

Abstract
Aim:
The objective of this study was to identify potential epigenetic mediating pathways linking early life social disadvantage (ELSD) to adulthood BMI.
Methods:
Sex-specific epigenome-wide two-stage mediation analyses were conducted in blood and adipose tissue, and mediation estimates were obtained using cross-product mediation analysis. Pathway analyses were conducted using GREAT software (Bejerano Lab, CA, USA).
Results:
Candidate mediation CpG sites were identified in adipose tissue, but not blood, and were sex-specific. Significant mediation sites in females included CpG loci in genes: PKHG1, BCAR3, ADAM5P, PIEZO1, FGFRL1, FASN and DPP9, among others. Pathway analyses revealed evidence of enrichment for processes associated with TFG-β signaling and immunologic signatures. In males, significant mediation loci included sites in MAP3K5 and RPTOR, which have previously been associated with adipogenesis, inflammation and insulin resistance.
Conclusion:
Our findings provide supportive evidence for the mediating role of epigenetic mechanisms in the effect of early life social disadvantage on adulthood BMI.
Keywords: : adipose tissue, adiposity, BMI, childhood adversity, DNA methylation, epigenetics, social disadvantage, social epidemiology
Epigenetic pathways, such as altered DNA methylation (DNAm), have been hypothesized as plausible mechanisms by which early life exposures and adult disease may be linked [1–4]. Evidence for the association between childhood social environments and epigenetic programming within genes related to stress reactivity and inflammation continues to grow [5–9]. Furthermore, as evidence for the ability of early life social environments to effect change on epigenetic pathways has increased, so too has the evidence for dysregulation of epigenetic processes in adiposity [10–14], suggesting a possible connection between social adversity and obesity vis-à-vis epigenetic mediators.
Few studies have directly interrogated the mediating role of epigenetic mechanisms in the impact of early life social disadvantage (ELSD) on adulthood adiposity. A previous mediation study identified sex-specific methylation loci in biologically plausible genes that were strong mediation candidates for the effect of childhood socioeconomic status (SES) on adulthood BMI in adipose tissue [15]. However, childhood SES does not explicitly include psychosocial or environmental measures of social disadvantage. Indeed, social adversity in the form of adverse childhood experiences has been shown to associate with adiposity independently of SES [16–22]. To this end, we conducted mediation analyses in biologically relevant adipose tissue to formally assess potential alterations in DNAm that might lie on the pathway between ELSD and adulthood adiposity. Given accumulating evidence of differential methylation patterns [15–17] as well as differential effects of socioeconomic factors on adiposity between males and females [15,18], mediation analyses were stratified by sex. As both psychosocial and environmental exposures have also been shown to associate with adiposity, we reasoned that an inclusive measure extending beyond only socioeconomic status would provide a more accurate and holistic measure of early life experiences. Thus, ELSD was conceived as a composite index of not only socioeconomic but also specific environmental and psychosocial measures of adversity. The objective of this study was to identify epigenetic-mediating mechanisms through which ELSD could influence adulthood BMI, utilizing a prospective study with directly assessed ELSD during childhood, as well as BMI and epigenetic methylation patterns in blood and adipose tissue during middle adulthood.
Materials & methods
Study sample
Study participants were recruited from the Longitudinal Effects on Aging Perinatal (LEAP) Project, a nested substudy of the New England Family Study (NEFS). The NEFS is a large prospectively assessed cohort of 17,921 offspring of pregnant women in the Collaborative Perinatal Project who were born in Providence, Rhode Island and Boston, MA (USA) between 1959 and 1966. The LEAP substudy enrolled and assessed 400 Providence-born participants who were not deceased, not incarcerated, had assessments taken at age 7 years, and were located within 100 miles of a clinical assessment site during 2010–2011. Of these, 316 had adequate adipose tissue biopsy performed, 68 refused adipose tissue biopsies and 16 had inadequate biopsy specimens. A final, representative sample of 143 of these 316 participants was selected for blood and adipose tissue methylation analyses. The study protocol was approved by the institutional review boards at Brown University and Memorial Hospital of Rhode Island.
Collection of covariates & tissue samples
Bodyweight and height were obtained by trained personnel using a calibrated stadiometer and weighing scale, and then converted into BMI as weight per height squared (kg/m2). ELSD was assessed prospectively at age 7 using a composite summary score by summing across ten component measures of social disadvantage (Gilman et al., Submitted). For each component measure, a score of 0 (low adversity), 0.5 (medium adversity) or 1 (high adversity) was assigned. The components comprising the summary score were: parental income (>150% poverty threshold, 100–150% poverty threshold, <poverty threshold); parental occupation (nonmanual, manual, welfare only); household crowding (<1 person/room, 1–1.5 persons/room, ≥1.5 persons/room); family structure (two parents at home, single parent, divorced or separated); changes in parent's marital status (0, 1, 2+); number of moves since birth (0–1, 2, 3+); parental employment history (employed, not employed in the past year, 1+ years unemployed); changes in primary caregiver (no, yes); death of a sibling (no, yes); and age 7 change in economic situation since birth (same or better, worse). All possible component category options above were presented from low to high adversity. Changes in primary caregiver, death of a sibling and change in economic situation since birth are binary covariates and thus were classified as either low or high. A maximum score of ten was possible. Other covariates of interest included age at time of clinical assessment, race (white or nonwhite), sex, prenatal maternal smoking (cigarettes/day) and BMI at age 7.
Tissue sample collection & methylation profiling
Subcutaneous adipose tissue samples were aspirated from the upper outer quadrant of the buttock via 16-gauge needle. Whole blood samples were centrifuged to obtain buffy coat. DNA extraction from the adipose tissue samples and the buffy coat was performed according to manufacturer protocol using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, CA, USA) and the Zymo Genomic DNA Clean & Concentrator Kit (Zymo Research, CA, USA). DNA sodium bisulfite conversion was conducted according to manufacturer protocol using the EZ-96 DNA Methylation-Direct and EZ DNA Methylation-Direct kits (Zymo Research). Blood and adipose tissue samples were distributed randomly across plates prior to analysis using the Infinium HumanMethylation450 BeadChip array (Illumina, CA, USA) at the UCSF Institute for Human Genetics, Genomics Core Facility (CA, USA), following standard Illumina protocols. Preprocessing of the methylation data was conducted as described in Huang et al. (2016) [19]. Briefly, background and dye-bias corrections were applied using the ‘methylumi’ package in R, normalization was performed using the β-Mixture Quantile Dilation approach and adjustments for batch effects were made using linear mixed models [20]. After quality control and probe filtering, 285,163 CpG sites were included in the analyses.
Statistical analyses
Epigenome-wide two-stage mediation analyses
We conducted sex-specific two-stage epigenome-wide association scans (EWAS) for significance testing of putative CpG candidates by which methylation might mediate the effect of ELSD on adult BMI [21]. In the first stage, we conducted EWAS analyses to detect association between CpG methylation and adult BMI using the following model:
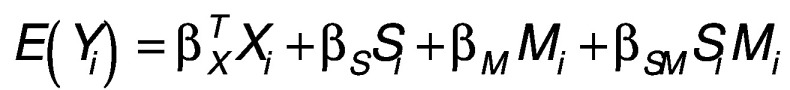 |
(Equation 1) |
where Y, S, M and X are adult BMI, ELSD, methylation M-value and the confounding covariates (including age, race, BMI at age 7, maternal smoking), for subject i respectively. Only those loci demonstrating sufficient evidence of association with BMI under a likelihood ratio test comparing [1] against a reduced model excluding all CpG effects and interaction terms were considered putative mediators. To screen the first-stage results, we applied a p-value threshold to identify significant CpG loci at p < 0.005.
For the second stage, we conducted EWAS analyses to detect the association between CpG methylation and ELSD among the CpG-BMI loci that met our first-stage criteria using the following model:
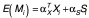 |
(Equation 2) |
where M, X and S are defined as in [1]. Only those candidate CpG loci, which met the first-stage p-value threshold, and also demonstrated association with ELSD in the second stage, again at p < 0.005, were reported.
The above two-stage mediation analyses were conducted for DNAm in blood and adipose tissue separately. To adjust for potential confounding arising from cellular heterogeneity in blood tissue, cell mixture deconvolution (CMD) was performed using the method developed by Houseman et al. [22], and admixture estimates were included as adjustment covariates in the blood EWAS. CMD was not conducted in adipose tissue due to the lack of reference libraries for adipose tissue-specific differentially methylated regions and an insufficient sample size for a reference-free CMD approach.
