

Abstract
Background/Aim:
NME/NM23 nucleoside dipho-sphate kinase 1 (NME1) is a metastasis suppressor gene, exhibiting reduced expression in metastatic cancers and the ability to suppress metastatic activity of cancer cells. We previously identified NME1-regulated genes with prognostic value in human melanoma. This study was conducted in melanoma cell lines aiming to elucidate the mechanism through which NME regulates one of these genes, aldolase C (ALDOC).
Materials and Methods:
ALDOC mRNA and protein expression was measured using qRT-PCR and immunoblot analyses. Promoter-luciferase constructs and chromatin immunoprecipitation were employed to measure the impact of NME1 on ALDOC transcription.
Results:
NME1 enhanced ALDOC transcription, evidenced by increased expression of ALDOC pre-mRNA and activity of an ALDOC promoter-luciferase module. NME1 was detected at the ALDOC promoter, and forced NME1 expression resulted in enhanced occupancy of the promoter by NME1, increased presence of epigenetic activation markers (H3K4me3 and H3K27ac), and recruitment of RNA polymerase II.
Conclusion:
This is the first study to indicate that NME1 induces transcription through its direct binding to the promoter region of a target gene.
Keywords: Melanoma, NME1, ALDOC, metastasis suppressor, ALDOC promoter, transcription, epigenetic markers
Melanoma that has progressed to metastatic stages is highly lethal, with a dismal 5-year survival rate of 15–20% in the United States (1). NME/NM23 nucleoside diphosphate kinase (NDPK) 1 (NME1) (also known as nucleoside diphosphate kinase-A/NDPK-A or Nm23-H1) was the first metastasis suppressor gene (MSG) to be identified, exhibiting the ability to inhibit the metastatic properties of melanoma cells in the absence of significant impacts on primary tumor growth (2). Moreover, expression of NME1 is associated with better overall clinical outcome in several cancer types (3). In addition, we have demonstrated that ablation of the Nme1-Nme2 locus in mice confers strong metastatic activity in a model of ultraviolet light-induced melanoma, providing in vivo validation of metastasis suppressor functions for the Nme1 and/or Nme2 genes (4). The metastasis-specific suppressor function of NME1 and other MSGs render them valuable for the discovery of molecular mechanisms that specifically regulate the metastatic process. Importantly, such mechanisms represent potential molecular targets for therapy of cancers in their advanced stages.
Early efforts revealed numerous molecular pathways in the cytosol and cell membrane that are regulated by NME1 and could mediate its inhibitory effects on cellular motility and invasion (5–7). In addition, previous studies have reported that NME1 regulates the expression of a number of genes in melanoma and breast carcinoma cell lines that could mediate its metastasis suppressor function (8, 9). One of these reports highlighted the expression of the mRNA encoding lysophosphatidic acid receptor (EDG2) as a target of NME1-mediated suppression in breast carcinoma cell lines (10). NME1 has been shown to bind and cleave single-stranded DNA in vitro (11, 12), suggesting potential direct participation in regulating gene transcription and DNA repair processes within the nucleus (13). Chromatin immunoprecipitation (ChIP) analyses have revealed binding of NME1 to DNA motifs in the promoter regions of the platelet-derived growth factor-A (PDGFA), TP53 and other genes (14, 15). Moreover, a number of studies have provided evidence that NME1 and NME2 exh.ibit indirect interactions with transcription-regulatory elements, suggesting a co-regulatory function (16–22). No study to date, however, has clearly demonstrated that NME proteins regulate transcription of genes via direct interactions with DNA.
Herein, the role of NME1 as a DNA-binding transcription factor was analyzed using the ALDOC gene as a prototype. NME1 was shown to upregulate the expression of un-spliced, pre-mRNA levels of ALDOC, as well as to trans-activate the ALDOC promoter. Moreover, it was demonstrated that NME1 is localized to both upstream and proximal promoter regions of ALDOC , and this binding is increased upon forced expression of NME1 protein. In parallel, forced NME1-expressing cells elicited an epigenetic signature in the 5’-flanking region of the ALDOC gene, evidenced by increased trimethylation of the lysine 4 residue on histone 3 (H3K4me3), acetylation of lysine 27 on histone 3 (H3K27ac), and recruitment of RNA polymerase II. The present study provided a novel and comprehensive demonstration of a transcription factor function for the metastasis suppressor, NME1.
Materials and Methods
Cell lines and culture conditions.
WM1158, and WM278 melanoma cell lines (kindly provided by Dr. Meenhard Herlyn, Wistar Institute, Philadelphia, PA, USA), were cultured in Tu2% media composed of MCDB:Leibovitz-15 medium (4:1, v/v), 2 mM CaCl2, 2.5 μg/ml insulin (all components of Tu2% media were from Sigma-Aldrich, St. Louis, MO, USA) and 2% fetal bovine serum (Life Technologies, Grand Island, NY, USA) and maintained at 37˚C in 5% CO2. MDA-MB-435s/M14 cells (referred to as M14 in this text) were obtained from Dr. Rina Plattner (University of Kentucky, Lexington, KY, USA) and maintained in complete DMEM media containing 4.5 g/l D-glucose, 584 mg/l L-glutamine, and 110 mg/l sodium pyruvate (all components from Life Technologies) and supplemented with 10% FBS. All cells were cultured at 37˚C in 10% CO2. M14 cells stably expressing NME1 were generated as previously described (23). Briefly, M14 cells were transfected with pEGFP-N1-NME1 or pEGFP-N1 plasmids and selected for stable integration using G418 (Life Technologies). Forced expression of NME1 in WM1158 cells was achieved by transduction with pSMPUW-IRES-EGFP-NME1 or pSMPUW-IRES-EGFP lentivirus at a multiplicity of infection (MOI) of 50. Fluorescence activated cell sorting (FACS; FACSAria Flow Cytometer, BD biosciences, Mountain View, CA, USA) was used to sort GFP-positive cells. Knockdown of NME1 in the WM278 cell line was achieved by transient lentiviral transduction with Mission Sigma (SHC002, Sigma-Aldrich) shNME1 TRCN0000010062 or pLKO.1 non-target shRNA at a MOI of 1000.
Quantitative real-time polymerase chain reaction (qRT-PCR).
