

Abstract
Background & Aims:
Activation of KRAS signaling and overexpression of the aurora kinase A (AURKA) are often detected in luminal gastrointestinal cancers. We investigated regulation of ribosomal protein S6 kinase B1 (RPS6KB1) by AURKA and the effects of alisertib, an AURKA inhibitor, in mice xenograft tumors grown from human gastrointestinal cancer cells with mutant, activated forms of KRAS.
Methods:
We tested the effects of alisertib, or AURKA overexpression or knock down, in 10 upper gastrointestinal or colon cancer cell lines with KRAS mutations or amplifications using the CellTiter-Glo luminescence and clonogenic cell survival assays. We used the proximity ligation in situ assay to evaluate protein co-localization and immunoprecipitation to study protein interactions. Nude mice with xenograft tumors grown from HCT116, SNU-601, SW480, or SNU-1 cells were given oral alisertib (40 mg/kg, 5 times/week) for 4 weeks. Tumor samples were collected and analyzed by immunoblots and immunohistochemistry. Tissue microarrays from 151 paraffin-embedded human colon tumors, with adjacent normal and adenoma tissues, were analyzed by immunohistochemistry for levels of AURKA.
Results:
Alisertib reduced proliferation and survival of cell lines tested. AURKA knockdown or inhibition with alisertib reduced levels of phosphorylated RPS6KB1 (at T389), and increased levels of proteins that induce apoptosis including BIM, cleaved PARP, and cleaved caspase 3. AURKA co-localized and interacted with RPS6KB1, mediating RPS6KB1 phosphorylation at T389. We detected AURKA-dependent phosphorylation of RPS6KB1 in cell lines with mutations in KRAS, but not in cells with wild-type Ras. Administration of alisertib to mice with xenograft tumors significantly reduce tumor volumes (P < .001). The agent reduced phosphorylation of RPS6KB1 and Ki-67, and increased levels of cleaved caspase 3, in tumor tissues. In analyses of tissue microarrays, we found significant overexpression of AURKA in gastrointestinal tumor tissues compared with non-tumor tissues (P=.0003).
Conclusion:
In studies of gastrointestinal cancer cell lines with activated KRAS, we found AURKA to phosphorylate RPS6KB1 to promote cell proliferation and survival and growth of xenograft tumors in mice. Agents that inhibit AURKA might slow growth of gastrointestinal tumors with activation of KRAS.
Keywords: mutation, oncogenic, signal transduction, colon cancer
Graphical Abstract
Introduction
KRAS is the most frequently mutated isoform of the RAS protein family and its activation plays critical roles in cellular transformation and cancer development. Nearly eighty percent of KRAS mutations occur at codon 12 and are associated with poor clinical outcome 1. Activation of oncogenic KRAS by mutations or amplifications is well characterized in luminal gastrointestinal cancers that include colorectal, gastric, and esophageal adenocarcinomas 2–5. Tumors with activated KRAS signaling networks are particularly resistant to current available treatments6, 7. Although several attempts have been made to target oncogenic KRAS, to date, no specific therapeutic agents have proven clinical success against oncogenic KRAS 8. Understanding the interaction between oncogenic KRAS and its downstream signaling pathways can lead to the development of effective therapeutic strategies to target crucial signaling axes downstream of activated KRAS.
Ribosomal protein S6 kinase B1 (RPS6KB1, also known as P70S6K), a mitogen-activated serine/threonine protein kinase, has been linked to diverse cellular processes including glucose homeostasis, mRNA processing, protein synthesis, cell growth and survival 9. Activation of RPS6KB1 occurs through phosphorylation of serine or threonine residues where its phosphorylation at threonine 389 is essential for its catalytic activity 10. The commonly observed constitutive activation of RPS6KB1 in human malignancies suggests its potential role as a therapeutic target 11, 12.
