

Abstract
Background
Several antibiotics have been evaluated in Crohn's disease (CD), however randomised controlled trials (RCTs) have produced conflicting results.
Objectives
To assess the efficacy and safety of antibiotics for induction and maintenance of remission in CD.
Search methods
We searched MEDLINE, Embase, CENTRAL, the Cochrane IBD Group Specialized Register and Clinicaltrials.gov database from inception to 28 February 2018. We also searched reference lists and conference proceedings.
Selection criteria
RCTs comparing antibiotics to placebo or an active comparator in adult (> 15 years) CD patients were considered for inclusion.
Data collection and analysis
Two authors screened search results and extracted data. Bias was evaluated using the Cochrane risk of bias tool. The primary outcomes were failure to achieve clinical remission and relapse. Secondary outcomes included clinical response, endoscopic response, endoscopic remission, endoscopic relapse, histologic response, histologic remission, adverse events (AEs), serious AEs, withdrawal due to AEs and quality of life. Remission is commonly defined as a Crohn's disease activity index (CDAI) of < 150. Clinical response is commonly defined as a decrease in CDAI from baseline of 70 or 100 points. Relapse is defined as a CDAI > 150. For studies that enrolled participants with fistulizing CD, response was defined as a 50% reduction in draining fistulas. Remission was defined as complete closure of fistulas. We calculated the risk ratio (RR) and corresponding 95% confidence interval (95% CI) for dichotomous outcomes. We calculated the mean difference (MD) and corresponding 95% CI for continuous outcomes. GRADE was used to assess the certainty of the evidence.
Main results
Thirteen RCTs (N = 1303 participants) were eligible. Two trials were rated as high risk of bias (no blinding). Seven trials were rated as unclear risk of bias and four trials were rated as low risk of bias. Comparisons included ciprofloxacin (500 mg twice daily) versus placebo, rifaximin (800 to 2400 mg daily) versus placebo, metronidazole (400 mg to 500 mg twice daily) versus placebo, clarithromycin (1 g/day) versus placebo, cotrimoxazole (960 mg twice daily) versus placebo, ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four time daily) versus methylprednisolone (0.7 to 1 mg/kg daily), ciprofloxacin (500 mg daily), metronidazole (500 mg daily) and budesonide (9 mg daily) versus placebo with budesonide (9 mg daily), ciprofloxacin (500 mg twice daily) versus mesalazine (2 g twice daily), ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, ciprofloxacin (500 mg twice daily) with infliximab versus placebo with infliximab, clarithromycin (750 mg daily) and antimycobacterial versus placebo, and metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) versus placebo. We pooled all antibiotics as a class versus placebo and antibiotics with anti‐tumour necrosis factor (anti‐TNF) versus placebo with anti‐TNF.
The effect of individual antibiotics on CD was generally uncertain due to imprecision. When we pooled antibiotics as a class, 55% (289/524) of antibiotic participants failed to achieve remission at 6 to 10 weeks compared with 64% (149/231) of placebo participants (RR 0.86, 95% CI 0.76 to 0.98; 7 studies; high certainty evidence). At 10 to 14 weeks, 41% (174/428) of antibiotic participants failed to achieve a clinical response compared to 49% (93/189) of placebo participants (RR 0.77, 95% CI 0.64 to 0.93; 5 studies; moderate certainty evidence). The effect of antibiotics on relapse in uncertain. Forty‐five per cent (37/83) of antibiotic participants relapsed at 52 weeks compared to 57% (41/72) of placebo participants (RR 0.87, 95% CI 0.52 to 1.47; 2 studies; low certainty evidence). Relapse of endoscopic remission was not reported in the included studies. Antibiotics do not appear to increase the risk of AEs. Thirty‐eight per cent (214/568) of antibiotic participants had at least one adverse event compared to 45% (128/284) of placebo participants (RR 0.87, 95% CI 0.75 to 1.02; 9 studies; high certainty evidence). The effect of antibiotics on serious AEs and withdrawal due to AEs was uncertain. Two per cent (6/377) of antibiotic participants had at least one adverse event compared to 0.7% (1/143) of placebo participants (RR 1.70, 95% CI 0.29 to 10.01; 3 studies; low certainty evidence). Nine per cent (53/569) of antibiotic participants withdrew due to AEs compared to 12% (36/289) of placebo participants (RR 0.86, 95% CI 0.57 to 1.29; 9 studies; low certainty evidence) is uncertain. Common adverse events in the studies included gastrointestinal upset, upper respiratory tract infection, abscess formation and headache, change in taste and paraesthesia
When we pooled antibiotics used with anti‐TNF, 21% (10/48) of patients on combination therapy failed to achieve a clinical response(50% closure of fistulas) or remission (closure of fistulas) at week 12 compared with 36% (19/52) of placebo and anti‐TNF participants (RR 0.57, 95% CI 0.29 to 1.10; 2 studies; low certainty evidence). These studies did not assess the effect of antibiotics and anti‐TNF on clinical or endoscopic relapse. Seventy‐seven per cent (37/48) of antibiotics and anti‐TNF participants had an AE compared to 83% (43/52) of anti‐TNF and placebo participants (RR 0.93, 95% CI 0.76 to 1.12; 2 studies, moderate certainty evidence). The effect of antibiotics and anti‐TNF on withdrawal due to AEs is uncertain. Six per cent (3/48) of antibiotics and anti‐TNF participants withdrew due to an AE compared to 8% (4/52) of anti‐TNF and placebo participants (RR 0.82, 95% CI 0.19 to 3.45; 2 studies, low certainty evidence). Common adverse events included nausea, vomiting, upper respiratory tract infections, change in taste, fatigue and headache
Authors' conclusions
Moderate to high quality evidence suggests that any benefit provided by antibiotics in active CD is likely to be modest and may not be clinically meaningful. High quality evidence suggests that there is no increased risk of adverse events with antibiotics compared to placebo. The effect of antibiotics on the risk of serious adverse events is uncertain. The effect of antibiotics on maintenance of remission in CD is uncertain. Thus, no firm conclusions regarding the efficacy and safety of antibiotics for maintenance of remission in CD can be drawn. More research is needed to determine the efficacy and safety of antibiotics as therapy in CD
Plain language summary
Antibiotics for the treatment of Crohn's disease
What is Crohn's disease?
Crohn's disease (CD) is an inflammatory disorder that can affect any segment of the gastrointestinal tract from the mouth to the anus. Common symptoms of CD include fever, diarrhea, abdominal pain and weight loss. CD is characterized by periods of relapse when people experience symptoms and periods of remission when the symptoms stop.
What are antibiotics?
Antibiotics are medications used to treat bacterial infections. Antibiotics are designed to target specific bacterial populations and have different mechanisms of action to stop a bacterial population from growing or eradicate the bacteria.
What is the purpose this study?
Antibiotics are commonly used for managing patients with CD because the inflammatory process in the bowel was believed to be triggered by a specific bacterial pathogen. Elimination of this bacterial target would allow the inflammatory process to resolve. However, current clinical guidelines do not recommend use of antibiotic agents to induce or maintain clinical remission in patients with CD because there is no definitive evidence to suggest a benefit to using antibiotics in this way.
How was this study performed?
A systematic review of current literature was performed to determine whether antibiotic therapy is effective to induce or maintain remission in CD. An electronic search of several databases was performed and studies that met our inclusion criteria were selected for further evaluation. Statistical analyses were performed to determine which specific antibiotics had an overall benefit.
What were the results?
Several antibiotics, including ciprofloxacin, metronidazole, clarithromycin, rifaximin and cotrimoxazole, have been studied in CD. Most of the included studies were small in size. When we pooled antibiotics as a class, these drugs provided a modest benefit over placebo (i.e. a fake drug such as a sugar pill) for induction of remission and improvement of CD symptoms. For example, remission rates were 45% (253/542) in participants who received antibiotics compared to 36% (82/231) in participants who received placebo. We rated the quality of evidence supporting this outcome as high. Few studies assessed the use of antibiotics for maintenance of remission in CD. The impact of antibiotics on preventing relapse in CD is uncertain. Antibiotics do not appear to increase the risk of side effects when compared to placebo. Common side effects reported in the studies included gastrointestinal upset, upper respiratory tract infection, abscess formation, headache, change in taste and paraesthesia (pins and needles in the extremities). Serious side effects were not well reported in the studies and the impact of antibiotics on the risk of serious side effects is uncertain.
Conclusions
Moderate to high quality evidence suggests that any benefit provided by antibiotics in active CD is likely to be very modest. High quality evidence suggests that there is no increased risk of side effects with antibiotics compared to placebo. The effect of antibiotics on the risk of serious side effects is uncertain. The effect of antibiotics on preventing relapse in CD is uncertain. Thus, no firm conclusions regarding the benefits and harms of antibiotics for maintenance of remission in CD can be drawn. More research is needed to determine the harms and benefits of antibiotic therapy in CD.
Summary of findings
Background
Description of the condition
Crohn's disease (CD) is an inflammatory disorder of the gastrointestinal tract that most commonly affects the ileum and the colon. Characteristic histologic features of the disease include transmural inflammation and mucosal ulceration. The exact etiology of CD is unclear, however both genetic and environmental factors are important contributors (Elson 2005; Scribano 2013). In this regard, the human microbiome is considered to be a key environmental risk factor.
In animal models, interactions between the mucosal immune system and commensal bacteria contribute to the observed pathological changes seen in CD (Elson 1995; Elson 2005; Rath 1999). Subsequent human studies have demonstrated that patients with CD have higher concentrations of intestinal and colonic bacteria (Scribano 2013), and higher populations of specific bacteria (Gevers 2014), compared to healthy controls. Patients with CD may also have impaired barrier function that facilitates translocation of microbes into the mucosa (Marks 2006).
Pathogenic bacterial strains, including Escherichia coli (Mylonaki 2005), have been isolated in the mucosal and mesenteric lymph nodes of these patients (Ambrose 1984). Furthermore, there is a change in the microbial composition with fewer species overall and a relative overrepresentation of Enterobacteriaceae,Proteobacteria,Actinobacteria (Sartor 2008), and Bacteroides (Barnich 2007). These observations support the notion that the pathological response in CD is driven by an abnormal response to the host microbiome and that manipulation of the flora though antibiotic treatment might be a potential therapy (Sartor 2008).
Description of the intervention
Given the proposed link between increased intestinal bacterial concentrations and chronic inflammation, antibiotics have been considered for the treatment of CD (Swidsinski 2002). Studies have suggested Escherichia coli as specific bacterial targets, among others (Mylonaki 2005; Sartor 2008).
How the intervention might work
Several antibiotics have been evaluated for the treatment of CD. Reduction of the bacterial load in the intestinal mucosa might reduce the pathological immune response in the intestinal mucosa (Scribano 2013; Swidsinski 2002). Furthermore, antibiotics also act to limit bacterial translocation and reduce the concentration of adherent bacteria to the lumen and mucosa (Scribano 2013). In patients who have high levels of Escherichia in their microbiome, treatment with mesalamine showed a decrease in intestinal inflammation. This further suggests the crucial role the gut microbiome may have in IBD pathophysiology and the potential use for antimicrobial agents (Kostic 2014). Cumulatively, these data have raised the possibility that alteration of the mucosal flora may have a therapeutic role in CD by inhibiting the stimulus for pathogenic immune responses (Ott 2004; Swidsinski 2002).
Why it is important to do this review
Given the extensive animal and human data that support the role of bacteria in the pathogenesis of CD, it is reasonable to postulate that antibiotic therapy might be effective for either induction or maintenance of remission in CD. However, several potential problems exist with this approach. First, use of broad‐spectrum antibiotics is a very blunt strategy that may aggravate the aforementioned dysbiosis. Second, the resident flora are determined by both genetic and dietary factors that may be difficult or impossible to modify on a chronic basis. Therefore, treatment, if effective, might have to be continued indefinitely. Finally broad‐spectrum antibiotic therapy is associated with important adverse effects, notably an increased risk of Clostridium difficile infection. For these reasons evidence from high quality randomized controlled trials (RCTs) is necessary before antibiotics are accepted as effective and safe for the treatment of CD.
No current recommendations exist regarding the antibiotic of choice, dose, or duration for treatment of CD. The most recent guidelines published by the World Gastroenterology Organisation support the use of antibiotics in perianal disease, fistulizing disease, and bacterial overgrowth secondary to stricturing disease, despite limited supporting evidence (Bernstein 2016). There is evidence regarding antibiotic use in post‐operative CD management (Bernstein 2016).
Objectives
To determine whether antibiotic therapy is safe and effective for induction or maintenance of remission in CD.
Methods
Criteria for considering studies for this review
Types of studies
RCTs of adult patients (> 15 years of age) were considered for inclusion. Induction of remission studies needed to have a minimum duration of at least four weeks to be considered for inclusion. Maintenance of remission studies needed to have a minimum duration of at least six months to be considered for inclusion.
Types of participants
Patients with active or quiescent CD (as defined by the original studies) were considered for inclusion.
Types of interventions
Trials that compare oral antibiotic therapy to a placebo or an active comparator were considered for inclusion.
Types of outcome measures
Primary outcomes
The primary outcome measure for induction of remission studies was the proportion of patients who failed to achieve remission, as defined by the original studies. The primary outcome for maintenance of remission studies was the proportion of patients who relapsed, as defined by the included studies.
Secondary outcomes
Secondary efficacy outcomes, as defined by the original studies, were the proportion of patients:
1. Who failed to achieve clinical response (as defined by the original studies);
2. Who failed to achieve endoscopic response (as defined by the original studies);
3. Who failed to achieve endoscopic remission (as defined by the original studies);
4. Who failed to achieve histological response (as defined by the original studies);
5. Who failed to achieve histological remission (as defined by the original studies);
6. Who had an endoscopic relapse (as defined by the original studies);
7. Who failed to achieve both clinical and endoscopic response (as defined by the original studies);
8. Who failed to achieve both clinical and endoscopic remission (as defined by the original studies); and
9. Health‐related quality of life (as measured by a validated quality of life instrument). Safety outcomes were the proportion of patients:
10. With any adverse event (AE);
11. With serious adverse events (SAE); and
12. Who withdrew from the study due to adverse events.
Search methods for identification of studies
Electronic searches
We searched the following databases for relevant studies:
1. MEDLINE (Ovid, 1946 to present);
2. Embase (Ovid, 1984 to present);
3. CENTRAL; and
4. The Cochrane IBD Group Specialized Register.
5. Clinicaltrials.gov
The search strategies are listed in Appendix 1.
Searching other resources
We also searched the references listed in relevant studies and review articles for additional citations not identified in the search. Furthermore, conference proceedings from major meetings (Digestive Disease Week, the European Crohn's and Colitis Organisation congress, and the United European Gastroenterology Week conference) from the last five years were searched for studies published in abstract form only.
Data collection and analysis
Selection of studies
Two authors (CMT and CEP) screened the search results independently for eligible studies based on the inclusion criteria as listed. Disagreements were discussed until a consensus is reached. Any disagreements were brought to a third author (JKM) for resolution.
Data extraction and management
Data were extracted from included studies by two independent authors (CMT and CEP). Any disagreements over extracted data were first discussed and then brought to a third author (JKM) for resolution if deemed necessary.
Assessment of risk of bias in included studies
The methodological quality of included studies was independently assessed by two authors (CMT and CEP) using the Cochrane risk of bias tool (Higgins 2011). We assessed several factors including sequence generation, allocation sequence concealment, blinding, incomplete outcome data, selective outcome reporting and other potential sources of bias. Studies were judged to be at high, low or unclear risk of bias. Any disagreements regarding risk of bias were first discussed and then brought to a third author (JKM) for resolution.
We used the GRADE approach to determine the overall certainty of evidence supporting both primary and selected secondary outcomes (Guyatt 2008; Schünemann 2011). For the 'Summary of findings' tables, we included the following outcomes: failure to achieve clinical remission (at study endpoint), failure to maintain clinical remission (or relapse at study endpoint), failure to achieve clinical response (at study endpoint), failure to maintain endoscopic remission (or endoscopic relapse at study endpoint), adverse events, adverse events, serious adverse events and study withdrawal due to adverse events. Evidence from RCTs was considered high certainty. However, the certainty of the evidence could have been downgraded after considering the following factors:
1. Risk of bias;
2. Indirect evidence;
3. Inconsistency (unexplained heterogeneity);
4. Imprecision; and
5. Publication bias.
Each outcome was reviewed to determine the overall certainty of evidence supporting the outcome. The outcome was classified as high certainty (the estimate of effect is very unlikely to be changed despite further research); moderate certainty (the estimate of effect is unlikely to be changed despite further research); low certainty (the estimate of effect may be changed despite further research) or very low certainty (the estimate of effect likely will be changed with further research).
Measures of treatment effect
Review Manager (RevMan 5.3.5) was used to analyse the data on an intention‐to‐treat (ITT) basis. We calculated the risk ratio (RR) and corresponding 95% confidence interval (95% CI) for dichotomous outcomes and the mean difference (MD) and corresponding 95% CI for continuous outcomes.
Unit of analysis issues
To deal with repeated observations on participants, we determined appropriate fixed intervals for follow‐up for each outcome. Cross‐over trials were included if data was available for the first phase of the trial prior to cross‐over. To deal with events that may re‐occur (e.g. adverse events), we reported on the proportion of participants who experience at least one event. Separate comparisons were performed for studies that compared antibiotics to placebo and for studies that compared antibiotics to other active therapies. We also performed separate comparisons for each type of antibiotic. If we encountered multiple treatment groups (e.g. different dose groups of antibiotics), we divided the placebo group across the treatment groups or we combined groups to create a single pair‐wise comparison as appropriate.
Dealing with missing data
An intention‐to‐treat analysis was used for dichotomous outcomes whereby patients with missing treatment outcomes were assumed to be treatment failures. Sensitivity analyses were performed to assess the impact of this assumption on the effect estimate.
Assessment of heterogeneity
Heterogeneity was assessed using the Chi² test (a P value of 0.10 was considered statistically significant) and the I² statistic. We considered an I² statistic> 75% to indicate high heterogeneity among study data, > 50% indicated moderate heterogeneity and > 25% will indicated low heterogeneity (Higgins 2003). Sensitivity analysis were conducted to explore possible explanations for heterogeneity.
Assessment of reporting biases
We initially compared outcomes listed in the protocol to those reported in the published manuscript. If we did not have access to the protocol, we used the outcomes listed in the methods sections of the published manuscript compared to what was reported in the results section. If any pooled analyses included 10 or more studies, we investigated potential publication bias using funnel plots (Egger 1997).
Data synthesis
Data for meta‐analysis from individual trials were combined when the interventions, patient groups and outcomes were similar, as deemed by author consensus. We calculated the pooled RR and corresponding 95% CI for dichotomous outcomes and the pooled MD and corresponding 95% CI for continuous outcomes. The standardized mean difference (SMD) and 95% CI was calculated when different scales were used to measure the same outcome. A fixed‐effect model was used to pool data unless significant heterogeneity existed between the studies. A random‐effects model was used if heterogeneity existed (I² = 50 to 75%). We did not pool data for meta‐analysis if a high degree of heterogeneity (I² ≥ 75%) was found.