Total RNA was isolated based on manufacturer instructions using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA). A total of 1 μg of isolated RNA was converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA). Analyses were performed on a CFX Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). PCR was conducted with 40 cycles of: 15 s at 95˚C (denaturation), 30 s at 60˚C (primer annealing), and 5 s at 65˚C (extension). Relative fold-changes in expression of genes of interest were determined by the 2 –ΔΔCT method using SYBR green qPCR master mix (Applied Biosystems) with normalization to internal control RPL13a. Specified primer sequences (Table I) were included in reactions at a final concentration of 200 nM. Total RNA was collected for each cell line from three independent replicates.
Table I.
Oligodeoxynucleotide primers used for polymerase chain reaction analyses
| Gene/mRNA | Forward primer | Reverse primer |
|---|---|---|
| ALDOA | CGTCCACGGACTCTCCGTTA | GCGATGTCAGACAGCTCCTT |
| ALDOB | CGGCAGTCCCGAGAAATCCT | CATCAAGCCCTTGAATGGTGG |
| ALDOC | GCTGTCCCAGGAGTGACCTT | CATTCACCTCAGCCCGCTT |
| ALDOC Intron | CTGAGAGAAGAGGGTGGCAA | CCAATCAAGGATGCCAACCC |
| RPL13a | CATAGGAAGCTGGGAGCAAG | GCCCTCCAATCAGTCTTCTG |
| ChIP Primers (−542/−389) | AAACTCTGGGCACTGGACTG | TTCAGGGCAGGAAGTAATGAA |
| ChIP Primers (−188/−4) | GAGGGCAGTCCCTAACAGC | GTAAATGAGGCTGCGGATGT |
| ChIP Primers (+31/+244) | GGATCAGAACCCGAGCTGT | GAGCCATGGGTTTCAAGTA |
Immunoblot analysis.
Whole cell lysates of M14, WM1158, and WM278 cells were generated using RIPA lysis buffer (10mM Tris-HCl pH 7.5, 1mM EDTA, 0.5mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 140mM NaCl) supplemented with Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL, USA). SDS-polyacrylamide gel electrophoresis (AnyKD Criterion Precast Midi Protein Gel, Bio-Rad) was performed on 5–20 μg of protein lysates as quantified by the BCA assay (Thermo Scientific), followed by transfer to nitrocellulose membranes (BioRad). Membranes were incubated with specific antibodies against NME1 (1:2,000, 16h at 4˚C; 610247, BD Biosciences, San Jose, CA, USA) and ALDOC (1:1,000, 48h at 4˚C; SC-12065, Santa Cruz Biotechnologies, Santa Cruz, CA, USA), diluted in 5% non-fat dry milk in Tris buffered saline with 0.1% Tween-20 (TBST). Antibody anti-Histone 3 (1:5,000, 16 h at 4˚C; clone D1H12, Cell Signaling, Danvers, MA, USA) was used to verify equal loading of lysate protein. Three washes with TBST buffer were performed, followed by 1h incubation at room temperature with one of the following isotype specific-HRP-conjugated secondary antibodies: ECL-conjugated anti-mouse IgG for anti-NME1 blots (1:20,000; GE Healthcare, Chicago, IL, USA), anti-goat-IgG-HRP for anti-ALDOC blots (1:10,000; sc-2020, Santa Cruz,) and ECL-conjugated anti-rabbit IgG for anti-Histone 3 blots (1:50,000; GE Healthcare). Proteins were detected on immunoblots by chemiluminescence after addition of Amersham ECL Prime Western Blot Detection Reagent (GE Healthcare) and exposure to Amersham Hyperfilm ECL (GE Healthcare).
Promoter-luciferase assay.
M14 cells were transiently transfected with 100 ng of a Renilla luciferase pLightSwitch promoter plasmid containing the transcriptional regulatory region of ALDOC (−880 to +118) (SwitchGear Genomics, Menlo Park, CA, USA). Additionally, 50 ng of a pcDNA3.1-based Firefly luciferase plasmid (24) (Addgene, Cambridge, MA, USA), was co-transfected to serve as a transfection efficiency control. Cells were transfected using Fugene 6 (Promega, Madison, WI, USA) as previously described (25) , with luciferase activity measured in cell lysates 48 h post-transfection (Dual Luciferase Reporter Assay Kit, Promega, Madison, WI, USA). The Renilla luciferase promoter signal was normalized to Firefly luciferase activity, and relative fold-change in relative luminescence units (RLU) was quantified.
Chromatin immunoprecipitation (ChIP).
Sheared chromatin for ChIP analysis was generated using the ChIP-IT Express Kit (Active Motif, Carlsbad, CA, USA). Fifteen micrograms of chromatin were pre-cleared with Protein G magnetic beads (Cell Signaling) and isotype control antibodies, mouse IgG (clone G3A1, Cell Signaling) or rabbit IgG (clone DA1E, Cell Signaling), for 1 h at 4˚C. Immunoprecipitation with 5 μg of specified antibodies against NME1 (SC-NM301, Santa Cruz Biotechnologies), Histone 3 lysine 27 acetylation (H3K27ac, clone D5E4, Cell Signaling), Histone 3 lysine 4 trimethylation (H3K4, Cell Signaling), and RNA Polymerase II (Active Motif) was performed overnight at 4˚C on an end-to-end rotator. Eluted DNA was purified using the QiaQuick PCR Purification Kit (Qiagen) prior to qPCR analysis, using specified primer sequences found in Table I. Quantification was achieved by normalization to input DNA, and expressed as % input.
Gene expression profiling.
Gene expression data from the GSE46517 dataset published by Kabbarah et al. was obtained from the Gene Expression Omnibus (26). Normalized log2 expression for ALDOC was evaluated in 17 normal skin and benign nevi, 31 primary melanomas, and 73 metastatic melanoma specimens. Genomic and clinical data from the Skin Cutaneous Melanoma collection through The Cancer Genome Atlas (TCGA-SKCM) was obtained via cBioPortal (27–29). Patient identifiers, and normalized log2 mRNA expression of ALDOC were downloaded from cBioPortal and aligned with corresponding clinical attributes (e.g. Breslow depth). For Breslow depth correlation to ALDOC expression, primary melanoma specimens were assigned into one of two groups corresponding to depths of less than or equal to 4 mm, or greater than 4 mm.
