

Abstract
Cigarette smoke, a source of numerous oxidants, produces oxidative stress and exaggerated inflammatory responses that lead to irreversible lung tissue damage. It is the single, most significant risk factor for chronic obstructive pulmonary disease (COPD). Although an intrinsic defense system that includes both enzymatic and non-enzymatic modulators exists to protect lung tissues against oxidative stress, impairment of these protective mechanisms has been demonstrated in smokers and COPD patients. The antioxidant enzyme GSH peroxidase (GPx) is an important part of this intrinsic defense system. Although cigarette smoke has been shown to downregulate its expression and activity, the underlying mechanism is not known. Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear hormone receptor with antioxidant effects. PPARγ activation has demonstrated protective effects against cigarette smoke-induced oxidative stress and inflammation. Molecular mechanisms for PPARγ’s antioxidant function likewise remain to be elucidated. This study explored the link between PPARγ and GPx3 and found a positive association in cigarette smoke extract (CSE)-exposed human bronchial epithelial cells. Moreover, we provide evidence that identifies GPx3 as a PPARγ transcriptional target. Attenuation of antioxidant effects in the absence of GPx3 highlights the antioxidant’s prominent role in mediating PPARγ’s function. We also demonstrate that ligand-mediated PPARγ activation blocks CSE-induced reactive oxygen species and hydrogen peroxide production via upregulation of GPx3. In summary, our findings describing the molecular mechanisms involving GPx3 and PPARγ in CSE-induced oxidative stress and inflammation may provide valuable information for the development of more effective therapeutics for COPD.
Keywords: nuclear hormone receptor, glutathione (GSH), GSH peroxidases, antioxidant, reactive oxygen species (ROS), hydrogen peroxide (H2O2), oxidative stress, cigarette smoke, PPAR response element (PPRE), human bronchial epithelial cells, inflammation, NF-κB
Graphical Abstract:
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is characterized by persistent and progressive airflow limitation and impaired gas exchange [1, 2]. It is also associated with chronic inflammation and airway remodeling [1, 2]. Affecting millions of people worldwide, it not only poses a major health threat but represents a significant socioeconomic burden [1]. Furthermore, the prevalence, mortality, and burden of COPD are expected to rise in the coming years, partly due to the failure of current therapeutic strategies to prevent disease progression and exacerbations [1]. Cumulative exposure to cigarette smoke is the single, most outstanding risk factor for COPD; cigarette smoke, which contains and produces numerous oxidants, causes cellular and tissue oxidative stress and thus further leads to exaggerated inflammatory responses and irreversible lung tissue damage [3–5]. Activated inflammatory and structural cells also produce reactive oxygen species (ROS), contributing to the vicious cycle of oxidative stress and inflammation in this disease [2, 4, 5]. Signs of elevated oxidative stress are evident in both smokers and COPD patients [5].
The lung is equipped with defense mechanisms against oxidative stress that may result from the normal immune responses to external irritants or the metabolic activity of structural cells [2, 4]. Glutathione (GSH) is one of the notable non-enzymatic players in such antioxidant processes, and its homeostasis, crucial for normal cellular functions, is regulated by a large network of enzymes that includes GSH peroxidases (GPxs). GPxs protect cells and tissues from oxidative damage primarily by reducing hydrogen peroxide and related lipid hydroperoxides. Studies showing reduced GSH concentrations and GPx activity in the lungs of cigarette smoke condensate-exposed rats [6, 7] indicate the involvement of these molecules in cigarette smoke-associated pathologies.
Peroxisome proliferator-activated receptor γ (PPARγ) belongs to the nuclear hormone receptor superfamily of transcription factors. Upon ligand-mediated activation, it displays a wide range of biological functions including anti-inflammatory and antioxidant effects [8–10] that are executed via regulation of other transcription factors, notably nuclear factor-κB (NF-κB). PPARγ can be stimulated by various endogenous compounds such as saturated and polyunsaturated fatty acids, eicosanoid derivatives, and nitrated fatty acids, as well as by synthetic molecules including the thiazolidinediones used to treat type 2 diabetes [11, 12]. Rosiglitazone and pioglitazone reduced cytokine production by alveolar macrophages from COPD patients [13]. These agonists also downregulated pulmonary inflammation in the subchronic tobacco smoke mouse model. Another PPARγ agonist, ciglitazone, showed similar anti-inflammatory effects on lung myeloid dendritic cells isolated from cigarette smokers with emphysema as well as in the mouse model of cigarette smoke-induced emphysema [14]. The well-characterized natural PPARγ ligand 15-deoxy-Δ12,14-PGJ2 also decreased the cigarette smoke extract (CSE)-induced production of inflammatory mediators by rat alveolar macrophages [15]. These studies substantiate the roles of PPARγ and its agonists in alleviating cigarette smoke-associated COPD.
We previously reported that CSE triggers inflammatory responses and oxidative stress in human lung epithelial cells by downregulating PPARγ [16]. However, molecular mechanisms underlying PPARγ’s antioxidant function remain unclear. In this study, we explored the link between PPARγ and GPx3 in CSE-exposed human bronchial epithelial (HBE) cells. We found that expression and activity of GPx3, as well as GSH concentration, were downregulated in COPD lung tissues and HBE cells from COPD patients (COPD HBE cells), whereas oxidative stress, as indicated by increased ROS/hydrogen peroxide (H2O2) production, was increased. CSE exposure similarly caused oxidative stress in normal HBE (NHBE) cells, decreasing GPx3 expression and activity and inducing oxidative enzyme expression. Our study further revealed that GPx3 is a direct transcriptional target of PPARγ and mediates the receptor’s antioxidant effects in CSE-exposed HBE cells. We accordingly found that PPARγ activation by rosiglitazone blocked CSE-induced GPx3 downregulation and ROS/H2O2 production in these cells. By revealing a molecular mechanism underlying PPARγ’s antioxidant effects, these findings support the therapeutic application of PPARγ agonists in COPD.