Joint mediation analysis
The two-stage joint testing approach has been shown to be more powerful in identifying mediation effects via hypothesis testing [21], but it fails to provide estimates for the mediated effect of ELSD on adulthood BMI. We therefore implemented cross-product-based mediation analyses to quantify and test the direct effect (DE) and the indirect effect (IE) for the set of CpG loci that survived both levels of the two-stage approach [23–26]. Under the framework of causal inference, using potential outcomes and counterfactuals, the point estimates of the indirect (i.e., the mediation effect) and direct effects can be expressed by a combination of regression parameters of the above two models: 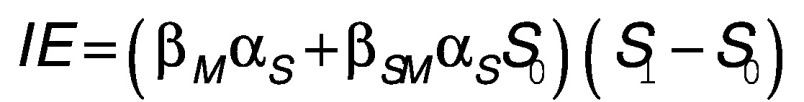 and
and 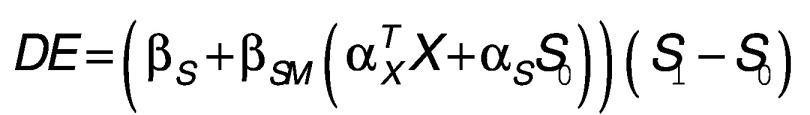 , where S
0 and S
1 represent two different counterfactual realizations of ELSD. The corresponding variance and CIs were obtained via a bootstrap procedure. p-values were also calculated using the joint significance test (JST) as the maximum of the two stage p-values for each site [27]. For loci with DE and IE estimates in the same direction, proportion of mediation was calculated as IE/(IE + DE) to characterize the proportion of the total effect of ELSD on BMI mediated by methylation per site.
, where S
0 and S
1 represent two different counterfactual realizations of ELSD. The corresponding variance and CIs were obtained via a bootstrap procedure. p-values were also calculated using the joint significance test (JST) as the maximum of the two stage p-values for each site [27]. For loci with DE and IE estimates in the same direction, proportion of mediation was calculated as IE/(IE + DE) to characterize the proportion of the total effect of ELSD on BMI mediated by methylation per site.
Pathway analysis
To investigate the enrichment of signals within genes belonging to common biological pathways, processes or gene sets, a broad base of genes inclusive of those with weaker signals is needed. In order to conduct pathway analyses, all CpG sites surviving a relaxed p-value threshold of p < 0.01 in the both stages of the two-stage mediation analyses were included for males and females. Because methylation data are comprised of specific sites, not all of which map to genes, a pathway approach which focuses on regulatory regions, rather than specific gene constructs, was implemented using the Genomic Regions Enrichment of Annotations Tool (GREAT) version 3.0 software (Bejerano Lab, CA, USA) using default parameters [28]. Input regions for each CpG site were specified as ±2 bp on either side of the genomic location of the CpG. Enrichment terms significant at p < 0.005 by both the binomial and hypergeometric enrichment tests and with binomial fold enrichment greater than two were obtained, and those with false discovery rate Q < 0.1 were reported. As before, males and females were analyzed separately.
Results
Clinical & demographic characteristics of the sample
The mean age at assessment in the final analytic sample (n = 143) was 46.9 years, and study subjects were mostly white (66.4%) and female (51.8%). Distributions of the different characteristics across both males and females were consistent with distributions in the full LEAP sample (n = 400). The clinical and demographic features of the sample are summarized in Table 1.
Table 1. . Clinical and demographic characteristics of the Longitudinal Effects on Aging Perinatal sample.
| Characteristics | Total (n = 143) | Males (n = 69) | Females (n = 74) |
|---|---|---|---|
| Age in years: | |||
| – Mean (SD) | 46.9 (1.7) | 47.1 (1.6) | 46.8 (1.8) |
| Race % (no.): | |||
| – White | 66.4 (95) | 63.8 (44) | 68.9 (51) |
| – Non white | 33.6 (48) | 36.2 (25) | 31.1 (23) |
| Age 7 BMI: | |||
| – Mean (SD) | 16.3 (2.5) | 16.5 (2.8) | 16.1 (2.1) |
| Maternal cigarettes/day: | |||
| – Median (IQR) | 5 (0–20) | 4.5 (0–20) | 6 (0–20) |
| – Range | 0–50 | 0–40 | 0–50 |
| ELSD: | |||
| – Median (IQR) | 2.5 (1.5–4.0) | 2.5 (1.375–4.0) | 2.5 (1.5–4.0) |
| – Range | 0–8 | 0–8 | 0–6.5 |
| Adult BMI: | |||
| – Mean (SD) | 31.5 (7.4) | 31.7 (5.8) | 31.3 (8.7) |
| – Range | 19.4–65.5 | 21.2–46.1 | 19.4–65.5 |
BMI: Body mass index; ELSD: Early life social disadvantage (age 7); IQR: Interquartile range; SD: Standard deviation.
ELSD-BMI candidate-mediating loci are sex & tissue specific
In the two-stage EWAS analyses, adipose tissue methylation at 32,100 and 5757 out of 285,163 CpG sites were identified as associated with BMI after surviving the first-stage p-value threshold in males and females, respectively. Of the surviving first-stage CpG loci, 28 sites in males and 131 sites in females were identified as jointly significant with ELSD on BMI at p < 0.005 (Supplementary Tables 1 & 2) in the second stage. Mediating methylation loci were unique in females and males; no candidate-mediating sites common to both sexes were present. Cross-product-based mediation analyses revealed 22 candidate CpG sites in males and 100 in females with evidence of mediating the effect of ELSD on adulthood BMI (p-value for indirect effect [pindirect] < 0.05) (Tables 2, 3 & Supplementary Table 3), providing additional support for the intermediary role played by these loci. In the two-stage analysis of peripheral blood leukocytes, 1882 and 895 loci survived the first stage in males and females, respectively. No loci survived second-stage thresholding in males. Three loci survived in females but none were identified as significant mediators in cross-product-based analyses (Supplementary Table 3). Thus, no candidate-mediating loci were further investigated in peripheral blood leukocytes.
Table 2. . Mediation effects for top 22 loci in male adipose tissue surviving the two-stage mediation analysis thresholds and with indirect p-value <0.05.
| CpG ID | Chr | Gen Loc | Gene | Gene group | Re: CGI | p(S->Mj) | p(Mj->BMI) | Indirect | 95% CI | pindirect | Direct | 95% CI | pdirect | R2 | Prop. Med |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cg08996506 | 16 | 16070868 | ABCC1 | Body | 1.22E-04 | 0.001‡ | 0.922 | (0.397–1.5) | <0.001 | 0.196 | (-0.853–1.28) | 0.808 | 0.054 | 0.825 | |
| cg23644736 | 1 | 55544866 | USP24 | Body | 0.003‡ | 1.13E-04 | 0.817 | (0.319–1.38) | <0.001 | 0.253 | (-0.645–1.48) | 0.678 | 0.208 | 0.764 | |
| cg15230985 | 17 | 78753887 | RPTOR | Body | 0.004‡ | 2.34E-06 | 0.997 | (0.436–1.68) | 0.002 | 0.478 | (-0.507–1.69) | 0.402 | 0.159 | 0.676 | |
| cg18573842 | 6 | 30139421 | TRIM15 | Body | N_Shore | 0.001 | 0.001‡ | -0.662 | (-1.26 to -0.229) | 0.002 | 0.691 | (-0.234–1.79) | 0.148 | 0.123 | |
| cg26106417 | 4 | 3425381 | RGS12 | Body | 0.002 | 0.004‡ | 0.790 | (0.261–1.42) | 0.002 | 0.159 | (-0.847–1.28) | 0.896 | 0.108 | 0.832 | |
| cg26390400 | 5 | 10556455 | 0.002 | 0.002‡ | 0.850 | (0.323–1.42) | 0.002 | 0.391 | (-0.568–1.27) | 0.470 | 0.099 | 0.685 | |||
| cg03657040 | 5 | 175083981 | HRH2 | TSS1500 | N_Shore | 0.003‡ | 0.001 | 0.745 | (0.198–1.51) | 0.004 | 0.064 | (-0.908–1.18) | 0.982 | 0.108 | 0.921 |
| cg10893483 | 1 | 156712783 | HDGF | 3′UTR | S_Shore | 0.003 | 0.005‡ | 0.675 | (0.142–1.77) | 0.004 | 0.398 | (-0.464–1.59) | 0.428 | 0.066 | 0.629 |
| cg23588713 | 12 | 56523270 | ESYT1 | Body | Island | 0.003‡ | 0.001 | 0.603 | (0.175–1.15) | 0.004 | -0.495 | (-1.48–0.308) | 0.152 | 0.131 | |
| cg25277723 | 6 | 137072948 | MAP3K5 | Body | 0.001‡ | 4.86E-04 | -0.744 | (-1.48 to -0.16) | 0.004 | 0.422 | (-0.643–1.37) | 0.394 | 0.117 | ||
