

Abstract
Background
We describe the virulence factors of a methicillin-sensitive Staphylococcus aureus sequence type (ST) 45 strain, MCRF184, (spa type t917), that caused severe necrotizing fasciitis in a 72-year-old diabetic male. The genome of MCRF184 possesses three genomic islands: a relatively large type III νSaα with 42 open reading frames (ORFs) that includes superantigen- and lipoprotein-like genes, a truncated νSaβ that consists mostly of the enterotoxin gene cluster (egc), and a νSaγ island with 18 ORFs including α-toxin. Additionally, the genome has two phage-related regions: phage φSa3 with three genes of the immune evasion cluster (IEC), and an incomplete phage that is distinct from other S. aureus phages. Finally, the region between orfX and orfY harbors a putative efflux pump, acetyltransferase, regulators, and mobilization genes instead of genes of SCCmec.
Results
Virulence factors included phenol soluble modulins (PSMs) α1 through α4 and PSMs β1 and β2. Ten ORFs identified in MCRF184 had not been reported in previously sequenced S. aureus strains.
Conclusion
The dire clinical outcome in the patient and the described virulence factors all suggest that MCRF184, a ST45 strain is a highly virulent strain of S. aureus.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5394-2) contains supplementary material, which is available to authorized users.
Keywords: Staphylococcus aureus; Virulence factors; Necrotizing fasciitis, ST45, enterotoxin gene cluster
Background
The ability of S. aureus to colonize and infect humans comes from a large arsenal of virulence genes including genes for proteins to attach to host tissue, tissue-degrading enzymes, leukocidins, antibiotic-resistance, pyrogenic toxins, and immunomodulating proteins [1]. A number of S. aureus genomes have been sequenced to identify potential new virulence genes or novel combinations of known virulence genes [2]. These studies have led to the identification of new genomic islands and genetic elements, which harbor known and putative toxins, phenol-soluble modulins, and accessory genes to virulence [3–6]. Differences in virulence of S. aureus strains, however, may be due to even small differences in genome sequence: Kennedy et al [7] studied genetic variation in USA300 MRSA strains and found that large differences in virulence in a mouse sepsis model occurred among strains with relatively few genetic differences. Single SNP differences have recently been demonstrated to underpin the virulence of some strains [8, 9]. Similarly, the insertion of IS256 (a transposable element) into the promotor of the rot gene increased virulence [10]. Panton-Valentine leukocidin (PVL), a major virulence factor of S. aureus has been shown to have a direct role in necrotizing fasciitis [5]. We describe here the virulence traits of MCRF184, a methicillin-sensitive, ST45 strain that caused a debilitating necrotizing fasciitis in a diabetic man, necessitating the amputation of the patient’s leg to save his life.
Results
Overview of antimicrobial resistance and virulence gene content
MCRF184 is a methicillin-susceptible strain that belongs to sequence type (ST) 45 and spa type t917. This strain was recovered during both the early and late stages of the infection of the leg [11]. Among some of the known virulence factors of S. aureus, the genome of this strain harbored clumping factors genes clfA and clfB, fibronectin binding protein gene fnbA but not fnbB, collagen binding adhesion gene cna, intracellular adhesion gene icaA and newly identified toxin genes – bsa, staphylococcal superantigen-like gene 1 (ssl1), and lpl110 (Table 1). The staphylococcal enterotoxins, staphylococcal superantigen-like (ssl) genes, and genes involved in immune evasion were present on mobile genetic elements. MCRF184 was negative for toxic shock syndrome toxin (tst), and the Panton-Valentine leucocidin (lukSF-PV).
Table 1.