MATERIALS AND METHODS
Cells, tissue samples, and treatments
HBE cells obtained from Lonza (Walkersville, MD) were grown and maintained in bronchial epithelial cell media (Lonza) supplemented with growth factors and hormones, according to the manufacturer’s instructions. These cells were cultured at 37°C in a humidified atmosphere of 5% CO2. Monolayer cultures at 90% confluence were deprived of serum for 24 h prior to treatment.
Human lung tissue sampling, agonist and antagonist treatments, transfection, measurement of cytokines, chemokines (ELISA-based kits, R&D Systems, Minneapolis, MN), and ROS (Total ROS Activity Assay Kit, AAT Bioquest, Sunnyvale, CA), H2O2 (Amplex Red Hydrogen Peroxide Assay Kit, Thermo Fisher Scientific) and assessment of transcription factor-DNA binding (ELISA-based kits, Active Motif, Carlsbad, CA) have been described previously [16, 17].
CSE preparation
CSE was prepared as described previously [16]. Briefly, smoke from research-grade cigarettes (3R4F; Kentucky Tobacco Research and Development Center, University of Kentucky, Lexington, KY) was slowly bubbled into the medium. The extract, defined as 100% CSE, was diluted to the indicated concentrations and used within 10 minutes of preparation. For control experiments, air was bubbled into the medium.
GPx3 and GSH assay
Levels of GPx3 (AG-45A-0020YEK-KI01; AdipoGen LIFE SCIENCES; San Diego, CA) and GSH (703002; Cayman Chemical; Ann Arbor, MI) were quantified with commercially available kits according to the manufacturers’ instructions.
Western blotting
Western blotting was performed as described previously [18]. Primary antibodies against GPx3 (58361), NF-κB p65 (372), PPARγ (7196), cyclooxygenase-2 (COX2 [23983]), NADPH oxidase 4 (NOX4 [21860]), Lamin B1 (20682), and β-Actin (1616) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against pIKKα (2697) and pIκBα (9246) were from Cell Signaling Technology (Beverly, MA). The secondary antibodies, IR-680RD (926–68072 and 73) and IR-800CW (926–32212, 13 and 14), were obtained from LI-COR Biosciences (Lincoln, NE). The infrared signal was detected using an Odyssey Infrared Imager (LI-COR Biosciences).
DNA affinity precipitation
DNA affinity precipitation was performed as described previously [17]. Briefly, Dynabeads M-280 Streptavidin (Invitrogen; Carlsbad, CA) were mixed with biotinylated oligonucleotides containing the indicated PPAR response element (PPRE) (Supplemental Table 1) and incubated at room temperature for 30 min. Next, the biotinylated oligonucleotide-coupled beads were added to nuclear proteins and incubated for 30 min at room temperature with rotation. Samples were washed three times with cold binding and washing buffer. Bead-bound biotinylated oligonucleotide-protein complexes were eluted and subjected to Western blotting as described above with specific antibodies as indicated. An aliquot of the nuclear sample was set aside without streptavidin bead incubation to be used as an input control.
Chromatin immunoprecipitation (ChIP) assay
SimpleChIP Enzymatic Chromatin Immunoprecipitation kit with magnetic beads (Cell Signaling Technology, Beverly, MA) was used as described previously [19]. Antibody-bound DNA-protein complexes were eluted and subjected to real-time PCR with specific primers that amplified the GPx3 PPRE and β-Actin (Supplemental Table 1).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts from NHBE cells were incubated with 50 nM of the indicated double-stranded oligonucleotides (Supplemental Table 1) end-labeled with infrared dye IRDye 700 in binding buffer (100 mM Tris, 500 mM KCl, 10 mM DTT [pH 7.5]), poly(deoxyinosinic-deoxycytidylic) (1 µg/µl in 10 mM Tris, 1 mM EDTA), 25 mM DTT, and 2.5% Tween 20. Samples were then separated on 5% non-denaturing polyacrylamide gels in 1× Tris-Borate EDTA buffer (130 mM Tris [pH 8.3], 45 mM boric acid, 2.5 mM EDTA). In the supershift assay, the reaction mixture was incubated with anti-PPARγ antibody. The infrared signal was detected using an Odyssey Infrared Imager.
Statistical analysis
Data are presented as the mean ± SD. Differences between groups were analyzed using unpaired t-test or analysis of variance, followed by a Bonferroni multiple comparison correction. Analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA). A P value < 0.05 was considered significant.
RESULTS
GPx3 downregulation in COPD is associated with a reduction in cellular GSH and an increase in oxidative stress
To evaluate the role of GPx3 in the pathophysiology of COPD, we determined the expression and activity levels of GPx3 in COPD patients. Both expression and activity of GPx3 were reduced in COPD lung tissues (Fig. 1A) and COPD HBE cells (Fig. 1A and 1B) compared to their normal counterparts (Fig. 1A and 1B). The expression of the anti-inflammatory transcription factor PPARγ was decreased while pro-inflammatory NF-κB p65 expression was elevated (Fig. 1A). We also found lower GSH content in COPD HBE cells than in NHBE cells (Fig. 1C). GSH has been proposed to be one of the primary antioxidants in the respiratory tract [20]. COPD HBE cells accordingly displayed an increased level of oxidative stress evidenced by enhanced ROS (Fig. 1D and 1E) and H2O2 (Fig. 1F) production, suggesting impaired antioxidant and anti-inflammatory mechanisms. These results suggest a relationship between downregulated GPx3 and enhanced oxidative state in COPD.