Subgroup analysis and investigation of heterogeneity
Planned subgroup analysis (data allowing) included:
a) Patient baseline characteristics (i.e. sex, age, weight, disease duration, disease severity, time since diagnosis, concomitant medication, objective markers of inflammation such as C‐reactive protein, and previous exposure to anti‐tumour necrosis factor‐alpha therapy); and
b) Different antibiotic doses.
Sensitivity analysis
We planned to use sensitivity analysis to assess the impact of random‐effects and fixed‐effect modelling, risk of bias, type of report (full manuscript, abstract or unpublished data) and loss to follow‐up on the pooled effect estimate.
Results
Description of studies
Results of the search
The literature search conducted on 28 February 2018 retrieved 2334 records for consideration. We removed all duplicate records, which left 1803 records for screening. Two authors (CMT and CEP) reviewed the titles and abstracts independently and in duplicate. Forty‐eight articles were selected for full text review (see Figure 1). Thirty reports of 25 studies were excluded with reasons (See Characteristics of excluded studies). Seventeen reports of 13 trials met the inclusion criteria and were included in the review (See Characteristics of included studies). One ongoing study was identified (NCT02240108).
1.

Study flow diagram.
Included studies
Of the 13 eligible RCTs identified (N = 1303), five different antibiotics (ciprofloxacin, metronidazole, clarithromycin, rifaximin and cotrimoxazole) were evaluated. Eleven of these trials were placebo‐controlled (Ambrose 1985; Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009; West 2004), and two were active comparator trials (Colombel 1999; Prantera 1996). Two of the placebo‐controlled trials also included active comparator arms (Ambrose 1985; Thia 2009). Patients were adults with active CD at the time of randomisation. The majority of the included studies defined an adult as 18 years of age or older, however, Steinhart 2002, Ambrose 1985 and Thia 2009 included patients 14, 15 and 16 years of age or older, respectively.
Four placebo‐controlled RCTs (Arnold 2002; Dewint 2014; Thia 2009; West 2004) evaluated ciprofloxacin. One active comparator trial randomised patients to ciprofloxacin or oral mesalamine (Colombel 1999). In two studies, ciprofloxacin was administered in conjunction with an anti‐TNF agent (Dewint 2014; West 2004). In the Dewint 2014 study, all patients were treated with self‐administered adalimumab at an induction dose of 160 mg at day 0 and 80 mg at week 2, followed by maintenance of 40 mg every 4 weeks until week 24. In the West 2004 study, all participants received infliximab at a dose of 5 mg/kg at weeks 6, 8 and 12. Rifaximin was evaluated in two placebo‐controlled RCTs (Prantera 2006; Prantera 2012). Metronidazole was studied in three induction trials (Ambrose 1985; Steinhart 2002; Thia 2009) and one maintenance trial (Sutherland 1991). Two studies evaluated metronidazole in combination with other therapeutic agents. In Prantera 1996, patients were assigned to metronidazole combined with ciprofloxacin or methylprednisolone, while patients enrolled in Steinhart 2002 received metronidazole combined with ciprofloxacin or placebo. All participants in the Steinhart 2002 study received budesonide (9 mg/day). One study compared a combination of cotrimoxazole and metronidazole with placebo (Ambrose 1985). Two trials compared clarithromycin to placebo (Lieper 2008; Selby 2007). In Lieper 2008 patients were randomised to placebo or clarithromycin and followed for three months. Selby 2007 assigned patients to clarithromycin, oral rifabutin, oral clofazimine or placebo, in addition to a tapering course of prednisolone.
Excluded studies
Twenty‐five studies were excluded with reasons after the full text review was performed. In ten studies, data on outcomes of interest were not available in the manuscript, (Allan 1997; Biancone 1998; Goodgame 2001; Gui 1997; Hartley‐Asp 1981; Jigaranu 2014; Laudage 1983; Lee 2018; Mitelman 1982; Turunen 1995) Two of these studies were cross‐over trials that did not report on outcomes pre‐crossover (Blichfeldt 1978; Ursing 1982). Nine trials were not RCTs (Bernstein 1992; Gilat 1982; Jaworski 2016; Koretz 1997; Leiper 2000; Melmed 2009; Ronge 2007; Steele 2009; To 1995). Three trials was terminated and data were not available (Koch 2007; Rogler 2014; Steinhart 2008). One study evaluated rectal therapy, which was beyond the scope of this review (Maeda 2010).
Risk of bias in included studies
The risk of bias for the included studies is summarized in Figure 2. Overall, most studies received low or unclear risk of bias ratings for the for seven domains.
2.
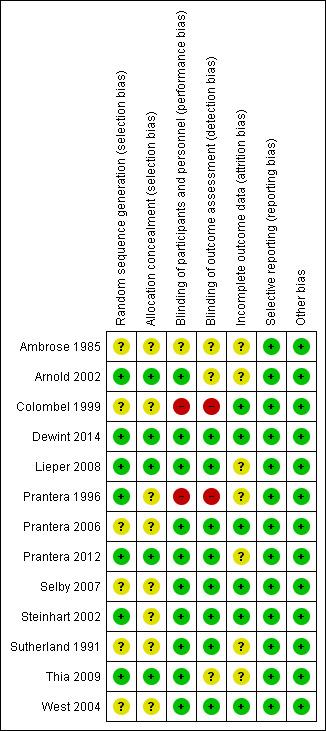
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Six of the included studies did not adequately describe the methods used to for random sequence generation and therefore received an unclear risk of bias assessment for this domain (Ambrose 1985; Colombel 1999; Prantera 2006; Selby 2007; Sutherland 1991; West 2004). The remaining seven studies were rated as low risk of bias for this item (Arnold 2002; Dewint 2014; Lieper 2008; Prantera 1996; Prantera 2012; Steinhart 2002; Thia 2009).
Eight of the included studies did not adequately describe the methods used to conceal allocation and therefore received an unclear risk of bias rating for this domain (Ambrose 1985; Colombel 1999; Prantera 1996; Prantera 2006; Selby 2007; Steinhart 2002; Sutherland 1991; West 2004) . The remaining five studies received a low risk of bias rating for this item (Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2012; Thia 2009) .
Blinding
One study did not adequately describe whether participants and personnel were blinded, and therefore received an unclear risk of bias rating for this domain (Ambrose 1985). A total of 10 studies were rated as low risk of bias for blinding of participants and personnel (Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009; West 2004). Two studies were rated as high risk of bias for this domain (Colombel 1999; Prantera 2006). In Colombel 1999, participants and investigators were not blinded. In Prantera 1996, patients were blinded but some investigators were unblinded.
It was unclear whether outcome assessors were blinded in three studies (Ambrose 1985; Arnold 2002; Thia 2009). Eight of the included studies were rated as low risk of bias for blinded of outcome assessment (Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; West 2004), while two studies did not employ blinded outcome assessment and received high risk of bias ratings (Colombel 1999; Prantera 1996).
Incomplete outcome data
Seven of the included studies were rated as unclear risk of bias with regard to incomplete outcome data (Ambrose 1985; Arnold 2002; Lieper 2008; Prantera 1996; Prantera 2012; Sutherland 1991; Thia 2009). The remaining six studies were rated as low risk of bias (Colombel 1999; Dewint 2014; Prantera 2006; Selby 2007; Steinhart 2002; West 2004).
Selective reporting
All studies were rated as low risk of bias for selective reporting (Ambrose 1985; Arnold 2002; Colombel 1999; Dewint 2014; Lieper 2008; Prantera 1996; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009; West 2004).
Other potential sources of bias
All studies were rated as low risk of bias for other sources of bias (Ambrose 1985; Arnold 2002; Colombel 1999; Dewint 2014; Lieper 2008; Prantera 1996; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009; West 2004).
Effects of interventions
Summary of findings for the main comparison. Antibiotic compared to placebo for induction and maintenance of remission in Crohn's disease.
| Antibiotic compared to placebo for induction and maintenance of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Antibiotic Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Antibiotic | |||||
| Failure to enter clinical remission Follow‐up: 6‐10 weeks |
645 per 1,000 | 555 per 1,000 (490 to 632) | RR 0.86 (0.76 to 0.98) | 773 (7 RCTs) | ⊕⊕⊕⊕ HIGH | Clinical remission was defined as CDAI ≤150 Antibiotics included Cotrimoxazole, Metronidazole, Ciprofloxacin, Clarithromycin, and Rifaximin |
| Failure to maintain clinical remission Follow‐up: 52 weeks |
569 per 1,000 | 495 per 1,000 (296 to 837) | RR 0.87 (0.52 to 1.47) | 155 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | Clinical remission was defined as CDAI ≤150 Antibiotics included Cotrimoxazole and Clarithromycin |
| Failure to achieve clinical response Follow‐up: 10‐14 weeks |
492 per 1,000 | 379 per 1,000 (315 to 458) | RR 0.77 (0.64 to 0.93) | 617 (5 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | Clinical response was defined as a reduction in CDAI score of 100 points and/or a 50% or greater reduction in perianal fistulas Antibiotics included Ciprofloxacin and Rifaximin |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 6‐52 weeks |
451 per 1,000 | 392 per 1,000 (338 to 460) | RR 0.87 (0.75 to 1.02) | 852 (9 RCTs) | ⊕⊕⊕⊕ HIGH | Adverse events included gastrointestinal upset, upper respiratory tract infection, abscess formation, headache and paraesthesia Antibiotics included Cotrimoxazole, Metronidazole, Ciprofloxacin, Clarithromycin, and Rifaximin |
| Serious adverse events Follow‐up: 6‐52 weeks |
7 per 1,000 | 12 per 1,000 (2 to 70) | RR 1.70 (0.29 to 10.01) | 520 (3 RCTs) | ⊕⊕⊝⊝ LOW4 | Serious adverse events were not well described in the studies. Reported serious adverse events included one scrotal edema and one death Antibiotics included Rifaximin, Ciprofloxacin and Metronidazole |
| Withdrawal due to adverse events Follow‐up: 6‐52 weeks |
125 per 1,000 | 107 per 1,000 (71 to 161) | RR 0.86 (0.57 to 1.29) | 858 (9 RCTs) | ⊕⊕⊝⊝ LOW 5 | Adverse events leading to withdrawal included worsening CD, gastrointestinal symptoms,headache, abscess, rash, arthralgia, nausea, vomiting, arthropathy and infusion reaction Antibiotics included Cotrimoxazole, Metronidazole, Ciprofloxacin, Clarithromycin, and Rifaximin |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to sparse data (78 events)
2 Downgraded one level due to heterogeneity (I2 = 63%)
3 Downgraded one level due to sparse data (267 events)
4 Downgraded two levels due to very sparse data (8 events)
5 Downgraded one level due to sparse data (89 events)
Summary of findings 2. Antibiotic with anti‐TNF compared to placebo with anti‐TNF for induction of remission in Crohn's disease.
| Antibiotic with anti‐TNF compared to placebo with anti‐TNF for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Antibiotic with anti‐TNF Comparison: Placebo with anti‐TNF | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo with anti‐TNF | Risk with Antibiotic with anti‐TNF | |||||
| Failure to enter clinical remission | Not reported | This outcome was reported in one study. We decided to pool this study with the other anti‐TNF study below (failure to achieve clinical response or remission Antibiotics included Ciprofloxacin |
||||
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to achieve clinical response or remission Follow‐up: 12 weeks |
365 per 1,000 | 208 per 1,000 (106 to 402) | RR 0.57 (0.29 to 1.10) | 100 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 | Clinical response was defined as a 50% reduction in perianal fistulas. Remission was defined as a closure of fistulas Antibiotics included Ciprofloxacin |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12 weeks |
827 per 1,000 | 769 per 1,000 (628 to 926) | RR 0.93 (0.76 to 1.12) | 100 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 2 | Adverse events included nausea, vomiting, upper respiratory tract infections, fatigue and headache Antibiotics included Ciprofloxacin |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 12 weeks |
77 per 1,000 | 63 per 1,000 (15 to 265) | RR 0.82 (0.19 to 3.45) | 100 (2 RCTs) | ⊕⊕⊝⊝ LOW 3 | Adverse events leading to withdrawal included gastrointestinal symptoms, transfusion reaction and herpes simplex virus infection Antibiotics included Ciprofloxacin |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded one level due to sparse data (80 events)
2 Downgraded two levels due to very sparse data (29 events)
3 Downgraded two levels due to very sparse data (7 events)
Ciprofloxacin versus placebo
Failure to enter clinical remission at week 10 or 12
Two placebo‐controlled trials involving a total of 65 patients reported on the proportion of patients who failed to enter clinical remission at week 10 or 12 (Arnold 2002; Thia 2009). Forty‐five per cent (17/38) of patients who received ciprofloxacin (500 mg twice daily) failed to achieve clinical remission compared with 74% (20/27) of patients assigned to placebo (RR 0.61, 95% CI 0.41 to 0.92). No heterogeneity was detected for this comparison (I² = 31%). The GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 3).
1. Ciprofloxacin (500 mg twice daily) compared to placebo for induction of remission in Crohn's disease.
| Ciprofloxacin compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Ciprofloxacin | |||||
| Failure to enter clinical remission Follow‐up:10‐12 weeks |
741 per 1,000 | 489 per 1,000 (311 to 770) | RR 0.66 (0.42 to 1.04) | 65 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission Follow‐up: 24 weeks |
842 per 1,000 | 320 per 1,000 (185 to 573) |
RR 0.38 (0.22 to 0.68) |
47 (1 RCT) |
⊕⊕⊝⊝ LOW 2 | Clinical remission was defined as CDAI ≤150 |
| Failure to have clinical response Follow‐up: 10 weeks |
875 per 1,000 | 403 per 1,000 (175 to 893) |
RR 0.46 (0.20 to 1.02) |
18 (1 RCT) |
⊕⊕⊝⊝ LOW 3 | Clinical response was defined as at least a 50% reduction in baseline draining fistulas |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 10‐24 weeks |
407 per 1,000 | 407 per 1,000 (232 to 717) | RR 1.00 (0.57 to 1.76) | 65 (2 RCTs) | ⊕⊕⊝⊝ LOW 4 | Adverse events included upper respiratory tract infection, abscess, open fistula, arthralgias and unpleasant taste/sore mouth |
| Serious adverse events | 0 per 1,000 | 0 per 1,000 (0 to 0) |
not estimable | 18 (1 RCT) | No serious adverse events were observed | |
| Withdrawal due to adverse events Follow‐up: 10‐24 weeks |
148 per 1,000 | 50 per 1,000 (10 to 247) | RR 0.34 (0.07 to 1.67) | 65 (2 RCTs) | ⊕⊕⊝⊝ LOW 5 | Withdrawals were due to worsening Crohn's disease |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (37 events)
2 Downgraded two levels due to very sparse data (25 events)
3 Downgraded two levels due to very sparse data (11 events)
4 Downgraded two levels due to very sparse data (26 events)
5 Downgraded two levels due to very sparse data (6 events)
Failure to maintain clinical remission at week 24
One study (Arnold 2002, N = 48) reported on failure to maintain clinical remission at 24 weeks. Thirty‐two per cent (9/28) of patients receiving ciprofloxacin (500 mg twice daily) relapsed at 24 weeks compared with 84% (16/19) of patients assigned to placebo (RR 0.38. 95% CI 0.22 to 0.68) (Arnold 2002). The GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 3).
Failure to have a clinical response at week 10
One study (Thia 2009) evaluated failure to achieve clinical response at week 10. Forty per cent (4/10) of patients assigned to ciprofloxacin (500 mg twice daily) failed to have a clinical response at week 10 compared with 88% (7/8) of placebo patients (RR 0.46. 95% CI 0.20 to 1.02). The GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data ( See Table 3).
Adverse events
Two studies that enrolled a total of 38 patients provided data on the proportion of patients with AEs (Arnold 2002; Thia 2009). Thirty‐nine per cent (15/38) of patients receiving ciprofloxacin (500 mg twice daily) experienced an AEcompared with 41% (11/27) of placebo patients (RR 1.0, 95% CI 0.57 to 1.76). No heterogeneity was detected for this comparison (I² = 0%). The GRADE analysis indicated that the certainty of evidence for this outcome was low due to very sparse data (See Summary of findings table 1). AEs in the ciprofloxacin group included Clostridium difficile infection, upper respiratory tract infection and abscess or open fistula. AEs in the placebo group included arthralgias, unpleasant taste/sore mouth and upper respiratory tract infections.
Serious adverse events
No patients in Thia 2009 reported serious SAEs. Arnold 2002 did not report on SAEs.
Withdrawal due to adverse events
Two studies (Arnold 2002; Thia 2009; N = 65) provided data on the proportion of patients who withdrew due to AEs. Seven per cent (2/38) of ciprofloxacin participants withdrew due to an AEcompared to 15% (4/27) placebo participants (RR 0.34, 95% CI 0.07 to 1.67). The overall certainty of evidence for this outcome was rated as low due very sparse data (SeeTable 3). Patients in both ciprofloxacin and placebo groups withdrew due to flare of disease.
Rifaximin versus placebo
Failure to enter clinical remission at week 12 or 14
Two placebo‐controlled trials enrolling a total of 489 patients reported on the proportion of patients who failed to enter clinical remission at week 12 or 14 (Prantera 2006; Prantera 2012). A total of 48% (174/360) of patients receiving rifaximin (800 mg to 2400 mg daily) failed to achieve remission compared with 60% (77/129) of those patients who received placebo (RR 0.82, 95% CI 0.69 to 0.98). No heterogeneity was detected (I² = 0%). The GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 4).
2. Rifaximin (800 mg to 2400 mg daily) compared to placebo for induction of remission in Crohn's disease.
| Rifaximin compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Rifaximin Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Rifaximin | |||||
| Failure to enter clinical remission Follow‐up 12‐14 weeks |
597 per 1,000 | 489 per 1,000 (412 to 585) |
RR 0.82 (0.69 to 0.98) |
489 (2 RCTs) |
⊕⊕⊕⊝ MODERATE1 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response Follow‐up: 12‐14 weeks |
519 per 1,000 | 426 per 1,000 (348 to 525) | RR 0.82 (0.67 to 1.01) | 489 (2 RCTs) | ⊕⊕⊕⊝ MODERATE2 | Clinical response was defined as reduction of CDAI ≥ 70 points and reduction in CDAI score of 100 points |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12‐14 weeks |
473 per 1,000 | 392 per 1,000 (312 to 492) | RR 0.83 (0.66 to 1.04) | 489 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | Adverse events included gastrointestinal disorders, infections, headache and ocular disorders |
| Serious adverse events Follow‐up: 12‐14 weeks |
8 per 1,000 | 9 per 1,000 (2 to 35) | RR 1.11 (0.27 to 4.54) | 489 (2 RCTs) | ⊕⊕⊝⊝ LOW 4 | The types of serious adverse events were not described by study authors |
| Withdrawal due to adverse events Follow‐up: 12 ‐14 weeks |
62 per 1,000 | 78 per 1,000 (37 to 164) | RR 1.25 (0.59 to 2.64) | 489 (2 RCTs) | ⊕⊕⊝⊝ LOW 5 | The adverse events leading to withdrawal were not described by study authors |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to sparse data (251 events)
2Downgraded one level due to sparse data (221 events)
3 Downgraded one level due to sparse data (201 events)
4 Downgraded two levels due to very sparse data (7 events)
5 Downgraded two levels due to very sparse data (37 events)
Planned subgroup analyses performed according to dose demonstrated 51% (67/131) of patients who received rifaximin 1600 mg once‐daily (OD) failed to enter clinical remission at week 12 or 14 compared with 62% (29/47) of patients who received placebo (RR 0.68, 95% CI 0.50 to 0.93). Forty per cent (51/126) of patients who received rifaximin 800 mg OD failed to enter clinical remission at week 12 or 14 compared with 60% (29/48) of patients who received placebo (RR 0.85, 95% CI 0.65 to 1.12) and 54% (56/103) of patients who received 2400 mg OD failed to enter clinical remission at week 12 or 14 compared with 56% (19/34)of placebo (RR 0.97, 95% CI 0.69 to 1.38). For dose, the test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 2.40, df = 2, P = 0.30, I² = 16.7%).