Major virulence-related genes in S. aureus strain, MCRF184
| Locus | Location | |
|---|---|---|
| Enterotoxins | CKU_1443 SE | core |
| CKU_1636 seg | vSaβ | |
| CKU_1637 sen | vSaβ | |
| CKU_1638 seu | vSaβ | |
| CKU_1639 sei | vSaβ | |
| CKU_1640 sem | vSaβ | |
| CKU_1641 seo | vSaβ | |
| Exotoxins | CKU_0360 ssl1 | vSaα |
| CKU_0361 ssl2 | vSaα | |
| CKU_0362 ssl4 | vSaα | |
| CKU_0363 ssl3 | vSaα | |
| CKU_0365 ssl5 | vSaα | |
| CKU_0366 ssl9 | vSaα | |
| CKU_0367 ssl10 | vSaα | |
| CKU_0370 ssl11 | vSaα | |
| CKU_0998 ssl12 | vSaγ | |
| CKU_0999 ssl13 | vSaγ | |
| CKU_1000 ssl14 | vSaγ | |
| Exfoliative toxin | CKU_1005 eta | vSaγ |
| Alpha-hemolysin | CKU_0995 hla | vSaγ |
| Beta-hemolysin | CKU_1753 hlb | фSa3 |
| Delta-hemolysin (RNAIII) | CKU_2494 hld | core |
| Gamma-hemolysin Component | CKU_2175 hlgA | core |
| Gamma-hemolysin Component | CKU_2176 hlgC | core |
| Gamma-hemolysin Component | CKU_2177 hlgB | core |
| Adhesins | ||
| Collagen-binding protein | CKU_2442 cna | core |
| Fibronectin-adhesin | CKU_2253 fnbA | core |
| Elastin adhesin | CKU_1327 ebpS | core |
| Laminin-adhesin | CKU_0713 eno | core |
| Fibrinogen | CKU_0723 clfA | core |
| Fibrinogen | CKU_2384 clfB | core |
| Fibrinogen | CKU_0989 fib | core |
| Fibrinogen | CKU_0496 sdrC | core |
| Exoenzymes | ||
| Serine protease | CKU_0857 htrA | core |
| Serine V8 protease | CKU_0881 sspA | core |
| Cysteine protease | CKU_0880 sspB | core |
| Cysteine protease | CKU_0879 sspC | core |
| Lipase precursor | CKU_0273 geh | core |
| Lipase precursor | CKU_2426 gehC | core |
| Lipase | CKU_0588 lipA | core |
| Esterase | CKU_2106 | core |
| Hyaluronate lyase | CKU_1961 hysA2 | core |
| Termonuclease | CKU_1173 nucH | core |
| Cell wall hydrolase | CKU_1081 lytN | core |
| Zinc metalloprotease | CKU_1096 | core |
| Clp protease proteolytic subunit | CKU_0704 clpP | core |
| Clp protease ATP binding subunit | CKU_0813 clpB | core |
| Clp protease ATP binding subunit | CKU_1509 clpX | core |
| Clp protease ATP binding subunit | CKU_2300 clpL | core |
| Phenol Soluble Modulins | ||
| PSMα1 | 426,966 to 426,902 | core |
| PSMα2 | 426,870 to 426,805 | core |
| PSMα3 | 426,752 to 426,685 | core |
| PSMα4 | 426,620 to 426,559 | core |
| PSMβ1 | CKU_1007 | core |
| PSMβ2 | CKU_1008 | core |
| PSMδ | 1,955,755 to 1,955,676 | core |
| Immunomodulators | ||
| Staphylokinase | CKU_1760 sak | ф Sa3 |
| Chemotaxis inhibiting protein | CKU_1758 chp | ф Sa3 |
| Complement inhibitor | CKU_1757 | ф Sa3 |
| Immunoglobulin G binding protein A | CKU_0065 spa | core |
| Immunoglobulin G binding protein | CKU_2174 sbi | core |
| Lipoprotein like gene products | CKU_2474 lpl1 | vSaα |
| Lipoprotein like gene products | CKU_0373 lpl2 | vSaα |
| Lipoprotein like gene products | CKU_0374 lpl3 | vSaα |
| Lipoprotein like gene products | CKU_0375 lpl4 | vSaα |
| Lipoprotein like gene products | CKU_0376 lpl5 | vSaα |
| Lipoprotein like gene products | CKU_0377 lpl6 | vSaα |
| Lipoprotein like gene products | CKU_0378 lpl | vSaα |
| Virulence related genes | ||
| Biofilm genes | CKU_2420 icaR | core |
| CKU_2421 icaA | core | |
| CKU_2422 icaD | core | |
| CKU_2423 icaB | core | |
| CKU_2424 icaC | core | |
| Leukocidin GH | CKU_1786 lukGH | core |
| Regulatory genes | ||
| S. aureus exotoxin (SaeRS) | CKU_0640 saeS | core |
| CKU_0641 saeR | core | |
| Staphylococcal accessory regulator (sarA) | CKU_0551 sarA | core |
| Sigma factor B | CKU_1825 sigB | core |
| Repressor of Toxins | CKU_1594 rot | core |
Mobile genetic elements
The MCRF184 strain harbored six MGEs (Fig. 1): vSaα, vSaβ, vSaγ, φSa3, an incomplete phage, and a newly identified region between orfX and orfY, named MGEXY that also harbored mobilization genes. The incomplete phage has not been previously described, and the MGEXY harbored novel combinations of sequences. The MCRF184 genome did not include intact pathogenicity islands [12], plasmids, or integrative conjugative elements (ICE6013, Tn916/Tn5801) [13].
Fig. 1.