Aurora kinase A (AURKA), a serine/threonine kinase that plays a critical role in regulating cell cycle and mitosis in normal cells, is frequently amplified and/or overexpressed in GI malignancies (including esophageal, gastric, and colorectal cancers) 13-15. Overexpression of AURKA can serve as an independent prognostic marker for poor clinical outcomes in cancer patients 16. We and others have reported that aberrant overexpression of AURKA mediates activation of key oncogenic signaling pathways in cancer cells such as NF-κB and β-catenin/WNT signaling13. Furthermore, AURKA can suppress p53 by several mechanisms that include phosphorylation and activation of HDM217, 18. Of note, AURKA can regulate c-MYC levels by several mechanisms that include transcription, phosphorylation, and translation19-22. Alisertib, an AURKA inhibitor, as a single agent or in combination with other drugs attenuated cancer cell survival in several pre-clinical studies23, 24 In the last few years, several phase I/II clinical trials that utilize alisertib have emerged in hematologic and solid tumor malignancies25, 26.
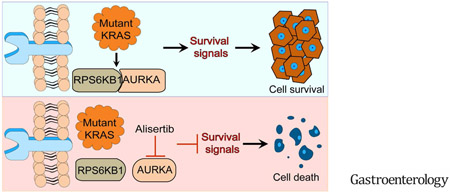
In this study, we demonstrate that AURKA plays a crucial role in phosphorylating RPS6KB1 in KRAS-mutant cancer cells. Targeting AURKA-RPS6KB1 axis using alisertib was effective in reducing tumor growth in tumor xenograft mouse models, suggesting that this approach could be a novel therapeutic strategy in KRAS-driven luminal GI cancers.
Materials and Methods
Reagents
Alisertib was prepared and stored according to the manufacturer’s instructions (Millennium Pharmaceuticals, Inc., Cambridge, MA). Specific antibodies against p-MTOR(Ser2448), p-MEK1/2(Ser217/221), p-AKT(Ser473), p-AURKA(T288), mTOR, MEK, AKT, AURKA, RPS6KB1, p-RPS6KB1(T389), Phospho-(Ser/Thr) Phe, and β-Actin were purchased from Cell Signaling Technology (Beverly, MA). Recombinant human AURKA and RPS6KB1 proteins were obtained from Cell Sciences (Canton, MA). Specific antibodies against KRAS were purchased from Santa Cruz Biotechnology (Dallas, TX). KRAS-G12D lentiviral vector was purchased from Applied Biological Materials (Richmond, BC, Canada). Tet-One™ Inducible Expression System and Tet System Approved FBS were purchased from Clontech (Palo Alto, CA). Transfection reagent LipoJet was purchased from SignaGen Laboratories (Gaithersburg, MD). Tet-on expression system, doxycycline-free fetal bovine serum, and human cell cycle arrays were purchased from Clontech (Palo Alto, CA).
Cell lines and cell culture
Human esophageal adenocarcinoma cell line ESO26 was obtained from Sigma-Aldrich (St. Louis, MO), human colorectal cancer (CRC) cell lines (SW480, HCT116, SW620, RKO, SK-CO-1, LS180, and LS153) and human gastric cancer cell lines (AGS, SNU-1, MKN45) were obtained from ATCC (Manassas, VA) and Riken Cell Bank (Tsukuba, Japan); gastric cancer cell line STKM2 was a generous gift from Dr. Alexander Zaika, University of Miami; gastric cancer cell line SNU-601 was purchased from the Korean Cell Line Bank (Jongno-gu, Seoul). AGS and STKM2 were maintained in F12 medium (GIBCO, Carlsbad, CA); CRC cell lines were maintained in RPMI-1640 medium (GIBCO); SNU-1, MKN45 and ESO26 were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO). All cell lines were supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin (GIBCO). All cell lines were ascertained to conform to the original in vitro morphologic characteristics and were authenticated by Genetica DNA Laboratories using short tandem repeat profiling (Genetica DNA Laboratories).