Statistical analysis.
Unless otherwise stated, statistical analyses were performed using t-test in Microsoft Excel v 1809. For RT-qPCR analysis, results represent the mean of three independent experiments, with experiment analyzed in triplicate. Relative luciferase activity was determined from the average of 2 independent experiments, with each experiment conducted with triplicate wells. A representative experiment from two independent experiments is displayed for ChIP analysis. Analysis of the GSE46517 dataset was performed using SigmaPlot software (Sysat Software, San Jose, CA, USA) by one-way-analysis of variance (ANOVA) using post-hoc: Holm-Sidak method. Significance of log2 mRNA expression of ALDOC in the TCGA dataset was determined by t- test. Resulted p-values <0.05 were considered statistically significant.
Results
NME1 up-regulated the expression of ALDOC in melanoma cell lines.
qRT-PCR analysis confirmed our prior observation (9) of strong NME1-mediated upregulation of ALDOC mRNA expression (800-fold, p< 0.05) in M14 cells (Figure 1A, left). Immunoblot analysis demonstrated a NME1-induced increase (4.9-fold) in ALDOC protein levels (Figure 1A, right). To further corroborate this observation, forced NME1 expression was also conducted in the melanoma cell line WM1158, which exhibits low endogenous expression of NME1. Forced NME1 expression was accomplished with a lentiviral expression vector containing the NME1 cDNA, followed by an IRES motif and a GFP-coding module. NME1-overexpressing cells (GFP-positive) obtained by fluorescent-activated cell sorting (FACS) exhibited greatly increased expression of ALDOC mRNA (6-fold, p<0.05) and protein (3.8-fold) compared to vector-transfected controls (Figure 1B). Finally, to further confirm the regulatory axis between NME1 and ALDOC, the impact of silencing NME1 expression was examined in the melanoma cell line WM278, which expresses NME1 at normal levels. Cells were infected with a lentiviral construct expressing a shRNA, which effectively silences the NME1 transcript and results in decreased ALDOC mRNA expression as we have previously shown (9). Treatment with the anti-NME1 shRNA sequence indeed elicited a significant decrease (62%) in expression of ALDOC protein (Figure 1C).
Figure 1.
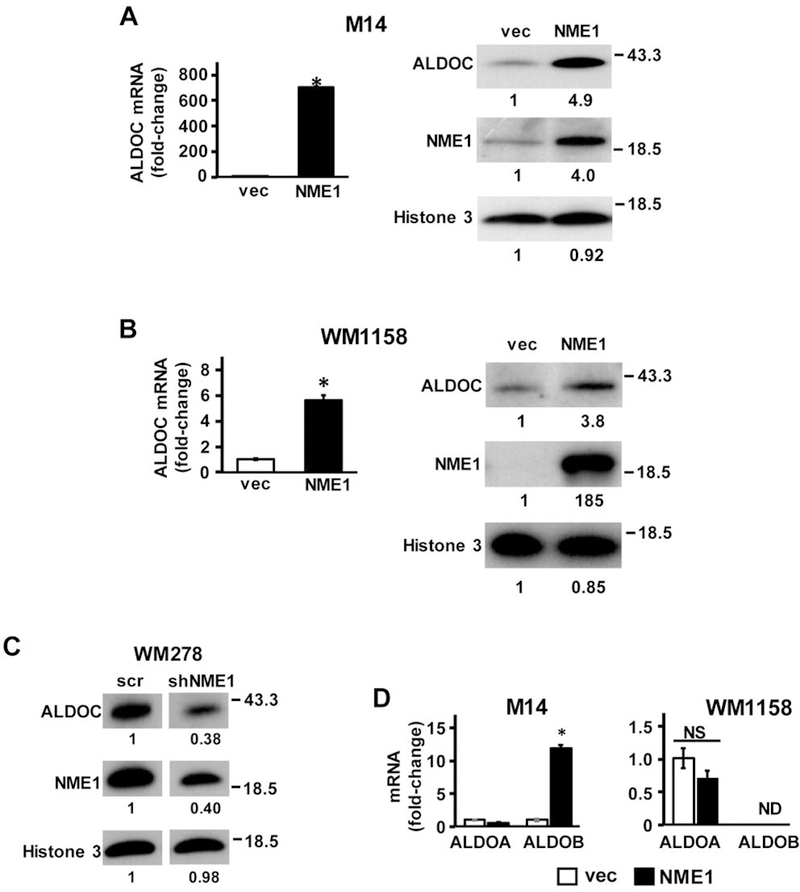
NME1 induces the expression of ALDOC mRNA and protein. (A left) ALDOC mRNA was measured by qRT-PCR in M14 cells stably transfected with empty expression vector (vec, unfilled bar) or NME1 expression vector (NME1, black filled bar). (A right) Expression of ALDOC protein was determined by immunoblot analysis in vec and NME1 variants of M14 cells. (B) ALDOC mRNA and protein expression in vec- and NME1-expressing variants of WM1158 melanoma cells. (C) WM278 cells were infected with lentiviruses expressing a scrambled shRNA or an shRNA sequence directed to NME1. ALDOC protein expression was determined by immunoblot analysis as in (A). Asterisks in panels A and B indicate statistically significant differences (p<0.05) in mRNA expression compared to the control vec, as determined by t-test. (D) Impact of NME1 overexpression on ALDOA and ALDOB mRNAs in M14 (left) and WM1158 (right) cells was determined by qRT- PCR. *p<0.05. Fold-changes in protein expression are expressed relative to controls and displayed below corresponding lanes in all immunoblots. Relative protein expression was determined by densitometric analysis of immunoreactive bands using ImageJ software. ALDOC, aldolase C.