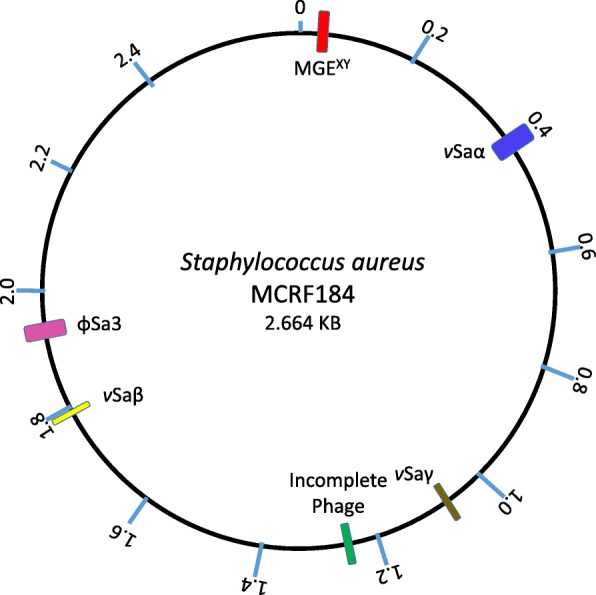
Circular representation of the MCRF184 genome. Virulence genomic islands are marked: vSaα [blue], vSaβ [yellow], vSaγ [brown], Incomplete phage [green], φSa3 [pink] and MGXY [red]
Genomic islands of MCRF184
vSaα
Genomic islands, generally 10 to 200 kb long, are a cluster of genes acquired by horizontal transfer [14]. The vSaα was a type III genomic island (Fig. 2) and harbored alleles of eight ssl and seven lipoprotein-like (lpl) genes. The vSaα region was nearly identical to vSaα of two other ST45 strains, CA-347 [15] and an unpublished genome, CFSAN007835 (GenBank # CP017685.1). In vSaα, eleven SNPs accounted for the differences between MCRF184 and CA347 (Table 2), eight of which were in protein coding regions—five of which would result in amino acid substitutions and one in a truncated protein in both MCRF184 and CA347. All but two of these changes in coding regions were in hypothetical proteins; of the two other changes, one was in an exotoxin gene and the other in the host specificity gene, hsdS (CKU_0369) of the restriction modification system.
Fig. 2.

vSaα genomic island showing lipoprotein-like and staphylococcal superantigen like genes along with mobile genetic elements
Table 2.
Comparison of vSaα of MCRF184 with the strain, CA347 identifying SNP differences between them
| Nucleotide position | AA Change | Region | CDS | CDS Position | Change | Codon Change | Polymorphism Type | Protein Effect |
|---|---|---|---|---|---|---|---|---|
| 392,566 | A - > T | HP | CKU_2476 | 292 | C - > T | GCT - > ACT | SNP (transition) | Substitution |
| 395,313 | non-coding | A - > C | SNP (transversion) | n/a | ||||
| 398,723 | HP | CKU_0352 | 161 | A - > T | SNP (transversion) | Truncation | ||
| 399,587 | A - > T | HP | CKU_0355 | 52 | C - > T | GCA - > ACA | SNP (transition) | Substitution |
| 401,239 | non-coding | T - > C | SNP (transition) | n/a | ||||
| 401,244 | non-coding | A - > G | SNP (transition) | n/a | ||||
| 404,499 | I - > K | exotoxin | CKU_0361 | 473 | T - > A | ATA - > AAA | SNP (transversion) | Substitution |
| 412,382 | I - > V | hsdS | CKU_0369 | 76 | A - > G | ATT - > GTT | SNP (transition) | Substitution |
| 418,429 | G - > W | HP | CKU_0374 | 658 | G - > T | GGG - > TGG | SNP (transversion) | Substitution |
| 423,848 | HP | CKU_0381 | 168 | C - > T | GGC - > GGT | SNP (transition) | None | |
| 423,933 | F - > V | HP | CKU_0381 | 253 | T - > G | TTT - > GTT | SNP (transversion) | Substitution |
vSaβ
The vSaβ of MCRF184 was truncated compared to vSaβ in MW2 and USA300FPR3757. It harbored eleven ORFs including the enterotoxin gene cluster (egc) genes: seg, sen, seu sei, sem and seo (Fig. 3), and was nearly identical in genes present in all three ST45 strains. Four genetic differences were noted in the vSaβ islands between MCRF184 and CA347 strain (Table 3), three of which were single nucleotide polymorphisms (SNPs). A significant additional difference was the deletion of two transposases in MCRF184, but present in CA347 strain. Furthermore, one of the SNPs in sen would lead to a truncated protein in MCRF184. The region containing two genes – a rep gene coding for a helicase and a second gene coding for a hypothetical protein, between positions 1,785,972 and 1787, 688 were unique to the three CC45 strains and not found in other S. aureus vSaβ islands. The observation that a hypothetical protein and the helicase were found in the three ST45 strains but absent from the other vSaβ islands sequenced could be of significance for the ST45 strains’ pathogenicity.
Fig. 3.

vSaβ genomic island showing genes of the enterotoxin gene cluster (egc)
Table 3.
Comparison of vSaβ of MCRF184 with strain, CA347 identifying SNP differences between them
| Nucleotide Position | AA Change | Region | CDS | CDS Position | Change | Codon Change | Polymorphism Type | Protein Effect |
|---|---|---|---|---|---|---|---|---|
| 1787,882 | CDS | 1223 bases | n/a | deletion | loss of two transposases, CA347_RS09315 and CA347_RS09320 | |||
| 1,789,959 | noncoding | C - > T | SNP (transition) | |||||
| 1,791,116 | M- > STOP | sen | CKU_1637 | 756 | T - > A | SNP (transversion) | truncation | |
| 1,795,127 | N - > D | seo | CKU_1641 | 235 | T - > C | AAT - > GAT | SNP (transition) | Substitution |
The genomic islands, vSaα and vSaβ generally exist in four allelic forms in S. aureus strains and their specificity is determined by the structural differences in hsdS (host specificity determinant), a rapidly evolving gene with amino acid sequence level identity across the S. aureus genomes of less than 66% [3]. vSaβ lacked the hsdS (Fig. 3).
vSaγ
Comparison of the vSaγ sequence between the two other ST45 strains, MCRF184 and CA-347, revealed conserved gene order and no amino acid differences. Comparing nucleotide and amino acid sequences between them (Table 4), there were only three differences in protein-coding regions, none of which resulted in an amino acid change. A comparison with other ST types S. aureus strains showed conservation of gene composition.
