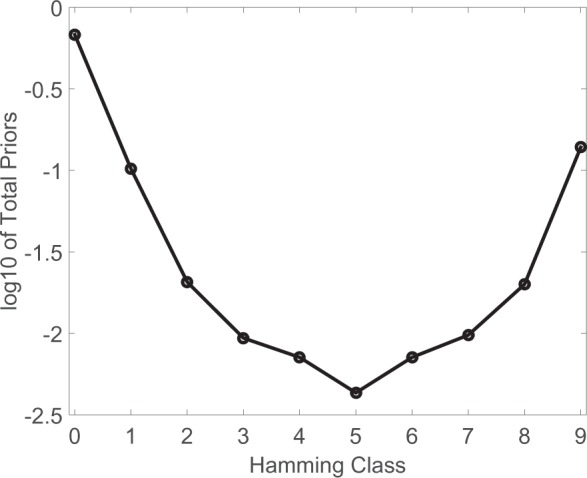
Summary of the estimated eQTL probabilities from the cis-eQTL analysis of the GTEx
data. Each circle represents the log (base 10) of the probability of a gene–SNP pair
having eQTL in 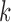 out of 9 tissues, where
out of 9 tissues, where
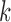 ranges from
ranges from 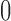 to
to
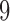 .
.
4.3. Results
Applied to the full 9D model with FDR threshold 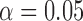 , the local FDR step-up
procedure identified roughly 1.28 million gene–SNP pairs (roughly
12% of the total) with an eQTL in at least
one tissue. We subsequently applied the MAP rule to each significant discovery in order to
assess tissue specificity. To validate the discoveries, we also applied the Meta-Tissue
method to the same data set. Meta-Tissue produces a p value for each gene–SNP pair for
testing the existence of eQTL in any tissue. We further adjusted the p values (Benjamini and Yekutieli, 2001) to control the FDR. About
80% of the MT-eQTL discoveries (i.e. 1.03 million) are replicated in Meta-Tissue,
providing a highly concordant result. We further investigated the unique discoveries of
each method (about 250 thousand from MT-eQTL, and 177 thousand from Meta-Tissue). The left
panel of Figure 3 shows the Meta-Tissue p values of
the unique discoveries from MT-eQTL. Small p values are enriched, indicating the unique
MT-eQTL discoveries are well supported by Meta-Tissue. The right panel of Figure 3 presents the MT-eQTL local FDRs of the unique
discoveries from Meta-Tissue. The unique Meta-Tissue discoveries are only moderately
supported by MT-eQTL. The MT-eQTL provides a systematic way to leverage information across
gene–SNP pairs, and offers explicit estimates of model parameters with critical biological
interpretation.
, the local FDR step-up
procedure identified roughly 1.28 million gene–SNP pairs (roughly
12% of the total) with an eQTL in at least
one tissue. We subsequently applied the MAP rule to each significant discovery in order to
assess tissue specificity. To validate the discoveries, we also applied the Meta-Tissue
method to the same data set. Meta-Tissue produces a p value for each gene–SNP pair for
testing the existence of eQTL in any tissue. We further adjusted the p values (Benjamini and Yekutieli, 2001) to control the FDR. About
80% of the MT-eQTL discoveries (i.e. 1.03 million) are replicated in Meta-Tissue,
providing a highly concordant result. We further investigated the unique discoveries of
each method (about 250 thousand from MT-eQTL, and 177 thousand from Meta-Tissue). The left
panel of Figure 3 shows the Meta-Tissue p values of
the unique discoveries from MT-eQTL. Small p values are enriched, indicating the unique
MT-eQTL discoveries are well supported by Meta-Tissue. The right panel of Figure 3 presents the MT-eQTL local FDRs of the unique
discoveries from Meta-Tissue. The unique Meta-Tissue discoveries are only moderately
supported by MT-eQTL. The MT-eQTL provides a systematic way to leverage information across
gene–SNP pairs, and offers explicit estimates of model parameters with critical biological
interpretation.
Fig. 3.

The left panel is the histogram of the Meta-Tissue p values for the 250 thousand unique discoveries from MT-eQTL, from the GTEx analysis of eQTL in at least one tissue; the right panel is the histogram of the MT-eQTL local FDRs for the 177 thousand unique discoveries in Meta-Tissue.
A unique advantage of MT-eQTL over Meta-Tissue is the ease of eQTL tissue specificity
assessment. To facilitate the visualization of eQTL discoveries, let us focus on a
two-tissue MT-eQTL model. As an example, Figure 1b
shows scatter plots of z-statistics for lung and thyroid. The upper panel shows the
density plot of the raw z-statistics (MT-eQTL input); the lower panel only shows the
discoveries with eQTL in at least one of the tissues (MT-eQTL output). The z-statistic
vectors deemed insignificant are omitted, leading to the white space at the center of the
plot. The remaining points are colored according to their assessed tissue specificity
based on the MAP approach: dark gray represents the configuration
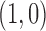 in which there is an eQTL in tissue 1
but not tissue 2; black represents the configuration
in which there is an eQTL in tissue 1
but not tissue 2; black represents the configuration 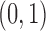 in
which there is an eQTL in tissue 2 but not tissue 1; and light gray represents the
configuration
in
which there is an eQTL in tissue 2 but not tissue 1; and light gray represents the
configuration 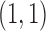 in which there is an eQTL in both
tissues. The overall shape of each plot is a tilted ellipse, with extreme values along the
main diagonal and, to a lesser extent, along the coordinate axes. As expected, significant
points close to one of the coordinate axes show evidence for an eQTL in a single tissue
(tissue specific eQTL), while those along the positive diagonal show evidence for eQTL in
both tissues (common eQTL). We remark that this analysis easily extends to an arbitrary
number of tissues.