Aldolase enzymes are well-known for their function in glycolysis, where they catalyze the reversible conversion of fructose 1,6 bisphosphate to glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone (DHAP) (30). The family of aldolases are comprised of 3 different isotypes (A, B, and C). While each of the isotypes perform the same primary enzymatic function, they are expressed in a tissue specific manner. Aldolase A (ALDOA) is ubiquitously expressed and is the most widely studied of the aldolase isozymes, with highest expression in muscle, while aldolase B (ALDOB) is generally localized to liver, and kidney cortex tissues (30). ALDOC is expressed primarily in brain and other neural crest-derived cells, including melanocytes (31). To determine the specificity with which NME1 upregulates expression of ALDOC, qRT-PCR analysis was performed to measure steady-state concentrations of ALDOA and ALDOB mRNA in M14 and WM1158 cells. No changes in ALDOA expression were observed in either of the cell lines in response to NME1 overexpression (Figure 1D). ALDOB mRNA was detected at very low levels in the vector-transfected control M14 cells and its expression was induced in NME1-overexpressing M14 cells (12-fold, p<0.05; Figure 1B). However, even after induction by NME1, ALDOB mRNA expression was much lower in M14 cells than that of ALDOC mRNA (average Ct values: ALDOC mRNA in control cells, 31.0±0.1; ALDOB mRNA in control cells, 39.5±0.5 ALDOB mRNA in NME1-overexpressing cells, 37.4±0.2). In the WM1158 cell line, ALDOB mRNA was not detected in any group (control vec or NME1-overexpressing). Together, these findings demonstrate that the ALDOC transcript is the predominant isoform in this cell line. The results highlight the tissue specificity of the expression of these isozymes and confirm the strong NME1-mediated up-regulation of ALDOC gene expression.
Recruitment of NME1 to the ALDOC promoter is associated with epigenetic activation and increased occupancy by RNA polymerase II.
The robust upregulation of ALDOC mRNA expression conferred by NME1 across multiple melanoma cell lines suggested that this induction might be mediated via ALDOC transcription enhancement. To this end, qRT-PCR analysis was performed using primers directed at the intronic region of ALDOC to detect precursor mRNA (pre-mRNA) levels (Table I). The pre-mRNA reflects the unprocessed RNA that is synthesized from the DNA template directly, thus providing an indirect index of transcriptional activity. A significant increase in ALDOC pre-mRNA levels was observed in both the M14 and WM1158-NME1 expressing cells (Figure 2A), strongly suggesting that NME1-dependent induction of ALDOC occurred at the transcriptional level.
Figure 2.
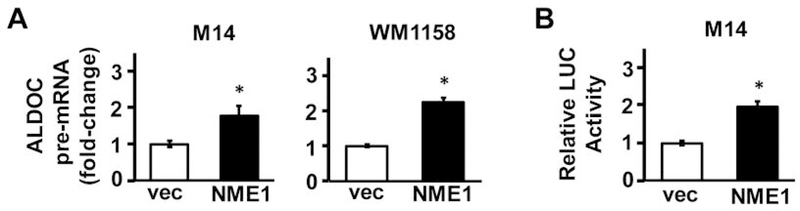
NME1 induces transcription of the ALDOC gene. (A) Impact of NME1 overexpression on nascent ALDOC transcripts in M14 (left) and WM1158 (right) cells was determined by qRT-PCR using primers containing intronic sequences (see Materials and Methods). (B) A luciferase promoter plasmid containing a −880 to +118 fragment of the ALDOC gene was used for transient transfection of vec and NME1 variants of M14 cells. Luciferase activity is expressed relative to that obtained in vec control cells. *p<0.05. ALDOC, aldolase C.
Given the up-regulation of ALDOC expression by NME1 in multiple melanoma cell lines, and the observed increase in the ALDOC pre-mRNA, we next intended to determine whether NME1 activates the ALDOC promoter. M14 cell line was selected for further analysis since it had the strongest induction of ALDOC by NME1. A plasmid containing a region of the ALDOC promoter (–880 to +118) located upstream of the Renilla luciferase reporter gene was used for the transfection of M14 cells, which were stably transfected with either empty or NME1 expression vector. Luciferase activity was significantly higher in the presence of forced NME1 expression (Figure 2B), consistent with NME1-mediated activation of ALDOC transcription via interactions with the −880 to +118 region of the ALDOC gene. The observed increased in relative luciferase activity provides evidence that NME1 activates the ALDOC promoter.
To further assess the impact of NME1 on transcription of the ALDOC gene, chromatin immunoprecipitation (ChIP) analysis was conducted with three different DNA amplicons located upstream and downstream of the transcription start site of ALDOC gene (–542 to −398, −188 to −4, and +31 to +244). Likely targets of epigenetic modification within the ALDOC gene were identified prior to the study using the University of California, Santa Cruz (USCS) genome browser and ChIPseq data from the Encyclopedia of DNA Elements (ENCODE) project (32–33). ENCODE contains ChIP-seq data from several different cell types that localizes transcription factor and RNA polymerase binding, as well as histone marker presentation across the genome (33). The epigenetic profile for the ALDOC gene provided by ENCODE was characterized by enrichment of the epigenetic marker of transcriptional activation, histone 3-lysine27Ac (H3K27ac), within the −630 to −200 region (Figure 3A) upstream of the transcription start site. In addition, ENCODE identified enrichment of the activation marker (H3K4Me3) within the 5’ UTR of the ALDOC gene. While ChIP analysis conducted in control M14 cells failed to detect the H3K27ac modification, forced NME1 expression resulted in a significant increase of H3K27ac across all three regions analyzed (Figure 3B), consistent with NME1-mediated activation of the ALDOC promoter. NME1 expression also resulted in significant enrichment of the transcription activation mark H3K4me3 within the +31 to +244 region of the ALDOC gene (Figure 3B), consistent with predictions from the ENCODE site. In a similar fashion, NME1 was not detected at the queried locations of the ALDOC gene in vector-transfected M14 cells, but was detected at all three regions when NME1 was overexpressed (Figure 3B). Finally, RNA polymerase II was detected at only low levels in vector-transfected cells across the three regions studied, while its recruitment was significantly enhanced by NME1 overexpression. Association of RNA polymerase II with amplicons adjacent to the transcription start site can be attributed to a lack of precision of the ChIP procedure and has previously been described (34). Taken together, the ChIP analyses demonstrate that NME1 expression results in the activation of ALDOC promoter, with inducible recruitment of NME1 to the promoter region strongly suggesting its participation in the activation mechanism.
Figure 3.