Failure to have a clinical response at weeks 12 or 14
In these same studies (Prantera 2006; Prantera 2012), 43% (154/360) patients receiving rifaximin (800 mg to 2400 mg daily)failed to respond at weeks 12 or 14 compared with 52% (67/129) of patients receiving placebo (RR 0.82, 95% CI 0.67 to 1.01). No heterogeneity was seen for this comparison (I² = 0%). The overall certainty of evidence for this outcome was moderate due to sparse data (See Table 4).
Planned subgroup analyses performed according to dose demonstrated a difference between the rifaximin 1600 mg once‐daily (OD) and placebo. However, no difference between the rifaximin 800 mg OD or 2400 mg OD group and the placebo group was observed. In patients who received rifaximin 800 mg daily, 46% (60/131) of patients treated with rifaximin failed to achieve response at 12 or 14 weeks compared with 51% (24/47) of patients treated with placebo (RR 0.91, 95% CI 0.65 to 1.28). No heterogeneity was seen for this comparison (I² = 0%). In patients who received 1600 mg of rifaximin daily, 32% (41/126) of patients on the study drug failed to respond, compared with 52% (25/48) of patients receiving placebo (RR 0.63, 95% CI 0.43 to 0.91). No heterogeneity was seen in this comparison (I² = 0%). In patients who received 2400 mg of rifaximin daily, 51% (53/103) of patients failed to respond compared with 53% (18/34) in the placebo group (RR 0.97, 95% CI 0.67 to 1.40). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 3.16, df = 2, P = 0.30, I² = 36.7%).
Adverse events
In total, 39% (140/360) patients who received rifaximin (800 mg to 2400 mg daily) reported an AE compared to 47% (61/129) of those who received placebo (RR 0.83, 95% CI 0.66 to 1.04). No heterogeneity was seen for this comparison (I² = 0%). The overall certainty of evidence for this outcome was moderate due to sparse data (See Table 4). AEs in the rifaximin group included gastrointestinal disorders, headache and skin and subcutaneous tissue disorders. AEs in the placebo group included gastrointestinal disorders, ocular disorders and headache.
Subgroup analysis by dose showed 34% (45/131) of patients taking rifaximin 800 mg had AEs compared with 49% (23/47) of patients who received placebo (RR 0.72, 95% CI 0.49 to 1.05). Forty per cent (50/126) of patients taking rifaximin 1600 mg had AEs compared with 48% (23/48) of patients who received placebo (RR 0.84, 95% CI 0.58 to 1.21). Forty four per cent (45/103) of patients taking rifaximin 2400 mg had AEs compared with 44% (15/34) of patients who received placebo (RR 0.99, 95% CI 0.64 to 1.53) (Prantera 2006; Prantera 2012). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 1.20, df = 2, P = 0.55, I² = 0%).
Serious adverse events
Two per cent (6/360) of patients who received rifaximin (800 mg to 2400 mg daily) reported a SAE compared with 1% (1/129) of patients in the placebo group (RR 1.11, 95% CI 0.27 to 4.54). No heterogeneity was seen in this comparison (I² = 0%). The overall certainty of evidence for this outcome was low due to very sparse data (See Table 4). The one SAE reported in Prantera 2006 included scrotal edema. SAEs were not well described in Prantera 2012. However, one death was reported in the rifaximin group. The investigators felt that this death was not related to treatment,
Subgroup analysis by dose showed 2% (2/131) of patients taking rifaximin 800 mg experienced SAEs compared with 0% (0/47) of patients who received placebo (RR 1.64, 95% CI 0.08 to 33.26). Two per cent (2/126) of patients taking rifaximin 1600 mg had a SAEs compared with 0% (0/48) of patients who received placebo (RR 1.31, 95% CI 0.14 to 12.08). Two per cent (2/103) of patients taking rifaximin 2400 mg experienced SAEss compared with 3% (1/34) of patients who received placebo (RR 0.66, 95% CI 0.06 to 7.05) (Prantera 2006; Prantera 2012). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 0.27, df = 2, P = 0.87, I² = 0%).
Withdrawal due to adverse events
Eight per cent (29/360) of patients receiving rifaximin (800 mg to 2400 mg daily) withdrew from studies due to AEs, compared to 6% (8/129) of patients receiving placebo (RR 1.25, 95% CI 0.59 to 2.64). No heterogeneity was seen in this comparison (I² = 0%). The overall certainty of evidence for this outcome was low due to very sparse data (See Table 4). A summary AEs that led to withdrawal was not reported by study authors.
Subgroup analysis by dose showed 4% (5/131) of patients taking rifaximin 800 mg withdrew due to AEs compared with 6% (3/47) of patients who received placebo (RR 0.56, 95% CI 0.14 to 2.16). Six per cent (8/126) of patients taking rifaximin 1600 mg withdrew due to AEs compared with 6% (3/48) of patients who received placebo (RR 1.07, 95% CI 0.30 to 3.83). Sixteen per cent (16/103) of patients taking rifaximin 2400 mg withdrew due to AEs compared with 6% (2/34) of patients who received placebo (RR 2.64, 95% CI 0.64 to 10.90)(Prantera 2006; Prantera 2012). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 2.42, df = 2, P = 0.30, I² = 17.5%).
Metronidazole versus placebo
Failure to enter clinical remission at week 6 or 10
Two placebo controlled trials (Ambrose 1985; Thia 2009), that comprised a total of 50 patients, reported on the number of patients who failed to enter clinical remission at weeks 6 or 10. One of these studies had failure of clinical remission as a primary end point (Ambrose 1985) and another had failure of clinical remission as a secondary end point (Thia 2009) at weeks 6 or 10. Two therapeutic doses of metronidazole (400 mg to 500 mg twice daily) were used in these studies. Sixty per cent (15/25) of patients who received metronidazole failed to enter clinical remission at week 6 or 10 compared with 68% (17/25) of patients who received placebo (RR 0.91, 95% CI 0.62 to 1.33). No heterogeneity was seen for this comparison (I² = 45%). A GRADE analysis indicated that the overall certainty of the evidence for the this outcome was low due to very sparse data (See Table 5).
3. Metronidazole (400 mg to 500 mg twice daily) compared to placebo for induction of remission in Crohn's disease.
| Metronidazole compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Metronidazole Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Metronidazole | |||||
| Failure to enter clinical remission Follow‐up: 6‐10 weeks |
680 per 1,000 | 619 per 1,000 (422 to 904) | RR 0.91 (0.62 to 1.33) | 50 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was defined as closure of all open actively draining fistulas at baseline |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response Follow‐up: 10 weeks |
875 per 1,000 | 604 per 1,000 (341 to 1,000) |
RR 0.69 (0.39 to 1.21) |
18 (1 RCT) |
⊕⊕⊝⊝ LOW 2 | Clinical response was defined as at least a 50% reduction in baseline draining fistulas |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 6‐10 weeks |
274 per 1,000 | 233 per 1,000 (88 to 633) | RR 0.85 (0.32 to 2.31) | 149 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 3 4 | Adverse events include gastrointestinal upset, abscess formation and arthropathy.paraesthesias and sore mouth. |
| Serious adverse events | 0 per 1,000 | 0 per 1,000 (0 to 0) | not estimable | 15 (1 RCT) | No serious adverse events were observed | |
| Withdrawal due to adverse events Follow‐up: 6‐10 weeks |
148 per 1,000 | 114 per 1,000 (53 to 248) | RR 0.77 (0.36 to 1.68) | 149 (3 RCTs) | ⊕⊕⊝⊝ LOW 5 | Withdrawal due to adverse events was most often due to headache, gastrointestinal symptoms, abscess formation, rash and arthralgia |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (32 events)
2 Downgraded two levels due to very sparse data (13 events)
3Downgraded one level due to serious inconsistency (I2 = 71%)
4 Downgraded two levels due to very sparse data (33 events)
5 Downgraded two levels due to very sparse data (19 events)
Failure to enter clinical remission at week 16
An additional study evaluated failure to achieve clinical remission at week 16 in 99 patients (Sutherland 1991). Remission in this case was defined by improvement in the patients Crohn's Disease Activity Index (CDAI) score to less than 150. The pooled analysis showed no difference between metronidazole (400 mg to 500 mg twice daily) and placebo for induction of clinical remission. Sixty‐eight per cent (43/63) of patients receiving metronidazole failed to achieve remission at week 16 compared with 67% (24/36) of patients receiving placebo (RR 1.03, 95% CI 0.77 to 1.36). No heterogeneity was seen for this comparison (I² = 0%).
Planned subgroup analysis according to dose showed no difference in clinical remission rates. Two different doses of metronidazole were used in this study. In the group that received 10 mg/kg of metronidazole, 64% (21/33) of patients failed to achieve remission at week 16, compared with 67% (12/18) in the group that received placebo (RR 0.95, 95% CI 0.63 to 1.45). In group that received 20 mg/kg of metronidazole, 73% (22/30) of patients failed to achieve remission compared with 67% (12/18) of patients assigned to placebo (RR 1.10, 95% CI 0.74 to 1.63). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 0.24, df = 1, P = 0.63, I² = 0%).
Failure to have clinical response at week 10
Thia 2009 evaluated failure to achieve clinical response at 10 weeks in 19 patients. Sixty per cent (6/10) of patients assigned to metronidazole failed to achieve clinical response at week 10 compared with 88% (7/8) of patients who received placebo (RR 0.69, 95% CI 0.39 to 1.21). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 5).
Adverse events
Eighteen per cent (16/87) of metronidazole patients reported AEs compared to 27% (17/62) of those assigned to placebo (RR 0.80, 95% CI 0.48 to 1.33). A GRADE analysis indicated that the overall certainty of the evidence for AEs was very low due to very serious inconsistency and very sparse data (See Summary of findings table 3). AEs in the metronidazole group included gastrointestinal upset, abscess formation and arthropathy. AEs in the placebo group included gastrointestinal upset, paraesthesias and sore mouth.
Serious adverse events
Thia 2009 reported no SAEs.
Withdrawal due to adverse events
Eleven per cent (10/88) of patients assigned to metronidazole withdrew from the study due to AEscompared with 15% (9/61) of patients on placebo (RR 0.77, 95% CI 0.36 to 1.68) (Ambrose 1985; Sutherland 1991; Thia 2009). A GRADE analysis indicated that the overall certainty of the evidence for withdrawal due to AEs was low due to very sparse data (See Table 5). Withdrawal due to AEs in the metronidazole group was most often due to headache, gastrointestinal symptoms and abscess formation and in the placebo group was most commonly due to rash and arthralgia.
Clarithromycin versus placebo
Failure to enter clinical remission at 12 weeks
One study that evaluated a total of 41 patients used clarithromycin as an induction agent (Lieper 2008). The primary end point of this study was clinical remission at 12 weeks as defined by CDAI ≤ 150. Eighty‐four per cent (16/19) of patients who received clarithromycin (1 g daily) failed to enter clinical remission at 12 weeks compared to 81% (18/22) of patients assigned to receive placebo (RR 1.03, 95% CI 0.78 to 1.36). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 6).
4. Clarithromycin (1 g/day) compared to placebo for induction of remission in Crohn's disease.
| Clarithromycin compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Clarithromycin Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Clarithromycin | |||||
| Failure to enter clinical remission Follow‐up:12 weeks |
818 per 1,000 | 843 per 1,000 (638 to 1,000) | RR 1.03 (0.78 to 1.36) | 41 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response Follow‐up: 12 weeks |
818 per 1,000 | 736 per 1,000 (532 to 1,000) |
RR 0.90 (0.65 to 1.26) |
41 (1 RCT) |
⊕⊕⊝⊝ LOW 2 | Clinical response was defined by CDAI reduction by ≥ 70 from baseline |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12 weeks |
45 per 1,000 | 210 per 1,000 (26 to 1,000) | RR 4.63 (0.57 to 37.96) | 41 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Adverse events included gastrointestinal symptoms |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 12 weeks |
500 per 1,000 | 370 per 1,000 (180 to 760) | RR 0.74 (0.36 to 1.52) | 41 (1 RCT) | ⊕⊕⊝⊝ LOW 4 | Withdrawal due to adverse events was most often due to gastrointestinal symptoms |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (34 events)
2 Downgraded two levels due to very sparse data (32 events)
3Downgraded two levels due to very sparse data (5 events)
4 Downgraded two levels due to very sparse data (18 events)
Failure to have clinical response at 12 weeks
Seventy‐four per cent (14/19) of clarithromycin (1 g daily) patients failed to have a clinical response at week 12, compared with 82% (18/22) of patients assigned to placebo (RR 0.90, 95% CI 0.65 to 1.26) (Lieper 2008).The certainty of evidence for this outcome was rated as low due to very sparse data (See Table 6).
Adverse events
Twenty‐one per cent (4/19) of patients who received clarithromycin (1 g daily) reported an AE compared with 5% (1/22) of patients in the placebo group (RR 4.63, 95% CI 0.57 to 37.96) (Lieper 2008). Most common AEs seen in both clarithromycin and placebo group were gastrointestinal symptoms..A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 6).
Serious adverse events
Lieper 2008 did not report on this outcome.
Withdrawal due to adverse events
Thirty‐seven per cent (7/19) of patients who received 1 g daily clarithromycin withdrew due to AEs, compared to 50% (11/22) of those in the placebo group (RR 0.74, 95% CI 0.36 to 1.52) (Lieper 2008). The most common reason for withdrawal due to AEs seen in both the clarithromycin and placebo groups was gastrointestinal symptoms. The overall certainty of the evidence for this outcome was low due to very sparse data (See Table 6).
Cotrimoxazole versus placebo
Failure to enter clinical remission at week 12
One study that evaluated 33 patients assessed the efficacy of cotrimoxazole (960 mg twice daily) induction therapy (Ambrose 1985). The primary end point of this study was an improvement in a clinical assessment score created by the Authors at week 12. This score was defined by the authors. Sixteen patients were randomised to the cotrimoxazole arm of this study and 17 received placebo. Sixty‐nine per cent (11/16) of patients who received cotrimoxazole failed to enter clinical remission at 12 weeks compared to 59% (10/17) of patients assigned to placebo (RR 1.17, 95% CI 0.70 to 1.96). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 7).
5. Cotrimoxazole (960 mg twice daily) compared to placebo for induction of remission in Crohn's disease.
| Cotrimoxazole compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Cotrimoxazole Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Cotrimoxazole | |||||
| Failure to enter clinical remission Follow‐up: week 12 |
588 per 1,000 | 688 per 1,000 (412 to 1,000) | RR 1.17 (0.70 to 1.96) | 33 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was defined as improvement in clinical assessment and laboratory indices |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have a clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12 weeks |
176 per 1,000 | 125 per 1,000 (25 to 653) | RR 0.71 (0.14 to 3.70) | 33 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Adverse events included nausea, vomiting and arthropathy |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 12 weeks |
59 per 1,000 | 125 per 1,000 (12 to 1,000) | RR 2.13 (0.21 to 21.22) | 33 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Withdrawal due to adverse events was most often due to nausea, vomiting and arthropathy |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (21 events)
2 Downgraded two levels due to very sparse data (5 events)
3 Downgraded two levels due to very sparse data (3 events)
Adverse events
One per cent (2/16) of patients in the cotrimoxazole (960 mg twice daily) group reported an AE compared with 18% (3/17) of patients who received placebo (RR 0.71, 95% CI 0.14 to 3.70) (Ambrose 1985). AEs in cotrimoxazole group included nausea, vomiting and arthropathy. AEs in the placebo group were not mentioned by study authors. The overall certainty of evidence for this outcome is low due to very sparse data (See Table 7).
Serious adverse events
Ambrose 1985 did not report on this outcome.
Withdrawal due to adverse events
Thirteen per cent (2/16) of patients receiving 960 mg twice daily cotrimoxazole withdrew due to AEs, compared to 6% (1/17) of patients in the placebo group (RR 2.13, 95% CI 0.21 to 21.22) (Ambrose 1985). AEs leading to withdrawal in the cotrimoxazole group included nausea, vomiting and arthropathy. AEs leading to study withdrawal in the placebo group were not described by the study authors.The overall certainty of evidence for this outcome was low due to very sparse data (See Table 7).
Ciprofloxacin and metronidazole versus methylprednisolone
Failure to enter clinical remission at week 12
Prantera 1996 (N=41) compared a combination of ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) to methylprednisolone (0.7‐1 mg/kg daily) induction therapy. The primary end point of this study was clinical remission at 12 weeks as defined by CDAI ≤ 150. Fifty‐five per cent (12/22) of patients in the antibiotic group failed to enter clinical remission at week 12, compared with 37% (7/19) of patients receiving methylprednisolone (RR 1.48, 95% CI 0.73 to 2.99). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 8).
6. Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) compared to methylprednisolone (0.7‐1 mg/kg daily) for induction and maintenance of remission in Crohn's disease.
| Ciprofloxacin and metronidazole compared to methylprednisone for induction and maintenance of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin and metronidazole Comparison: Methylprednisone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with methylprednisone | Risk with Ciprofloxacin and metronidazole | |||||
| Failure to enter clinical remission Follow‐up: 12 weeks |
368 per 1,000 | 545 per 1,000 (269 to 1,000) | RR 1.48 (0.73 to 2.99) | 41 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1 2 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission Follow‐up: 52 weeks |
684 per 1,000 | 773 per 1,000 (527 to 1,000) | RR 1.13 (0.77 to 1.65) | 41 (1 RCT) | ⊕⊕⊝⊝ VERY LOW2 3 | Clinical remission was defined as CDAI ≤150 |
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12‐52 weeks |
105 per 1,000 | 273 per 1,000 (62 to 1,000) | RR 2.59 (0.59 to 11.36) | 41 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Adverse events include nausea, metallic taste, reflux symptoms, Cushingoid facies and acne |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 12‐52 weeks |
105 per 1,000 | 273 per 1,000 (62 to 1,000) | RR 2.59 (0.59 to 11.36) | 41 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Withdrawal due to adverse events was most often due to nausea, vomiting, reflux symptoms, hypertension, elevated amylase, acne and tremor |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (19 events)
2 Downgraded one level due to high risk bias (blinding)
3 Downgraded two levels due to very sparse data (30 events)
4 Downgraded two levels due to very sparse data (8 events)
Failure to maintain clinical remission at week 52
Seventy‐seven per cent (17/22) of patients assigned to antibiotics failed to maintain clinical remission at week 52 compared to 68% (13/19) of patients who received methylprednisolone (0.7‐1 mg/kg daily) (RR 1.13, 95% CI 0.77 to 1.65) (Prantera 1996). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 8)
Adverse events
Twenty‐seven per cent (6/22) of patients who received combination antibiotic therapy reported an AE compared with 11%(2/19) of patients who received methylprednisolone (RR 2.59, 95% CI 0.59 to 11.36) (Prantera 1996). AEs in patients receiving the combination of ciprofloxacin and metronidazole included nausea, metallic taste and reflux symptoms. AEs in steroid group included Cushingoid facies, acne and reflux. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 8).