in which there is an eQTL in both
tissues. The overall shape of each plot is a tilted ellipse, with extreme values along the
main diagonal and, to a lesser extent, along the coordinate axes. As expected, significant
points close to one of the coordinate axes show evidence for an eQTL in a single tissue
(tissue specific eQTL), while those along the positive diagonal show evidence for eQTL in
both tissues (common eQTL). We remark that this analysis easily extends to an arbitrary
number of tissues.
MT-eQTL also effectively leverages information in multiple tissues to improve eQTL detection in a single or a subset of tissues. To investigate how the use of auxiliary tissues increases statistical power, we studied a sequence of nested MT-eQTL models and focused on eQTL discoveries in a single tissue. For each of the nine tissues, we first fitted the 1-dimensional model with just the primary tissue and then added other tissues one by one alphabetically to get a sequence of super-models. For each considered model, we applied the adaptive thresholding procedure to the marginal local FDR for the primary tissue, and recorded the number of significant discoveries in that tissue. Figure 4 shows the number of significant discoveries versus the dimension of a model. Each curve corresponds to a case where one of the nine tissues is set to be the primary tissue. The number of eQTL discoveries in each primary tissue increases with the dimension of a model.
Fig. 4.
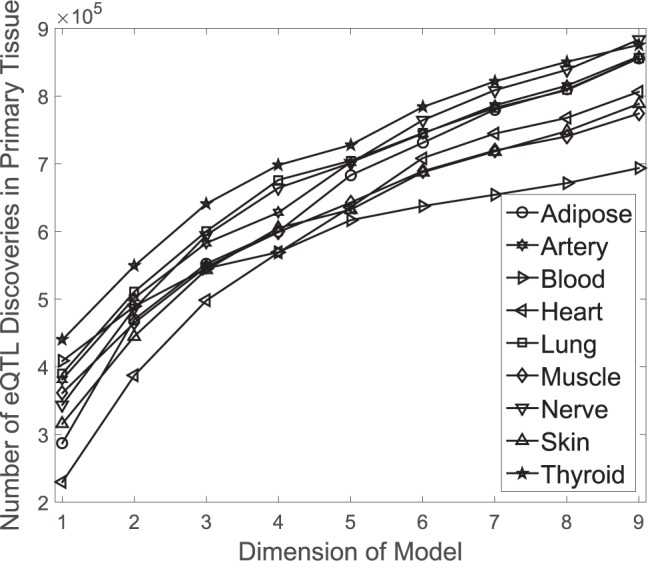
The number of significant discoveries in a primary tissue versus the dimension of a
MT-eQTL model. Each curve corresponds to a case where one of the nine tissues is set
to be the primary tissue. The FDR threshold is fixed to be 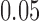 .
.
5. Conclusion
In this article, we proposed a hierarchical Bayesian model, MT-eQTL, for multi-tissue eQTL analysis. We adopted an empirical Bayes approach to estimate the model and to perform inferences. We also proved a substantial theoretical property to support the method in a realistic setting. The proposed methodology greatly enhances classical single-tissue eQTL analysis methods by accounting for the information shared among tissues.
There are a number of interesting directions for future research. Perhaps the most
important is to extend the proposed framework to a large number (e.g.
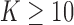 ) of tissues. The large tissue
setting poses real challenges as the total number of configurations grows exponentially in
the number of tissues, making the current implementation excessively slow and
computationally costly. Another direction is to relax the assumption that the covariance
matrix
) of tissues. The large tissue
setting poses real challenges as the total number of configurations grows exponentially in
the number of tissues, making the current implementation excessively slow and
computationally costly. Another direction is to relax the assumption that the covariance
matrix 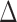 in Model (2.5) is constant across gene–SNP pairs.
Different genes may have distinct correlation patterns between tissues, which might warrant
the use of gene-specific covariance matrices in setting where the number of samples is
large. Lastly, it is of interest to extend the method to the identification of trans-eQTLs,
which exhibit higher levels of tissue-specificity than (Jo
and others, 2016).
in Model (2.5) is constant across gene–SNP pairs.
Different genes may have distinct correlation patterns between tissues, which might warrant
the use of gene-specific covariance matrices in setting where the number of samples is
large. Lastly, it is of interest to extend the method to the identification of trans-eQTLs,
which exhibit higher levels of tissue-specificity than (Jo
and others, 2016).