| cg06852605 | 17 | 76422640 | DNAH17 | Body | S_Shore | 0.004‡ | 0.001 | 0.752 | (0.179–1.49) | 0.006 | 0.16 | (-0.651–1.06) | 0.816 | 0.126 | 0.825 |
| cg08062812 | 1 | 91191232 | Island | 0.001‡ | 0.001 | -0.769 | (-1.65 to -0.223) | 0.006 | 0.329 | (-0.659–1.82) | 0.496 | 0.103 | |||
| cg17334453 | 17 | 14479244 | 0.004‡ | 1.29E-04 | 0.799 | (0.312–1.400) | 0.006 | -0.172 | (-0.896–0.678) | 0.628 | 0.096 | ||||
| cg24162251 | 14 | 32420205 | 0.003‡ | 0.002 | -0.618 | (-1.100 to -0.143) | 0.006 | 0.308 | (-0.502–1.290) | 0.426 | 0.140 | ||||
| cg14319235 | 1 | 79472282 | ELTD1 | Body | Island | 0.004 | 0.005‡ | -0.591 | (-1.340 to -0.113) | 0.008 | 0.217 | (-0.799–1.170) | 0.660 | 0.087 | |
| cg15056348 | 10 | 134350107 | INPP5A | TSS1500 | N_Shore | 3.13E-04‡ | 6.17E-05 | -0.949 | (-1.880 to -0.309) | 0.010 | 0.508 | (-0.538–1.800) | 0.334 | 0.213 | |
| cg09730801 | 17 | 48048875 | DLX4 | Body; TSS1500 | N_Shore | 0.003‡ | 0.003 | -0.574 | (-1.270 to -0.094) | 0.014 | 0.414 | (-0.693–1.500) | 0.462 | 0.151 | |
| cg10793301 | 20 | 56064194 | HMGB1L1 | TSS200 | 0.005‡ | 0.003 | 0.606 | (0.121–1.380) | 0.016 | 0.256 | (-0.737–1.210) | 0.686 | 0.067 | 0.703 | |
| cg25100475 | 1 | 227750970 | ZNF678 | TSS1500 | N_Shore | 0.004‡ | 0.002 | 0.679 | (0.098–1.670) | 0.018 | 0.586 | (-0.347–1.540) | 0.270 | 0.067 | 0.537 |
| cg01021682 | 3 | 101926162 | 0.002‡ | 0.002 | 0.644 | (0.084–1.300) | 0.022 | -0.235 | (-1.170–0.654) | 0.546 | 0.074 | ||||
| cg01956154 | 14 | 94423399 | ASB2 | 5′UTR; 1stExon | 0.004 | 0.005‡ | -0.523 | (-1.110 to -0.058) | 0.024 | 0.381 | (-0.617–1.340) | 0.392 | 0.131 | ||
| cg24243753† | 6 | 97972900 | 0.005‡ | 2.59E-04 | -0.553 | (-1.410 to -0.018) | 0.044 | 0.603 | (-0.334–1.720) | 0.204 | 0.170 | ||||
†Denotes sites that were also identified as mediating candidates in prior mediation analyses for socioeconomic status and BMI in the same cohort.
‡Denotes the joint significance test p-values, i.e., the maximum p-value from each stage of the two-stage mediation testing analyses.
Chr: Chromosome number; Direct: Direct effect; FDR(Mj->BMI): False discovery rate for association of methylation mediator with BMI; Gen Loc: Genetic location; Indirect: Indirect effect of methylation mediator in association of early life social disadvantage with BMI; pdirect: p-value for direct effect; pindirect: p-value for indirect effect; p(Mj->BMI): p-value for association of methylation mediator with BMI; p(S->Mj): p-value for association of early life social disadvantage with methylation mediator; Prop.Med: Proportion of total association between early life social disadvantage and BMI explained by DNA methylation (calculated indirect effect/[indirect effect + direct effect]; applicable only if indirect and direct effects are in the same direction); R2: Marginal coefficient of variation; Re: CGI: Relationship to canonical CpG island; S: Early life social disadvantage at age 7 years.
Table 3. . Mediation effects for 27 selected top loci in female adipose tissue surviving the two-stage mediation analysis thresholds and with indirect p-value < 0.05.
| CpG ID | Chr | Gen Loc | Gene | Gene group | Re: CGI | p(S->Mj) | p(Mj->BMI) | Indirect | 95% CI | pindirect | Direct | 95% CI | pdirect | R2 | Prop. Med |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cg04145890 | 4 | 1007657 | FGFRL1 | Body | S_Shore | 1.62E-04‡ | 2.07E-05 | 1.350 | (0.557–2.320) | <0.001 | 1.150 | (-0.096–2.300) | 0.068 | 0.366 | 0.540 |
| cg26750548† | 10 | 88441152 | LDB3 | Body | 0.001 | 0.001‡ | 0.930 | (0.281–1.920) | <0.001 | 1.150 | (-0.063–2.260) | 0.056 | 0.322 | 0.447 | |
| cg11950105† | 17 | 80050646 | FASN | Body | Island | 0.002‡ | 0.001 | 0.682 | (0.248–1.350) | <0.001 | 0.790 | (-0.269–1.970) | 0.134 | 0.308 | 0.463 |
| cg21103829 | 5 | 140054320 | DND1; HARS | TSS1500; Body | S_Shore | 0.001 | 0.002‡ | 1.160 | (0.385–2.240) | <0.001 | 1.340 | (0.012–2.660) | 0.048 | 0.298 | 0.464 |
| cg17977470 | 17 | 7758854 | TMEM88 | Body | Island | 0.004‡ | 0.001 | 0.729 | (0.262–1.400) | 0.002 | 1.160 | (0.034–2.280) | 0.046 | 0.301 | 0.386 |
| cg18410680† | 17 | 7758397 | TMEM88 | 1stExon | N_Shore | 0.002 | 0.005‡ | 0.878 | (0.143–1.840) | 0.022 | 1.260 | (0.174–2.370) | 0.024 | 0.345 | 0.411 |
| cg16641055 | 16 | 88846292 | PIEZO1§ | Body | S_Shore | 0.002 | 0.003‡ | 0.851 | (0.275–1.650) | 0.002 | 1.300 | (-0.037–2.540) | 0.066 | 0.277 | 0.396 |
| cg27630153† | 16 | 88845038 | PIEZO1§ | Body | Island | 0.001 | 0.003‡ | 0.598 | (0.066–1.390) | 0.018 | 0.747 | (-0.419–1.910) | 0.184 | 0.304 | 0.445 |
| cg17274827† | 1 | 94075555 | BCAR3 | Body | 0.001 | 0.003‡ | 1.070 | (0.316–2.030) | 0.002 | 1.910 | (0.796–2.960) | 0.002 | 0.167 | 0.359 | |
| cg15925478 | 1 | 94081080 | BCAR3 | Body | 0.002‡ | 3.55E-04 | 0.600 | (0.006–1.350) | 0.050 | 0.766 | (-0.133–1.740) | 0.110 | 0.370 | 0.439 | |
| cg02655351 | 1 | 3498101 | MEGF6 | Body | N_Shore | 0.001‡ | 1.96E-04 | 0.825 | (0.249–1.750) | 0.004 | 0.771 | (-0.354–1.820) | 0.158 | 0.384 | 0.517 |
| cg01993576 | 6 | 44187674 | SLC29A1 | 5′UTR | S_Shore | 0.001 | 0.003‡ | 0.764 | (0.214–1.490) | 0.004 | 1.030 | (-0.373–2.210) | 0.134 | 0.271 | 0.426 |
| cg07568841 | 7 | 30362781 | ZNRF2 | Body | 3.55E-05 | 0.005‡ | 0.986 | (0.302–1.890) | 0.004 | 1.010 | (-0.729–2.410) | 0.254 | 0.334 | 0.494 | |
| cg12793803 | 17 | 62084217 | ICAM2 | 5′UTR;1stExon | 0.003‡ | 8.82E-04 | 0.652 | (0.188–1.270) | 0.004 | 0.911 | (-0.128–1.930) | 0.088 | 0.334 | 0.417 | |
| cg26422861 | 7 | 56160737 | PHKG1 | TSS200 | 0.002‡ | 3.68E-04 | 1.150 | (0.254–2.200) | 0.010 | 1.210 | (0.012–2.200) | 0.050 | 0.388 | 0.487 | |
| cg08370546 | 7 | 56160770 | PHKG1 | TSS200 | 0.002‡ | 0.002‡ | 0.811 | (0.042–1.790) | 0.042 | 1.070 | (-0.071–2.180) | 0.074 | 0.385 | 0.431 | |
| cg10780949 | 15 | 67418316 | SMAD3 | Body; 1stExon; 5′UTR | 0.002‡ | 0.002 | 0.730 | (0.155–1.570) | 0.012 | 1.010 | (-0.283–2.060) | 0.138 | 0.316 | 0.420 | |
| cg11199639 | 8 | 39172111 | ADAM5P | TSS200 | 0.001 | 0.002‡ | 0.975 | (0.183–1.940) | 0.014 | 1.200 | (0.254–2.310) | 0.020 | 0.344 | 0.448 | |
| cg19659741 | 8 | 39172099 | ADAM5P | TSS200 | 0.002 | 0.003‡ | 0.567 | (0.040–1.480) | 0.020 | 0.941 | (0.008–2.080) | 0.046 | 0.341 | 0.376 | |
| cg07387286 | 8 | 39172120 | ADAM5P | TSS200 | 0.003 | 0.004‡ | 0.699 | (-0.024–1.860) | 0.066 | 1.100 | (0.042–2.310) | 0.038 | 0.330 | 0.389 | |
| cg11981599 | 8 | 11566526 | GATA4 | Body | Island | 2.20E-04 | 2.49E-04‡ | 0.888 | (0.144–1.820) | 0.016 | 0.674 | (-0.505–1.880) | 0.226 | 0.365 | 0.569 |
| cg25840926† | 2 | 20647987 | RHOB | 1stExon; 3′UTR | Island | 3.86E-05 | 0.005‡ | 0.984 | (0.155–1.760) | 0.018 | 1.010 | (-0.135–2.060) | 0.092 | 0.329 | 0.493 |
| cg25490145† | 17 | 80358850 | C17orf101 | Body | N_Shelf | 3.40E-05 | 0.001‡ | 0.789 | (0.144–1.550) | 0.020 | 0.413 | (-0.895–1.540) | 0.512 | 0.399 | 0.656 |
| cg02422603† | 13 | 114890566 | RASA3 | Body | 0.001 | 0.003‡ | 0.900 | (0.155–1.820) | 0.022 | 1.110 | (-0.192–2.490) | 0.084 | 0.303 | 0.448 | |
| cg02907425 | 19 | 4688820 | DPP9 | Body | Island | 0.003 | 0.005‡ | 0.874 | (0.067–2.040) | 0.034 | 1.390 | (0.288–2.430) | 0.016 | 0.380 | 0.386 |
| cg16582779 | 14 | 29237202 | FOXG1 | 1stExon | Island | 0.001 | 0.003‡ | 0.535 | (0.040–1.330) | 0.036 | 0.725 | (-0.536–1.870) | 0.230 | 0.389 | 0.425 |
| cg16145324 | 6 | 36020012 | MAPK14 | Body | 0.002‡ | 6.38E-04 | 0.809 | (0.270–1.580) | 0.002 | 1.060 | (-0.025–1.990) | 0.052 | 0.367 | 0.433 | |
†Denote sites that were also identified as mediating candidates in prior mediation analyses for socioeconomic status and BMI in the same cohort.