Serious adverse events
Prantera 1996 did not report on SAES.
Withdrawal due to adverse events
Twenty‐seven per cent (6/22) of patients receiving antibiotics withdrew from the study due to AEs, compared with 11% (2/19) of patients on steroid (RR 2.59, 95% CI 0.59 to 11.36) (Prantera 1996). AEs leading to withdrawal in patients receiving ciprofloxacin and metronidazole included nausea, vomiting and reflux symptoms. AEs in steroid group resulting in withdrawal included hypertension, elevated amylase, acne and tremor. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 8).
Ciprofloxacin and metronidazole and budesonide versus placebo and budesonide
Failure to enter clinical remission at week 8
One study (N = 134) compared a combination of ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) to placebo (Steinhart 2002). Both groups received oral budesonide (9 mg daily) induction therapy. The primary end point of this study was clinical remission at eight weeks as defined by CDAI < 150. Sixty‐eight per cent (45/66) of patients in the antibiotic group failed to achieve clinical remission at week 12, compared with 62% (43/69) of patients who received placebo (RR 1.08, 95% CI 0.84 to 1.38). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 9).
7. Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg/daily) compared to placebo with budesonide (9 mg/daily) for induction of remission in Crohn's disease.
| Ciprofloxacin and metronidazole and budesonide compared to placebo with budesonide for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin and metronidazole Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Ciprofloxacin and metronidazole | |||||
| Failure to enter clinical remission Follow‐up: 8 weeks |
632 per 1,000 | 683 per 1,000 (531 to 873) | RR 1.08 (0.84 to 1.38) | 134 (1 RCT) | ⊕⊕⊕⊝ MODERATE 1 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 8 weeks |
0 per 1,000 | 0 per 1,000 (0 to 0) | RR 27.81 (1.69 to 458.44) | 134 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Adverse events included taste disturbance, dizziness, gastrointestinal upset and vaginitis |
| Serious adverse events Follow‐up: 8 weeks |
29 per 1,000 | 46 per 1,000 (8 to 263) | RR 1.55 (0.27 to 8.95) | 134 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | The types of serious adverse events were not reported |
| Withdrawal due to adverse events Follow‐up: 8 weeks |
0 per 1,000 | 0 per 1,000 (0 to 0) | RR 27.81 (1.69 to 458.44) | 134 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Withdrawals due to adverse events were most often due to nausea, taste disturbance and rash |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to sparse data (88 events)
2 Downgraded two levels due to very sparse data (13 events)
3 Downgraded two levels due to very serious imprecision (5 events)
Adverse events
Twenty per cent (13/66) of patients receiving antibiotics reported an AE compared with 0% (0/68) of patients who received placebo (RR 27.81, 95% CI 1.69 to 458.44) (Steinhart 2002). Common AEs in the antibiotics group included taste disturbance, dizziness and gastrointestinal upset. Common AEs in the placebo group included gastrointestinal upset, dizziness and vaginitis.The overall certainty of evidence for this outcome was low due to very sparse data (See Table 9).
Serious adverse events
Five per cent (3/66) of patients receiving antibiotics experienced a SAE compared with 0% (0/68) of patients who received placebo (RR1.55, 95% CI 0.27 to 8.95) (Steinhart 2002). Specific SAEs were not described by the study authors.The overall certainty of evidence for this outcome was low due to very sparse data (See Table 9).
Withdrawal due to adverse events
Twenty per cent (13/66) of patients receiving antibiotics withdrew from the study due to AEs, compared with 0% (0/68) of patients who received placebo (RR 27.81, 95% CI 1.69 to 458.44) (Steinhart 2002). Patients in the antibiotic group withdrew due to nausea, taste disturbance and rash. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 9).
Ciprofloxacin versus mesalazine
Failure to enter clinical remission at week 6
Colombel 1999 (N = 40) compared ciprofloxacin (500 mg twice daily) with mesalamine (2 g twice daily) induction therapy. The primary end point of this study was clinical remission at six weeks defined by a CDAI ≤ 150. Forty‐four per cent (8/18) of patients who received ciprofloxacin failed to enter clinical remission at week six, compared with 45% (10/22) of patients assigned to mesalamine (RR 0.98, 95% CI 0.49 to 1.95). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was very low due to very sparse data and high risk of bias (lack of blinding) (See Table 10).
8. Ciprofloxacin (500 mg twice daily) compared to mesalazine (2 g twice daily) for induction of remission in Crohn's disease.
| Ciprofloxacin compared to mesalazine for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin Comparison: Mesalazine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with mesalazine | Risk with Ciprofloxacin | |||||
| Failure to enter clinical remission Follow‐up: 6 weeks |
455 per 1,000 | 445 per 1,000 (223 to 886) | RR 0.98 (0.49 to 1.95) | 40 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse Events Follow‐up: 6 weeks |
0 per 1,000 | 0 per 1,000 (0 to 0) | RR 3.63 (0.16 to 84.11) | 40 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 3 | Adverse events included abdominal pain |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 6 weeks |
0 per 1,000 | 0 per 1,000 (0 to 0) | RR 3.63 (0.16 to 84.11) | 40 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 3 | The withdrawal due to an adverse event was due to abdominal pain |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to high risk of bias (no blinding)
2 Downgraded two levels due to very sparse data (18 events)
3 Downgraded two levels due to very sparse data (1 event)
Adverse events
Six per cent (1/18) of patients who received ciprofloxacin reported an AE compared with 0% (0/22) of patients assigned to mesalamine (RR 3.63, 95% CI 0.16 to 84.11) (Colombel 1999). The AE in the ciprofloxacin group was abdominal pain. The overall certainty of evidence for this outcome was very low due to very sparse data and risk of bias (lack of blinding) (See Table 10).
Serious adverse events
Colombel 1999 did not report on SAEs.
Withdrawal due to adverse events
Six per cent (1/18) of patients who received ciprofloxacin withdrew from the study due to AEs, compared with 0% (0/22) of patients assigned to mesalamine (RR 3.63, 95% CI 0.16 to 84.11) (Colombel 1999). The AE that led to withdrawal in the ciprofloxacin group was abdominal pain.The overall certainty of evidence for this outcome was very low due to very sparse data and risk of bias (lack of blinding) (See Table 10).
Ciprofloxacin with adalimumab versus placebo with adalimumab
Failure to enter clinical remission at week 12
Dewint 2014 compared ciprofloxacin (500 mg twice daily) to placebo in 76 patients who also received concomitant adalimumab induction therapy (Dewint 2014). Secondary end points included clinical remission at twelve weeks as defined by CDAI. Twenty‐four per cent (9/37) of patients in the ciprofloxacin group failed to enter clinical remission at week 12, compared with 36% (14/39) of patients who received placebo (RR 0.68, 95% CI 0.33 to 1.37). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very serious imprecision (See Table 11).
9. Ciprofloxacin (500 mg twice daily) with adalimumab compared to placebo with adalimumab for induction and maintenance of remission in Crohn's disease.
| Ciprofloxacin with adalimumab compared to placebo with adalimumab for induction and maintenance of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin with adalimumab Comparison: Placebo with adalimumab | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo with adalimumab | Risk with Ciprofloxacin with adalimumab | |||||
| Failure to enter clinical remission Follow‐up: 12 weeks |
359 per 1,000 | 244 per 1,000 (118 to 492) | RR 0.68 (0.33 to 1.37) | 76 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was defined as the closure of all fistulas All patients were treated with self‐administered adalimumab (patients were given induction dosing of 160 mg at day 0 and 80 mg at week 2, followed by maintenance of 40 mg every 4 weeks until week 24) |
| Failure to maintain clinical remission Follow‐up: 24 weeks |
See comments | Although the authors reported on this outcome, we did not include it as participants only received ciprofloxacin up to week 12. All participants received maintenance adalimumab after week 12 | ||||
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse Events Follow‐up: 12‐24 weeks |
872 per 1,000 | 837 per 1,000 (697 to 1,000) | RR 0.96 (0.80 to 1.16) | 76 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | Adverse events included respiratory tract infection, fatigue and headache |
| Serious adverse events Follow‐up: 12‐24 weeks |
77 per 1,000 | 81 per 1,000 (18 to 377) | RR 1.05 (0.23 to 4.90) | 76 (1 RCT) | ⊕⊕⊝⊝ LOW 4 | Serious adverse events included sagittal sinus thrombosis, severe disease flares, herpes simplex infection and parastomal herniation |
| Withdrawal due to adverse events Follow‐up: 12‐24 weeks |
77 per 1,000 | 54 per 1,000 (9 to 305) | RR 0.70 (0.12 to 3.97) | 76 (1 RCT) | ⊕⊕⊝⊝ LOW 5 | Specific adverse events causing withdrawal were not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (23 events)
2 Downgraded two levels due to very sparse data (29 events)
3 Downgraded one level due to sparse data (65 events)
4 Downgraded two levels due to very sparse data (6 events)
5 Downgraded two levels due to very sparse data (5 events)
Adverse events
Eighty‐four per cent (31/37) of ciprofloxacin (500 mg twice daily) patients reported an AE compared with 87% (34/39) of patients assigned to placebo (RR 0.96, 95% CI 0.80 to 1.16) (Dewint 2014). Common AEs included upper respiratory tract infection, fatigue and headache.The overall certainty of evidence for this outcome is moderate due to sparse data (See Table 11).
Serious adverse events
Eight per cent (3/37) of patients who received ciprofloxacin (500 mg twice daily) had a SAE compared with 8% (3/39) of patients assigned to placebo (RR 1.05, 95% CI 0.23 to 4.90) (Dewint 2014). SAEs in patients who received ciprofloxacin included sagittal sinus thrombosis and severe disease flares. SAEs in patients who received placebo included herpes simplex infection, parastomal herniation and severe disease flares. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 11).
Withdrawal due to adverse events
Five per cent (2/37) of patients who received ciprofloxacin (500 mg twice daily) withdrew from the study due to AEs, compared with 8% (3/39) of patients assigned to placebo (RR 0.70, 95% CI 0.12 to 3.97) (Dewint 2014). The specific AEs causing withdrawal were not described by the study authors. The overall certainty of evidence for this outcome is low due to very sparse data (See Table 11).
Ciprofloxacin with infliximab versus placebo with infliximab
Failure to have clinical response at week 12
West 2004 (N = 24) compared ciprofloxacin (500 mg twice daily) to placebo in patients who also received infliximab therapy for the treatment of fistulizing CD. The primary endpoint was clinical response defined as 50% or greater reduction in draining fistulae confirmed by no drainage despite firm finger compression. Secondary end points included AEs and withdrawal due to AEs. Twenty‐four patients were enrolled in this trial. Nine per cent (1/11) of patients in the ciprofloxacin group failed to have a clinical response at week 12, compared with 38% (5/13) of patients who received placebo (RR 0.24, 95% CI 0.03 to 1.73). GRADE analysis indicated that the overall certainty of the evidence for the this outcome was low due to very sparse data (See Table 12).
10. Ciprofloxacin (500 mg twice daily) with infliximab compared to placebo with infliximab for induction of remission in Crohn's disease.
| Ciprofloxacin with infliximab compared to placebo with infliximab for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Ciprofloxacin with infliximab Comparison: Placebo with infliximab | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo with infliximab | Risk with Ciprofloxacin with infliximab | |||||
| Failure to enter clinical remission | Not reported | This outcome was not reported | ||||
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response Follow‐up: 12 weeks |
385 per 1,000 | 92 per 1,000 (12 to 665) | RR 0.24 (0.03 to 1.73) | 24 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Clinical response was defined as 50% or greater reduction in draining fistulae confirmed by no drainage despite firm finger compression |
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 12 weeks |
692 per 1,000 | 547 per 1,000 (284 to 1,000) | RR 0.79 (0.41 to 1.51) | 24 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Adverse events included nausea, rash and diarrhea and metallic taste |
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal due to adverse events Follow‐up: 12 weeks |
77 per 1,000 | 91 per 1,000 (6 to 1,000) | RR 1.18 (0.08 to 16.78) | 24 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Adverse events leading to withdrawal included infusion reaction and disease exacerbation |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (6 events)
2 Downgraded two levels due to very sparse data (15 events)
3 Downgraded two levels due to very sparse data (2 events)
Adverse events
Fifty‐five per cent (6/11) of patients who received ciprofloxacin (500 mg twice daily) reported an AE compared with 69% (9/13) of those who received placebo (RR 0.79, 95% CI 0.41 to 1.51) (West 2004). Common AEs in the ciprofloxacin group included nausea, rash, diarrhea, arthralgias, infusion reactions, and perianal abscess. Common AEs in the placebo group included metallic taste, exacerbation of CD, arthralgias, infusion reaction, headache, nausea and perianal abscess. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 12).
Serious adverse events
West 2004 did not report on SAEs.
Withdrawal due to adverse events
Nine per cent (1/11) of patients who received ciprofloxacin (500 mg twice daily) withdrew from the study due to an AE compared with 8% (1/13) of placebo patients (RR 1.18, 95% CI 0.08 to 16.78) (West 2004). AEs leading to withdrawal included an infusion reaction in the ciprofloxacin group and disease exacerbation in the placebo group. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 12).
Clarithromycin and antimycobacterial versus placebo
Failure to enter clinical remission at week 16
One study performed a controlled trial that compared combination antimycobacterial therapy with clarithromycin (750 mg daily), clofazimine (50 mg daily) and rifabutin (450 mg daily) to placebo as long term therapy for CD (Selby 2007). Primary end points were failure to maintain clinical remission at weeks 52, 104 or 156 weeks. Secondary end points were induction of remission at week 16 (CDAI ≤ 150), failure to achieve endoscopic remission and withdrawal due to AEs. Two‐hundred and thirteen patients were enrolled in the induction phase and one hundred and twenty‐two in the maintenance phase. Thirty‐four per cent (35/102) of patients in the clarithromycin group failed to have a clinical response at week 16, compared with 50% (56/111) of patients receiving placebo (RR 0.68, 95% CI 0.49 to 0.94). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 13).
11. Clarithromycin and antimycobacterial compared to placebo for maintenance of remission in Crohn's disease.
| Clarithromycin and antimycobacterial compared to placebo for maintenance of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Clarithromycin and antimycobacterial Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Clarithromycin and antimycobacterial | |||||
| Failure to enter clinical remission Follow‐up: 16 weeks |
505 per 1,000 | 343 per 1,000 (247 to 474) | RR 0.68 (0.49 to 0.94) | 213 (1 RCT) | ⊕⊕⊕⊝ MODERATE 1 | Clinical remission was defined as CDAI ≤150 |
| Failure to maintain clinical remission Follow‐up: 1 year |
564 per 1,000 | 389 per 1,000 (265 to 569) | RR 0.69 (0.47 to 1.01) | 122 (1 RCT) | ⊕⊕⊕⊝ MODERATE 2 | Clinical remission was defined as CDAI ≤150 |
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission Follow‐up: 3 years |
800 per 1,000 | 864 per 1,000 (736 to 1,000) | RR 1.08 (0.92 to 1.27) | 122 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | The definition of endoscopic remission was not reported |
| Adverse events | Not reported | This outcome was not reported | ||||
| Serious adverse events | Not reported | This outcome was not reported | ||||
| Withdrawal of adverse events Follow‐up: 16 weeks‐3 years |
73 per 1,000 | 75 per 1,000 (21 to 265) | RR 1.03 (0.29 to 3.64) | 122 (1 RCT) | ⊕⊕⊝⊝ LOW 4 | Adverse events leading to withdrawal included abdominal distention, abnormal liver enzymes, vaginal candidiasis and myalgia |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to sparse data (91 events)
2 Downgraded one level due to sparse data (57 events)
3 Downgraded one level due to sparse data (102 events)
4 Downgraded two levels due to very sparse data (9 events)
Failure to maintain clinical remission at 1 year
Thirty‐nine per cent (26/67) of patients taking clarithromycin (750 mg daily) failed to maintain clinical remission at one year compared to 56% (31/55) of patients receiving placebo (RR 0.69, 95% CI 0.47 to 1.01) (Selby 2007). The overall certainty of evidence for this outcome was moderate due to sparse data (SeeTable 13).
Failure to maintain endoscopic remission at 3 years
Eighty‐seven per cent (58/67) of patients who received clarithromycin (750 mg daily) failed to maintain endoscopic remission at three years compared to 80% (44/55) of patients who received placebo (RR 1.08, 95% CI 0.92 to 1.27) (Selby 2007). The overall certainty of evidence for this outcome was moderate due to sparse data (See Table 13).
Adverse events
Selby 2007 did not report on the proportion of participants who experienced at least one AE.
Serious adverse events
Selby 2007 did not report on the proportion of participants who had a SAE.
Withdrawal due to adverse events
Seven per cent (5/67) of patients who received clarithromycin withdrew from the study due to an AE, compared with 7% (4/55) of patients who received placebo (RR 1.03, 95% CI 0.29 to 3.64) (Selby 2007). AEs leading to withdrawal in the clarithromycin group included abdominal distention, abnormal liver enzymes and vaginal candidiasis. AEs leading to withdrawal in the placebo group included abdominal distention, vaginal candidiasis and myalgia. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 13).
Metronidazole and cotrimoxazole versus placebo
Failure to enter clinical remission at week 6
Ambrose 1985 (N=38) evaluated a combination of metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) induction therapy. The primary end point of this study was clinical remission at six weeks as defined by the authors' own scoring system. The secondary end points were AEs, SAEs or withdrawal due to AEs. Sixty‐two per cent (13/21) of patients in the antibiotic group failed to enter clinical remission at week six, compared with 59% (10/17) of patients assigned to placebo (RR 1.05, 95% CI 0.63 to 1.77). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 14).
12. Metronidazole and cotrimoxazole compared to placebo for induction of remission in Crohn's disease.
| Metronidazole and cotrimoxazole compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Participants with active CD Setting: Outpatient Intervention: Metronidazole and cotrimoxazole Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Metronidazole and cotrimoxazole | |||||
| Failure to enter clinical remission Follow‐up: 6 weeks |
588 per 1,000 | 618 per 1,000 (371 to 1,000) | RR 1.05 (0.63 to 1.77) | 38 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Clinical remission was based on clinical assessment and laboratory indices |
| Failure to maintain clinical remission | Not reported | This outcome was not reported | ||||
| Failure to have clinical response | Not reported | This outcome was not reported | ||||
| Failure to maintain endoscopic remission | Not reported | This outcome was not reported | ||||
| Adverse events Follow‐up: 6 weeks |
235 per 1,000 | 47 per 1,000 (5 to 388) | RR 0.20 (0.02 to 1.65) | 38 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Adverse events included nausea, vomiting, esophagitis and erythema nodosum |
| Serious adverse events Follow‐up: 6 weeks |
59 per 1,000 | 16 per 1,000 (1 to 371) | RR 0.27 (0.01 to 6.30) | 38 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | Serious adverse events included surgery abscess, nausea, vomitting and erythema nodosum |
| Withdrawal due to adverse events Follow‐up: 6 weeks |
59 per 1,000 | 48 per 1,000 (3 to 706) | RR 0.81 (0.05 to 12.01) | 38 (1 RCT) | ⊕⊕⊝⊝ LOW 4 | Adverse events leading to withdrawal included surgery abscess, nausea, vomiting and erythema nodosum |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very sparse data (23 events)
2 Downgraded two levels due to very sparse data (5 events)
3 Downgraded two levels due to very sparse data (1 event)
4 Downgraded two levels due to very sparse data (2 events)
Adverse events
Five per cent (1/21) of patients who received antibiotics reported an AE compared with 24% (4/17) of patients assigned to placebo (RR 0.20, 95% CI 0.02 to 1.65) (Ambrose 1985). Common AEs in the treatment group included nausea, vomiting, esophagitis and erythema nodosum. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 14).