Supplementary Material
Acknowledgements
We would like to thank all the members of the GTEx consortium. We also thank Dereje Jima for conducting the Meta-Tissue analysis on the GTEx data. Conflict of Interest: None declared.
Supplementary material
Supplementary material is available at http://biostatistics.oxfordjournals.org.
Funding
National Institute of Health (NIH) (Grant R01 MH090936 and MH101819-0); National Science Foundation (NSF) (Grant DMS 0907177 and DMS 1310002); and Environmental Protection Agency (EPA) (Grant STAR RD83580201), in part.
References
- Benjamini Y. and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300. [Google Scholar]
- Benjamini Y. and Yekutieli D. (2001). The control of the false discovery rate in multiple testing under dependency. Annals of Statistics 29, 1165–1188. [Google Scholar]
- Brown C. D., Mangravite L. M. and Engelhardt B. E. (2013). Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genetics 9, e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T. T. and Sun W. (2009). Simultaneous testing of grouped hypotheses: finding needles in multiple haystacks. Journal of the American Statistical Association 104, 1467–1481. [Google Scholar]
- Dawson J. A. and Kendziorski C. (2012). An empirical Bayesian approach for identifying differential coexpression in high-throughput experiments. Biometrics 68, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas A. S., Deutsch S., Stranger B. E., Montgomery S. B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M.. and others (2009). Common regulatory variation impacts gene expression in a cell type–dependent manner. Science 325, 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. (2007). Size, power and false discovery rates. The Annals of Statistics 35, 1351–1377. [Google Scholar]
- Efron B. (2008). Microarrays, empirical Bayes and the two-groups model. Statistical Science, 1–22. [Google Scholar]
- Efron B., Tibshirani R., Storey J. D. and Tusher V. (2001). Empirical Bayes analysis of a microarray experiment. Journal of the American Statistical Association 96, 1151–1160. [Google Scholar]
- Flutre T., Wen X., Pritchard J. and Stephens M. (2013). A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genetics 9, e1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J., Wolfs M. G., Deelen P., Westra H. J., Fehrmann R. S., Te Meerman G. J., Buurman W. A., Rensen S. S., Groen H. J., Weersma R. K.. and others (2012). Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genetics 8, e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits A., Li Y., Tesson B. M., Bystrykh L. V., Weersing E., Ausema A., Dontje B., Wang X., Breitling R., Jansen R. C. and de Haan G. (2009). Expression quantitative trait loci are highly sensitive to cellular differentiation state. PLoS Genetics 5, e1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo B., He Y., Strober B. J., Parsana P., Aguet F., Brown A. A., Castel S. E., Gamazon E. R., Gewirtz A., Gliner G.. and others (2016). Distant regulatory effects of genetic variation in multiple human tissues. bioRxiv, 074419. [Google Scholar]
- Kendziorski C. and Wang P. (2006). A review of statistical methods for expression quantitative trait loci mapping. Mammalian Genome 17, 509–517. [DOI] [PubMed] [Google Scholar]
- Leek J. T. and Storey J. D. (2007). Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genetics 3, e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton M. A., Kendziorski C. M., Richmond C. S., Blattner F. R. and Tsui K.-W. (2001). On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. Journal of Computational Biology 8, 37–52. [DOI] [PubMed] [Google Scholar]
- Newton M. A., Noueiry A., Sarkar D. and Ahlquist P. (2004). Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics 5, 155–176. [DOI] [PubMed] [Google Scholar]
- Nica A. C., Parts L., Glass D., Nisbet J., Barrett A., Sekowska M., Travers M., Potter S., Grundberg E., Small K.. and others (2011). The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genetics 7, e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E., Bottolo L., Langley S. R., Heinig M., McDermott-Roe C., Sarwar R., Pravenec M., Hübner N., Aitman T. J., Cook S. A. and Richardson S. (2010). New insights into the genetic control of gene expression using a Bayesian multi-tissue approach. PLoS Computational Biology 6, e1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3, 3. [DOI] [PubMed] [Google Scholar]
- Stegle O., Parts L., Piipari M., Winn J. and Durbin R. (2012). Using probabilistic estimation of expression residuals (peer) to obtain increased power and interpretability of gene expression analyses. Nature Protocols 7, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. D. and Tibshirani R. (2003). Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul J. H., Han B., Ye C., Choi T. and Eskin E. (2013). Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genetics 9, e1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. (2015). The genotype-tissue expression (gtex) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbottom A. (1979). A note on the derivation of fisher’s transformation of the correlation coefficient. The American Statistician 33, 142–143. [Google Scholar]
- Wright F. A., Shabalin A. A. and Rusyn I. (2012). Computational tools for discovery and interpretation of expression quantitative trait loci. Pharmacogenomics 13, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. A., Sullivan P. F., Brooks A. I., Zou F., Sun W., Xia K., Madar V., Jansen R., Chung W., Zhou Y. H.. and others (2014). Heritability and genomics of gene expression in peripheral blood. Nature Genetics 46, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.