‡Italicized p-values denote the joint significance test p-values, i.e., the maximum p-value from each stage of the two-stage mediation testing analyses.
§PIEZO1 was previously known as FAM38A.
Chr: Chromosome number; Direct: Direct effect; FDR(Mj->BMI): False discovery rate for association of methylation mediator with BMI; Gen Loc: Genetic Location; Indirect: Indirect effect of methylation mediator in association of early life social disadvantage with BMI; pdirect: p-value for direct effect; pindirect: p-value for indirect effect; p(Mj->BMI): p-value for association of methylation mediator with BMI; p(S->Mj): p-value for association of early life social disadvantage with methylation mediator; Prop.Med: Proportion of total association between early life social disadvantage and BMI explained by DNA methylation (calculated indirect effect/[indirect effect + direct effect]; applicable only if indirect and direct effects are in the same direction); R2: Marginal coefficient of variation; Re: CGI: Relationship to canonical CpG island; S: Early life social disadvantage at age 7 years.
The CpG sites with the strongest evidence for mediation in female adipose tissue (i.e., those with the most significant cross-product-based indirect effects) were: cg04145890 (pindirect < 0.001, p-value for joint significance test [pJST] = 1.62 × 10-4) in FGFRL1, cg26750548 (pindirect < 0.001, pJST = 0.001) LDB3, and cg1195015 (pindirect < 0.001, pJST = 0.002) in FASN. Several genes included multiple top hits. The multihit genes included: ADAM5P with cg11199639 (pindirect = 0.014, pJST = 0.002), cg19659741 (pindirect = 0.02, pJST = 0.003), cg07387286 (pindirect = 0.066, pJST = 0.004); BCAR3 with cg17274827 (pindirect = 0.002, pJST = 0.003) and cg15925478 (pindirect = 0.05, pJST = 0.002); PHKG1 with cg26422861 (pindirect = 0.01, pJST = 0.002) and cg08370546 (pindirect = 0.042, pJST = 0.002); PIEZO1 with cg16641055 (pindirect = 0.002, pJST = 0.003) and cg27630153 (pindirect = 0.018, pJST = 0.003); and TMEM88 with cg17977470 (pindirect = 0.002, pJST = 0.004) and cg18410680 (pindirect = 0.022, pJST = 0.005).
In males, notable CpG sites with the strongest evidence for mediation in adipose tissue were: cg08996506 (pindirect < 0.001, pJST = 6.44E-04) in ABCC1; cg23644736 (pindirect < 0.001, pJST = 0.003) in USP24; cg15230985 (pindirect = 0.002, pJST = 0.004) in RPTOR; and cg25277723 (pindirect = 0.004, pJST = 0.001) in MAP3K5.
ELSD, methylation, BMI & associated biological processes in adipose tissue
After relaxing the p-value thresholds in the two-stage analyses, a total of 109 and 397 CpG sites were selected for pathway analysis in male and female adipose tissue, respectively. GREAT analyses in males yielded only two nonspecific cancer-related enrichment terms (Supplementary Table 5). In females, the top pathway analysis results identified 32 enrichment terms (Table 4). These terms largely fell into categories associated with immunologic signatures of gene regulation in various leukocytes including macrophages, dendritic and T cells. Notable identified pathways with high fold-enrichment scores included MAPK and bone morphogenetic protein receptor signaling pathways.
Table 4. . Female adipose tissue Genomic Regions Enrichment of Annotations Tool pathway analysis results.
| GREAT v3.0.0 (hg19) | AR: Basal + extension: 5 Kb upstream, 1 Kb downstream, 1 Mb maximum extension, curated regulatory domains included | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enrichment | Terms | Binomial test | Hypergeometric test | |||||||||
| p-value | FDR Q | FE | Obs. R. hits | RSC | p-value | FDR Q | FE | Obs. G hits | Tot. genes | GSC | ||
| GO MF | RNA polymerase II transcription factor binding | 3.25E-04 | 0.092 | 2.865 | 15 | 0.038 | 1.36E-06 | 0.002 | 4.721 | 14 | 88 | 0.023 |
| MSigPW | Map kinase inactivation of SMRT corepressor | 1.16E-04 | 0.031 | 8.133 | 6 | 0.015 | 3.49E-04 | 0.077 | 10.790 | 4 | 11 | 0.007 |
| MSigPW | Genes involved in activated TAK1 mediates p38 MAPK activation | 1.46E-04 | 0.028 | 7.789 | 6 | 0.015 | 1.29E-05 | 0.017 | 10.473 | 6 | 17 | 0.010 |
| MSigPW | BMP receptor signaling | 6.95E-04 | 0.057 | 3.519 | 10 | 0.025 | 6.81E-05 | 0.022 | 5.652 | 8 | 42 | 0.013 |
| MSigPW | Genes involved in YAP1- and WWTR1 (TAZ)-stimulated gene expression | 8.15E-04 | 0.060 | 4.745 | 7 | 0.018 | 1.14E-04 | 0.030 | 7.418 | 6 | 24 | 0.010 |
| MSigPW | Genes involved in nucleotide-binding domain, leucine rich repeat containing receptor (NLR) signaling pathways | 1.18E-03 | 0.078 | 4.448 | 7 | 0.018 | 1.91E-05 | 0.013 | 5.806 | 9 | 46 | 0.015 |
| MSigPb | Genes upregulated in DO11.10 cells (hybridoma) by expression of transcriptionally activating form of HDAC7 [GeneID = 51564] and downregulated by its transcriptionally repressing form | 1.47E-08 | 0.000 | 4.026 | 24 | 0.060 | 2.22E-05 | 0.011 | 2.983 | 19 | 189 | 0.031 |
| MSigPb | Genes upregulated in luminal-like breast cancer cell lines compared with the mesenchymal-like ones | 3.21E-06 | 0.005 | 2.443 | 33 | 0.083 | 3.85E-05 | 0.014 | 2.195 | 31 | 419 | 0.051 |
| MSigPb | Genes downregulated in polysomal and total RNA samples from SW480 cells (primary colorectal carcinoma) compared with the SW620 cells (lymph node metastasis from the same individual) | 5.60E-06 | 0.006 | 3.389 | 19 | 0.048 | 3.32E-06 | 0.006 | 3.709 | 17 | 136 | 0.028 |
| MSigPb | Upregulated genes in B-cell chronic lymphocytic leukemia patients expressing high levels of ZAP70 and CD38 [GeneID = 7535;952], which are associated with poor survival | 6.74E-06 | 0.006 | 2.638 | 27 | 0.068 | 9.31E-05 | 0.026 | 2.473 | 22 | 264 | 0.036 |
| MSigPb | Transcripts depleted from pseudopodia of NIH/3T3 cells (fibroblast) in response to haptotactic migratory stimulus by fibronectin, FN1 [GeneID = 2335] | 9.47E-06 | 0.006 | 2.063 | 42 | 0.106 | 9.11E-04 | 0.093 | 1.714 | 38 | 658 | 0.063 |
| MSigPb | Extracellular matrix (ECM) related genes upregulated early (within 30 min) in dermal fibroblasts after addition of TGFB1 [GeneID = 7040] | 1.69E-05 | 0.007 | 4.606 | 12 | 0.030 | 2.18E-04 | 0.043 | 4.307 | 9 | 62 | 0.015 |
| MSigPb | Genes downregulated in liver tumor compared with the normal adjacent tissue | 1.80E-05 | 0.006 | 2.742 | 23 | 0.058 | 6.57E-04 | 0.082 | 2.248 | 20 | 264 | 0.033 |
| MSigPb | Genes upregulated in T98 cells (glioma) 48 h after treatment with IFN-β | 5.51E-05 | 0.015 | 3.795 | 13 | 0.033 | 6.78E-04 | 0.081 | 3.709 | 9 | 72 | 0.015 |
| MSigPb | Genes that cooperate with MYC and TBX2 [GeneID = 4609;6909] to transform MEF cells (mouse embryonic fibroblasts) | 1.34E-04 | 0.028 | 15.912 | 4 | 0.010 | 5.09E-04 | 0.075 | 9.891 | 4 | 12 | 0.007 |