Fig. 1. GPx3 expression and activity are decreased in COPD patients.
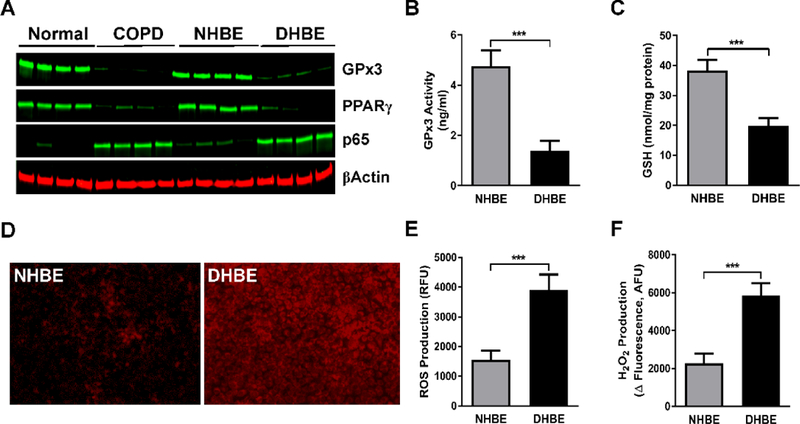
(A)Western blotting was performed to determine the expression of GPx3, PPARγ, and NF-κB p65 in COPD lung tissues (left panel) and DHBE (diseased HBE; also described as COPD HBE in the main text) cells (right panel) as well as in normal lung tissues and NHBE cells. β-Actin served as a loading control. (B and C) GPx3 activity (B) and GSH level (C) were assessed in NHBE and DHBE cells, as described in Materials and Methods. (D-F) Level of intracellular oxidative stress was examined in NHBE and DHBE cells by ROS fluorescent staining (D), fluorescence-based assays of ROS (E) and H2O2 (F) production. A representative image is shown. The data are expressed as the mean ± SD with n = 4; ***P < 0.001.
CSE induces inflammatory responses in NHBE cells by downregulating PPARγ and upregulating NF-κB
Bronchial epithelial cells lining the respiratory tract are the first to encounter inhaled hazardous materials, including the numerous gaseous and particulate substances present in cigarette smoke. While providing physical, chemical, and immunological barriers, these cells are also the targets of direct and second-hand cigarette smoke and thus can facilitate COPD pathogenesis [21–23]. Because cigarette smoking, the major risk factor for COPD, produces lung inflammation and oxidative stress [5, 17], we sought to assess HBE cells’ response to CSE exposure. We found that CSE downregulated PPARγ’s expression (Fig. 2A) and its DNA binding activity (Fig. 2B) in HBE cells. Conversely, NF-κB p65 expression (Fig. 2A) and its transcriptional activity (Fig. 2C) were enhanced by CSE treatment. Stimulatory phosphorylation of IKKα (NF-κB activator) and suppressive phosphorylation of IκBα (NF-κB inhibitor) were also increased (Fig. 2D), attesting to the activation of the pro-inflammatory NF-κB pathway. Accordingly, the levels of the inflammatory molecules TNF-α, IL-6, and IL-8 were also elevated (Fig. 2E). Thus, CSE induces inflammatory responses in HBE cells.
Fig. 2. CSE induces inflammatory responses in NHBE cells.

NHBE cells were treated with air (negative control) or 10% CSE for 6 h. Following treatment: (A) Whole cell extracts were obtained and subjected to Western blotting for PPARγ and NF-κB p65 expression. β-Actin served as a loading control. (B and C) Nuclear extracts were obtained and the DNA-binding activities of PPARγ (B) and NF-κB (C) were examined. (D) Phosphorylation of IKKα and IκBα in whole cell extracts was assessed by Western blotting. β-Actin served as a loading control. (E) Cell culture media was collected and CSE-induced release and concentrations of TNF-α, IL-6 and IL-8 were then determined by an ELISA-based assay. The data are expressed as the mean ± SD with n = 3. The results were reproduced independently at least two times; **P < 0.01, ***P < 0.001.
CSE-induced oxidative stress in NHBE cells is associated with downregulation of GPx3
Cigarette smoke is implicated in causing an oxidant-antioxidant imbalance in cells and tissues [5, 24]. GPx3 is an antioxidant enzyme that provides protection against oxidative stress/damage by catalyzing the reduction of H2O2. To evaluate whether a decrease in GPx3 expression and activity we observed in COPD samples (Fig. 1A and 1B) is a manifestation of disrupted oxidant-antioxidant balance, we treated NHBE cells with CSE and assessed GPx3 expression and activity as well as the oxidative state. CSE induced the expression of the oxidative enzymes COX2 and NOX4 (Fig. 3A). Conversely, GPx3 expression (Fig. 3A) and activity (Fig. 3B) were decreased by CSE exposure. Higher intracellular ROS (Fig. 3C) and H2O2 (Fig. 3D) production indicated an elevated level of oxidative stress in CSE-treated NHBE cells. Thus, our results suggest that CSE can cause oxidative stress via suppression of GPx3 in addition to other mechanisms such as induction of oxidative enzymes.