Serious adverse events
Zero per cent (0/21) of patients receiving antibiotics reported a SAE compared with 6% (1/17) of patients in the placebo group (RR 0.27, 95% CI 0.01 to 6.30) (Ambrose 1985). The SAE in placebo group was described as a severe clinical deterioration requiring hospital admission. The overall certainty of evidence for this outcome was low due to very sparse data (See Table 14).
Withdrawal due to adverse events
Five per cent (1/21) of patients who received antibiotics withdrew from the study due to AEs, compared with 6% (1/17) of patients assigned to placebo (RR 0.81, 95% CI 0.05 to12.01) (Ambrose 1985). The specific AEs leading to withdrawal were not reported . The overall certainty of evidence for this outcome is low due to very sparse data (See Table 14).
Antibiotic versus placebo
Failure to enter clinical remission at weeks 6 to 10
Data from seven studies including 773 participants were pooled for failure to enter clinical remission at weeks 6 to 10 (Ambrose 1985; Arnold 2002; Lieper 2008; Prantera 2006; Prantera 2012; Sutherland 1991; Thia 2009). Fifty‐five per cent (289/524) of patients who received an antibiotic failed to achieve remission at weeks 6 to 10 compared with 65% (149/231) of patients assigned to placebo (RR 0.86, 95% CI 0.76 to 0.98). No heterogeneity was detected for this comparison (I² = 11%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was high (See Table 1).
Failure to enter clinical response at weeks 10, 12 or 14
Data from five studies including 617 participants were pooled for failure to have a clinical response at weeks 10, 12 or 14 endpoint (Dewint 2014; Prantera 2006; Prantera 2012; Thia 2009; West 2004). Forty‐one per cent (174/428) of patients who received antibiotic failed to achieve a clinical response at weeks 10, 12 or 14 compared with 49% (93/189) of patients assigned to placebo (RR 0.77, 95% CI 0.64 to 0.93). No heterogeneity was detected for this comparison (I² = 0%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 1).
Failure to maintain clinical remission at 52 weeks
Data from two studies including 155 participants were pooled for failure to maintain clinical remission at 52 weeks (Ambrose 1985; Selby 2007). Forty‐five per cent (37/83) of patients who received antibiotic failed to maintain clinical remission at 52 weeks compared with 57% (41/72) of patients who received placebo (RR 0.87, 95% CI 0.52 to 1.47). Moderate to high heterogeneity was detected for this comparison (I² = 63%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 1).
Adverse events
AEs were reported in nine studies that included 852 participants (Ambrose 1985; Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Sutherland 1991; Thia 2009; West 2004). Thirty‐eight per cent (214/568) of patients on antibiotics experienced an AE compared with 45% (128/284) of patients who received placebo (RR 0.87, 95% CI 0.75 to 1.02). No heterogeneity was detected for this comparison (I² = 6%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was high (See Table 1). Common AEs in the antibiotic group included gastrointestinal upset, upper respiratory tract infection, abscess formation and headache. Common AEs in placebo group included gastrointestinal upset, change of taste and paraesthesia.
Serious adverse events
SAEs were reported in three studies that included 520 participants (Ambrose 1985; Prantera 2006; Prantera 2012; Thia 2009). Two per cent (6/395) of patients who received antibiotic experienced a SAEs compared with 1%(2/160) of patients assigned to placebo (RR 1.12, 95% CI 0.26 to 4.76). No statistically heterogeneity was detected for this comparison (I² = 0%). GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 1). Many of the SAEs were not described by the study authors. One scrotal edema was reported in the Prantera 2006 study and one death was reported in the Prantera 2012 study.
Withdrawal due to adverse events
Study withdrawal due to AEs was reported in nine studies including 858 participants (Ambrose 1985; Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Sutherland 1991; Thia 2009; West 2004). Nine per cent (53/569) of patients who received antibiotic withdrew due to AEs compared with 12% (36/289) of patients assigned to placebo (RR 0.86, 95% CI 0.57 to 1.29). No heterogeneity was detected for this comparison (I² = 0%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to sparse data (See Table 1). Common AEs leading to withdrawal included worsening CD, gastrointestinal symptoms, headache, abscess, rash, arthralgia, nausea, vomiting, arthropathy and infusion reaction.
Antibiotic with anti‐TNF versus placebo with anti‐TNF
Clinical response at week 12
Two studies including 100 patients were pooled to evaluate the effect of antibiotics (500 mg twice daily) on failure to achieve clinical response at week 12 in the presence of a TNF antagonist (Dewint 2014; West 2004). Twenty‐one per cent (10/48) of patients who received an antibiotic failed to achieve a clinical response at week 12 compared with 37% (19/52) of patients assigned to placebo (RR 0.57, 95% CI 0.29 to 1.10). No heterogeneity was detected for this comparison (I² = 0%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 2).
Adverse events
AEs were reported in two studies that included 100 participants (Dewint 2014; West 2004). Seventy‐seven per cent (37/48) of patients who received antibiotics (500 mg twice daily) experienced an AE compared with 83% (43/52) of patients assigned to placebo (RR 0.93, 95% CI 0.76 to 1.12). No heterogeneity was detected for this comparison (I² = 0%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was moderate due to sparse data (See Table 2). AEs in the antibiotic group included nausea, vomiting and upper respiratory tract infections. AEs in the placebo group included change of taste, upper respiratory infections, fatigue and headache.
Serious adverse events
SAEs were only reported by Dewint 2014. These data are reported for the comparison Ciprofloxacin with adalimumab versus placebo with adalimumab.
Withdrawal due to adverse events
Withdrawal due to AEs was reported in two studies that included 100 participants (Dewint 2014; West 2004). Six per cent (3/48) of patients who received antibiotic withdrew due to AEs compared with 8% (4/52) of patients assigned to placebo (RR 0.82, 95% CI 0.19 to 3.45). No heterogeneity was detected for this comparison (I² = 0%). A GRADE analysis indicated that the overall certainty of the evidence for this outcome was low due to very sparse data (See Table 2). AEs leading to withdrawal in the antibiotic group included gastrointestinal symptoms and infusion reactions. AEs leading to withdrawal in the placebo group included gastrointestinal symptoms and herpes simplex virus infection.
Discussion
Summary of main results
This review includes 11 randomised, placebo‐controlled trials (Ambrose 1985; Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009; West 2004) and two active comparator trials (Colombel 1999; Prantera 1996), which examined the efficacy and safety of antibiotics for inducing and maintaining remission in patients with CD. Two of the placebo‐controlled trials also included active comparator arms (Ambrose 1985; Thia 2009). The included studies compared antibiotics to placebo, other antibiotics, methylprednisolone and mesalamine. In two studies, antibiotics were also given in conjunction with the TNF‐alpha inhibitors infliximab and adalimumab (Dewint 2014; West 2004).
One study did not use a validated outcome measure for their study end points (Ambrose 1985). Ambrose 1985 used overall general health, abdominal pain, number of liquid stools on the previous day and disease complications to assess efficacy. Eleven studies defined clinical remission as a CDAI score less than or equal to 150 (Arnold 2002; Colombel 1999; Dewint 2014; Lieper 2008; Prantera 1996; Prantera 2006; Prantera 2012; Selby 2007; Steinhart 2002; Sutherland 1991; Thia 2009). Overall, many studies had different durations of therapy. West 2004 used clinical response, Peritoneal Disease Activity Score (PDAI) and endoanal ultrasonography as study endpoints. The variety of outcome measures made our analysis challenging.
Two studies focused specifically on rifaximin therapy (800 mg to 2400 mg daily) (Prantera 2006; Prantera 2012). These studies reported on the proportion of patients who failed to enter clinical remission at week 12 or 14. Subgroup analyses based on dose showed a benefit for rifaximin 1600 mg once‐daily over placebo (RR 0.68, 95% CI 0.50 to 0.93), but not for the 800 mg once daily or 2400 mg once daily subgroups. We are hesitant to recognize this as a treatment effect. There was no consistent effect across dose groups and no consistent effect of a higher dose of medication when compared with a lower dose of medication. Any benefit provided by rifaximin appears to be modest. Overall, 48% (174/360) of rifaximin patients failed to enter clinical remission at weeks 12 to 14 compared to 60% (77/129) of placebo patients (RR 0.82, 95% CI 0.69 to 0.98; moderate certainty evidence). Moderate certainty evidence suggests there was no difference in AEs between the subgroups. Thirty‐nine per cent (140/360) of patients in the rifaximin group experienced an AE compared to 47% (61/129) of placebo patients. Low certainty evidence suggests there was no difference in serious AEs. Two per cent (6/360) of rifaximin patients experienced a SAE compared to 1% (1/129) of placebo patients.
The effects of metronidazole, clarithromycin, and cotrimoxazole on CD are mostly uncertain. Three studies (N = 149) assessed metronidazole (400 mg to 500 mg twice daily) in participants with active CD (Ambrose 1985; Sutherland 1991; Thia 2009). At 6 to 10 weeks, 60% (15/25) of participants receiving metronidazole failed to achieve remission compared to 68% (17/25) of placebo participants (RR 0.91, 95% CI 0.62 to 1.33; low certainty evidence). Similar results were found for a study that assessed remission at 16 weeks (Sutherland 1991). One study (N = 41) assessed clarithromycin therapy (1 g daily) in participants with active CD (Lieper 2008). Low certainty evidence suggests clarithromycin provides no benefit over placebo for induction of clinical remission or improvement in participants with active CD. At 12 weeks 84% (16/19) of clarithromycin participants failed to achieve clinical remission compared to 82% (18/22) of placebo participants (RR 1.03, 95% CI 0.78 to 1.36). One study (N = 33) assessed cotrimoxazole (960 mg twice daily) in participants with active CD (Ambrose 1985). Low certainty evidence suggests that cotrimoxazole does not provide any benefit over placebo for induction of remission in CD. At 12 weeks, 69% (11/16) of cotrimoxazole participants failed to achieve remission compared to 59% (10/17) of placebo participants (RR 1.17, 95% CI 0.70 to 1.96). Ambrose 1985 (N = 38) also assessed combination therapy with metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) in participants with active CD. The effect of combination therapy with these two antibiotics on active CD are uncertain. At 6 weeks, 62% (13/21) of participants who received combination antibiotics failed to achieve remission compared to 59% (10/17) of placebo participants (RR 1.05, 95% CI 0.63 to 1.77; low certainty evidence). Selby 2007 assessed combination therapy with clarithromycin (750 mg daily) and antimycobacterial drugs in participants with active CD. Moderate quality evidence suggests that combination therapy with clarithromycin and antimycobacterial drugs is superior to placebo for induction of remission in active CD. At 16 weeks, 34% (35/102) of participants who had combination therapy failed to achieve remission compared to 50% (56/111) of placebo participants.
Prantera 1996 compared combination therapy with ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) to methylprednisolone (0.7 to 1 mg/kg/day) in participants with active CD. The effect of treatment with ciprofloxacin and metronidazole on active CD is uncertain, although antibiotics do not appear to provide a benefit over corticosteroids. At 12 weeks, 54% (12/22) of participants who received combination therapy with antibiotics failed to achieve remission compared to 37% (7/19) of participants who received corticosteroids (RR 1.48, 95% CI 0.73 to 2.99, very low certainty evidence). Colombel 1999 compared ciprofloxacin (500 mg twice daily) to mesalazine (2 g twice daily) in participants with active CD. The effect of ciprofloxacin on active CD is uncertain. Forty‐four per cent (8/18) of participants in the ciprofloxacin group failed to achieve remission at week 6 compared to 45% (10/22) of mesalazine participants (RR 0.98, 95% CI 0.49 to 1.95; very low certainty evidence).
Although the evidence supporting individual types of antibiotics is uncertain, when we look at antibiotics as a class, moderate to high certainty evidence suggests that any benefit provided by antibiotics in active CD is modest and may not be clinically meaningful. For example, in a pooled analysis of 7 studies including 773 participants, 47% (253/542) of participants receiving antibiotics achieved remission at 6 to 10 weeks compared to 36% (82/231) of placebo participants (Ambrose 1985; Arnold 2002; Lieper 2008; Prantera 2006; Prantera 2012; Sutherland 1991; Thia 2009). The certainty of the evidence for this outcome was high. In a pooled analysis of 5 studies including 617 participants, 59% (254/428) of participants receiving antibiotics improved clinically at 10 to 14 weeks compared to 51% (96/189) of placebo participants ( Dewint 2014; Prantera 2006; Prantera 2012; Thia 2009; West 2004). The certainty of the evidence for this outcome was moderate. High certainty evidence suggests there was little difference in the rate of AEsin patients who received antibiotic compared to placebo (Ambrose 1985; Arnold 2002; Dewint 2014; Lieper 2008; Prantera 2006; Prantera 2012; Sutherland 1991; Thia 2009; West 2004). Thirty‐eight per cent (214/568) of participants in the antibiotic group experienced at least one AEcompared to 45% of placebo participants.
Two studies (N = 155) looked at maintenance of remission at 52 weeks as a clinical end point (Ambrose 1985; Selby 2007). Both of these studies used CDAI as their outcome. The impact of antibiotics on maintenance of remission is uncertain. At 52 weeks, 45% 37/83) of antibiotic participants relapsed compared to 57% (41/72) of placebo participants (RR 0.87, 95% CI 0.52 to 1.47; low certainty evidence). Two studies looked at antibiotics compared with placebo in the presence of anti‐TNF therapy in both treatment and placebo groups (Dewint 2014; West 2004). One of these studies used CDAI as their clinical end point (Dewint 2014). The impact of antibiotics on clinical improvement in CD patients being treated with concurrent TNF therapy is uncertain. At 12 weeks, 21% (10/48) of antibiotic participants failed to have a response to therapy compared to 36% (19/52) of placebo participants (RR 0.57, 95% CI 0.29 to 1.10; low certainty evidence).
Overall completeness and applicability of evidence
The main focus of this review was to determine whether antibiotics are effective at inducing and maintaining remission in patients with CD. There were a few challenges that we came across regarding the data that were collected. One of the main challenges of this review was the certainty of the evidence found in the literature. Many of the studies had small patient samples and few outcomes were assessed. We were unable to collect data for many of our pre‐specified outcomes. For example, only one study had endoscopic outcomes as one of the secondary outcomes (Selby 2007). Pooled data of antibiotics used in conjunction with anti‐TNF was of low certainty. Very few studies looked at our maintenance of remission end point. Two studies had this end point and the data was pooled (Ambrose 1985, Selby 2007) .
One other challenge was the wide range of antibiotics and comparisons assessed in the included studies. Ciprofloxacin was compared to placebo in two studies (Arnold 2002; Thia 2009). Ciprofloxacin was also administered in conjunction with metronidazole and compared with methylprednisolone therapy in one study (Prantera 1996). Another study looked at ciprofloxacin and metronidazole compared to placebo in participants receiving budesonide therapy(Steinhart 2002). Ciprofloxacin was also compared to mesalamine in one study (Colombel 1999). Two studies looked at ciprofloxacin therapy compared with placebo but started both groups on biologic therapy as part of their trial. Dewint 2014 used adalimumab and West 2004 used infliximab. Ciprofloxacin was used most often in studies when compared with the other antibiotics likely because of its activity against common gastrointestinal bacteria. Rifaximin was studied in two studies using several different doses (Prantera 2006; Prantera 2012). Metronidazole was studied in two studies (Ambrose 1985; Thia 2009). Clarithromycin was studied in one study (Lieper 2008). Cotrimoxazole was studied in one study (Ambrose 1985). Ambrose 1985 also looked at a combination of metronidazole and cotrimoxazole when compared with placebo. Overall, the duration of therapy varied across the studies.
Due to the overall small sample size, we suspect this body of research does not accurately represent the CD population and further study in this area is needed . This research also highlights the potential for adverse effects using long‐term antibiotic therapy despite the low rate of adverse effects seen in the treatment groups.
Quality of the evidence
We have included 13 studies with 1303 patients in this review. The studies were rated at unclear risk of bias for several important certainty indicators. Mainly in regards to randomisation practices and concealment of the study group allocation. Two studies were deemed to have a high risk of bias because they were unblinded. Five studies did not report all outcomes and in two studies it was unclear whether there was a reporting bias implicating their results. GRADE analysis indicated that overall certainty of evidence for the two pooled analyses was overall moderate for antibiotics compared to placebo for induction and maintenance of CD and low for the pooled analysis of antibiotics compared to placebo in the presence of an anti‐TNF for induction of and maintenance of remission in CD. This was largely due to sparse data in these studies.
Potential biases in the review process
We performed a comprehensive literature review in an effort to ensure that all relevant studies were included. We also had two independent authors assess studies for inclusion, extract data and assess risk of bias. The main limitations to this review are that many of the included studies had small sample sizes. Some studies were excluded because data on outcomes were not available and attempts made to contact trialists for the data were unsuccessful. Outcomes were measured at several different time points which made comparisons difficult. Lastly, the overall certainty of the evidence in this review ranged from very low for some outcomes to high for others.
Agreements and disagreements with other studies or reviews
One other systematic review has looked at the effect of antibiotic therapy on the induction and maintenance of CD but antimycobacterial therapy was also included in this analysis (Khan 2011). There is a separate Cochrane review that assesses the effect of anti‐tuberculosis therapy on maintaining remission in patients with CD (Patton 2016). Our basis for differentiating between antibiotic therapy and antimycobacterial therapy was determined using the World Health Organization definition of antimycobacterial therapy.
Authors' conclusions
Implications for practice.
Moderate to high quality evidence suggests that any benefit provided by antibiotics in active CD is likely to be modest and may not be clinically meaningful. High quality evidence suggests that there is no increased risk of AEs with antibiotics compared to placebo. The effect of antibiotics on the risk of SAEs is uncertain. The effect of antibiotics on maintenance of remission in CD is uncertain. Thus, no firm conclusions regarding the efficacy and safety of antibiotics in maintenance of CD can be drawn. More research is needed to determine the efficacy and safety of antibiotics as induction and maintenance therapy in CD.
Implications for research.
More research is needed to determine the efficacy and safety of antibiotics as induction and maintenance therapy in CD. At this point, we do not have a clear bacterial treatment target that has been shown to be beneficial in inducing or maintaining disease remission in CD patients. Studies suggest a rationale for alteration in the microbiome in the pathogenesis of CD based on animal models but this idea is currently not echoed in the patient population studied in the literature. Perhaps there eventually will be a specific bacterial target identified but at this point, our bacterial agents are likely not targeted at the right microbe in the right dose for the right duration to have a clinically significant treatment effect.