| MSigPb | Genes downregulated in freshly isolated CD31- [GeneID = 5175] (stromal stem cells from adipose tissue) versus the CD31+ (nonstem) counterparts | 1.54E-04 | 0.029 | 2.321 | 24 | 0.060 | 3.68E-05 | 0.015 | 2.786 | 20 | 213 | 0.033 |
| MSigPb | Cluster P7 of genes with similar expression profiles in peripheral T lymphocytes after FOXP3 [GeneID = 50943] loss of function | 2.32E-04 | 0.034 | 3.102 | 14 | 0.035 | 1.29E-04 | 0.029 | 3.840 | 11 | 85 | 0.018 |
| MSigPb | Cluster 2: ECM-related genes upregulated in dermal fibroblasts within 30 min after TGFB1 [GeneID = 7040] addition; reached a plateau after that | 2.54E-04 | 0.030 | 5.789 | 7 | 0.018 | 2.86E-04 | 0.051 | 6.358 | 6 | 28 | 0.010 |
| MSigPb | Downregulated genes in both rectal and colon carcinoma compared with normal mucosa samples | 5.87E-04 | 0.055 | 2.963 | 13 | 0.033 | 3.83E-04 | 0.061 | 3.400 | 11 | 96 | 0.018 |
| MSigOS | Genes upregulated in a panel of epithelial cell lines by TGFB1 [Gene ID = 7040] | 6.19E-06 | 0.001 | 2.650 | 27 | 0.068 | 1.43E-08 | 0.000 | 3.870 | 24 | 184 | 0.039 |
| MSigIS | Genes upregulated in comparison of dendritic cells stimulated with R848 at 2 h vs DCs stimulated with R848 for 8 h | 4.83E-07 | 0.001 | 3.688 | 21 | 0.053 | 1.80E-04 | 0.049 | 2.727 | 17 | 185 | 0.028 |
| MSigIS | Genes upregulated in comparison of monocytes cultured for 0 days vs those cultured for 1 day | 1.28E-06 | 0.001 | 3.234 | 23 | 0.058 | 3.78E-04 | 0.056 | 2.561 | 17 | 197 | 0.028 |
| MSigIS | Genes upregulated in comparison of macrophages exposed to 5 worms/well Brugia malayi vs macrophages exposed to Mycobacterium tuberculosis | 6.40E-06 | 0.003 | 3.019 | 22 | 0.055 | 1.11E-04 | 0.053 | 2.739 | 18 | 195 | 0.030 |
| MSigIS | Genes downregulated in comparison of macrophages treated with IL25 [GeneID = 64806] versus neutrophils treated with IL25 [GeneID = 64806] | 6.95E-06 | 0.003 | 2.833 | 24 | 0.060 | 8.86E-07 | 0.002 | 3.314 | 22 | 197 | 0.036 |
| MSigIS | Genes downregulated in comparison of dendritic cells stimulated with Pam3Csk4 (TLR1/2 agonist) at 12 h vs dendritic cells stimulated with Gardiquimod (TLR7 agonist) at 12 h | 7.50E-06 | 0.002 | 2.988 | 22 | 0.055 | 2.59E-06 | 0.002 | 3.212 | 21 | 194 | 0.035 |
| MSigIS | Genes upregulated in comparison of macrophages exposed to Toxoplasma gondii vs macrophages exposed to M. tuberculosis | 2.10E-05 | 0.004 | 2.971 | 20 | 0.050 | 5.98E-04 | 0.076 | 2.461 | 17 | 205 | 0.028 |
| MSigIS | Genes downregulated in comparison of dendritic cells exposed to T. gondii vs macrophages exposed to T. gondii | 4.52E-05 | 0.008 | 2.649 | 22 | 0.055 | 1.11E-04 | 0.053 | 2.739 | 18 | 195 | 0.030 |
| MSigIS | Genes upregulated in comparison of type 2 myeloid (T2M) cells treated with IL25 [GeneID = 64806] vs neutrophils treated with IL25 [GeneID = 64806] | 9.63E-05 | 0.014 | 2.656 | 20 | 0.050 | 3.36E-04 | 0.058 | 2.587 | 17 | 195 | 0.028 |
| MSigIS | Genes upregulated in comparison of dendritic cells stimulated with poly(I:C) (TLR3 agonist) at 24 h vs dendritic cells stimulated with Pam3Csk4 (TLR1/2 agonist) at 24 h | 9.96E-05 | 0.014 | 2.649 | 20 | 0.050 | 1.11E-04 | 0.053 | 2.739 | 18 | 195 | 0.030 |
| MSigIS | Genes upregulated in comparison of untreated CD4 [GeneID = 920] T cells at 0 h vs the untreated cells at 4 h | 1.22E-04 | 0.015 | 2.690 | 19 | 0.048 | 3.32E-04 | 0.063 | 2.682 | 16 | 177 | 0.026 |
| MSigIS | Genes upregulated in comparison of peripheral blood mononuclear cells from healthy donors vs peripheral blood mononuclear cells from infants with acute RSV infection | 1.66E-04 | 0.019 | 2.811 | 17 | 0.043 | 3.77E-04 | 0.060 | 2.652 | 16 | 179 | 0.026 |
| MSigIS | Genes upregulated in comparison of dendritic cells stimulated with lipopolysaccharide (LPS) (TLR4 agonist) at 16 h vs dendritic cells stimulated with Pam3Csk4 (TLR1/2 agonist) at 16 h | 3.00E-04 | 0.027 | 2.668 | 17 | 0.043 | 3.16E-04 | 0.067 | 2.600 | 17 | 194 | 0.028 |
FDR Q: False discovery rate Q statistic; FE: Fold enrichment; G: Genes; GO MF: Gene ontology molecular function; GSC: Gene set coverage; MSigIS: MSigDB immunologic signature; MSigOS: MSigDB oncogenic signature; MSigPB: MSigDB perturbation; MSigPW: MSigDB pathway; Obs.: Observed; R: Region; RSC: Region set coverage; Tot.: Total.
Discussion
This study identified a number of novel CpG loci with statistical evidence of mediating the association between ELSD and adult adiposity as measured by BMI. These loci were tissue and sex specific; significant CpG sites were identified only in fat tissue and were unique between males and females. It is unclear why such a greater number of CpG loci were associated with BMI in stage one of our mediation analyses among males (77,245 CpGs) versus females (8868 CpGs). However, this may be in part due to sex specificity of epigenetic pathways contributing to adiposity: male adiposity might be influenced by a larger, more varied range of epigenetic pathways than female adiposity, which might be influenced by more specific mechanisms such as those affected by ELSD.
Notably, one of our top mediating sites in females was cg04145890, a CpG site mapping to FGFRL1 which was also recently implicated in a Finnish monozygotic twin study of the association between methylation and BMI in adipose tissue [11], though at a different site. Interestingly, a large number of mediating loci identified in female fat tissue analyses localized to genes known to have associations with adipogenesis in obesity, insulin sensitivity and diabetes. For example, DPP9 inhibition was shown to impair adipocyte differentiation by inhibiting PPARγ induction [29]. Increased expression of FASN has been linked to higher visceral fat accumulation and insulin resistance in humans [30], and obesogenic feeding in mice has been show to upregulate FASN expression and to associate with specific methylation signatures [31]. Among the multihit genes, ADAM5P belongs to the disintegrin and metalloproteinase family of genes, which are regulators of cellular adhesion, migration and signaling. ADAMs have been implicated in a number of human diseases, and play a role in both normal and pathogenic inflammatory responses (Figure 1) [32,33]. However, the specific literature for ADAM5P in adipogenic or inflammatory processes is limited. With respect to the other multisite genes: BCAR3 has been implicated in genome wide association (GWA) loci interactions for diabetes [34] and is involved in the TGF-β signaling pathway, which plays a key role in both insulin resistance and adipogenesis [35,36]; and PHKG1, which is involved in the encoding of a catalytic subunit of phosphorylase kinase thus contributing to the cascade activation of glycogen breakdown, and in which mutations can cause a rare form of glycogen storage disease [37].