Table 4.
Comparison of vSaγ of MCRF184 with strain, CA347 identifying SNP differences between them
| Nucleotide Position | AA Change | Region | CDS | CDS Position | Change | Codon Change | Polymorphism Type | Protein Effect |
|---|---|---|---|---|---|---|---|---|
| 1,086,484 | No | XTP/dITP diphosphatase | CKU_0983 | 87 | T > C | TAT > TAC | SNP (transition) | None |
| 1,071,553 | A | HP | CKU_0988 | 1 | A > T | ATG > TAG | SNP (transversion) | None |
| 1,077,630 | C | ssl12 | CKU_0998 | 145 | C > G | ACA > AGA | SNP (transversion) | None |
This genomic island contains the IEC2 cluster, including the α-haemolysin (Hla) and the prototype βPFT of S. aureus. The vSaγ (Fig. 4) was flanked by the genes murI (glutamic racemase) and argF (ornithine transcarbamoylase subunit F). It additionally contained three more ssls.
Fig. 4.

vSaγ genomic island. Included in this island is the IEC2 group of genes
The phages of MCRF184
φSa3
The φSa3 (Fig. 5) was inserted into the hlb gene, making it a β-hemolysin-converting bacteriophage (βC-φ). This phage is known to carry IEC1, which is variable in gene content among strains [16]. In MCRF184, IEC1 consists of sak – (truncated amidase) – chp – scn suggesting that it is an IEC type B [17]. The truncated amidase is not unique to MCRF184, and an intact amidase is upstream of sak and forms part of the endolysin-holin lytic module of the phage. There was also lukGH genes located downstream of the phage element (Fig. 5), representing a core genome virulence factor in MCRF184.
Fig. 5.

φSa3 phage-derived region showing genes of the immune evasion cluster (IEC)
An incomplete phage
A novel incomplete phage was located between nucleotide positions 1,242,209 to 1,258,118 (Fig. 6). PHASTER analysis found it to be an incomplete prophage (PHASTER score 40; < 70 considered incomplete). Twenty of the 27 proteins were identified as phage proteins. Three of the 27 proteins matched staphylococcal phage φNM3. The complete sequence of this incomplete phage had a > 99% sequence identity with ST45 strains CA-347 and CFSAN007835. The gene content was unusual in having a terminase large subunit gene (terL) instead of a small subunit gene (terS), and in having a phage head morphogenesis gene. Interestingly, SaPIbov5 is known to have terL but not terS, and is mobilized by both pac- and cos-type helper phages [16]. The glutamine synthetase gene is not known to be used as an integration site by SaPIs, but it is used by an unrelated 30 kb phage, ϕ909 described in S. epidermidis [18]. The integrase of this incomplete phage was distinct from the groups defined for S. aureus phage [19] and SaPIs [20].
Fig. 6.

Incomplete phage showing phage-derived genes
MGEXY
The SCCmec cassette in MRSA is usually present at the 3′ end of the conserved gene orfX, an rRNA methyltransferase at a position ~ 34,000 base pairs from the origin of the replication [21]. The region between orfX and orfY, a tRNA dihydrouridine synthetase is known to be highly variable in gene content among S. aureus strains [22, 23]. In MCRF184, this region has a series of restriction-modification genes (hsdR, hsdM, and R-M type III) and a unique combination of putative antimicrobial resistance genes (emrB/qacA, tetR) located near the mobilization genes, int and tnp for transposon and integrase (Fig. 7). The putative efflux pump, emrB/qacA, is among those known for S. aureus [24]. The position of the hsdR and hsdM genes and the R-M type III system in this location of the S. aureus genome appears to be well conserved (Fig. 7). However, the presence of emrB/qacA, tetR and int and tnp in this region appear unique to MCRF184, CA-347, CFSAN007835 (all ST45 types) and an ST508 S. aureus isolated from a Buruli ulcer [25].
Fig. 7.

MGEXY (Mobile Genetic Element between orfX and orfY) is a region on the genome showing unique mix of antibiotic-resistance genes and mobile genetic elements. Other S. aureus genome regions are shown for comparison. orfX is also known as rlmH
The phenol soluble modulins of MCRF184
Phenol soluble modulins (PSMs) are a family of amphipathic, alpha-helical peptides that have multiple roles in pathogenesis and are critical determinants of staphylococcal virulence [26]. In MCRF184, we identified all four α peptides, the two β peptides and the δ-peptide (Additional file 1: Figure S1A and B). We further confirmed the PSMs by determining their predicted structures: characteristic α-helical secondary structures that were amphipathic—hydrophilic on one side and hydrophobic on the other—using PEP-FOLD [27, 28] for the alpha PSMs and SWISS-MODEL Workspace [29] for the beta PSMs (Additional file 1: Figure S1C).