Acknowledgements
Funding for the Cochrane IBD Group (May 1, 2017 ‐ April 30, 2022) has been provided by Crohn's and Colitis Canada (CCC).
Appendices
Appendix 1. Search strategies
Embase
1. random$.tw. 2. factorial$.tw. 3. (crossover$ or cross over$ or cross‐over$).tw. 4. placebo$.tw. 5. single blind.mp. 6. double blind.mp. 7. triple blind.mp. 8. (singl$ adj blind$).tw. 9. (double$ adj blind$).tw. 10. (tripl$ adj blind$).tw. 11. assign$.tw. 12. allocat$.tw. 13. crossover procedure/ 14. double blind procedure/ 15. single blind procedure/ 16. triple blind procedure/ 17. randomised controlled trial/ 18. or/1‐17 19. (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) 20. 18 not 19 21. Exp Crohn disease/ 22. Crohn*.mp. 23. inflammatory bowel disease*.mp. 24. IBD.mp. 25. Or/21‐24 26. Exp antibiotics/ 27. antibiotic*.mp. 28. Exp anti‐bacterial agents/ 29. anti*bacter*.mp. 30. bacteriocid*.mp. 31. bactericid*.mp. 32. anti*microbial.mp. 33. (ciprofloxacin or metronidazole or levamisole or ornidazole or fusidin or rifaximin or vancomycin or fusidic acid or nitazoxanide or teicoplanin or rifampicin or bacitracin or fidaxomicin or amoxicillin or azithromycin or cephalosporin* or cephalexin or clarithromycin or clindamycin or doxycycline or erythromycin or flouroquinolone* or levofloxacin or macrolide* or nitrofurantoin or penicillin or tetracycline or trimethoprim).mp. 34. or/26‐33 35. 20 and 25 and 34
MEDLINE
1. random$.tw. 2. factorial$.tw. 3. (crossover$ or cross over$ or cross‐over$).tw. 4. placebo$.tw. 5. single blind.mp. 6. double blind.mp. 7. triple blind.mp. 8. (singl$ adj blind$).tw. 9. (double$ adj blind$).tw. 10. (tripl$ adj blind$).tw. 11. assign$.tw. 12. allocat$.tw. 13. randomised controlled trial/ 14. or/1‐13 15. (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) 16. 14 not 15 17. Exp Crohn disease/ 18. Crohn*.mp. 19. inflammatory bowel disease*.mp.
20. IBD.mp. 21. Or/17‐20 22. Exp antibiotics/ 23. antibiotic*.mp. 24. Exp anti‐bacterial agents/ 25. anti*bacter*.mp. 26. bacteriocid*.mp. 27. bactericid*.mp. 28. anti*microbial.mp. 29. (ciprofloxacin or metronidazole or levamisole or ornidazole or fusidin or rifaximin or vancomycin or fusidic acid or nitazoxanide or teicoplanin or rifampicin or bacitracin or fidaxomicin or amoxicillin or azithromycin or cephalosporin* or cephalexin or clarithromycin or clindamycin or doxycycline or erythromycin or flouroquinolone* or levofloxacin or macrolide* or nitrofurantoin or penicillin or tetracycline or trimethoprim).mp.
30. or/22‐29 31. 16 and 21 and 30
Cochrane CENTRAL Register of Controlled Trials
#1 MeSH descriptor: [Crohn Disease] explode all trees #2 Crohn #3 inflammatory bowel disease #4 IBD #5 MeSH descriptor: [Anti‐Bacterial Agents] explode all trees #6 antibiotic*.mp. #7 bacteriocid*.mp. #8 bactericid*.mp. #9 anti*microbial.mp. #10 (ciprofloxacin or metronidazole or levamisole or ornidazole or fusidin or rifaximin or vancomycin or fusidic acid or nitazoxanide or teicoplanin or rifampicin or bacitracin or fidaxomicin or amoxicillin or azithromycin or cephalosporin* or cephalexin or clarithromycin or clindamycin or doxycycline or erythromycin or flouroquinolone* or levofloxacin or macrolide* or nitrofurantoin or penicillin or tetracycline or trimethoprim).mp. #11 #1 or #2 or #3 or #4 #12 #5 or #6 or #7 or #8 or #9 or #10 #13 #11 and #12
Cochrane IBD Group Specialized register 1. Antibiotics and Crohn’s Disease
Clinical trials.gov 1.Antibiotics and Crohn’s Disease 2.Antibiotics and Inflammatory bowel disease
Data and analyses
Comparison 1. Ciprofloxacin (500 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 10 or 12 | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.41, 0.92] |
| 2 Failure to maintain clinical remission at week 24 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Failure to have a clinical response at week 10 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.57, 1.76] |
| 5 Serious adverse events | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Withdrawal due to adverse events | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.07, 1.67] |
1.1. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 1 Failure to enter clinical remission at week 10 or 12.
1.2. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 2 Failure to maintain clinical remission at week 24.
1.3. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 3 Failure to have a clinical response at week 10.
1.4. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 4 Adverse events.
1.5. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 5 Serious adverse events.
1.6. Analysis.
Comparison 1 Ciprofloxacin (500 mg twice daily) versus placebo, Outcome 6 Withdrawal due to adverse events.
Comparison 2. Rifaximin (800 mg to 2400 mg daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission week 12 or 14 | 2 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.69, 0.98] |
| 1.1 Dose 800 mg daily | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.65, 1.12] |
| 1.2 Dose 1600 mg daily | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.50, 0.93] |
| 1.3 Dose 2400 mg daily | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.69, 1.38] |
| 2 Failure to have clinical response at week 12 or 14 | 2 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.67, 1.01] |
| 2.1 Dose 800 mg daily | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.65, 1.28] |
| 2.2 Dose 1600 mg daily | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.43, 0.91] |
| 2.3 Dose 2400 mg daily | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.67, 1.40] |
| 3 Adverse events | 2 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.66, 1.04] |
| 3.1 Dose 800 mg daily | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.49, 1.05] |
| 3.2 Dose 1600 mg daily | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.58, 1.21] |
| 3.3 Dose 2400 mg daily | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.64, 1.53] |
| 4 Serious adverse events | 2 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.27, 4.54] |
| 4.1 Dose 800 mg daily | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.08, 33.26] |
| 4.2 Dose 1600 mg daily | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.14, 12.08] |
| 4.3 Dose 2400 mg daily | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.06, 7.05] |
| 5 Withdrawal due to adverse events | 2 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.59, 2.64] |
| 5.1 Dose 800 mg daily | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.14, 2.16] |
| 5.2 Dose 1600 mg daily | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.30, 3.83] |
| 5.3 Dose 2400 mg daily | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.64 [0.64, 10.90] |
2.1. Analysis.
Comparison 2 Rifaximin (800 mg to 2400 mg daily) versus placebo, Outcome 1 Failure to enter clinical remission week 12 or 14.
2.2. Analysis.
Comparison 2 Rifaximin (800 mg to 2400 mg daily) versus placebo, Outcome 2 Failure to have clinical response at week 12 or 14.
2.3. Analysis.
Comparison 2 Rifaximin (800 mg to 2400 mg daily) versus placebo, Outcome 3 Adverse events.
2.4. Analysis.
Comparison 2 Rifaximin (800 mg to 2400 mg daily) versus placebo, Outcome 4 Serious adverse events.
2.5. Analysis.
Comparison 2 Rifaximin (800 mg to 2400 mg daily) versus placebo, Outcome 5 Withdrawal due to adverse events.
Comparison 3. Metronidazole (400 mg to 500 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission week 6 or 10 | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.62, 1.33] |
| 2 Failure to enter clinical remission at week 16 | 1 | 99 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.77, 1.36] |
| 2.1 Dose 10 mg/kg | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.63, 1.45] |
| 2.2 Dose 20 mg/kg | 1 | 48 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.1 [0.74, 1.63] |
| 3 Failure to have clinical response at week 10 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 3 | 149 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.32, 2.31] |
| 5 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Withdrawal due to adverse events | 3 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.36, 1.68] |
| 6.1 Dose 10 mg/kg or 20 mg/kg | 3 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.36, 1.68] |
3.1. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 1 Failure to enter clinical remission week 6 or 10.
3.2. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 2 Failure to enter clinical remission at week 16.
3.3. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 3 Failure to have clinical response at week 10.
3.4. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 4 Adverse events.
3.5. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 5 Serious adverse events.
3.6. Analysis.
Comparison 3 Metronidazole (400 mg to 500 mg twice daily) versus placebo, Outcome 6 Withdrawal due to adverse events.
Comparison 4. Clarithromycin (1 g/day) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at 12 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Failure to have clinical response at 12 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
4.1. Analysis.
Comparison 4 Clarithromycin (1 g/day) versus placebo, Outcome 1 Failure to enter clinical remission at 12 weeks.
4.2. Analysis.
Comparison 4 Clarithromycin (1 g/day) versus placebo, Outcome 2 Failure to have clinical response at 12 weeks.
4.3. Analysis.
Comparison 4 Clarithromycin (1 g/day) versus placebo, Outcome 3 Adverse events.
4.4. Analysis.
Comparison 4 Clarithromycin (1 g/day) versus placebo, Outcome 4 Withdrawal due to adverse events.
Comparison 5. Cotrimoxazole (960 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 12 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
5.1. Analysis.
Comparison 5 Cotrimoxazole (960 mg twice daily) versus placebo, Outcome 1 Failure to enter clinical remission at week 12.
5.2. Analysis.
Comparison 5 Cotrimoxazole (960 mg twice daily) versus placebo, Outcome 2 Adverse events.
5.3. Analysis.
Comparison 5 Cotrimoxazole (960 mg twice daily) versus placebo, Outcome 3 Withdrawal due to adverse events.
Comparison 6. Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) versus methylprednisolone (0.7‐1 mg/kg daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 12 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Failure to maintain clinical remission at week 52 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
6.1. Analysis.
Comparison 6 Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) versus methylprednisolone (0.7‐1 mg/kg daily), Outcome 1 Failure to enter clinical remission at week 12.
6.2. Analysis.
Comparison 6 Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) versus methylprednisolone (0.7‐1 mg/kg daily), Outcome 2 Failure to maintain clinical remission at week 52.
6.3. Analysis.
Comparison 6 Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) versus methylprednisolone (0.7‐1 mg/kg daily), Outcome 3 Adverse events.
6.4. Analysis.
Comparison 6 Ciprofloxacin (500 mg twice daily) and metronidazole (250 mg four times daily) versus methylprednisolone (0.7‐1 mg/kg daily), Outcome 4 Withdrawal due to adverse events.
Comparison 7. Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg daily) versus placebo and budesonide (9 mg daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 8 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
7.1. Analysis.
Comparison 7 Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg daily) versus placebo and budesonide (9 mg daily), Outcome 1 Failure to enter clinical remission at week 8.
7.2. Analysis.
Comparison 7 Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg daily) versus placebo and budesonide (9 mg daily), Outcome 2 Adverse events.
7.3. Analysis.
Comparison 7 Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg daily) versus placebo and budesonide (9 mg daily), Outcome 3 Serious adverse events.
7.4. Analysis.
Comparison 7 Ciprofloxacin (500 mg twice daily) and metronidazole (500 mg twice daily) and budesonide (9 mg daily) versus placebo and budesonide (9 mg daily), Outcome 4 Withdrawal due to adverse events.
Comparison 8. Ciprofloxacin (500 mg twice daily) versus mesalazine (2 g twice daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 6 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse Events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
8.1. Analysis.
Comparison 8 Ciprofloxacin (500 mg twice daily) versus mesalazine (2 g twice daily), Outcome 1 Failure to enter clinical remission at week 6.
8.2. Analysis.
Comparison 8 Ciprofloxacin (500 mg twice daily) versus mesalazine (2 g twice daily), Outcome 2 Adverse Events.
8.3. Analysis.
Comparison 8 Ciprofloxacin (500 mg twice daily) versus mesalazine (2 g twice daily), Outcome 3 Withdrawal due to adverse events.
Comparison 9. Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 12 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Failure to maintain clinical remission at week 24 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
9.1. Analysis.
Comparison 9 Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, Outcome 1 Failure to enter clinical remission at week 12.
9.2. Analysis.
Comparison 9 Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, Outcome 2 Failure to maintain clinical remission at week 24.
9.3. Analysis.
Comparison 9 Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, Outcome 3 Adverse events.
9.4. Analysis.
Comparison 9 Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, Outcome 4 Serious adverse events.
9.5. Analysis.
Comparison 9 Ciprofloxacin (500 mg twice daily) with adalimumab versus placebo with adalimumab, Outcome 5 Withdrawal due to adverse events.
Comparison 10. Ciprofloxacin (500 mg twice daily) with infliximab versus placebo with infliximab.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to achieve clinical response at week 12 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
10.1. Analysis.
Comparison 10 Ciprofloxacin (500 mg twice daily) with infliximab versus placebo with infliximab, Outcome 1 Failure to achieve clinical response at week 12.
10.2. Analysis.
Comparison 10 Ciprofloxacin (500 mg twice daily) with infliximab versus placebo with infliximab, Outcome 2 Adverse events.
10.3. Analysis.
Comparison 10 Ciprofloxacin (500 mg twice daily) with infliximab versus placebo with infliximab, Outcome 3 Withdrawal due to adverse events.
Comparison 11. Clarithromycin (750 mg daily) and antimycobacterial versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 16 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Failure to maintain clinical remission at 1 year | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Endoscopic relapse at 3 years | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Withdrawal of adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
11.1. Analysis.
Comparison 11 Clarithromycin (750 mg daily) and antimycobacterial versus placebo, Outcome 1 Failure to enter clinical remission at week 16.
11.2. Analysis.
Comparison 11 Clarithromycin (750 mg daily) and antimycobacterial versus placebo, Outcome 2 Failure to maintain clinical remission at 1 year.
11.3. Analysis.
Comparison 11 Clarithromycin (750 mg daily) and antimycobacterial versus placebo, Outcome 3 Endoscopic relapse at 3 years.
11.4. Analysis.
Comparison 11 Clarithromycin (750 mg daily) and antimycobacterial versus placebo, Outcome 4 Withdrawal of adverse events.
Comparison 12. Metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at week 6 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
12.1. Analysis.
Comparison 12 Metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) versus placebo, Outcome 1 Failure to enter clinical remission at week 6.
12.2. Analysis.
Comparison 12 Metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) versus placebo, Outcome 2 Adverse events.
12.3. Analysis.
Comparison 12 Metronidazole (400 mg twice daily) and cotrimoxazole (960 mg twice daily) versus placebo, Outcome 3 Withdrawal due to adverse events.
Comparison 13. Antibiotic versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to enter clinical remission at 6 to 10 weeks | 7 | 773 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.76, 0.98] |
| 2 Failure to achieve clinical response at week 10 or 12 or 14 | 5 | 617 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.64, 0.93] |
| 3 Failure to maintain clinical remission at 52 weeks | 2 | 155 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.52, 1.47] |
| 4 Adverse events | 9 | 852 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.75, 1.02] |
| 5 Serious adverse events | 3 | 520 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.70 [0.29, 10.01] |
| 6 Withdrawal due to adverse events | 9 | 858 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.57, 1.29] |
13.1. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 1 Failure to enter clinical remission at 6 to 10 weeks.
13.2. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 2 Failure to achieve clinical response at week 10 or 12 or 14.
13.3. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 3 Failure to maintain clinical remission at 52 weeks.
13.4. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 4 Adverse events.
13.5. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 5 Serious adverse events.
13.6. Analysis.
Comparison 13 Antibiotic versus placebo, Outcome 6 Withdrawal due to adverse events.
Comparison 14. Antibiotic (500 mg twice daily) with anti‐TNF versus placebo with anti‐TNF.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to achieve clinical response at week 12 | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.29, 1.10] |
| 2 Adverse events | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.76, 1.12] |
| 3 Withdrawal due to adverse events | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.19, 3.45] |
14.1. Analysis.
Comparison 14 Antibiotic (500 mg twice daily) with anti‐TNF versus placebo with anti‐TNF, Outcome 1 Failure to achieve clinical response at week 12.
14.2. Analysis.
Comparison 14 Antibiotic (500 mg twice daily) with anti‐TNF versus placebo with anti‐TNF, Outcome 2 Adverse events.
14.3. Analysis.
Comparison 14 Antibiotic (500 mg twice daily) with anti‐TNF versus placebo with anti‐TNF, Outcome 3 Withdrawal due to adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ambrose 1985.
| Methods | Randomized, placebo‐controlled trial Trial duration was 6 weeks |
|
| Participants | Patients aged 15‐70 with documented histological or radiological diagnosis of CD on stable therapy for 28 days prior to study entry (N = 72) | |
| Interventions | Oral cotrimoxazole 960 mg twice daily (BID) with oral placebo (n = 16) Oral metronidazole 400 mg BID with oral placebo (n = 18) Oral cotrimoxazole 960 mg BID and oral metronidazole 400 mg BID (n = 21) Oral placebo (n = 17) |
|
| Outcomes |
Primary outcome: improvement in clinical score (defined by the authors: general health yesterday, abdominal pain yesterday, number of liquid stools yesterday, abdominal mass, complications) Investigators also looked at changes in fecal flora and hematologic measures |
|
| Notes | Patients were 15 years and older Trial did not mention whether it was blinded Intention to treat not performed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 16 patients did not complete the study More patients in the cotrimoxazole and metronidazole group (8/21) failed to complete the trial compared to the other groups (placebo: 4/17; cotrimoxazole: 4/16; metronidazole 4/18) Study did not quantify adverse events based on treatment group (7/72) |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Arnold 2002.
| Methods | Randomized, placebo‐controlled trial Trial duration was 6 months |
|
| Participants | Patients age 18‐70 with moderately active CD (CDAI > 150) (N = 47) |
|
| Interventions | Oral ciprofloxacin (500 mg BID) (n = 28) Oral placebo (n = 19) |
|
| Outcomes |
Primary outcome: clinical remission (CDAI < 150) at 6 months Secondary outcome: clinical remission (CDAI < 150) at 1 month and 3 month intervals, adverse event data |
|
| Notes | Initially 84 patients were screened for trial, 43/84 to ciprofloxacin and 41/84 to receive placebo. 37 patients were then excluded. Reasons outlined. Left 47 patients who were used in analysis. Ten (10/47) patients were lost to follow‐up. Details regarding blinding of participants and investigators not included |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The hospital pharmacy controlled computer randomisation, medication distribution, and unblinding procedures |
| Allocation concealment (selection bias) | Low risk | The hospital pharmacy controlled computer randomisation, medication distribution, and unblinding procedures |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind: identical placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 37 patients were excluded with reasons following randomisation process (28 lost to follow up, 8 had CDAI score < 150, 1 had psychologic disorder that would interfere with study participant requirements) 47 patients were reported in data, 10 then lost to follow up during trial All patients excluded were reported |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Colombel 1999.