Figure 1. . Mediation analysis of methylation profiles across PHKG1 CpG sites in female adipose tissue.

Top mediating sites in males did not include multihit genes; however, the analyses revealed several biologically plausible genes associated with inflammation and insulin resistance. In particular, these included RPTOR, which when disrupted in macrophages was shown to reduce inflammation and insulin resistance in mice experiments [38,39].
In the methylation profile of PHKG1 (Figure 1), robust associations between ELSD and DNAm (blue line), and DNAm and BMI (green line), and significant indirect effect (red line) were observed in the CpG sites located within 200–1500 bp of the transcription start site, the 5′UTR or the first exon. In total, 7 of 14 methylation sites in PHKG1 demonstrated significant mediation at p < 0.05. In MAP3K5 (Figure 2), 2 of 21 methylation sites were significant for a mediation effect, and at each of these sites, a correspondingly robust signal was observed in the ELSD-DNAm and DNAm-BMI effects. Similar analyses for the other genes revealed: 3 of 25 sites in FGFRL1, 2 of 9 sites in ADAM5P, 2 of 14 sites in BCAR3, 1 of 16 sites in DPP9, 1 of 6 sites in MAPK14, and 2 of 286 sites in RPTOR.
Figure 2. . Mediation analysis of methylation profiles across MAP3K5 CpG sites in male adipose tissue.

The top sites presented in this study also included several loci which were consistent with sites identified in previous work exploring sex-specific adipose tissue epigenetic mediation between childhood SES and adulthood adiposity in the same cohort [40]. Specifically, among our top findings, 1 CpG site in men and 11 CpG sites in women were also observed in the top hits from the childhood SES analyses (Tables 2 & 3). As our measure of ELSD was inclusive of measures of childhood SES, while encompassing additional measures of environmental and psychosocial adversity (e.g., changes in parent's marital status, death of a sibling), overlapping findings were not unexpected. Indeed, the fact that some methylation sites were common to different, but related, early life constructs lend further credibility to the results presented here. Sensitivity analyses adjusting for childhood socioeconomic index in the association between ELSD, DNAm and BMI, can be viewed in Supplementary Table 6.
MAPK signaling in adipose tissue
Unique MAPK-related signals were discovered in both male and female adipose mediation analyses, and MAPKs were also involved in significant enrichment terms in the female pathway analyses. Specifically, MAP3K5 and MAPK14 were identified as potential sites of ELSD-BMI mediation in males and females, respectively. In a recent study, MAP3K5 mRNA and protein expression was shown to be upregulated in adipose tissue by transcription factor E2F1 vis-à-vis JNK-mediated sensitization to activation at the MAP3K5 promoter [41].MAPK14 modulates glucose metabolism and limits autophagy in nutrient-deprived conditions [42]. Genetic variants in MAPK14 were recently associated with diabetic foot ulcers [43], and a low-frequency variant in MAPK14 was also found to associate with myeloperoxidase, a biomarker for cardiovascular events [44]. More generally, MAPK signaling pathways and their constituents have been found to associate with a number of stress-induced obesogenic processes in adipose tissue including: regulation of adipose tissue inflammation and insulin resistance [45], and adipogenesis [46].
Biological pathways from ELSD to obesity
A number of plausible pathways linking early life social environments and DNAm can be found in existing literature, including the classic findings of Weaver et al. [47] on the effect of early life maternal care on the epigenetic profiles of rat pups, and the reversibility of these differences through cross fostering to effect a stable change lasting into adulthood. This study and several that followed successfully established the notion of persistent epigenetic modifications induced by social programming [2,3,48,49]. In human studies, the more specific mechanisms by which the early life social environment might impact epigenetic patterns span hypotheses that posit the role of nutrition [50], modified stress responses [1,5–9,51] and altered immune system function [52,53]. Meanwhile, the connection between adiposity and altered methylation profiles has been extensively explored in blood tissue [54–59], and more recently several studies have also been conducted in the more biologically relevant adipose tissue [11,13,19,60]. The common thread connecting all of these studies is the likely role of epigenetic mechanisms as a mediator between early life social environments and health outcomes later in life.
Our findings in adipose tissue were sex specific. This is consistent with previous studies suggesting that epigenetic mechanisms and patterns differ by sex [13,15,61], including a recent study which identified within-pair differential DNAm patterns comparing monozygotic male and female pairs, suggesting differential sensitivity to methylation by sex [17]. The effects of socioeconomic gradients also have been shown to have stronger associations with adiposity in females than males [18]. We identified CpG loci in males and females that mapped to genes associated with adipogenesis, inflammation, diabetes and insulin resistance. That both male and female candidate mediation loci implicated adipogenesis, inflammatory and insulin signaling pathways, either via a gene hit or through pathway enrichment, is compelling – a rich body of literature has consistently connected ELSD, maladaptive stress responses and proneness to inflammation [53,62–70], all of which have well-established associations with adiposity. More specifically, the prevailing ‘toxic stress’ hypothesis suggests stress induced by a social environment can become ‘toxic’ by negatively modifying the epigenetic landscape in ways that increase disease risk and persist across time [71]. For example, social stressors in early life have been found to induce changes in the functioning of the hypothalamic–pituitary–adrenal axis, thereby reducing glucocorticoid and increasing proinflammatory signaling [9,53,72]. Such stress-related hormonal changes in the action of the hypothalamic–pituitary–adrenal axis could potentially modify epigenetic regulation of adipose tissue in a systemic manner, facilitating adipose dysregulation and contributing to obesity and cardiovascular risk. Indeed, very small changes in hormonal levels can have profound effects, and we cannot rule out either very small alterations that are not yet detectable with our current instruments or regulatory changes in CpGs associated with regulatory action that are not interrogated on the platform employed.
Strengths & limitations
This study sought to identify epigenetic mediators of ELSD on adult adiposity by using a rich source of existing data and biosamples from clinically relevant tissue sites: adipose tissue. Most existing studies of epigenetic alterations associated with ELSD and adiposity have been conducted in blood tissue, which has been shown to serve as precarious surrogate for adipose tissue [19]. Nonetheless, our findings must be interpreted with some caution as we were not able to adjust for potential cell admixture in the adipose tissue due to inadequate statistical power. Although methods for reference-free cell type adjustment exist [22], they require a large number of degrees of freedom that could not be accommodated with our sample size. Thus, it remains unclear whether the mediating CpG sites reported in this analysis were identified due to differential methylation patterns, or due to potential differences in adipose tissue composition between subjects with higher versus lower levels of ELSD. However, it is worth noting that there is an increasing body of literature that argues for assessing epigenetic effects in tissues that are relevant to the disease of interest. Adipose tissue is complex, and also includes blood in its composition. We reasoned that the blood cells in adipose tissue may exhibit completely different characteristics and concentrations in fat than in blood, and these features might themselves be important indicators of disease-associated processes.
The exposure-outcome data were obtained from a prospective, observational study, where methylation and BMI were measured simultaneously, leading to some limitations with respect to causal interpretations of our findings. Specifically, causal mediation analyses depend on certain assumptions regarding confounding: no unmeasured confounding between ELSD and methylation, methylation and BMI, and ELSD and BMI, and finally, no ELSD-induced confounder of the methylation and BMI relationship. Biologically, we are unable to distinguish with certainty the causal directionality of the BMI and methylation alterations with our current data. However, loss of methylation may arise either actively, via Tet enzyme-mediated action or through suppression of the DNA methyltransferase. The addition of methylation is likely DNA methyltransferase mediated, although the precise inducer of this action remains unknown. Since these biochemical dynamics take longer to evolve than the body weight changes, the assumption that BMI is affected by the DNAm may be more plausible than the reverse. Future exploration of these individual mechanisms is feasible and should be quite informative.
Although we followed conventional p-value thresholds, the selection of such cutoffs is generally arbitrary with limited theoretical justification in literature. Our two-stage p-value thresholds, however, were within range of similar studies [73–80]. The application of mediation analyses in a high-dimensional setting also increases the difficulty of multiple correction adjustment, which necessitates downstream review of the identified candidate sites for biological plausibility through, for example, support by prior literature or enrichment analysis.