Discussion
The whole genome sequence analysis of MCRF184, a clinically virulent and aggressive strain showed virulence features in common with two other ST45 strains, CA-347 and CFSAN007835 available in GenBank. However, these virulence features of the other genomes have not been described. Our analysis of the genomic islands of MCRF184 points to several distinctive virulence features: a streamlined νSaβ that mostly consists of the egc, and an MGEXY that appears to be unique to ST45 strains of S. aureus.
With regards to the virulence factors of the νSaα, Nguyen [30] showed that deletion of the lpl cluster, which is also present in the νSaα genomic island of MCRF184, prevents the stimulation of the production of proinflammatory cytokines in human monocytes, macrophages, and keratinocytes. They further demonstrated that purified lipoprotein, Lpl1 was able to elicit a TLR2-dependent response and that heterologous expression enhanced their immune stimulatory activity, particularly contributing to the invasion of S. aureus into human keratinocytes and mouse skin, compared to cells without these virulence genes. Thus, the lpl cluster of MCRF184 vSaα may help stimulate virulence by stimulating a host inflammatory response that can cause symptoms of pain, swelling, erythema and fever.
The egc in MCRF184 encodes six genes, which belong to a superantigen family that are capable of triggering a massive toxic shock response [31]. Proteins encoded by egc are not reported to be highly immunogenic, but they can evade immune response due to lack of neutralization by the human sera [32]. In a comprehensive study done by Roetzer et al [33], it has been shown that 1) supernatants from a strain harboring egc were sufficient for a lethal outcome in rabbits, 2) different quantities of egc encoded enterotoxins are produced by S. aureus isolates, 3) 10 nanograms of expressed and purified recombinant SEI and SEN was lethal at 24 h and 48 h, and 4) sei and sen appear to play a more important role in virulence compared to the other egc genes. Stach et al [34], in a rabbit model of infective endocarditis, investigated the role of tstH and individual genes of egc and in a USA200 genetic background and noted that proteins from both genes independently contributed to development of vegetation and infective endocarditis. Proteins made by sem, seo, and seu contributed to the vegetation formation, and deletions of tstH and egc decreased the vegetation size. Furthermore, Johler et al [35] reported outbreaks of staphylococcal food poisoning and emetic activity from egc-harboring S. aureus belonging to clonal complex CC9 and CC45. These observations suggest that even though MCRF184 had a truncated vSaβ island, the virulence imparted by the egc genes alone could account for significant virulence through their modulation of the immune system, particularly in the 72-year-old diabetic male with the life and limb-threating necrotizing fasciitis. Furthermore, the presence of an IEC in φSa3 could have contributed to evading phagocytosis of the pathogen. Another interesting aspect of the MCRF184 genome is that it had three ferrichrome-binding proteins—fhuA, fhuB, and fhuD—important for growth under iron-restricted conditions [25]. The MCRF184 φSa3 was integrated into the hlb and extended to position 2 genes upstream of groEL.
The MGEXY region of MCRF184 was identical to the ones found in CA-347 and the S. aureus Buruli ulcer isolate [25]. The region encodes a restriction-modification system (hsdR/hsdM) and included an ermB/qacA drug resistance transporter gene of the major facilitator superfamily (MFS) including an integrase, a transposase, a tetR/acR family transcriptional regular [36], and a flanking tRNA. The presence of all three types of PSMs—four α-types, two β-types and one δ-type—and their ability to enhance virulence through cytolysis of cells of the immune system and biofilm formation suggest further mechanisms for the enhanced virulence of MCRF184.
Wang et al [6] showed that psmα mutants were severely attenuated in their ability to cause subcutaneous abscesses in the skin of mice compared with the wild-type strain. Thus, the psmα toxins in MCRF184 could have contributed significantly to their virulence in causing necrotizing fasciitis and in their ability to cause soft tissue infections in a mouse model studied. PSMs in S. aureus contribute to the formation of biofilms and detachment of biofilm clusters for dissemination. The presence of the PSMs in MCRF184 and the biofilm genes (Additional file 1: Figure S1), CKU_2420 through CKD_ 2424 may again enhance the necrotizing fasciitis capability of the strain. PSMs of the α-type are known to be cytolytic, and the δ-toxin has been shown to lead to mast cell degranulation. The δ-toxin of MCRF184 (Table 1) is found within the RNAIII gene (CKU_2494) downstream of the agrB gene (CKU_1795). The RNAIII is the effector of the Agr system [6]. An interesting role for the PSMα3 of MCRF184 is their formation of amyloids [37] that are cross-α-fibrils, a newly discovered mode of self-assembly characterized by the piling of α-helices (Additional file 1: Figure S1C) perpendicular to the fibril axis. Similarly, PSMα1 promotes biofilm stability by preventing disassembly by matrix degrading enzymes and mechanical stress [38].