| Methods | Randomized active comparator trial. Study participants and investigators were not blinded Intention to treat not performed 6 week trial |
|
| Participants | Patients aged >18 with acute ileal or colonic CD with no need for steroid treatment and a CDAI between 150 and 300 (N = 40) | |
| Interventions | Oral ciprofloxacin 500 mg BID (n = 18) or oral mesalazine 2g BID (n = 22) for 6 weeks | |
| Outcomes |
Primary outcome: Clinical remission (CDAI ≤ 150) at 6 weeks Secondary outcomes: Partial remission (CDAI ≤ 150 but change in CDAI between 50‐75), treatment failure (increase in CDAI value or insufficient improvement at week 3 with change in CDAI > 50 or absence of complete or partial remission at week 6) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not discussed |
| Allocation concealment (selection bias) | Unclear risk | Not discussed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Trial was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Trial was not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two patients stopped treatment in ciprofloxacin group (one for intolerance, one for personal reasons) One patient was lost to follow up in mesalamine group |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Dewint 2014.
| Methods | Multicenter, randomised, double‐ blind, placebo controlled trial Modified intention to treat Study duration was 24 weeks |
|
| Participants | Patients aged 18‐70 with active CD and perianal activity (N = 76) | |
| Interventions | Patient were randomised to oral ciprofloxacin 500 mg BID (n = 37) or placebo (n = 39) for 12 weeks and followed for a total of 24 weeks All patients were treated with self‐administered adalimumab (patients were given induction dosing of 160 mg at day 0 and 80 mg at week 2, followed by maintenance of 40 mg every 4 weeks until week 24) |
|
| Outcomes |
Primary outcome: Clinical response defined as a 50% reduction in perianal fistulas Secondary outcomes: Clinical remission, improvement in CDAI, Pediatric Crohn's Disease Activity Index (PCDAI) and Inflammatory Bowel Disease Questionnaire (IBDQ) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed through a centralised randomisation schedule in a 1:1 ratio |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation was performed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Clinical exam was performed at week 0, 12 and 24 by physician blinded for treatment allocation. Physician counted number of draining fistulas and excluded presence of abscesses |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Three patients from each treatment group were excluded from analysis. Reasons included (4 had no fistulas at baseline, 2 had a drain removed that was not according to protocol) |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias Note: Ciprofloxacin group lost 3 patients after initial randomisation (34/37). Placebo group lost 3 patients after initial randomisation (36/39). Reasons reported |
Lieper 2008.
| Methods | Randomized, placebo‐controlled trial. Investigators were blinded. Intention to treat Twelve week trial |
|
| Participants | Patients age 18 and older with active CD (CDAI > 200 and CRP ≥ 10 mg/L) (N = 41) | |
| Interventions | Patients were randomised to oral clarithromycin 1g daily (n = 19) or oral placebo (n = 22) for 3 months | |
| Outcomes |
Primary outcome: Clinical remission (CDAI ≤ 150) or clinical response (fall in CDAI by ≥70 from pre‐treatment level) at 3 months Secondary outcomes: Decrease in van Hees Activity Index, remission as per Harvey Bradshaw Index (≤ 4), improvement in IBDQ and decrease in serum CRP |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was carried out by pharmacist independent of the trial. Randomization was performed by a random allocation sequence in blocks of four (two active and two placebo) |
| Allocation concealment (selection bias) | Low risk | Centralized randomization by pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Patients and investigators were blinded to treatment allocation Placebo was identical in size, colour and taste |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were blinded to randomisation assignments |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More patients withdrawn from placebo group (7/19 from clarithromycin group, 12/22 of placebo group). More patients had worsening baseline disease in placebo group (4 in treatment group versus 9 in placebo group) |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Prantera 1996.
| Methods | Randomized active comparator trial, some investigators were blinded. Investigators who were evaluating clinical status and results were blinded Investigators who were administering study medication or monitoring compliance were not blinded. Patients were not blinded Intention to treat not performed Twelve week trial If achieved remission continued therapy for 1 year |
|
| Participants | Patients with current active colonic or ileal CD with CDAI > 200 (N = 41) | |
| Interventions | Oral ciprofloxacin 500 mg BID and metronidazole 250 mg QID (n = 22) or methylprednisolone 0.7‐1 mg/kg/day with a variable taper to 40 mg daily, followed by a taper of 4 mg weekly (n = 19) | |
| Outcomes |
Primary outcome: Clinical remission defined by CDAI ≤ 150 Secondary outcomes: Recovery of abdominal pain, diarrhea, fever, extraintestinal disease, improvement of general well being and weight gain |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization performed using a random number table |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Physicians who supervised randomisation and monitored compliance and side effects were aware of treatment assignment All other investigators were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Physicians who supervised randomisation and monitored compliance and side effects were unblinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Many more patients discontinued in the antibiotic group (11/22) compared to steroid group (5/19) Specific reasons for withdrawal not reported |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Prantera 2006.
| Methods | Randomized placebo controlled trial. Investigators and patients were blinded Intention to treat Twelve week trial |
|
| Participants | Patients aged 18‐70 with mild to moderate CD (CDAI 200‐400) (N = 83) | |
| Interventions | Randomized to receive oral rifaximin 800 mg daily (n = 25) and oral placebo, oral rifaximin 800 mg BID (n = 29) or oral placebo BID (n = 29). Other immunosuppressant’s kept at same dose of during study period | |
| Outcomes |
Primary outcome: Clinical remission defined by CDAI ≤ 150 Secondary outcomes: Reduction of CDAI ≥ 70 points from baseline, treatment failure and changes in quality of life (Inflammatory Bowel Disease questionnaire) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and investigators |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators blinded to participant assignment. CDAI and the Inflammatory Bowel Disease Questionnaire (IBDQ) scores were calculated by blinded investigators |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Discontinuation and reasons for withdrawal were fairly even between each group. (rifaximin 800 mg daily and placebo 8/25, rifaximin 800 mg BID 12/29 or placebo BID 13/29) |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Prantera 2012.
| Methods | Randomized, double blind, placebo controlled Intention to treat not performed Twenty‐four week trial |
|
| Participants | Patients aged 18‐75, active ileal or colonic CD (CDAI 220‐400) (N = 402) | |
| Interventions | Randomized to oral rifaximin 400 mg BID (n = 104), oral rifaximin 800 mg BID (n = 98), oral rifaximin 1200 mg BID (n = 99) or oral placebo (n = 101) | |
| Outcomes |
Primary outcome: Clinical remission (CDAI < 150) at 12 weeks Secondary outcomes: Number of patients who achieved clinical response at week 12 (reduction in CDAI score of 100 points), number of patients who maintained clinical remission at week 14 and 24 and number of patients with treatment failure (failure to achieve loss of at least 70 in CDAI score after 1 month, or an increase in CDAI score of >100 points from the baseline) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerized randomisation procedure |
| Allocation concealment (selection bias) | Low risk | Centralized randomization using an Interactive Voice Response System |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Questionaires used to collect outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | High rate of withdrawal from the study in each treatment group. (rifaximin 400 mg BID (41/106), rifaximin 800 mg BID (33/99), rifaximin 1200 mg BID (41/103) or placebo (43/102) |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Selby 2007.
| Methods | Randomized, placebo controlled trial with blinded investigators and patients Intention to treat not performed Induction phase 16 weeks in duration, maintenance phase 104 weeks in duration, trial 156 weeks total; all patients were started on a 16‐week tapering course of prednisolone starting at 40 mg/day |
|
| Participants | Patients over age 18 with active CD (CDAI >200) (N = 213 for induction phase and N = 122 for maintenance phase) |
|
| Interventions | Randomized to oral clarithromycin 750 mg/day, oral rifabutin 450 mg/ day, oral clofazimine 50 mg/day (n = 102) or oral placebo (three placebo pills were given to placebo group) (n = 111) Maintenance phase: antibiotics (n = 67), placebo (n = 55) |
|
| Outcomes |
Primary outcomes: The number of patients who had at least 1 relapse by 52, 104 or 156 weeks Secondary outcomes: Clinical remission at week 16 (CDAI ≤ 150), relapses within each study phase, time to first relapse, adverse events, endoscopic remission, need for surgery and quality of life |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind: matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators and participants were blinded to treatment allocation. Investigators were more likely to identify those on active treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Ninety‐one subjects were withdrawn during the induction phase (56 placebo group, 35 antibiotic group). Reasons included |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Steinhart 2002.
| Methods | Randomized, placebo controlled with blinded investigators Intention to treat not performed Eight week trial |
|
| Participants | Patients aged 14 years or older with active CD (CDAI 200‐400) (N = 134) | |
| Interventions | Patients were randomised to receive oral ciprofloxacin 500 mg BID and oral metronidazole 500 mg BID (n = 66) or oral placebo (n = 68) All patients received oral budesonide 9 mg once daily |
|
| Outcomes |
Primary outcome:The number of patients in remission at week 8, defined by CDAI < 150 Secondary outcomes: The mean changes in scores on the CDAI, quality of life, and adverse drug reactions |
|
| Notes | Patients were 14 years and older | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Investigators and participants were blinded to treatment assignment Double blind: identical placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were unaware of treatment assignments |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Sixteen patients in the placebo group and 22 patients in the antibiotic group did not complete the treatment course |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Sutherland 1991.
| Methods | Randomized, double‐blind, placebo controlled Intention to treat Sixteen week trial |
|
| Participants | N = 105, Patients with active CD with CDAI between 180‐450 (age not mentioned) | |
| Interventions | Patients were randomised to oral metronidazole 20 mg/kg (n = 33) or oral metronidazole 10 mg/kg (n = 30) or oral placebo (n = 36) | |
| Outcomes |
Primary outcome: Clinical remission (CDAI < 150) at week 16 Secondary outcomes: Changes in plasma orosomucoid and C reactive protein levels Authors also looked at the effect of metronidazole on disease location |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both patients and investigators were blinded to the treatment assignment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Both patients and investigators were blinded to the treatment assignment. No clinical measures were part of outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Equal discontinuation rate among study groups, metronidazole 10 mg/kg 11/33, metronidazole 20 mg/kg 9/30. But higher attrition rate in placebo group 20/36 |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Thia 2009.
| Methods | Randomized placebo controlled trial. Blinded participants and investigators Intention to treat Ten week trial |
|
| Participants | Patients aged 16 and older with confirmed CD with 1 or more actively draining perianal fistulas (N = 25) | |
| Interventions | Patients were randomised to receive oral ciprofloxacin 500 mg BID (n = 10), oral metronidazole 500 mg BID (n = 7) or oral placebo (n = 8) | |
| Outcomes |
Primary outcome: Closure of fistulas, meaning that they were no longer draining Secondary outcomes:Clinical remission at weeks 2 and 6, improvement in fistula symptoms at weeks 2, 6, and 10 (defined as 50% reduction in draining fistulas from baseline), short‐term durability of fistula closure defined as maintaining remission for at least 4 weeks, patient and physician global assess‐ ment measured at weeks 2, 6, and 10, PDAI at weeks 2, 6, and 10, IBDQ at weeks 2, 6, and 10; and CDAI at weeks 2, 6, and 10 |
|
| Notes | Patients 16 years and older | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was done using computer randomisation |
| Allocation concealment (selection bias) | Low risk | Medication distrubution was done by the investigational pharmacist service |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both patients and investigators were blinded to treatment assignments Patients took tablets with similar appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Specific efforts to maintain blinding among study physicians not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Seven subjects had left the study early, more in the metronidazole group (5/7) compared to others (1/10 in ciprofloxacin group and 1/8 in placebo group) Reasons for discontinuation included lost to follow‐up, withdrawal of consent, adverse events, and other reasons |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
West 2004.
| Methods | Randomized controlled trial with blinded investigators and participants Intention to treat Eighteen week trial |
|
| Participants | Patients aged with a diagnosis of CD that was complicated by at least one perianal fistula (N = 24) | |
| Interventions | Patients were randomised to receive oral ciprofloxacin 500 mg twice daily (n = 11) or oral placebo (n = 13) All patients also received 5 mg/kg infliximab at weeks 6, 8, and 12. Three patients received intravenous hydrocortisone prior to infliximab infusion |
|
| Outcomes |
Primary outcome: Clinical response (50% or greater reduction in draining fistulae confirmed by no drainage despite firm finger compression) Secondary outcomes: PDAI and 3D endoscopic ultrasound with 3% hydrogen peroxide used as a contrast medium |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both patients and investigators were blinded to treatment assignments |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Both patients and investigators were blinded to treatment assignments |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Rate of discontinuation was similar in both treatment groups 2/11 in ciprofloxacin group and 2/13 in placebo group |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No other apparent sources of bias |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Allan 1997 | Randomised controlled trial (N = 11) that studied oral metronidazole 20 mg/kg compared with oral placebo (number of patients in each group not reported ). Preliminary data suggested “one patient in each group improved”. Data unable to be extracted. Unable to reach authors |
| Bernstein 1992 | Not a randomised controlled trial. Commentary on Sutherland 1991 |
| Biancone 1998 | Patients (N = 26) had inactive CD at time of randomisation (CDAI < 150). Patients were randomised to receive oral rifaximin (800 mg/day) or placebo for seven days. Primary end point fecal alpha 1‐antitripsin clearance |
| Blichfeldt 1978 | Cross‐over study with blinded patients and investigators. Patients with active CD (N = 22) who were treated with salzosulfapyridin or prednisone were treated with metronidazole 1000 mg daily for 14 days. Primary end was a clinical score that was defined by investigators. Unable to extract data from first phase of crossover study. Unable to reach authors |
| Gilat 1982 | Not a randomised controlled trial |
| Goodgame 2001 | Patients were treated with combination therapy of both clarithromycin and ethambutol. Could not extract data for patients treated with clarithromycin alone. Antimycobacterial therapy was outside scope of this review |
| Gui 1997 | Not a randomised controlled trial. All patients (N = 46) received rifabutin and a macrolide antibiotic (clarithromycin or azithromycin) |
| Hartley‐Asp 1981 | Cross‐over trial with blinded investigators and patients. Patients with CD (N = 22) were randomised to start in metronidazole group (0.8g/day) or sulphasalazine group (3g/day). Study lasted for eight months. Patients were treated with each study drug for four months. Primary end point was chromosomal abnormalities. Study did not look at induction of remission or maintenance of remission as end point. Unable to extract data from first phase of crossover |
| Jaworski 2016 | Not a randomised controlled trial |
| Jigaranu 2014 | Patients were randomised to receive rifaximin or placebo in presence of standard therapy. Authors stated patients were on infliximab, adalimumab and 5‐aminosalicylate agents. Data could not be extracted. Could not reach authors |
| Koch 2007 | Trial terminated and no data were available |
| Koretz 1997 | Not a randomised controlled trial |
| Laudage 1983 | Unable to extract data for analysis. Unable to reach authors |
| Lee 2018 | Randomized controlled trial comparing rifaximin and placebo. Data could not be extracted. Unable to reach authors |
| Leiper 2000 | Not a randomised controlled trial. Open label study where patients with active CD (N = 25) were treated with oral clarithromycin 250 mg BID |
| Maeda 2010 | Patients randomised to receive perianal metronidazole ointment or placebo ointment. Ointment was 0.7 g applied perianally three times daily for 4 weeks. Systemic metronidazole was studied in another study. Did not meet criteria for oral therapy |
| Melmed 2009 | Not a randomised trial. Commentary of Leiper 2008 |
| Mitelman 1982 | Endpoints were chromosomal damage in response to metronidazole. Did not look at disease activity as an endpoint |
| Rogler 2014 | Study terminated due to inadequate patient enrolment. No data available |
| Ronge 2007 | Not a randomised trial |
| Steele 2009 | Not a randomised trial. Commentary regarding Thia 2009 |
| Steinhart 2008 | Trial was discontinued. Unable to recruit enough patients for study. No data available |
| To 1995 | Not a randomised trial |
| Turunen 1995 | No usable data. Unable to contact authors |
| Ursing 1982 | Cross over trial. Unable to extract data because response in treatment groups after first segment of trial was not included. Unable to contact authors |
Characteristics of ongoing studies [ordered by study ID]
NCT02240108.
| Trial name or title | A Double‐Blind, Placebo‐Controlled, Parallel‐Group, Multicenter, Multiregional, One Year Study to Assess the Efficacy and Safety of Twice Daily Oral Rifaximin Delayed Release Tablets for Induction of Clinical Remission With Endoscopic Response at 16 Weeks Followed by Clinical and Endoscopic Remission at 52 Weeks |
| Methods | A double‐blind, placebo‐controlled, parallel‐group, multicenter, multi‐regional, 52‐week study to assess the efficacy and safety of rifaximin DR tablets for the induction of clinical remission and endoscopic response at 16 weeks followed by clinical and endoscopic remission after 52 weeks of continuous therapy in subjects with active moderate Crohn's disease |
| Participants | Participants are 18 years and older with Crohn's disease |
| Interventions | Rifaximin delayed release (DR) oral tablets 800 mg BID administered continuously without dose adjustment for 52 weeks compared to a matching placebo |
| Outcomes |
Primary outcomes: 1) Clinical symptom remission at 16 weeks‐ Change from baseline in number of liquid/very soft stools and abdominal pain rating 2) Endoscopic response at 16 to 17 weeks‐ Change from baseline in simple endoscopic score for Crohn's Disease (SES‐CD) Secondary outcomes: 1) Clinical symptom remission at 52 weeks‐ Chnge from baseline in number of liquid/very soft stools and abdominal pain rating at 52 weeks 2) Endoscopic remission at 52 weeks‐ Second of the key co‐secondary efficacy measures: Chnage from baseline in SES‐CD at week 52 |
| Starting date | September 30, 2014 |
| Contact information | No contact information stated |
| Notes |
Differences between protocol and review
The protocol defined adult patients as those of 18 years of age or older. We chose to modify this definition to include patients of 15 years of age or older because many of these patients are treated using adult dosing of medications used to treat CD. Endoscopic relapse was not listed as a secondary outcome in the protocol but has been included in the full review. For the 'Summary of findings' tables, we included the following outcomes: failure to achieve clinical remission (at study endpoint), failure to maintain clinical remission (or relapse at study endpoint), failure to achieve clinical response (at study endpoint), failure to maintain endoscopic remission (or endoscopic relapse at study endpoint), adverse events, adverse events, serious adverse events and study withdrawal due to adverse events. We did not pre‐specify these outcomes in the published protocol.
Contributions of authors
All authors contributed to the development and writing of the protocol. All authors contributed to writing the final manuscript.
Declarations of interest
Cassandra M Townsend: None known
Claire E Parker: None known
John K MacDonald: None known
Tran M Nguyen: None known
Vipul Jairath has received has received consulting fees from AbbVie, Eli Lilly, GlaxoSmithKline, Arena pharmaceuticals, Genetech, Pendopharm, Sandoz, Merck, Takeda, Janssen, Robarts Clinical Trials, and Topivert, Celltrion; speaker’s fees from Takeda, Janssen, Shire, Ferring, Abbvie, and Pfizer. All of these activities are outside the submitted work.