CellTiter-Glo Luminescence Assay
CellTiter-Glo Luminescence Assay (Promega, Madison, WI) was used to determine IC50 and drug dose-response curves for each cell line following treatment with alisertib. Cells were seeded at 2,000 cells/well in a 96-well plate. Cells were treated with alisertib following a 12 × 2-fold serial dilution treatment in 5% FBS-DMEM medium. Five days later, cell viability was measured using the CellTiter-Glo reagent. The dose-response curves were fitted using the GraphPad Prism 5, following a non-linear regression (four parameter, least squares fit) method. IC50 values were determined by a fourparameter, non-linear regression method. Data was generated from at least three independent experiments.
Proximity ligation in situ assay (PLA)
To demonstrate the close distance (<40 nm) between two different proteins (AURKA and RPS6KB1) in GI cancer cells, PLA was performed using Duo-link In Situ-Fluorescence kits according to the manufacturer’s instructions (Sigma-Aldrich). The SW480 and AGS cells, grown on slides, were fixed in 4% paraformaldehyde for 30 min, and permeabilized using 0.5% Triton-X-100 for 20 min at room temperature. Cells were then incubated with blocking solution for 60 min and incubated overnight with primary antibodies at 4°C (anti-AURKA plus anti-RPS6KB1). The cells were subsequently incubated with PLA PLUS and MINUS probes for mouse and rabbit and incubated with ligation-ligase solution for 60 min at 37°C, subsequently with amplification-polymerase solution according to the manufacturer’s instructions. The slides were mounted with DAPI mounting solution. Each dot represents the close proximity of two interacting proteins within the cells. Cell images were acquired using an Olympus FV-1000 Inverted Confocal microscope (Olympus Co., Center Valley, PA).
Immunohistochemistry on human tissue arrays
Tissue microarrays containing cores from 151 paraffin-embedded de-identified human colon cancer tissue samples, with adjacent normal and adenomas, when available, were obtained from Vanderbilt Tissue Pathology Core Resource (TPSR). All tissue samples were coded and de-identified in accordance with Institutional Review Board-approved protocols. The histology of all tissue samples was verified using H&E staining. The stage of tumor samples ranged from 2A to 3C. The annotation of these tumors is provided in Supplementary Table S1. The tissue microarrays were used for immunohistochemical analysis using rabbit anti-AURKA (KR051; 1:100 dilution, TransGenic, Inc., Japan). For statistical analysis, a composite scoring system was developed to integrate the IHC signal intensity and the frequency of positive cells in the cytosol and nucleus. The immunoreactivity of the samples tested was assessed by a trained pathologist and scored for intensity (scaled 0-3) and frequency (scaled 0-4). A composite expression score (CES) with a full range from 0 to 12 was used; CES was calculated using the formula; CES = 4(intensity-1) + frequency, as previously described27.
Tumor xenografts
All animal work was approved by the Institutional Animal Care and Use Committee. HCT116, SNU-601, SW480, and SNU-1 cells (2-5×106) were suspended in 150 μl of PBS and Matrigel mixture and were injected into both flank regions of female 201 NIH-III nude mice (Charles River Laboratories, Wilmington, MA). To measure therapeutic efficacy and response, the tumors were allowed to grow to 150 – 200 mm3 in volume before starting treatment with alisertib (40 mg/kg, 5 times/week, orally) for four weeks. Tumor xenografts were measured every 3 days and tumor volumes were calculated according to the formula: Tvol = L × W2 × 0.5, in which Tvol is tumor volume, L is tumor length and W is tumor width. At the end of treatment, tumors were collected. Because the tumors almost disappeared at the end of the 4 week treatment, we included additional groups of mice that were treated for one week to measure the signaling effects. Western blot (p-AURKA (T288), AURKA, RPS6KB1) analysis was carried out using frozen tumors whereas immunohistochemical analysis was carried out on the formalin fixed paraffin-embedded tissues to measure Ki-67 (Cell Signaling) and cleaved caspase 3 (Cell Signaling) protein expression levels, following standard protocols. Immunostaining levels were evaluated by counting the immunostaining intensity in at least 200 cells from three independent areas of each tumor using ImageJ software. Relative integrated density indicates the quantification data of diaminobenzidine staining signal analyzed by the ImageJ IHC Toolbox plugin (https://imagej.nih.gov/ij/plugins/ihc-toolbox/index.html).