Fig. 3. CSE downregulates GPx3 and induces oxidative stress.
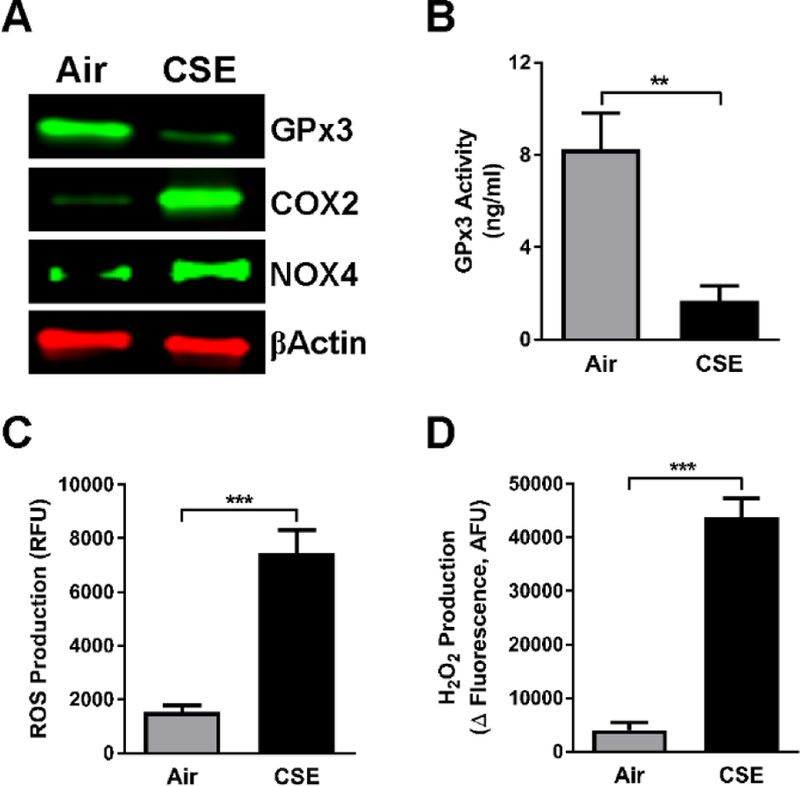
NHBE cells were treated with air or 10% CSE for 6 h. Following treatment; (A) Whole cell extracts were obtained and subjected to Western blotting for GPx3, COX2 and NOX4. β-Actin served as a loading control. (B) GPx3 activity was assessed in NHBE cells, as described in Materials and Methods. (C and D) Level of intracellular oxidative stress was examined in NHBE cells by fluorescence-based assays of ROS (C) and H2O2 (D) production. At least two independent experiments were performed. Data are expressed as the mean ± SD with n = 3; **P < 0.01, ***P < 0.001.
GPx3 is a PPARγ target gene
PPARγ elicits many of its biological effects by directly binding to its targets via specific DNA sequences termed PPREs and thus modulating gene expression. To determine whether GPx3 is a PPARγ transcriptional target, we first examined the relationship between GPx3 and PPARγ expression; specifically, GPx3 expression was measured in NHBE cells in which PPARγ was silenced by siRNA, overexpressed, or activated by the well-characterized agonist rosiglitazone. As shown in Fig. 4A, when PPARγ was knocked down, GPx3 expression decreased. Conversely, PPARγ overexpression or activation enhanced GPx3 expression (Fig. 4A). In each case, control treatment had no effect on GPx3 expression. The rosiglitazone-induced increase in GPx3 was PPARγ-dependent, as this effect was blocked by the absence of PPARγ (Fig. 4A). In addition, the enhancement of GPx3 expression by PPARγ overexpression was further increased by rosiglitazone (Fig. 4A). Thus, GPx3 and PPARγ expression exhibit a positive relationship in NHBE cells.
Fig. 4. GPx3 is a PPARγ transcriptional target.