Conclusion
MCRF184’s genome contained several distinguishing features, such as a truncated vSaβ, an incomplete phage and a MGEXY not seen other S. aureus STs. Virulence of this strain likely came from its unique genetic background and SNPs in regulatory elements of virulence genes including egc. It also highlights the fact that there are highly virulent S. aureus strains out there which despite lacking the known potent toxins such as Panton-Valentine leukocidin, alpha toxin, etc., are still capable of causing serious, debilitating disease in susceptible individuals.
Methods
The study was approved by the Marshfield Clinic Research Institute’s Institutional Review Board under the study number SHU10105 with waiver of documentation of informed consent.
Bacterial strain
The S. aureus strain MCRF184, was isolated multiple times from a 72-year-old male during the treatment of his necrotizing fasciitis [11], and we sequenced the first isolate’s genome.
Genome sequence and comparative analysis
The MCRF184 genome was sequenced by both a shotgun (single end) and a paired end libraries on a Roche 454 and assembled and annotated as described in Aswani et al 2016 [39] (BioProject PRJNA39571, BioSample SAMN02953006, GenBank CP014791). Its multilocus sequence type (MLST) and lack of SCCmec was deduced from the genome sequence and confirmed by Sanger sequencing and PCR.
Identification of genomic islands and other putative virulence genes
Genomic islands in the MCRF815 genome were confirmed using IslandFinder [40] and Zisland explorer [41]. Virulence factors were further identified using VirulenceFinder [42].
Prophage analysis
PHASTER (PHAge Search Tool - Enhanced Release) was used to analyze prophages in the genome [43]. This program is based on an earlier version called PHAST that detects prophage regions by examination prophage genes and their distance from each other [44].
Single nucleotide polymorphism (SNP) analysis
SNP Analysis was performed with Geneious 11.0.3 (https://www.geneious.com). To perform the analysis, DNA sequences of the three genomic island, vSaα, vSaβ, and vSaγ from MCRF184 were aligned with the corresponding island sequences of CA-347 using Geneious Alignment, a global alignment with free end gaps with a 65% similarity (5.0/− 4.0) cost matrix and gap open penalty of 12 and gap extension penalty of 3. Once aligned, Geneious called variants/SNPs and reported effect of the variants on protein translation using a Bacterial Genetic Code, and merging adjacent variations.
PSM peptide structure modelling
The predicted protein structure of the α-PSMs were determined using SWISS_MODEL Workplace [29] (https://swissmodel.expasy.org/interactive). The SWISS_MODEL accepted the peptide sequence as input, with no additional parameters required and it generated a PDB file formatted secondary structure, and a descriptive report. The protein structure of the β-PSMs was modelled using PEP-FOLD3 [27, 28] (http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3). The input was the PSM amino acid sequences to generate a 3-D structure of the peptide using sOPEP (structure Optimized Potential for Efficient structure Prediction) as the model sorter after 100 independent simulations.
Additional file
Figure S1. Phenol soluble modulins (PSM) of S. aureus MCRF184. Panel 1A shows the amino acid sequences of the alpha and beta PSMs arranged from the N-terminus to the C-terminus. Numbers at the right show the net charge of the peptides at pH 7.0, rounded to whole numbers, and considering N-formylation of the initial methionine residue. The highlighted text identifies the amphipathic α-helical domain. Panel 1B shows the location of the genes coding for these PSMs in the genome of MCRF184 core genome. Panel 1C shows the predicted structure of the PSMS using PEP-FOLD (for the alpha PSMs) and SWISS-MODEL Workspace (for the beta PSMs). The residues are color-coded by their position in the peptide chain. Each chain is drawn as a smooth spectrum from blue through green, yellow and orange to red. The N-terminus of the peptides is colored red and the C terminuses are drawn in blue. The structures show the characteristic α-helical structure of the C-terminus ends of the PSMs. (PDF 334 kb)
Acknowledgements
The authors would like to acknowledge Jennifer Kislow and Pravin Kaldhone for their technical assistance. Authors would also like to acknowledge Michigan State University’s Core Facility for generating the genome sequence data and initial assembly. Authors are indebted to D. Ashley Robinson from the Department of Microbiology and Immunology, University of Mississippi Medical Center, Jackson, Mississippi, USA who provided expert feedback on several versions of this manuscript. His incisive comments greatly enhanced the completeness of the manuscript.
Funding
The study was supported, in part, by Marshfield Clinic Research Institute.
Availability of data and materials
The datasets used in the study are available from the NCBI’s GenBank under the accession number CP014791.1.