Strengths of our analyses, beyond using appropriate tissue samples, include the prospective collection of ELSD measures during critical developmental periods in early childhood. We also used a composite measure of ELSD that encompassed parental socioeconomic indicators as well as childhood social environments and events, which directly captured multiple dimensions of the early life social experience. Additionally, the analytic methods included an efficient two-stage joint significance test for screening mediation effect in a genome-wide setting, an explicit estimation for the effect size of the mediation, and pathway analyses via GREAT. Standard approaches to pathway analysis are largely derived from gene expression data and are predicated on measuring the transcripts of biologically well-defined gene constructs. However, the analysis of methylation data, which typically amounts to specific sites, is likely better served by functional annotation analyses that focus on regulatory regions. By using GREAT, we were able to accommodate all candidate CpG sites regardless of whether they explicitly mapped to a gene, rather than discarding any nonmapped sites as in standard network analysis.
Conclusion
In summary, these results shed some light on potential epigenetic-mediating sites in adipose tissue which is likely the more relevant tissue for adiposity, and not peripheral blood leukocytes. Many of our mediation and pathway analyses results were consistent with findings in prior literature that have implicated inflammation and dysregulation of adipocytes in adipose tissue in obesogenic processes. Findings support the hypothesis that epigenetic pathways may play mediating roles in the associations of ELSD with adulthood adiposity.
Summary points.
Early life social disadvantage with respect to socioeconomic position, structural social environments and psychosocial measures of adversity, was shown to associate with methylation patterns and increased risk for adulthood adiposity through unique biological mechanisms for men versus women.
The effects of early life social disadvantage on adulthood adiposity were specific to adipose tissue; no mediation candidates were identified in peripheral blood.
The majority of the mediating loci identified in female adipose tissue mapped to genes known to have associations with adipogenesis, diabetes, insulin sensitivity, PPARγ and TGF-β signaling pathways. Notable genes of interest included FGFRL1, ADAM5P, BCAR3, DPP9 and PIEZO1.
Functional gene groupings were less apparent in candidate mediation CpG sites in males than in females, but several loci in males mapped to genes associated with inflammation and signaling pathways: RPTOR and MAP3K5.
Pathway analyses of mediation loci in female adipose tissue revealed compelling evidence of enrichment for immunologic signatures, and a number of MAPK-associated pathways.
These findings are consistent with those in prior literature which have linked patterns of increased inflammation and adipocyte dysregulation in adipose tissue, and thereby also alterations in resident leukocyte populations, with increasing adiposity.
Supplementary Material
Acknowledgements
The study protocol was approved by the institutional review boards at Brown University and Memorial Hospital of Rhode Island.
Footnotes
Financial & competing interests disclosure
This work was supported by the National Institute on Aging at the NIH (grant numbers RC2AG036666, R01AG048825), the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the Ministry of Science and Technology, Taiwan (grant number 105-2118-M-001-014-MY3). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Gudsnuk K, Champagne FA. Epigenetic influence of stress and the social environment. ILAR J. 2012;53(3–4):279–288. doi: 10.1093/ilar.53.3-4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szyf M, Weaver I, Meaney M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod. Toxicol. 2007;24(1):9–19. doi: 10.1016/j.reprotox.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Szyf M. DNA methylation, behavior and early life adversity. J. Genet. Genom. 2013;40(7):331–338. doi: 10.1016/j.jgg.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Tehranifar P, Wu H-C, Fan X, et al. Early life socioeconomic factors and genomic DNA methylation in mid-life. Epigenetics. 2013;8(1):23–27. doi: 10.4161/epi.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Needham BL, Smith JA, Zhao W, et al. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: the multi-ethnic study of atherosclerosis. Epigenetics. 2015;10(10):958–969. doi: 10.1080/15592294.2015.1085139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann A, Spengler D. The lasting legacy of social stress on the epigenome of the hypothalamic–pituitary–adrenal axis. Epigenomics. 2012;4(4):431–444. doi: 10.2217/epi.12.34. [DOI] [PubMed] [Google Scholar]
- 7.Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PLoS ONE. 2012;7(1):e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anacker C, O'Donnell KJ, Meaney MJ. Early life adversity and the epigenetic programming of hypothalamic–pituitary–adrenal function. Dialogues Clin. Neurosci. 2014;16(3):321–333. doi: 10.31887/DCNS.2014.16.3/canacker. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turecki G, Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol. Psychiatry. 2016;79(2):87–96. doi: 10.1016/j.biopsych.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv. Nutr. 2014;5(1):71–81. doi: 10.3945/an.113.004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietiläinen KH, Ismail K, Järvinen E, et al. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int. J. Obes. 2016;40(4):654–661. doi: 10.1038/ijo.2015.221. [DOI] [PubMed] [Google Scholar]
- 12.Huang R-C, Galati JC, Burrows S, et al. DNA methylation of the IGF2/H19 imprinting control region and adiposity distribution in young adults. Clin. Epigenet. 2012;4(1):21. doi: 10.1186/1868-7083-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agha G, Houseman EA, Kelsey KT, Eaton CB, Buka SL, Loucks EB. Adiposity is associated with DNA methylation profile in adipose tissue. Int. J. Epidemiol. 2015;44(4):1277–1287. doi: 10.1093/ije/dyu236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383(9933):1990–1998. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 15.Appleton AA, Armstrong DA, Lesseur C, et al. Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS ONE. 2013;8(9):e74691. doi: 10.1371/journal.pone.0074691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Liu J, Kaur M, Krantz ID. Characterization of DNA methylation and its association with other biological systems in lymphoblastoid cell lines. Genomics. 2012;99(4):209–219. doi: 10.1016/j.ygeno.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe M, et al. Osaka Twin Research Group. Yorifuji S, et al. Within-pair differences of DNA methylation levels between monozygotic twins are different between male and female pairs. BMC Med. Genomics. 2016;9(1):55. doi: 10.1186/s12920-016-0217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides evidence of sex differences in DNA methylations in monozygotic twins, suggesting that methylation variability is higher in males than females across 80% of autosomal CpG islands.
- 18.Senese LC, Almeida ND, Fath AK, Smith BT, Loucks EB. Associations between childhood socioeconomic position and adulthood obesity. Epidemiol. Rev. 2009;31(1):21–51. doi: 10.1093/epirev/mxp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y-T, Chu S, Loucks EB, et al. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016;11(3):227–236. doi: 10.1080/15592294.2016.1146853. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Explores the nonexchangeability of adipose and blood tissue with respect to differential global and local methylation profiles by tissue type.
- 20.Teschendorff AE, Marabita F, Lechner M, et al. A β-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacKinnon DP, Lockwood CM, Hoffman JM, West SG, Sheets V. A comparison of methods to test mediation and other intervening variable effects. Psychol. Methods. 2002;7(1):83–104. doi: 10.1037/1082-989x.7.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30(10):1431–1439. doi: 10.1093/bioinformatics/btu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–155. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 24.Rubin DB. Bayesian inference for causal effects: the role of randomization. Ann. Statist. 1978;6(1):34–58. [Google Scholar]
- 25.VanderWeele T, Vansteelandt S. Conceptual issues concerning mediation, interventions and composition. Stat. Interface. 2009;2:457–468. [Google Scholar]
- 26.Vanderweele TJ. A unification of mediation and interaction. Epidemiology. 2014;25(5):749–761. doi: 10.1097/EDE.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y-T. Joint significance tests for mediation effects of socioeconomic adversity on adiposity via epigenetics. Ann. Statist. 2018:1–24. [Google Scholar]
- 28.McLean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han R, Wang X, Bachovchin W, Zukowska Z, Osborn JW. Inhibition of dipeptidyl peptidase 8/9 impairs preadipocyte differentiation. Sci. Rep. 2015;5(12348):1–11. doi: 10.1038/srep12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berndt J, Kovacs P, Ruschke K, et al. Fatty acid synthase gene expression in human adipose tissue: association with obesity and Type 2 diabetes. Diabetologia. 2007;50(7):1472–1480. doi: 10.1007/s00125-007-0689-x. [DOI] [PubMed] [Google Scholar]
- 31.Gracia A, Elcoroaristizabal X, Fernández-Quintela A, et al. Fatty acid synthase methylation levels in adipose tissue: effects of an obesogenic diet and phenol compounds. Genes Nutr. 2014;9(4):29. doi: 10.1007/s12263-014-0411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol. Aspects Med. 2008;29(5):258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponnuchamy B, Khalil RA. Role of ADAMs in endothelial cell permeability: cadherin shedding and leukocyte rolling. Circ. Res. 2008;102(10):1139–1142. doi: 10.1161/CIRCRESAHA.108.177394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schierding W, O'Sullivan JM. Connecting SNPs in diabetes: a spatial analysis of meta-GWAS loci. Front. Endocrinol. 2015;6:102. doi: 10.3389/fendo.2015.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsurutani Y, Fujimoto M, Takemoto M, et al. The roles of transforming growth factor-Î2 and Smad3 signaling in adipocyte differentiation and obesity. Biochem. Biophys. Res. Comm. 2011;407(1):68–73. doi: 10.1016/j.bbrc.2011.02.106. [DOI] [PubMed] [Google Scholar]
- 36.Yadav H, Quijano C, Kamaraju AK, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 2011;14(1):67–79. doi: 10.1016/j.cmet.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehner M, Clemens PR, Engel AG, Kilimann MW. Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the α subunit. Hum. Mol. Genet. 1994;3(11):1983–1987. doi: 10.1093/hmg/3.11.1983. [DOI] [PubMed] [Google Scholar]
- 38.Jiang H, Westerterp M, Wang C, Zhu Y, Ai D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia. 2014;57(11):2393–2404. doi: 10.1007/s00125-014-3350-5. [DOI] [PubMed] [Google Scholar]
- 39.Lee PL, Tang Y, Li H, Guertin DA. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metabol. 2016;5(6):422–432. doi: 10.1016/j.molmet.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loucks EB, Huang Y-T, Agha G, et al. Epigenetic mediators between childhood socioeconomic disadvantage and mid-life body mass index: the New England Family Study. Psychosom. Med. 2016;78(9):1053–1065. doi: 10.1097/PSY.0000000000000411. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates association of early life socioeconomic indicators and DNA methylation with adulthood BMI, also using a two-stage mediation approach.