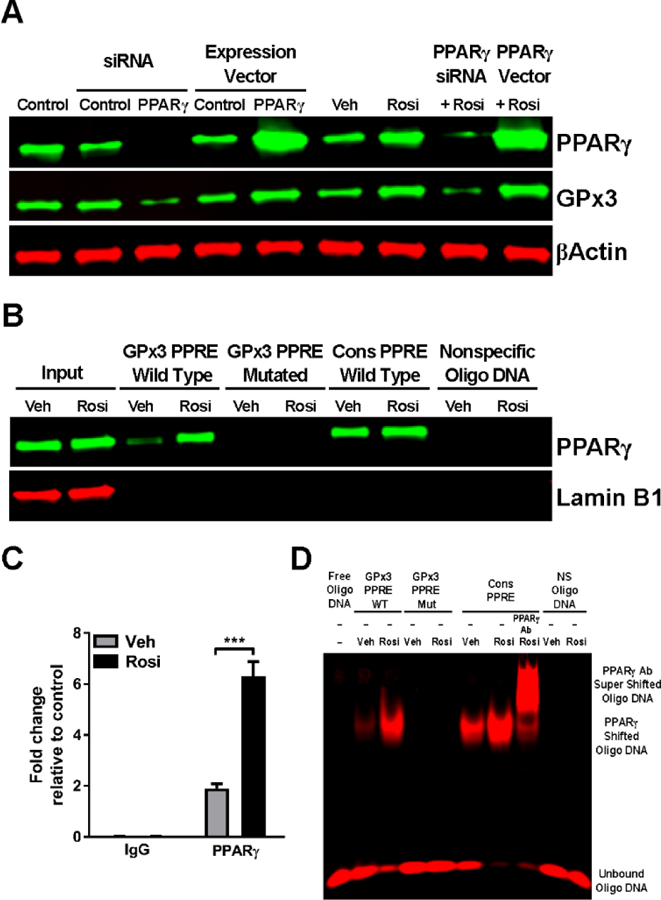
PPARγ in NHBE cells was silenced by PPARγ-specific siRNA, overexpressed via a PPARγ expression plasmid, or activated by 1 μM rosiglitazone; in each case, an appropriate control (scrambled siRNA, control expression plasmid, or vehicle, respectively) was included. In some experiments PPARγ-silenced or PPARγ-overexpressing cells were treated simultaneously with rosiglitazone. Following the treatment: (A) Whole cell extracts were obtained and subjected to Western blotting for PPARγ and GPx3. β-Actin served as a loading control. The results were reproduced independently at least two times. A representative image is shown. (B) Nuclear extracts were obtained and incubated with beads coupled to the biotinylated double-stranded oligonucleotides corresponding to the following: the wildtype (WT) or mutated (Mut) GPx3 PPRE, the consensus PPRE (positive control), or a non-specific sequence (negative control). Bead-bound oligonucleotide-protein complexes were eluted and subjected to Western blotting to determine the presence of PPARγ. Lamin B1 was used as a nuclear marker. The results were reproduced independently at least two times. A representative image is shown. (C) Chromatin was crosslinked and immunoprecipitated with antibodies to IgG (negative control) or PPARγ. Antibody-bound DNA-protein complexes were then subjected to real-time PCR with primers that specifically amplified the GPx3 PPRE. The data are expressed as the mean ± SD with n = 3 and the results were reproduced independently at least two times; ***P < 0.001. (D) Nuclear extracts were obtained and incubated with IR700 (red)-labeled double stranded oligonucleotides corresponding to the following: the WT or Mut GPx3 PPRE, the consensus PPRE, or a non-specific sequence. The supershift assay with anti-PPARγ antibody was performed to demonstrate the specificity of the interaction. The results were reproduced independently at least two times. A representative image is shown.
Next, the ability of activated PPARγ to physically interact with GPx3 was assessed by DNA-protein affinity assay. Oligonucleotides corresponding to the putative GPx3 PPRE were mixed with nuclear extracts of NHBE cells treated with or without rosiglitazone. Rosiglitazone treatment increased the GPx3 PPRE’s association with the cellular PPARγ-containing complexes compared to the control (Fig. 4B). As expected, the consensus PPRE used as a positive control recovered PPARγ, whereas non-specific oligonucleotides failed to do so (Fig. 4B). Importantly, mutations in the GPx3 PPRE (Supplemental Table 1) abrogated PPARγ binding (Fig. 4B), indicating the specificity of GPx3 PPRE-PPARγ interaction. These data suggest GPx3 is a direct PPARγ transcriptional target. We also examined the interaction of PPARγ with the GPx3 PPRE by ChIP assay with an anti-PPARγ antibody. The ChIP results recapitulated the PPARγ-GPx3 PPRE interaction seen with the DNA-protein affinity assay (Fig. 4C). The data from these two assays were further confirmed by an EMSA. GPx3 PPRE-bound PPARγ increased with rosiglitazone treatment (Fig. 4D). Conversely, PPARγ showed no binding to the mutated GPx3 PPRE or non-specific oligonucleotides (Fig. 4D). Consensus PPRE binding as well as an anti-PPARγ antibody-induced supershift of the PPARγ-PPRE complex, performed as controls, demonstrated the specificity of PPARγ-PPRE interaction (Fig. 4D). Together these results indicate that GPx3 contains a PPRE in its promoter region and is a direct PPARγ transcriptional target.
PPARγ activation can block CSE-induced downregulation of GPx3
We then investigated the implications of GPx3 as a PPARγ target in the context of our in vitro COPD system; i.e., the effect of PPARγ transcriptional activity on CSE-induced downregulation of GPx3. Consistent with the data in Fig. 2A and 3A, CSE exposure reduced both PPARγ and GPx3 expression (Fig. 5A–C). This GPx3 downregulation was accentuated by siRNA-mediated PPARγ knockdown (Fig. 5A) as well as by the PPARγ antagonist GW9662 (Fig. 5B). In contrast, rosiglitazone treatment blocked CSE-induced GPx3 downregulation (Fig. 5C). These results suggest CSE reduces GPx3 expression via downregulation of PPARγ expression/activity and that agonist-mediated PPARγ activation can circumvent such effect.
Fig. 5. CSE-induced downregulation of GPx3 correlates with PPARγ expression/activity.

(A)NHBE cells, transfected with PPARγ-specific siRNA or scrambled siRNA (negative control), were treated with air (negative control) or 10% CSE for 6 h. Following treatment, whole cell extracts were obtained and subjected to Western blotting for PPARγ and GPx3. (B) NHBE cells, treated with the PPARγ antagonist GW9662 (1 μM) or vehicle control, were exposed to air (negative control) or 10% CSE for 6 h. Following treatment, whole cell extracts were obtained and subjected to Western blotting for PPARγ and GPx3. (C) NHBE cells, treated with 1 μM rosiglitazone or vehicle control, were exposed to air (negative control) or 10% CSE for 6 h. Following treatment, whole cell extracts were obtained and subjected to Western blotting for PPARγ and GPx3. In all the experiments, β-Actin served as a loading control. The results were reproduced independently at least two times. A representative image is shown.