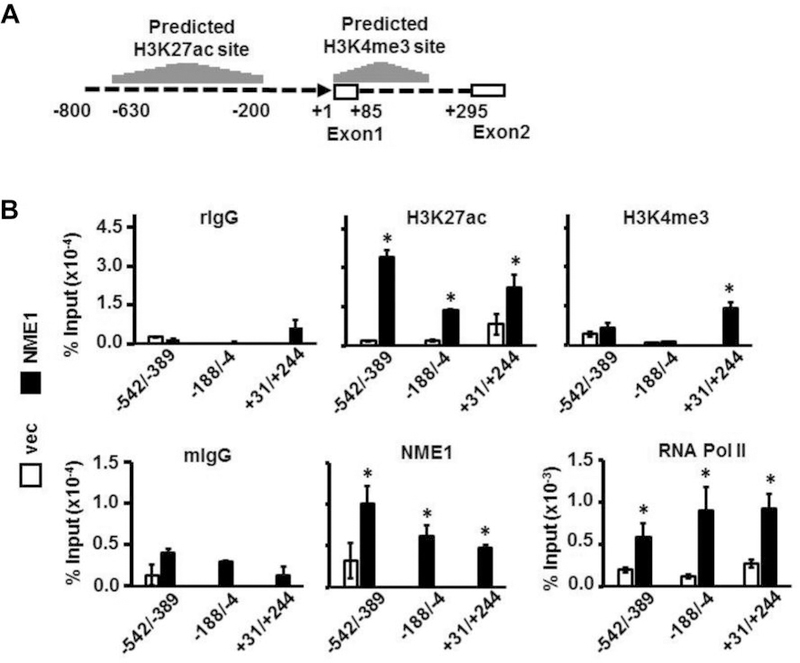
Recruitment of NME1 to the ALDOC promoter region is associated with a transcriptionally-active state. (A) The schematic displays 5’-flanking (−800 to −1) and 5’-untranslated (+1 to +295) regions of the ALDOC gene. Predicted acetylation of lysine 27 on histone 3 (H3K27Ac) and trimethylation of the lysine 4 residue on histone 3 (H3K4me3) sites are presented as histograms above the DNA sequence, and were derived from ChIP-seq data obtained from the ENCODE database using the UCSC genome browser. (B, upper panel) ChIP analysis was conducted as indicated with non-immune anti-rabbit IgG (rIgG) or rabbit antibodies directed to H3K27Ac or H3K4me3. (B, lower panel) ChIP analysis was conducted with non-immune anti-mouse IgG (rIgG) or mouse monoclonal antibodies directed to NME1 or RNA polymerase II (RNA Pol II). Immunoprecipitated DNA was analyzed by quantitative PCR using primer pairs yielding the three amplicons shown (−542 to −389, −188 to −4, or +31 to +244). *p<0.05. ALDOC, aldolase C.
ALDOC expression is reduced in metastatic and invasive melanoma.
The Genome Expression Omnibus (GEO) database repository was examined to assess the potential association between ALDOC expression and melanoma progression. Analysis of the GSE46517 dataset (26) revealed significantly lower expression of ALDOC mRNA in metastatic melanoma compared to normal skin and early stages of melanoma (Figure 4A). In addition to this dataset, ALDOC expression was analyzed in The Cancer Genome Atlas Skin Cutaneous Melanoma (TCGA-SKCM) (27). The TCGA-SKCM dataset is comprised of primary and metastatic melanoma samples along with clinical attributes of the samples collected. Normalized log2 mRNA values were downloaded from the c-BioPortal website (28, 29). While no significant difference in the expression of ALDOC mRNA was observed between primary and metastatic melanomas (data not shown), invasive melanomas (Breslow index >4 mm) indeed exhibited a significant reduction in ALDOC mRNA expression (Figure 4B). Breslow depth is an independent predictor of melanoma progression and metastatic potential (35). It should be noted that expression of the NME1 transcript was not a predictor of outcome or stage of melanoma in either the GSE46517 or TCGA-SKCM databases. This is not surprising, in light of our prior observation that down-regulation of NME1 protein expression in human metastatic melanoma cell lines results from destabilization of the NME1 protein itself via cathepsin-mediated degradation in the lysosomal compartment, and not from alterations in steady-state concentrations of the NME1 transcript (36). Taken together, these observations strongly suggest that ALDOC represents a relevant effector of the metastasis suppressor activity of NME1.
Figure 4.
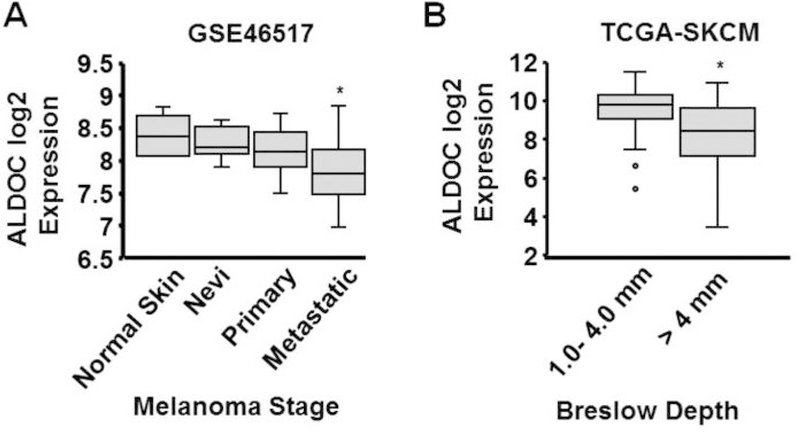
ALDOC mRNA expression is reduced in specimens of metastatic melanoma in humans, and is associated with significantly lower Breslow depth. (A) ALDOC mRNA expression was compared between biopsies of primary and metastatic melanoma (panel A, GEO dataset GSE46517). (B) Association of ALDOC mRNA expression with Breslow depth was measured in melanomas of The Cancer Genome Atlas, Skin and Cutaneous Melanoma (TCGA-SKCM). *p<0.05. ALDOC, aldolase C.