Brian Feagan has received fees for consulting from Abbott/AbbVie, Akebia Therapeutics, Allergan, Amgen, Applied Molecular Transport Inc., Aptevo Therapeutics, Astra Zeneca, Atlantic Pharma, Avir Pharma, Biogen Idec, BioMx Israel, Boehringer‐Ingelheim, Bristol‐Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, EnGene, Ferring Pharma, Roche/Genentech, Galapagos, GiCare Pharma, Gilead, Gossamer Pharma, GSK, Inception IBD Inc, JnJ/Janssen, Kyowa Kakko Kirin Co Ltd., Lexicon, Lilly, Lycera BioTech, Merck, Mesoblast Pharma, Millennium, Nestles, Nextbiotix, Novonordisk, ParImmune, Parvus Therapeutics Inc., Pfizer, Prometheus Therapeutics and Diagnostics, Progenity, Protagonist, Qu Biologics, Receptos, Salix Pharma, Shire, Sienna Biologics, Sigmoid Pharma, Sterna Biologicals, Synergy Pharma Inc., Takeda, Teva Pharma, TiGenix, Tillotts, UCB Pharma, Vertex Pharma, Vivelix Pharma, VHsquared Ltd., and Zyngenia; funds for research from AbbVie Inc., Amgen Inc., AstraZeneca/MedImmune Ltd., Atlantic Pharmaceuticals Ltd., Boehringer‐Ingelheim, Celgene Corporation, Celltech, Genentech Inc/Hoffmann‐La Roche Ltd., Gilead Sciences Inc., GlaxoSmithKline (GSK), Janssen Research & Development LLC., Pfizer Inc., Receptos Inc. / Celgene International, Sanofi, Santarus Inc., Takeda Development Center Americas Inc., Tillotts Pharma AG, UCB; fees for speaking from Abbott/AbbVie, JnJ/Janssen, Lilly, Takeda, Tillotts, and UCB Pharma; Scientific Advisory Board fees from Abbott/AbbVie, Allergan, Amgen, Astra Zeneca, Atlantic Pharma, Avaxia Biologics Inc., Boehringer‐Ingelheim, Bristol‐Myers Squibb, Celgene, Centocor Inc., Elan/Biogen, Galapagos, Genentech/Roche, JnJ/Janssen, Merck, Nestles, Novartis, Novonordisk, Pfizer, Prometheus Laboratories, Protagonist, Salix Pharma, Sterna Biologicals, Takeda, Teva, TiGenix, Tillotts Pharma AG, and UCB Pharma. All of these activities are outside the submitted work.
Reena Khanna has received honoraria from AbbVie, Jansen, Pfizer, Shire, Takeda, and Robarts Clinical Trials for consultancy. All of these activities are outside the submitted work.
New
References
References to studies included in this review
Ambrose 1985 {published data only}
- Ambrose NS, Allan RN, Keighley MR, Burdon DW, Youngs D, Barnes P, et al. Antibiotic therapy for treatment in relapse of intestinal Crohn's disease. A prospective randomized study. Diseases of the Colon & Rectum 1985;28(2):81‐5. [DOI] [PubMed] [Google Scholar]
Arnold 2002 {published data only}
- Arnold Gl, Beaves MR, Pryjdun VO, Mook WJ. Preliminary study of ciprofloxacin in active Crohn's disease. Inflammatory bowel diseases 2002;8(1):10‐5. [DOI] [PubMed] [Google Scholar]
Colombel 1999 {published data only}
- Colombel JF, Lemann M, Cassagnou M, Bouhnik Y, Duclos B, Dupas JL, et al. A controlled trial comparing ciprofloxacin with mesalazine for the treatment of active Crohn's disease. American Journal of Gastroenterology 1999;94(3):674‐78. [DOI] [PubMed] [Google Scholar]
- Colombel JF, Lémann M, Cassagnou M, Bouhnik Y, Duclos B, Dupas JL, et al. Ciprofloxacin vs mesalazine in the treatment of active Crohn's disease. Gut 1996;39 Suppl 3:A188. [DOI] [PubMed] [Google Scholar]
Dewint 2014 {published data only}
- Dewint P, Hansen BE, Verhey E, Oldenburg B, Hommes DW, Pierik M, et al. Adalimumab combined with ciprofloxacin is superior to adalimumab monotherapy in perianal fistula closure in Crohn's disease: a randomised, double‐blind, placebo controlled trial (ADAFI). Gut 2013;63:292‐9. [DOI] [PubMed] [Google Scholar]
- Dewint P, Hansen BE, Verhey E, Oldenburg B, Hommes DW, Pierik M, et al. Adalimumab combined with ciprofloxacin is superior to adalimumab monotherapy in perianal fistula closure in Crohn's disease: a randomised, double‐blind, placebo controlled trial (ADAFI). Gut 2014;63(2):292‐9. [DOI] [PubMed] [Google Scholar]
- Dewint P, Hansen BE, Verhey E, Oldenburg B, Hommes DW, Pierik MJ, et al. Adding ciprofloxacin to adalimumab results in a higher fistula closure rate in perianal fistulizing Crohn's disease. Gastroenterology 2012;142(5 suppl. 1):S212. [Google Scholar]
Lieper 2008 {published data only}
- Leiper K, Martin K, Ellis A, Watson AJ, Morris AI, Rhodes JM. Clinical trial: randomized study of clarithromycin versus placebo in active Crohn's disease. Alimentary pharmacology & therapeutics 2008;27(12):1233‐9. [DOI] [PubMed] [Google Scholar]
Prantera 1996 {published data only}
- Prantera C, Zannoni F, Scribano Ml, Berto E, Andreoli A, Kohn A, et al. An antibiotic regimen for the treatment of active Crohn's disease: a randomized, controlled clinical trial of metronidazole plus ciprofloxacin. American Journal of Gastroenterology 1996;91(2):328‐32. [PubMed] [Google Scholar]
Prantera 2006 {published data only}
- Prantera C, Lochs H, Campieri M, Scribano ML, Sturniolo GC, Castiglione F, et al. Antibiotic treatment of Crohn's disease: results of a multicentre, double blind, randomized, placebo‐controlled trial with rifaximin. Alimentary Pharmacology & Therapeutics 2006;23(8):1117‐25. [DOI] [PubMed] [Google Scholar]
Prantera 2012 {published data only}
- Lochs H, Prantera C, Gionchetti P, Danese S, Fogli MV, Grimaldi M. A new extended intestinal release formulation of rifaximin, 400 mg tablets, for the treatment of moderately active Crohn's disease. Gastroenterology 2011;1):S572. [DOI] [PubMed] [Google Scholar]
- Prantera C, Lochs H, Grimaldi M, Danese S, Scribano Ml, Gionchetti P. Rifaximin‐extended intestinal release induces remission in patients with moderately active Crohn's disease. Gastroenterology 2012;142(3):473‐81. [DOI] [PubMed] [Google Scholar]
Selby 2007 {published data only}
- Selby W, Pavli P, Crotty B, Florin T, Radford‐Smith G, Gibson P, et al. Two‐year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn's disease. Gastroenterology 2007;132(7):2313‐19. [DOI] [PubMed] [Google Scholar]
Steinhart 2002 {published data only}
- Steinhart AH, Feagan BG, Wong CJ, Vandervoort M, Mikolainis S, Croitoru K, et al. Combined budesonide and antibiotic therapy for active Crohn's disease: a randomized controlled trial. Gastroenterology 2002;123(1):33‐40. [DOI] [PubMed] [Google Scholar]
Sutherland 1991 {published data only}
- Sutherland L, Singleton J, Sessions J, Hanauer S, Krawitt E, Rankin G, et al. Double blind, placebo controlled trial of metronidazole in Crohn's disease. Gut 1991;32(9):1071‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thia 2009 {published data only}
- Thia KT, Mahadevan U, Feagan BG, Wong C, Cockeram A, Bitton A, et al. Ciprofloxacin or metronidazole for the treatment of perianal fistulas in patients with Crohn's disease: a randomized, double‐blind, placebo‐controlled pilot study. Inflammatory Bowel Diseases 2009;15(1):17‐24. [DOI] [PubMed] [Google Scholar]
West 2004 {published data only}
- West RL, Woude CJ, Hansen BE, Felt‐BersmaRJF, Tilburg AJP, Drapers JAG, et al. Clinical and endosonographic effect of ciprofloxacin on the treatment of perianal fistulae in Crohn's disease with infliximab: A double‐blind placebo‐controlled study. Alimentary Pharmacology and Therapeutics 2004;20(11‐12):1329‐36. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Allan 1997 {published data only}
- Allan R, Cooke WT. Evaluation of metronidazole in the management of Crohn's disease. Gut 1977;18:A422. [Google Scholar]
Bernstein 1992 {published data only}
- Bernstein CN, Shanahan F. Metronidazole in Crohn's disease: what's the score?. Gastroenterology 1992;102(4 Pt 1):1435‐6. [DOI] [PubMed] [Google Scholar]
Biancone 1998 {published data only}
- Biancone L, Ferrieri A, Silvestri M, Pallone F. Rifaximin in inactive Crohn's Disease: Effect on the intestinal protein loss as assessed by the fecal alpha1‐antitrypsin clearance. Journal of Clinical Research 1998;1(293‐301):289‐301. [Google Scholar]
Blichfeldt 1978 {published data only}
- Blichfeldt P, Blomhoff JP, Myhre E, Gjone E. Metronidazole in Crohn's disease. A preliminary report of a double blind cross over study. Scandinavian Journal of Gastroenterology 1976;11(Sup. 38):100. [DOI] [PubMed] [Google Scholar]
- Blichfeldt P, Blomhoff JP, Myhre E. Gjone E. Metronidazole in Crohn's disease. A double blind cross‐over clinical trial. Scandinavian journal of gastroenterology 1978;13(1):123‐7. [DOI] [PubMed] [Google Scholar]
Gilat 1982 {published data only}
- Gilat T. Metronidazole in Crohn's disease. Gastroenterology 1982;83(3):702‐4. [PubMed] [Google Scholar]
Goodgame 2001 {published data only}
- Goodgame RW, Kimball K, Akram S, Ike E, Ou CN, Sutton F, et al. Randomized controlled trial of clarithromycin and ethambutol in the treatment of Crohn's disease. Alimentary pharmacology & therapeutics 2001;15(12):1861‐6. [DOI] [PubMed] [Google Scholar]
Gui 1997 {published data only}
- Gui GPH, Thomas PRS, Tizard MLV, Lake J, Sanderson JD, Hermon‐Taylor J. Two‐year‐outcomes analysis of Crohn's disease treated with rifabutin and macrolide antibiotics. Journal of Antimicrobial Chemotherapy 1997;39(3):393‐400. [DOI] [PubMed] [Google Scholar]
Hartley‐Asp 1981 {published data only}
- Hartley‐Asp B, Mitelman F, Ursing B. The absence of clastogenic effect during long‐term and short‐term treatment with metronidazole. Mutation Research 1981;85(4):237‐8. [Google Scholar]
Jaworski 2016 {published data only}
- Jaworski A, Borody TJ, Ramrakha S, Sharma R, Leis S, Agrawal G, Gadalla S. Antibiotic therapy for treatment‐naive Crohn's disease patients. American Journal of Gastroenterology 2016;111(Supplement 1):S272. [Google Scholar]
Jigaranu 2014 {published data only}
- Jigaranu AO, Nedelciuc O, Blaj A, Badea M, Mihai C, Diculescu M, et al. Is rifaximin effective in maintaining remission in Crohn's disease?. Digestive Diseases 2014;32(4):378‐83. [DOI] [PubMed] [Google Scholar]
Koch 2007 {published data only}
- Koch TR. Treatment of Crohn's disease with an antibiotic regimen directed against mycobacterium avium paratuberculosis. Clinicaltrials.gov.
Koretz 1997 {published data only}
- Koretz RL. Maintaining remissions in Crohn's disease: A fat chance to please. Gastroenterology 1997;112(6):2155‐56. [DOI] [PubMed] [Google Scholar]
Laudage 1983 {published data only}
- Laudage G, Riemann JF, Schneider MU, Guggenmoos HI. Metronidazole in the treatment of ileocolitis Crohn's? Preliminary results in a prospective randomized study. Fortschr‐Med 1983;101(38):1704‐10. [Google Scholar]
Lee 2018 {published data only}
- Lee SD, Singla A, Rulyak SJ, Clark‐Snustad KD. A Randomized, prospective, double‐blind, placebo‐controlled, crossover study to evaluate the safety and efficacy of rifaximin for the treatment of moderate to severe Crohn's disease. Gastroenterology 2018;154(1 Supplement):S73‐4. [Google Scholar]
Leiper 2000 {published data only}
- Leiper K, Morris AI, Rhodes JM. Open label trial of oral clarithromycin in active Crohn's disease. Alimentary Pharmacology and Therapeutics 2000;14(6):801‐6. [DOI] [PubMed] [Google Scholar]
Maeda 2010 {published data only}
- Maeda Y, Ng SC, Durdey P, Burt C, Torkington J, Dhruva‐Rao P, et al. Metronidazole 10% ointment improves symptoms in perianal Crohn's disease with low systemic exposure. Gut 2009;58:A14. [Google Scholar]
- Maeda Y, Ng SC, Durdey P, Burt C, Torkington J, Rao PK, et al. Randomized clinical trial of metronidazole ointment versus placebo in perianal Crohn's disease. The British Journal of Surgery 2010;97(9):1340‐7. [DOI] [PubMed] [Google Scholar]
Melmed 2009 {published data only}
- Melmed GY. Clarithromycin for Crohn's: A flash in the pan?. Inflammatory Bowel Disease 2009;15(4):639‐40. [DOI] [PubMed] [Google Scholar]
Mitelman 1982 {published data only}
- Mitelman F, Strombeck B, Ursing B. Metronidazole exhibits no clastogenic activity in a double‐blind cross‐over study on Crohn's patients. Hereditas 1982;96(2):279‐86. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Strombeck B, Ursing B. No cytogenic effect of metronidazole. Lancet 1980;1(8180):1249‐50. [DOI] [PubMed] [Google Scholar]
Rogler 2014 {published data only}
- Rogler G. FIST‐MC‐09 study about the effect of doxycycline and acetylcystein in the treatment of CD‐associated fistulae. ClinicalTrials.gov. not published.
Ronge 2007 {published data only}
- Ronge R. Antibiotic therapy does not lower recurrence rate in Crohn's disease. Zeitschrift fur Gastroenterologie 2007;45(12):1218‐20. [Google Scholar]
Steele 2009 {published data only}
- Steele SR. Ciprofloxacin or metronidazole for the treatment of perianal fistulas in patients with Crohn's disease: A randomized, double‐blind, placebo‐controlled pilot study. Diseases of the Colon and Rectum 2009;52(8):1530‐31. [DOI] [PubMed] [Google Scholar]
Steinhart 2008 {published data only}
- Steinhart AH. Metronidazole and ciprofloxacin in the treatment of colonic Crohn's disease: The MACINTOCC trial. ClinicalTrials.gov..
To 1995 {published data only}
- To KW, Tsiaras WG, Thayer WR. Rifabutin use in inflammatory bowel disease. Archives of Ophthalmology 1995;113(11):1354. [DOI] [PubMed] [Google Scholar]
Turunen 1995 {published data only}
- Turunen U, Färkkilä M, Hakala K, Vuoristo M, Rahm V, Seppälä K, et al. Ciprofloxacin treatment combined with conventional therapy in Crohn's disease. A prospective, double blind, placebo controlled study. Gut 1995;37 Suppl 2:A193. [Google Scholar]
Ursing 1982 {published data only}
- Rosén A, Ursing B, Alm T, Bárány F, Bergelin I, Ganrot‐Norlin K, et al. A comparative study of metronidazole and sulfasalazine for active Crohn's disease: the cooperative Crohn's disease study in Sweden. I. Design and methodologic considerations. Gastroenterology 1982;83(3):541‐9. [PubMed] [Google Scholar]
- Ursing B. Treatment of Crohn disease with metronidazole. A Swedish multicentre study. Lakartidningen 1982;79(7):543‐5. [PubMed] [Google Scholar]
- Ursing B, Alm T, Barany F. A comparative study of metronidazole and sulfasalazine for active Crohn's disease: The cooperative Crohn's disease study in Sweden. II. Result. Gastroenterology 1982;83(3):550‐562. [PubMed] [Google Scholar]
References to ongoing studies
NCT02240108 {published data only}
- Valeant Pharmaceuticals International. A double‐blind, placebo‐controlled, parallel‐group, multicenter, multiregional, one year study to assess the efficacy and safety of twice daily oral rifaximin delayed release tablets for induction of clinical remission with endoscopic response at 16 weeks followed by clinical and endoscopic remission at 52 weeks in subjects with active moderate Crohn's disease. ClinicalTrials.gov.
Additional references
Ambrose 1984
- Ambrose NS, Johnson M, Burdon DW, Keighley MR. Incidence of pathogenic bacteria from mesenteric lymph nodes and ileal serosa during Crohn's disease surgery. British Journal of Surgery 1984;71:623‐5. [DOI] [PubMed] [Google Scholar]
Barnich 2007
- Barnich N, Darfeuille‐Michaud A. Role of bacterial in the etiopathogenesis of inflammatory bowel disease. World Journal of Gastroenterology 2007;13(42):5571‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bernstein 2016
- Bernstein C, Eliakim A, Fedail S, Fried M, Gearry R, Goh KL, et al. World gastroenterology organisation global guidelines inflammatory bowel disease: Update August 2015. Journal of Clinical Gastroenterology 2016;50(10):803‐18. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elson 1995
- Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344‐67. [DOI] [PubMed] [Google Scholar]
Elson 2005
- Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive and regulatory mechanisms of host dialogue with the microbiota. Immunological Reviews 2005;206:206‐76. [DOI] [PubMed] [Google Scholar]
Gevers 2014
- Gevers D, Kugathasan S, Denson LA, Vázquez‐Baeza Y, Treuren W, Ren B, et al. The treatment‐naive microbiome in new‐onset Crohn's disease. Cell Host Microbe 2014;15(3):382‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. British Medical Journal 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. British Medical Journal 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, GreenS, editors(s) editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Khan 2011
- Khan KJ, Ullman TA, Ford AC, Maria T, Abreu MT, Abadir A, et al. Antibiotic therapy in inflammatory bowel disease: A systematic review and meta‐analysis. American Journal of Gastroenterology 2011;106:661‐73. [DOI] [PubMed] [Google Scholar]
Kostic 2014
- Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel diseases: current status and the future ahead. Gastroenterology 2014;146(6):1489‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marks 2006
- Marks DJB, Harbord MWN, MacAllister R, Rahman FZ, Young J, Al‐Lazikani B, et al. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet 2006;367:668–78. [DOI] [PubMed] [Google Scholar]
Mylonaki 2005
- Mylonaki M, Rayment NB, Rampton DS, Hudspith BN, Brostoff J. Molecular characterization of rectal mucosa‐associated bacterial flora in inflammatory bowel disease. Inflammatory Bowel Disease 2005;11:481‐487. [DOI] [PubMed] [Google Scholar]
Ott 2004
- Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004;53:685‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Patton 2016
- Patton PH, Parker CE, MacDonald JK, Chande N. Anti‐tuberculous therapy for maintenance of remission in Crohn's disease. Cochrane Database of Systematic Reviews 2016, Issue 7. [DOI: 10.1002/14651858.CD000299.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rath 1999
- Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA‐B27 transgenic rats selectively colonized with bacteroides vulgatus or escherichia coli. Infection and Immunity 1999;67(6):2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sartor 2008
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008;134(2):577‐94. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Scribano 2013
- Scribano ML, Prantera C. Use of antibiotics in the treatment of Crohn’s disease. World Journal of Gastroenterology 2013;19(5):648‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Swidsinski 2002
- Swidsinski A, Ladhoff A, Pernthaler A, Swinsinski S, Loening‐Baucke V, Ortner M. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002;122:44‐54. [DOI] [PubMed] [Google Scholar]