GPx3 mediates PPARγ’s antioxidant activity
Because GPx3 performs important antioxidant functions, we hypothesized that GPx3 might mediate PPARγ’s antioxidant effects in CSE-exposed cells. To determine whether GPx3 is involved in this process, we silenced GPx3 in NHBE cells and assessed the outcome of CSE exposure. Activation of PPARγ by rosiglitazone reduced oxidative stress, as measured by intracellular ROS (Fig. 6A) and H2O2 (Fig. 6B) production, in CSE-exposed NHBE cells. However, GPx3 deficiency dampened this rosiglitazone effect on CSE-induced ROS and H2O2 production to less than 20%, whereas production in control siRNA-treated cells remained above 70% of the baseline (Fig. 6C and 6D). Taken together, our data argue that PPARγ counteracts CSE-incurred oxidative stress by modulating GPx3 expression.
Fig. 6. GPx3 mediates PPARγ’s antioxidant effects on CSE exposure.

Level of intracellular oxidative stress was examined by fluorescence-based assays of ROS (A and C) and H2O2 (B and D) production after the following treatments; (A and B) NHBE cells, treated with 1 μM rosiglitazone or vehicle control, were exposed to air (negative control) or 10% CSE for 6 h. (C and D) NHBE cells, transfected with GPx3-targeting or control scrambled siRNA, were treated with 1 μM rosiglitazone or vehicle control. All the test groups were then exposed to 10% CSE for 6 h. The data are expressed as the mean ± SD with n = 3 and the results were reproduced independently at least two times; *P < 0.05, ***P < 0.001.
DISCUSSION
Numerous oxidants found in cigarette smoke, a major risk factor in COPD, disturb the balanced redox state normally maintained by cells and tissues [25]. They also contribute to the oxidative burden indirectly by activating inflammatory and structural cells that produce ROS [2, 25]. The results of these events are chronic oxidative stress and inflammation, as seen in COPD.
PPARγ is a nuclear receptor with anti-inflammatory and antioxidant properties [8–10]. To elucidate the molecular mechanisms of PPARγ’s antioxidant functions, in the present study we investigated the link between PPARγ and the plasma/extracellular antioxidant GPx3. We found a positive association between PPARγ and GPx3 expression/activity in HBE cells and revealed that GPx3 is a PPARγ transcriptional target that mediates the receptor’s antioxidant effects (Fig. 7).
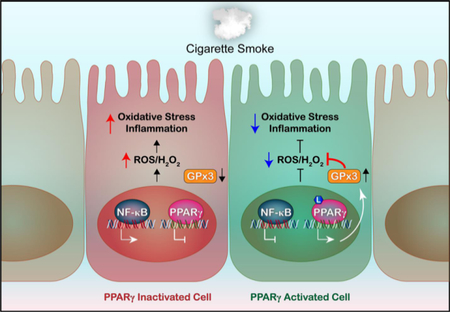
Fig. 7. PPARγ and GPx3 provide protection against cigarette smoke-induced oxidative stress and inflammation.

Cigarette smoke downregulates PPARγ and upregulates NF-κB signaling, leading to oxidative stress and exaggerated inflammatory responses in cells. In the presence of an activating ligand (L), however, PPARγ’s transcriptional activity is stimulated and GPx3 expression and activity are induced. Consequently, oxidative stress and inflammation are reduced.
Oxidative stress inhibits PPARγ transcription [26]. As this observation implies, the PPARG gene was found to be downregulated in smokers with emphysema, a pathological feature of COPD [14]. Conversely, in cigarette smoke-exposed mice, PPARγ and PPARγ agonists have displayed protective effects against the airspace enlargement indicative of emphysema pathology [14, 27]. Similar benefits have been reported in other diseases associated with increased oxidative burden [28–30]. Yet molecular mechanisms underlying these effects have so far remained unclear. We extend these earlier studies by identifying GPx3 as a direct transcriptional target of PPARγ that mediates its antioxidant effects in HBE cells. Similarly, in the context of obesity and diabetes, PPARγ has been shown to reduce the oxidative burden in skeletal muscle [31] and adipocytes/adipose tissues [32] via regulation of GPx3. It will thus be intriguing to investigate whether the PPARγ-GPx3 pathway plays a crucial role in other diseases linked with oxidative stress [33].
We showed a reduction in GPx3 expression in lung tissues from patients with advanced COPD. Its expression and activity were similarly downregulated in COPD HBE cells as well as in CSE-treated NHBE cells. Our findings are consistent with clinical data; a study reported reduced GPx activity in smokers with COPD compared to healthy controls (smokers or non-smokers) [34]. GPx activity was also lower in COPD patients compared to healthy controls [35], in severe COPD compared to moderate disease [35, 36], and in moderate COPD compared to mild disease [35, 37]. Moreover, Golpon et al. reported downregulation of the GPX3 gene in patients with severe emphysema [38]. Together, these data suggest that GPx3, especially since it is an extracellular enzyme that can be readily measured, can serve as a useful biomarker for COPD.