Additional experimental and statistical analysis
Additional details are provided in Supplementary Materials and Methods section. Quantification of Western blot data is summarized in Supplementary Figure S1.
Results
Inhibition of AURKA using alisertib inhibits survival of KRAS-driven GI cancer cells
To investigate the function of AURKA in promoting KRAS-driven oncogenesis, we evaluated the biological impact of inhibiting AURKA with alisertib, a selective AURKA inhibitor, in a panel of 10 luminal gastrointestinal cancer cell lines harboring KRAS mutations or amplifications (KRAS-driven). The characteristics of these cell lines are shown in Supplementary Table S2. The cell viability (5 days), in response to alisertib treatment, was determined by CellTiter-Glo Assay. Alisertib decreased cell viability in a dose-dependent manner in all tested upper GI cancer (UGC) cell lines (ESO26, AGS, SNU-1, SNU-601) and CRC cell lines (HCT116, SW480, SW620, SK-CO-1, LS180, LS153). The IC50 values ranged from 16 nM (HCT116) to 82 nM (SNU-601), with an average of 37.8 nM (Figure 1A, B). The long-term clonogenic survival assay (15 days), after a single overnight treatment with alisertib, demonstrated a significant reduction in the number of viable cell colonies in gastric cancer cells (ESO26, SNU-1, AGS, SNU-601) and CRC cells (HCT116, SK-CO-1 SW480, SW620, LS180, LS153) (Figure 1 C-F). Next, we examined the effect of alisertib (24, 48, and 72 hrs) on cell cycle progression in AGS, SNU-1 and SW480 cell lines. Treatment with alisertib also increased the percentage of polyploid cells, consistent with earlier reports23, (Supplementary Figure S2). Collectively, these results indicate that inhibition of AURKA using alisertib can effectively suppress cancer cell viability and induce polyploidy in KRAS-driven GI cancers.
Figure 1. Inhibition of AURKA with alisertib suppresses proliferation and survival of KRAS-driven GI cancer cells.

A & B, Upper GI cancer (UGC) cells (ESO26, AGS, SNU-1, SNU-601) & CRC cells (HCT116, SW480, SW620, SK-CO-1, LS180, LS153) were treated with alisertib for 5 days. Cell viability was measured using CellTiter-Glo assay, and dose response curves were generated following a four-parameter nonlinear regression method. C, Clonogenic cell survival assay of ESO26, AGS, SNU-1, and SNU-601, cells treated with alisertib for 24 hrs and cultured for an additional 15 days after drug withdrawal. D, An IC50 graph summarizing data shown in C. E, Clonogenic cell survival assay of CRC cells (HCT116, SW480, SW620, SK-CO-1, LS180, LS153). F, An IC50 graph summarizing data shown in E; the quantification is performed from three independent experiments.