Discussion
Although the ability of NME1 to regulate transcriptomic profiles in cancer cells is well-documented (8, 9), the molecular mechanisms underlying this activity have yet to be fully elucidated. An early study demonstrated a single-stranded DNA-binding activity of the NME2 isoform in vitro (11), as well as the ability to transactivate the c-MYC promoter (37). Our laboratory subsequently demonstrated the in vitro DNA-binding activity of the NME1 isoform toward single-stranded DNA motifs found in the PDGFA promoter region (12). Neither study determined whether these NME isoforms bound to these elements in living cells, nor whether DNA binding was associated with transcriptional activity of the putative target genes. Other studies employing ChIP analysis have indeed reported physical association of NME1 with DNA motifs in a number of genes with potential relevance to cancer phenotype (14, 15), but none has analyzed the impact of the DNA binding interactions on transcriptional activity. Another series of reports have identified potential co-regulator functions of NME1 that do not involve direct DNA binding (16–22). Our current study provides the novel observations of inducible recruitment of NME1 to the promoter region of its target gene, ALDOC, and a strong association between occupancy of NME1 and an epigenetically active state at the ALDOC promoter. The interaction of NME1 with the ALDOC promoter region appears to be direct, in light of its DNA-binding activity in vitro, and evidence obtained by ChIP analysis. The latter employs a formaldehyde crosslinking procedure favoring detection of DNA-protein contacts. No sequence motifs with homology to previously described NME-binding sequences in the c-MYC and PDGFA genes could be identified within the queried regions of the ALDOC gene. However, these regions do contain a multitude of GC-rich sequences likely to harbor single-stranded structures that could be recognized for NME1 binding. The possibility cannot be formally excluded; however, the procedure detected an indirect interaction of NME1 with the ALDOC gene via crosslinking to a primary DNA binding protein.
Recruitment of NME1 was associated with activation of ALDOC transcription, most likely by promoting assembly of a coactivator complex composed of histone acetylases and methylases that promote epigenetic activation of the locus. Another intriguing possibility is that the enzymatic functions of NME1 may play key roles in the transcriptional activation mechanism. For example, the NDPK activity of NME1 has been proposed to provide deoxyribonucleotides to effector molecules as diverse as AMPK and DNA polymerases via a “substrate channeling” mechanism (38, 39). The NDPK activity of NME1 recognizes ribonucleoside diphosphates and triphosphates for phosphate exchange as well (40), suggesting it could also promote RNA polymerase II activity by supplying rNTPs as needed. Alternatively, the 3’–5’ exonuclease activity of NME1 could promote acquisition of a transcriptionally active conformation for a given DNA motif by physical remodeling of the site (41). A potential precedent is provided by a key enhancer element in the vascular smooth muscle α-actin gene, whose activity is dependent upon the equilibrium between double-stranded DNA binding proteins that stabilize an active conformation and single-stranded DNA binding proteins that stabilize a transcriptionally-repressive, non-B-form DNA structure (42).
The robust up-regulation of ALDOC expression by NME1 strongly suggests that ALDOC contributes to metastasis suppressor activity. Indeed, the metastatic behavior of cancer cells can be influenced profoundly by alterations in cell metabolism (43, 44). The aldolase family enzymes (ALDOA, ALDOB, and ALDOC) predominantly function in glycolysis, catalyzing the reversible conversion of fructose 1,6 bisphosphate to glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) (45). This activity has been proposed to promote the Warburg effect and stem-like properties in cancer cells (46). Elevated ALDOA expression has been associated with poor overall outcome in colorectal cancer, pancreatic cancer, and lung squamous cell carcinoma (47–50). In striking contrast, however, both ALDOB and ALDOC have been shown to suppress metastatic potential in hepatocellular and oral squamous carcinomas (51–54). Moreover, some studies have suggested that the suppressor activity of ALDOC is independent of its glycolytic activity, mediated via direct binding to actin filaments and consequent inhibition of cytoskeletal remodeling and cell motility (55, 56). Herein, we observed that silencing of ALDOC mRNA expression in M14 melanoma cells with shRNA constructs fails to disrupt the motility-suppressing activity of NME1 (data not shown). This observation suggests that ALDOC expression may not be sufficient to mediate the metastasis-suppressor function of NME1, but does not exclude contributions in the context of an overall NME1-regulated profile of gene expression. We previously reported that NME1 regulates a signature of gene expression including ALDOC that collectively has prognostic value in melanoma (9). We herein report that the expression of ALDOC alone is lower in metastatic melanomas in one publicly available dataset (GSE46517), and is associated with reduced Breslow depth in melanomas of the TCGA-SKCM collection. Together, these observations suggest that ALDOC is a mediator of the metastasis suppressor function of NME1.
Overall, the studies outlined herein provide the novel observation that NME1 up-regulates the transcriptional activity of a target gene, ALDOC. Moreover, the induction was shown to involve direct interactions of NME1 with the promoter region of ALDOC , as well as conversion of the gene to an epigenetically-active condition. Future studies will be directed to addressing the mechanism through which NME1 induces the transcription of ALDOC and other target genes as a canonical DNA-binding transcription factor, the identification of NME1 response elements within those target genes, and the discovery of other factors (e.g. transcription factors and coregulators) with which NME1 interacts to regulate transcription.
Acknowledgements
These studies were supported by the National Institutes of Health/National Cancer Institute through research grants CA83237, CA159871 and CA159871-S1 (DMK), a training grant (T32 CA154274), and an education grant from the National Institutes of Health/National Institute of General Medical Sciences (R25 GM055036). We also wish to thank Drs. Thomas Hornyak, Dudley Strickland, Anthony Passaniti, Shawn Lupold, Stuart Martin, and Khavita Bhalla for helpful discussions and advice.
References
- 1.American Cancer Society website. Survival rates for melanoma skin cancer, by stage Available from: https://www.cancer.org/cancer/melanoma-skin-cancer/detection-diagnosis-staging/survival-rates-for-melanoma-skin-cancer-by-stage.html. Last accessed September 12, 2018.