- 41.Haim Y, Blüher M, Konrad D, et al. ASK1 (MAP3K5) is transcriptionally upregulated by E2F1 in adipose tissue in obesity, molecularly defining a human dys-metabolic obese phenotype. Mol. Metabol. 2017;6(7):725–736. doi: 10.1016/j.molmet.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desideri E, Vegliante R, Cardaci S, Nepravishta R, Paci M, Ciriolo MR. MAPK14/p38α-dependent modulation of glucose metabolism affects ROS levels and autophagy during starvation. Autophagy. 2014;10(9):1652–1665. doi: 10.4161/auto.29456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng W, Veluchamy A, Hébert HL, Campbell A, Colhoun HM, Palmer CNA. A genome-wide association study suggests that MAPK14 is associated with diabetic foot ulcers. Br. J. Dermatol. 2017;93:215–217. doi: 10.1111/bjd.15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waterworth DM, Li L, Scott R, et al. A low-frequency variant in MAPK14 provides mechanistic evidence of a link with myeloperoxidase: a prognostic cardiovascular risk marker. J. Am. Heart Assoc. 2014 doi: 10.1161/JAHA.114.001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Nguyen T, Tang P, et al. Regulation of adipose tissue inflammation and insulin resistance by MAPK phosphatase 5. J. Biol. Chem. 2015;290(24):14875–14883. doi: 10.1074/jbc.M115.660969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bost F, Aouadi M, Caron L, Binétruy B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie. 2005;87(1):51–56. doi: 10.1016/j.biochi.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 48.Champagne FA. Interplay between social experiences and the genome: epigenetic consequences for behavior. Adv. Genet. 2012;77:33–57. doi: 10.1016/B978-0-12-387687-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 49.Champagne FA, Curley JP. How social experiences influence the brain. Curr. Opin. Neurobiol. 2005;15(6):704–709. doi: 10.1016/j.conb.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 50.López-Jaramillo P, Silva SY, Rodríguez-Salamanca N, Duràn A, Mosquera W, Castillo V. Are nutrition-induced epigenetic changes the link between socioeconomic pathology and cardiovascular diseases? Am. J. Ther. 2008;15(4):362–372. doi: 10.1097/MJT.0b013e318164bf9c. [DOI] [PubMed] [Google Scholar]
- 51.Hoffmann A, Spengler D. DNA memories of early social life. Neuroscience. 2014;264:64–75. doi: 10.1016/j.neuroscience.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Bick J, Naumova O, Hunter S, et al. Childhood adversity and DNA methylation of genes involved in the hypothalamus–pituitary–adrenal axis and immune system: whole-genome and candidate-gene associations. Dev. Psychopathol. 2012;24(4):1417–1425. doi: 10.1017/S0954579412000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller GE, Chen E, Fok AK, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc. Natl Acad. Sci. USA. 2009;106(34):14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groom A, Potter C, Swan DC, et al. Postnatal growth and DNA methylation are associated with differential gene expression of the TACSTD2 gene and childhood fat mass. Diabetes. 2012;61(2):391–400. doi: 10.2337/db11-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60(5):1528–1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuehnen P, Mischke M, Wiegand S, et al. An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet. 2012;8(3):e1002543. doi: 10.1371/journal.pgen.1002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Relton CL, Groom A, St Pourcain B, et al. DNA methylation patterns in cord blood DNA and body size in childhood. PLoS ONE. 2012;7(3):e31821. doi: 10.1371/journal.pone.0031821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Almén MS, Jacobsson JA, Moschonis G, et al. Genome wide analysis reveals association of a FTO gene variant with epigenetic changes. Genomics. 2012;99(3):132–137. doi: 10.1016/j.ygeno.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Zhao J, Goldberg J, Vaccarino V. Promoter methylation of serotonin transporter gene is associated with obesity measures: a monozygotic twin study. Int. J. Obes. 2013;37(1):140–145. doi: 10.1038/ijo.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goni L, Milagro FI, Cuervo M, Martínez JA. Single-nucleotide polymorphisms and DNA methylation markers associated with central obesity and regulation of body weight. Nutr. Rev. 2014;72(11):673–690. doi: 10.1111/nure.12143. [DOI] [PubMed] [Google Scholar]
- 61.Zhang FF, Cardarelli R, Carroll J, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011;6(5):623–629. doi: 10.4161/epi.6.5.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol. Bull. 2011;137(6):959–997. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Danese A, Tan M. Childhood maltreatment and obesity: systematic review and meta-analysis. Mol. Psychiatry. 2013;19(5):544–554. doi: 10.1038/mp.2013.54. [DOI] [PubMed] [Google Scholar]
- 64.Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC. Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology. 2013;38(2):188–200. doi: 10.1016/j.psyneuen.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nazmi A, Oliveira IO, Horta BL, Gigante DP, Victora CG. Lifecourse socioeconomic trajectories and C-reactive protein levels in young adults: findings from a Brazilian birth cohort. Soc. Sci. Med. 2010;70(8):1229–1236. doi: 10.1016/j.socscimed.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loucks EB, Pilote L, Lynch JW, et al. Life course socioeconomic position is associated with inflammatory markers: the Framingham Offspring Study. Soc. Sci. Med. 2010;71(1):187–195. doi: 10.1016/j.socscimed.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pollitt RA, Kaufman JS, Rose KM, Diez-Roux AV, Zeng D, Heiss G. Early-life and adult socioeconomic status and inflammatory risk markers in adulthood. Eur. J. Epidemiol. 2007;22(1):55–66. doi: 10.1007/s10654-006-9082-1. [DOI] [PubMed] [Google Scholar]
- 68.Danese A, Moffitt TE, Harrington H, et al. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch. Pediatr. Adolesc. Med. 2009;163(12):1135–1143. doi: 10.1001/archpediatrics.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slopen N, Lewis TT, Gruenewald TL, et al. Early life adversity and inflammation in African Americans and whites in the midlife in the United States survey. Psychosom. Med. 2010;72(7):694–701. doi: 10.1097/PSY.0b013e3181e9c16f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Slopen N, Loucks EB, Appleton AA, et al. Early origins of inflammation: an examination of prenatal and childhood social adversity in a prospective cohort study. Psychoneuroendocrinology. 2015;51:403–413. doi: 10.1016/j.psyneuen.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gershon NB, High PC. Epigenetics and child abuse: modern-day darwinism – the miraculous ability of the human genome to adapt, and then adapt again. Am. J. Med. Genet. C Semin. Med. Genet. 2015;169(4):353–360. doi: 10.1002/ajmg.c.31467. [DOI] [PubMed] [Google Scholar]
- 72.Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33(6):693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in Type 1 diabetes mellitus. BMC Med. Genomics. 2010;3(1):33. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Z, Bolick SCE, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J. Natl Cancer Inst. 2013;105(10):694–700. doi: 10.1093/jnci/djt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cordoba R, Sanchez-Beato M, Herreros B, et al. Two distinct molecular subtypes of chronic lymphocytic leukemia give new insights on the pathogenesis of the disease and identify novel therapeutic targets. Leuk. Lymph. 2015;57(1):134–142. doi: 10.3109/10428194.2015.1034706. [DOI] [PubMed] [Google Scholar]
- 76.Rubicz R, Zhao S, Geybels M, et al. DNA methylation profiles in African American prostate cancer patients in relation to disease progression. Genomics. 2016 doi: 10.1016/j.ygeno.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Dam PA, van Dam P-JHH, Rolfo C, et al. In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget. 2016;7(3):2780–2795. doi: 10.18632/oncotarget.6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Houtepen LC, Vinkers CH, Carrillo-Roa T, et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat. Commun. 2016;7:10967. doi: 10.1038/ncomms10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448(7152):470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 80.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 2008;40(5):616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.