Abbreviations
- ICE
integrative conjugative elements
- IEC
immune evasion cluster
- lpl
lipoprotein-like
- MGE
mobile genetic element
- ORFs
open reading frames
- PSMs
phenol soluble modulins
- PVL
Panton-Valentine leukocidin
- SNP
single nucleotide polymorphisms
- Ssl
staphylococcal superantigen-like
- ST
sequence type
Authors’ contributions
SKS planned and arranged the study. SKS and WRS performed the experiments. VA, MP, FZN, BM, SKS, and WRS analyzed the data. SKS, VA, FZN, and WRS wrote the manuscript with support from all authors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Verbal consent from the patient was obtained by the physician to determine the unusual nature of the pathogen’s virulence as part of routine clinical care. This study was approved by the Marshfield Clinic Research Institute’s Institutional Review Board.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vijay Aswani, Email: vaswani@buffalo.edu.
Fares Najar, Email: fznajar@ou.edu.
Madhulatha Pantrangi, Email: madhu.pantrangi@preventiongenetics.com.
Bob Mau, Email: bmau@wisc.edu.
William R. Schwan, Email: wschwan@uwlax.edu
Sanjay K. Shukla, Phone: 715-389-5363, Email: shukla.sanjay@marshfieldresearch.org
References
- 1.Gordon RJ, Lowy FD. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(Suppl 5):S350–S359. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindsay JA, Holden MT. Staphylococcus aureus: superbug, super genome? Trends Microbiol. 2004;12(8):378–385. doi: 10.1016/j.tim.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359(9320):1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 4.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 5.Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A. 2004;101(26):9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13(12):1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy AD, Otto M, Braughton KR, Whitney AR, Chen L, Mathema B, et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci U S A. 2008;105(4):1327–1332. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald JR, Holden MT. Genomics of natural populations of Staphylococcus aureus. Annu Rev Microbiol. 2016;70:459–478. doi: 10.1146/annurev-micro-102215-095547. [DOI] [PubMed] [Google Scholar]
- 9.Sause WE, Copin R, O'Malley A, Chan R, Morrow BJ, Buckley PT, et al. Staphylococcus aureus strain Newman D2C contains mutations in major regulatory pathways that cripple its pathogenesis. J Bacteriol. 2017. [DOI] [PMC free article] [PubMed]
- 10.Benson MA, Ohneck EA, Ryan C, Alonzo F, 3rd, Smith H, Narechania A, et al. Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element. Mol Microbiol. 2014;93(4):664–681. doi: 10.1111/mmi.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan WR, Caldwell MD, Brady JM, Stemper ME, Reed KD, Shukla SK. Necrotizing fasciitis due to a methicillin-sensitive Staphylococcus aureus isolate harboring an enterotoxin gene cluster. J Clin Microbiol. 2007;45(2):668–671. doi: 10.1128/JCM.01657-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novick RP CG, Penadés JR. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol. 2010; 2010 Aug;8(8):11. [DOI] [PMC free article] [PubMed]
- 13.Sansevere EA, Robinson DA. Staphylococci on ICE: overlooked agents of horizontal gene transfer. Mob Genet Elements. 2017;7(4):1–10. doi: 10.1080/2159256X.2017.1368433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrindt U, Hochhut B, Hentschel U, Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. 2004;2(5):414–424. doi: 10.1038/nrmicro884. [DOI] [PubMed] [Google Scholar]
- 15.Stegger M, Driebe EM, Roe C, Lemmer D, Bowers JR, Engelthaler DM, et al. Genome Sequence of Staphylococcus aureus Strain CA-347, a USA600 Methicillin-Resistant Isolate. Genome Announc. 2013;1:4. doi: 10.1128/genomeA.00517-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Rubio R, Quiles-Puchalt N, Marti M, Humphrey S, Ram G, Smyth D, et al. Phage-inducible islands in the gram-positive cocci. ISME J. 2017;11(4):1029–1042. doi: 10.1038/ismej.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Wamel WJ, Rooijakkers SH, Ruyken M, van Kessel KP, van Strijp JA. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J Bacteriol. 2006;188(4):1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madhusoodanan J, Seo KS, Remortel B, Park JY, Hwang SY, Fox LK, et al. An enterotoxin-bearing Pathogenicity Island in Staphylococcus epidermidis. J Bacteriol. 2011;193(8):1854–1862. doi: 10.1128/JB.00162-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goerke C, Pantucek R, Holtfreter S, Schulte B, Zink M, Grumann D, et al. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J Bacteriol. 2009;191(11):3462–3468. doi: 10.1128/JB.01804-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novick RP, Christie GE, Penades JR. The phage-related chromosomal islands of gram-positive bacteria. Nat Rev Microbiol. 2010;8(8):541–551. doi: 10.1038/nrmicro2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su J, Liu X, Cui H, Li Y, Chen D, Li Y, Yu G. Rapid and simple detection of methicillin-resistance Staphylococcus aureus by orfX loop-mediated isothermal amplification assay. BMC Biotechnol. 2014;14:8. doi: 10.1186/1472-6750-14-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noto MJ, Kreiswirth BN, Monk AB, Archer GL. Gene acquisition at the insertion site for SCCmec, the genomic island conferring methicillin resistance in Staphylococcus aureus. J Bacteriol. 