- 2.Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA and Sobel ME: Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 80: 200–204, 1988. [DOI] [PubMed] [Google Scholar]
- 3.Hartsough MT and Steeg PS: Nm23/nucleoside diphosphate kinase in human cancers. J Bioenerg Biomembr 32: 301–308, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Jarrett SG, Novak M, Harris N, Merlino G, Slominski A and Kaetzel DM: NM23 deficiency promotes metastasis in a UV radiation-induced mouse model of human melanoma. Clin Exp Metastasis 30 : 25–36, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K and Sugimura H: Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc Natl Acad Sci USA 98: 4385–4390, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murakami M, Meneses PI, Lan K and Robertson ES: The suppressor of metastasis Nm23-H1 interacts with the Cdc42 Rho family member and the pleckstrin homology domain of oncoprotein Dbl-1 to suppress cell migration. Cancer Biol Ther 7: 677–688, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Li MQ, Shao J, Meng YH, Mei J, Wang Y, Li H, Zhang L, Chang KK, Wang XQ, Zhu XY and Li DJ: NME1 suppression promotes growth, adhesion and implantation of endometrial stromal cells via Akt and MAPK/Erk1/2 signal pathways in the endometriotic milieu. Human Reprod 28 : 2822–2831, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Horak CE, Lee JH, Elkahloun AG, Boissan M, Dumont S, Maga TK , Arnaud-Dabernat S, Palmieri D, Stetler-Stevenson WG, Lacombe ML, Meltzer PS and Steeg PS: Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res 67 : 7238–7246, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Leonard MK, McCorkle JR, Snyder DE, Novak M, Zhang Q, Shetty AC, Mahurkar AA and Kaetzel DM: Identification of a gene expression signature associated with the metastasis suppressor function of NME1: prognostic value in human melanoma. Lab Invest 98: 327–338, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horak CE, Mendoza A, Vega-Valle E, Albaugh M, Graff-Cherry C, McDermott WG, Hua E, Merino MJ, Steinberg SM, Khanna C and Steeg PS: Nm23-H1 suppresses metastasis by inhibiting expression of the lysophosphatidic acid receptor EDG2. Cancer Res 67: 11751–11759, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Postel EH, Berberich SJ, Flint SJ and Ferrone CA: Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 261 : 478–480, 1993. [DOI] [PubMed] [Google Scholar]
- 12.Ma D, Xing Z, Liu B, Pedigo N, Zimmer S, Bai Z, Postel E and Kaetzel DM: NM23-H1 cleaves and represses transcriptional activity of nuclease-hypersensitive elements in the PDGF-A promoter. J Biol Chem 277: 1560–1567, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Puts GS, Leonard MK, Pamidimukkala NV, Snyder DE and Kaetzel DM: Nuclear functions of NME proteins. Lab Invest 98: 211–218, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cervoni L, Egistelli L, Eufemi M, d’Abusco AS, Altieri F, Lascu I, Turano C and Giartosio A: DNA sequences acting as binding sites for NM23/NDPK proteins in melanoma M14 cells. J Cell Biochem 98 : 421–428, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Egistelli L, Chichiarelli S, Gaucci E, Eufemi M, Schinina ME, Giorgi A, Lascu I, Turano C, Giartosio A and Cervoni L: IFI16 and NM23 bind to a common DNA fragment both in the P53 and the cMYC gene promoters. J Cell Biochem 106: 666–672, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Su L, Qiu J, Xiao N, Lin J, Tan JH, Ou TM, Gu LQ, Huang ZS and Li D: Mechanistic studies for the role of cellular nucleic-acid-binding protein (CNBP) in regulation of c-myc transcription. Biochim et Biophys Acta 1830 : 4769–4777, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Roymans D, Willems R, Van Blockstaele DR and Slegers H: Nucleoside diphosphate kinase (NDPK/NM23) and the waltz with multiple partners: possible consequences in tumor metastasis. Clin Exp Metastasis 19: 465–476, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Choudhuri T, Murakami M, Kaul R, Sahu SK, Mohanty S, Verma SC, Kumar P and Robertson ES: Nm23-H1 can induce cell cycle arrest and apoptosis in B cells. Cancer Biol Ther 9: 1065–1078, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis CD, Likhite VS, McLeod IX, Yates JR and Nardulli AM: Interaction of the tumor metastasis suppressor nonmetastatic protein 23 homologue H1 and estrogen receptor alpha alters estrogen-responsive gene expression. Cancer Res 67: 10600–10607, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Rayner K, Chen YX, Hibbert B, White D, Miller H, Postel EH and O’Brien ER: Discovery of NM23-H2 as an estrogen receptor beta-associated protein: role in estrogen-induced gene transcription and cell migration. J Steroid Bioch Mol Biol 108: 72–81, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian C and Robertson ES: The metastatic suppressor Nm23-H1 interacts with EBNA3C at sequences located between the glutamine- and proline-rich domains and can cooperate in activation of transcription. J Virol 76 : 8702–8709, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhuri T, Verma SC, Lan K and Robertson ES: Expression of alpha V integrin is modulated by Epstein-Barr virus nuclear antigen 3C and the metastasis suppressor Nm23-H1 through interaction with the GATA-1 and Sp1 transcription factors. Virology 351: 58–72, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Novak M, Leonard MK, Yang XH, Kowluru A, Belkin AM and Kaetzel DM: Metastasis suppressor NME1 regulates melanoma cell morphology, self-adhesion and motility via induction of fibronectin expression. Exp Dermatol 24: 455–461, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safran M, Kim WY, O’Connell F, Flippin L, Gunzler V, Horner JW, Depinho RA and Kaelin WG Jr.: Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA 103: 105–110, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, McCorkle JR, Novak M, Yang M and Kaetzel DM: Metastasis suppressor function of NM23-H1 requires its 3’;−5’ exonuclease activity. Int J Cancer 128: 40–50, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C, Granter SR, Ramaswamy S, Golub T, Duncan LM, Wagner SN, Brennan C and Chin L: Integrative genome comparison of primary and metastatic melanomas. PLoS One 5: e10770, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Cancer Genome Atlas website. Available: https://cancergenome.nih.gov. Accessed September 12, 2018.