GPx3 knockdown attenuated rosiglitazone’s ability to suppress CSE-induced ROS and H2O2 production by more than 50%, which implies that GPx3 is the primary but probably not the sole mediator of PPARγ’s antioxidant function in HBE cells. PPARγ has been shown to directly regulate transcription of other antioxidants such as catalase [39], SOD1 [40, 41], and SOD2 [42] via PPREs found in their promoter regions. In addition, PPARγ and its ligands have been reported to negatively regulate NOX expression and downregulate oxidative burden [43–46]. Thus, while our data support GPx3’s prominent role in the PPARγ-mediated cellular response to CSE exposure, it will be interesting to determine whether any of the other redox pathways may be involved in PPARγ’s effect on COPD pathophysiology.
CONCLUSION
In this study, we have provided the first evidence that GPx3 plays a prominent role as a mediator of PPARγ’s antioxidant effects on the response to CSE exposure. Furthermore, we showed that PPARγ activation blocks CSE-induced GPx3 downregulation as well as ROS and H2O2 production. Currently, there are no treatments available to COPD patients that reverse the pathology or prevent disease progression. Thus, further understanding of the molecular mechanisms underlying the disease pathogenesis is of potential clinical value. Our findings support the therapeutic potential of PPARγ agonists to target inflammation and oxidative damage in COPD.
Supplementary Material
HIGHLIGHTS.
GPx3 expression is decreased in COPD patients’ lungs.
Cigarette smoke downregulates PPARγ and GPx3.
GPx3 is a PPARγ transcriptional target.
GPx3 deficiency attenuates PPARγ’s antioxidant effects.
Activated PPARγ blocks cigarette smoke-induced oxidative stress by increasing GPx3.
Acknowledgments
Funding
This work was supported by a merit review award from the U.S. Department of Veterans Affairs, Flight Attendant Medical Research Institute Clinical Innovator Award 16006, and National Institutes of Health Grants HL137842 and AI125338 (R.C.R).
Abbreviations
- COPD
chronic obstructive pulmonary disease
- ROS
reactive oxygen species
- GSH
glutathione
- GPx
GSH peroxidase
- PPARγ
Peroxisome proliferator-activated receptor γ
- NF-κB
nuclear factor-κB
- CSE
cigarette smoke extract
- HBE
human bronchial epithelial
- NHBE
normal HBE
- COX2
cyclooxygenase-2
- NOX4
NADPH oxidase 4
- H2O2
hydrogen peroxide
- PPRE
PPAR response element
- ChIP
chromatin immunoprecipitation
- EMSA
electrophoretic mobility shift assay
- WT
wild-type
- Mut
mutated
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare that they have no conflicts of interest with the contents of this article.
FOOTNOTES
Disclaimer
The contents in this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
REFERENCES
- 1.Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, et al. (2013) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 187, 347–65 [DOI] [PubMed] [Google Scholar]
- 2.Kirkham PA, Barnes PJ. (2013) Oxidative stress in COPD. Chest 144, 266–73 [DOI] [PubMed] [Google Scholar]
- 3.Eapen MS, Myers S, Walters EH, Sohal SS. (2017) Airway inflammation in chronic obstructive pulmonary disease (COPD): a true paradox. Expert Rev Respir Med 11, 827–39 [DOI] [PubMed] [Google Scholar]
- 4.Rahman I, Kinnula VL. (2012) Strategies to decrease ongoing oxidant burden in chronic obstructive pulmonary disease. Expert Rev Clin Pharmacol 5, 293–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacNee W (2000) Oxidants/antioxidants and COPD. Chest 117, 303S–17S [DOI] [PubMed] [Google Scholar]
- 6.Li XY, Rahman I, Donaldson K, MacNee W. (1996) Mechanisms of cigarette smoke induced increased airspace permeability. Thorax 51, 465–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahman I, Li XY, Donaldson K, Macnee W. (1995) Cigarette smoke, glutathione metabolism and epithelial permeability in rat lungs. Biochem Soc Trans 23, 235S. [DOI] [PubMed] [Google Scholar]
- 8.Nencioni A, Wesselborg S, Brossart P. (2003) Role of peroxisome proliferator-activated receptor gamma and its ligands in the control of immune responses. Crit Rev Immunol 23, 1–13 [DOI] [PubMed] [Google Scholar]
- 9.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82 [DOI] [PubMed] [Google Scholar]
- 10.Corona JC, Duchen MR. (2016) PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med 100, 153–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banno A, Reddy AT, Lakshmi SP, Reddy RC. (2018) PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl Receptor Res 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belvisi MG, Mitchell JA. (2009) Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol 158, 994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lea S, Plumb J, Metcalfe H, Spicer D, Woodman P, Fox JC, et al. (2014) The effect of peroxisome proliferator-activated receptor-gamma ligands on in vitro and in vivo models of COPD. Eur Respir J 43, 409–20 [DOI] [PubMed] [Google Scholar]
- 14.Shan M, You R, Yuan X, Frazier MV, Porter P, Seryshev A, et al. (2014) Agonistic induction of PPARgamma reverses cigarette smoke-induced emphysema. J Clin Invest 124, 1371–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin Y, Hou G, Li E, Wang Q, Kang J. (2014) PPARgamma agonists regulate tobacco smoke-induced Toll like receptor 4 expression in alveolar macrophages. Respir Res 15, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakshmi SP, Reddy AT, Zhang Y, Sciurba FC, Mallampalli RK, Duncan SR, et al. (2014) Down-regulated peroxisome proliferator-activated receptor gamma (PPARgamma) in lung epithelial cells promotes a PPARgamma agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem 289, 6383–93 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Lakshmi SP, Reddy AT, Reddy RC. (2017) Transforming growth factor beta suppresses peroxisome proliferator-activated receptor gamma expression via both SMAD binding and novel TGF-beta inhibitory elements. Biochem J 474, 1531–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy AT, Lakshmi SP, Muchumarri RR, Reddy RC. (2016) Nitrated Fatty Acids Reverse Cigarette Smoke-Induced Alveolar Macrophage Activation and Inhibit Protease Activity via Electrophilic S-Alkylation. PLoS One 11, e0153336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddy AT, Lakshmi SP, Zhang Y, Reddy RC. (2014) Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J 28, 5299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cantin AM, North SL, Hubbard RC, Crystal RG. (1987) Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol (1985) 63, 152–7 [DOI] [PubMed] [Google Scholar]