2008;190(4):1276–1283. doi: 10.1128/JB.01128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semmler T, Harrison EM, Lubke-Becker A, Ulrich RG, Wieler LH, Guenther S, et al. A look into the melting pot: the mecC-harboring region is a recombination hot spot in Staphylococcus stepanovicii. PLoS One. 2016;11(1):e0147150. doi: 10.1371/journal.pone.0147150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costa SS, Viveiros M, Amaral L, Couto I. Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol J. 2013;7:59–71. doi: 10.2174/1874285801307010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amissah NA, Chlebowicz MA, Ablordey A, Tetteh CS, Prah I, van der Werf TS, et al. Virulence potential of Staphylococcus aureus isolates from Buruli ulcer patients. Int J Med Microbiol. 2017;307(4–5):223–232. doi: 10.1016/j.ijmm.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38(4):698–719. doi: 10.1111/1574-6976.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen Y, Maupetit J, Derreumaux P, Tuffery P. Improved PEP-FOLD approach for peptide and Miniprotein structure prediction. J Chem Theory Comput. 2014;10(10):4745–4758. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 28.Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen MT, Kraft B, Yu W, Demircioglu DD, Hertlein T, Burian M, et al. The nuSaalpha Specific Lipoprotein Like Cluster (lpl) of S. aureus USA300 Contributes to Immune Stimulation and Invasion in Human Cells. PLoS Pathog. 2015;11(6):e1004984. doi: 10.1371/journal.ppat.1004984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotzin BL, Leung DY, Kappler J, Marrack P. Superantigens and their potential role in human disease. Adv Immunol. 1993;54:99–166. doi: 10.1016/S0065-2776(08)60534-9. [DOI] [PubMed] [Google Scholar]
- 32.Ferry T, Thomas D, Genestier AL, Bes M, Lina G, Vandenesch F, Etienne J. Comparative prevalence of superantigen genes in Staphylococcus aureus isolates causing sepsis with and without septic shock. Clin Infect Dis. 2005;41(6):771–777. doi: 10.1086/432798. [DOI] [PubMed] [Google Scholar]
- 33.Roetzer A, Gruener CS, Haller G, Beyerly J, Model N, Eibl MM. Enterotoxin Gene Cluster-Encoded SEI and SElN from Staphylococcus aureus Isolates are Crucial for the Induction of Human Blood Cell Proliferation and Pathogenicity in Rabbits. Toxins (Basel) 2016;8:11. doi: 10.3390/toxins8110314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stach CS, Vu BG, Merriman JA, Herrera A, Cahill MP, Schlievert PM, Salgado-Pabon W. Novel tissue level effects of the Staphylococcus aureus enterotoxin gene cluster are essential for infective endocarditis. PLoS One. 2016;11(4):e0154762. doi: 10.1371/journal.pone.0154762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johler S, Giannini P, Jermini M, Hummerjohann J, Baumgartner A, Stephan R. Further evidence for staphylococcal food poisoning outbreaks caused by egc-encoded enterotoxins. Toxins (Basel) 2015;7(3):997–1004. doi: 10.3390/toxins7030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuthbertson L, Nodwell JR. The TetR family of regulators. Microbiol Mol Biol Rev. 2013;77(3):440–475. doi: 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salinas N, Colletier JP, Moshe A, Landau M. Extreme amyloid polymorphism in Staphylococcus aureus virulent PSMalpha peptides. Nat Commun. 2018;9(1):3512. doi: 10.1038/s41467-018-05490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8(6):e1002744. doi: 10.1371/journal.ppat.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aswani V, Mau B, Shukla SK. Complete Genome Sequence of Staphylococcus aureus MCRF184, a Necrotizing Fasciitis-Causing Methicillin-Sensitive Sequence Type 45 Staphylococcus Strain. Genome Announc. 2016;4:3. doi: 10.1128/genomeA.00374-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015;43(W1):W104–W108. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei W, Gao F, Du MZ, Hua HL, Wang J, Guo FB. Zisland explorer: detect genomic islands by combining homogeneity and heterogeneity properties. Brief Bioinform. 2017;18(3):357–366. doi: 10.1093/bib/bbw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52(5):1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phenol soluble modulins (PSM) of S. aureus MCRF184. Panel 1A shows the amino acid sequences of the alpha and beta PSMs arranged from the N-terminus to the C-terminus. Numbers at the right show the net charge of the peptides at pH 7.0, rounded to whole numbers, and considering N-formylation of the initial methionine residue. The highlighted text identifies the amphipathic α-helical domain. Panel 1B shows the location of the genes coding for these PSMs in the genome of MCRF184 core genome. Panel 1C shows the predicted structure of the PSMS using PEP-FOLD (for the alpha PSMs) and SWISS-MODEL Workspace (for the beta PSMs). The residues are color-coded by their position in the peptide chain. Each chain is drawn as a smooth spectrum from blue through green, yellow and orange to red. The N-terminus of the peptides is colored red and the C terminuses are drawn in blue. The structures show the characteristic α-helical structure of the C-terminus ends of the PSMs. (PDF 334 kb)
Data Availability Statement
The datasets used in the study are available from the NCBI’s GenBank under the accession number CP014791.1.