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N: The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C and Schultz N: Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lebherz HG and Rutter WJ: Distribution of fructose diphosphate aldolase variants in biological systems. Biochemistry 8: 109–121, 1969. [DOI] [PubMed] [Google Scholar]
- 31.Silver DL and Pavan WJ: The origin and development of neural crest-derived melanocytes. In : From melanocytes to melanoma: the progression to malignancy (Hearing VJ and Leong SPL eds.). Totowan, NJ, USA: Humana Press, pp. 3–26, 2006. [Google Scholar]
- 32.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM and Haussler D: The human genome browser at UCSC. Genome Res 12: 996–1006, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ENCODE Project Consortium: An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinodoz M, Gobet C, Naef F and Gustafson KB: Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Genome Biol 15: R85, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurichi A, Miceli R, Camerini T, Mariani L, Patuzzo R, Ruggeri R, Gallino G, Tolomio E, Tragni G, Valeri B, Anichini A, Mortarini R, Moglia D, Pellacani G, Bassoli S, Longo C, Quaglino P, Pimpinelli N, Borgognoni L, Bergamaschi D, Harwood C, Zoras O and Santinami M: Prediction of survival in patients with thin melanoma: results from a multi-institution study. J Clin Oncol 32: 2479–2485, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Fiore LS, Ganguly SS, Sledziona J, Cibull ML, Wang C, Richards DL, Neltner JM, Beach C, McCorkle JR, Kaetzel DM and Plattner R: c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene 33: 4508–4520, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berberich SJ and Postel EH: PuF/NM23-H2/NDPK-B transactivates a human c-myc promoter-CAT gene via a functional nuclease hypersensitive element. Oncogene 10: 2343–2347, 1995. [PubMed] [Google Scholar]
- 38.Muimo R, Crawford RM and Mehta A: Nucleoside diphosphate kinase A as a controller of AMP-kinase in airway epithelia. J Bioenerg Biomembr 38: 181–187, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murthy S and Reddy GP: Replitase: complete machinery for DNA synthesis. J Cell Physiol 209: 711–717, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Niida H, Shimada M, Murakami H and Nakanishi M: Mechanisms of dNTP supply that play an essential role in maintaining genome integrity in eukaryotic cells. Cancer Sci 101: 2505–2509, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Postel E, Berberich SJ, Rooney JW and Kaetzel DM: Human NM23/nucleoside diphosphate kinase regulates gene expression through DNA binding to nuclease-hypersensitive elements. J Bioenerg Biomembr 32 : 277–284, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Knapp AM, Ramsey JE, Wang SX, Strauch AR and Kelm RJ Jr.,: Structure-function analysis of mouse Pur beta II. Conformation altering mutations disrupt single-stranded DNA and protein interactions crucial to smooth muscle alpha-actin gene repression. J Biol Chem 282 : 35899–35909, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Teoh ST and Lunt SY: Metabolism in cancer metastasis: bioenergetics, biosynthesis, and beyond. Wiley Interdiscip Rev Syst Biol Med 10: e1406, 2018. [DOI] [PubMed] [Google Scholar]
- 44.Pascual G, Dominguez D and Benitah SA: The contributions of cancer cell metabolism to metastasis. Dis Model Mech 11(8) pii: dmm032920, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penhoet EE, Kochman M and Rutter WJ: Molecular and catalytic properties of aldolase C. Biochemistry 8: 4396–4402, 1969. [DOI] [PubMed] [Google Scholar]
- 46.Chang YC, Yang YC, Tien CP, Yang CJ and Hsiao M: Roles of aldolase family genes in human cancers and diseases. Trends Endocrinol Metab 29: 549–559, 2018. [DOI] [PubMed] [Google Scholar]
- 47.Du S, Guan Z, Hao L, Song Y, Wang L, Gong L, Liu L, Qi X, Hou Z and Shao S: Fructose-bisphosphate aldolase a is a potential metastasis-associated marker of lung squamous cell carcinoma and promotes lung cell tumorigenesis and migration. PloS one 9: e85804, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawai K, Uemura M, Munakata K, Takahashi H, Haraguchi N, Nishimura J, Hata T, Matsuda C, Ikenaga M, Murata K, Mizushima T, Yamamoto H, Doki Y and Mori M: Fructose-bisphosphate aldolase A is a key regulator of hypoxic adaptation in colorectal cancer cells and involved in treatment resistance and poor prognosis. Int J Oncol 50: 525–534, 2017. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto T, Kudo M, Peng WX, Takata H, Takakura H, Teduka K, Fujii T, Mitamura K, Taga A, Uchida E and Naito Z: Identification of aldolase A as a potential diagnostic biomarker for colorectal cancer based on proteomic analysis using formalin-fixed paraffin-embedded tissue. Tumour Biol 37: 13595–13606, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grandjean G, de Jong PR, James B, Koh MY, Lemos R, Kingston J, Aleshin A, Bankston LA, Miller CP, Cho EJ, Edupuganti R, Devkota A, Stancu G, Liddington RC, Dalby K and Powis G: Definition of a Novel Feed-Forward Mechanism for Glycolysis-HIF1alpha Signaling in Hypoxic Tumors Highlights Aldolase A as a Therapeutic Target. Cancer Res 76: 4259–4269, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng SY, Lai PL, Pan HW, Hsiao LP and Hsu HC: Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol Rep 19: 1045–1053, 2008. [PubMed] [Google Scholar]
- 52.Tao QF, Yuan SX, Yang F, Yang S, Yang Y, Yuan JH, Wang ZG, Xu QG, Lin KY, Cai J, Yu J, Huang WL, Teng XL, Zhou CC, Wang F, Sun SH and Zhou WP: Aldolase B inhibits metastasis through Ten-Eleven Translocation 1 and serves as a prognostic biomarker in hepatocellular carcinoma. Mol Cancer 14: 170, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Kuramitsu Y, Takashima M, Yokoyama Y, Iizuka N, Tamesa T, Sakaida I, Oka M and Nakamura K: Identification of four isoforms of aldolase B down-regulated in hepatocellular carcinoma tissues by means of two-dimensional Western blotting. In Vivo 25: 881–886, 2011. [PubMed] [Google Scholar]
- 54.Li YJ, Huang TH, Hsiao M, Lin BR, Cheng SJ, Yang CN, Lai WT, Wu TS, Fan JR, Kuo MY and Chang CC: Suppression of fructose-bisphosphate aldolase C expression as a predictor of advanced oral squamous cell carcinoma. Head Neck 38(Suppl 1): E1075–1085, 2016. [DOI] [PubMed] [Google Scholar]
- 55.Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, Seth P, Lundquist MR, Tolan DR, Grant AK, Needleman DJ, Asara JM, Cantley LC and Wulf GM: Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 164: 433–446, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kusakabe T, Motoki K and Hori K: Mode of interactions of human aldolase isozymes with cytoskeletons. Arch Bioch Biophys 344: 184–193, 1997. [DOI] [PubMed] [Google Scholar]