- 21.Di Stefano A, Caramori G, Oates T, Capelli A, Lusuardi M, Gnemmi I, et al. (2002) Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur Respir J 20, 556–63 [DOI] [PubMed] [Google Scholar]
- 22.Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. (2018) Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am J Respir Cell Mol Biol 58, 157–6928933915 [Google Scholar]
- 23.Kode A, Yang SR, Rahman I. (2006) Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res 7, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernardo I, Bozinovski S, Vlahos R. (2015) Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol Ther 155, 60–79 [DOI] [PubMed] [Google Scholar]
- 25.Vlahos R, Bozinovski S. (2013) Glutathione peroxidase-1 as a novel therapeutic target for COPD. Redox Rep 18, 142–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. (2010) Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic Biol Med 48, 1618–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solleti SK, Simon DM, Srisuma S, Arikan MC, Bhattacharya S, Rangasamy T, et al. (2015) Airway epithelial cell PPARgamma modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am J Physiol Lung Cell Mol Physiol 309, L293–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, et al. (2006) Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol 530, 70–80 [DOI] [PubMed] [Google Scholar]
- 29.El-Naa MM, El-Refaei MF, Nasif WA, Abduljawad SH, El-Brairy AI, El-Readi MZ. (2015) In-vivo antioxidant and anti-inflammatory activity of rosiglitazone, a peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists in animal model of bronchial asthma. J Pharm Pharmacol 67, 1421–30 [DOI] [PubMed] [Google Scholar]
- 30.Lee KS, Kim SR, Park SJ, Park HS, Min KH, Jin SM, et al. (2006) Peroxisome proliferator activated receptor-gamma modulates reactive oxygen species generation and activation of nuclear factor-kappaB and hypoxia-inducible factor 1alpha in allergic airway disease of mice. J Allergy Clin Immunol 118, 120–7 [DOI] [PubMed] [Google Scholar]
- 31.Chung SS, Kim M, Youn BS, Lee NS, Park JW, Lee IK, et al. (2009) Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol Cell Biol 29, 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YS, Kim AY, Choi JW, Kim M, Yasue S, Son HJ, et al. (2008) Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Mol Endocrinol 22, 2176–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham-Huy LA, He H, Pham-Huy C. (2008) Free radicals, antioxidants in disease and health. Int J Biomed Sci 4, 89–96 [PMC free article] [PubMed] [Google Scholar]
- 34.Biljak VR, Rumora L, Cepelak I, Pancirov D, Popovic-Grle S, Soric J, et al. (2010) Glutathione cycle in stable chronic obstructive pulmonary disease. Cell Biochem Funct 28, 448–53 [DOI] [PubMed] [Google Scholar]
- 35.Mohammed A, Gutta V, Ansari MS, Saladi Venkata R, Jamil K. (2017) Altered antioxidant enzyme activity with severity and comorbidities of chronic obstructive pulmonary disease (COPD) in South Indian population. COPD Research and Practice 3, 4 [Google Scholar]
- 36.Kluchova Z, Petrasova D, Joppa P, Dorkova Z, Tkacova R. (2007) The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol Res 56, 51–6 [DOI] [PubMed] [Google Scholar]
- 37.Waseem SMA, Hussain M, Islam N. (2014) Oxidative Stress in Mild and Moderate COPD: Assessment of Oxidant Anti-Oxidant Imbalance. Biomed Res 25, 115–9 [Google Scholar]
- 38.Golpon HA, Coldren CD, Zamora MR, Cosgrove GP, Moore MD, Tuder RM, et al. (2004) Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol 31, 595–600 [DOI] [PubMed] [Google Scholar]
- 39.Girnun GD, Domann FE, Moore SA, Robbins ME. (2002) Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol 16, 2793–801 [DOI] [PubMed] [Google Scholar]
- 40.Yoo HY, Chang MS, Rho HM. (1999) Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene 234, 87– 91 [DOI] [PubMed] [Google Scholar]
- 41.Inoue I, Goto S, Matsunaga T, Nakajima T, Awata T, Hokari S, et al. (2001) The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 50, 3–11 [DOI] [PubMed] [Google Scholar]
- 42.Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, et al. (2007) Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res 76, 269–79 [DOI] [PubMed] [Google Scholar]
- 43.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, et al. (2015) Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 80, 111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hwang J, Kleinhenz DJ, Lassegue B, Griendling KK, Dikalov S, Hart CM. (2005) Peroxisome proliferator-activated receptor-gamma ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol 288, C899–905 [DOI] [PubMed] [Google Scholar]
- 45.Williams CR, Lu X, Sutliff RL, Hart CM. (2012) Rosiglitazone attenuates NF-kappaB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am J Physiol Cell Physiol 303, C213–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hwang J, Kleinhenz DJ, Rupnow HL, Campbell AG, Thule PM, Sutliff RL, et al. (2007) The PPARgamma ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul Pharmacol 46, 456–62 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.