Inhibition of AURKA promotes cell death and inhibits phosphorylation of RPS6KB1
To determine the role of AURKA in regulating oncogenic KRAS downstream effectors, we investigated if AURKA inhibition by alisertib can suppress downstream KRAS mediators, including phosphorylation and activation of MEK, AKT, MTOR, and RPS6KB1 in GI cancer cells (ESO26, AGS, SNU-601, SNU-1, HCT116, SW480, SW620). AURKA inhibition decreased expression of p-AKT (Ser473) in several cell lines (Supplementary Figure S3), whereas, we did not observe significant or consistent effects on p-MEK1/2 (Ser217/221) or p-MTOR (Ser2448). On the other hand, we found that RPS6KB1 phosphorylation at T389 was the most consistently decreased effector, following alisertib treatment across all GI cancer cell models (ESO26, AGS, SNU-601, SNU-1, HCT116, SW480, SW620) (Figure 2A, B). These findings suggest that AURKA may play a role in activation of RPS6KB1, independent of mTOR in KRAS-driven luminal GI cancers. In addition, treatment with alisertib also increased expression levels of common apoptosis markers such as BIM, cleaved caspase 3, and cleaved PARP, with enhanced expression of p21 (Figure 2A, B). As previously reported, p53 protein levels were induced by alisertib treatment in p53 wild-type cells (AGS, SNU-1, HCT116), but not in p53 mutant cells (ESO26, SNU-601, SW480, SW620) (Figure 2A, B), consistent with earlier studies showing that AURKA expression in cancer cells induces degradation of p5317, 28. Taken together, our data demonstrates that targeting AURKA can inhibit RPS6KB1 phosphorylation at T389.
Figure 2. AURKA inhibition downregulates RPS6KB1 protein phosphorylation at Thr-389, and upregulates protein expression of pro-apoptotic mediators in KRAS-driven GI cancer cells.

A, Upper GI cancer cells (ESO26, AGS, SNU-1, SNU-601) were treated with alisertib (0.2 μM) for 1, 2, 3, and 5 days, and cell lysates were subjected to Western blot analysis. The data showed that 1 or 2 days of alisertib treatment led to a decrease in the levels of p-RPS6KB1 (T389) and an increase in p53 (only in p53 WT cell lines), p21, BIM, cleaved PARP, and cleaved caspase 3. B, Colorectal cancer (CRC) cells (HCT116, SW480, SW620) were treated as described in A. Gel loading was normalized for equal β-Actin; representative blots from one of two independent experiments. The relative density of bands is shown under each immunoblot, after normalization to the levels of actin). Quantification of Western blot data is included in Supplementary Figure S1A. Black arrows indicate the molecular weight of proteins.
AURKA induces RPS6KB1 phosphorylation and activity in KRAS-driven GI cancer cells
To confirm the role of AURKA in regulating RPS6KB1 activity, we transiently overexpressed AURKA by using the Tet-One™ inducible expression system and examined the p-RPS6KB1. As shown, our data indicated that following doxycycline treatment, AURKA levels were induced with a notable increase in protein levels of p-RPS6KB1 (T389). The removal of doxycycline restored the protein expression of AURKA and p-RPS6KB1 (T389) back to their baseline levels (Figure 3A). Next, we knocked down endogenous AURKA and evaluated the expression of p-RPS6KB1 in GI cancer cell models (ESO26, AGS, SNU-601, SNU-1, HCT116, SW480, SW620). Western blot analysis indicated that downregulation of AURKA expression decreased the protein expression of p-RPS6KB1 (T389), without changing the total levels of RPS6KB1 protein in several tested cell lines (Figure 3B). Similar to alisertib results, the knockdown of AURKA induced protein expression of p53 (in p53-wild-type cell lines), p21, BIM, cleaved caspase 3, and cleaved PARP (Supplementary Figure S4A). The knockdown of AURKA also suppressed cell growth rates (Figure S4B). These data indicate that AURKA induces RPS6KB1 phosphorylation in KRAS-driven GI cancer cells.
Figure 3. Genetic knockdown of AURKA suppresses phosphorylation of RPS6KB1 in GI cancer cells.

A, AGS and HCT116, cells stably expressing Tet-One™ inducible AURKA, were cultured in the presence or absence of 1 μg/ml doxycycline for 72 hrs. Doxycycline-induced overexpression of AURKA led to an increase in p-RPS6KB1 (T389). B, Western blot analysis showing reduced protein levels of p-RPS6KB1 (T389), following siRNA knockdown of AURKA in GI cancer cells. C, Knockdown of RPS6KB1 using specific siRNA in GI cancer cells (AGS, SNU-601, SNU-1, HCT116, SW480, SW620). Western blot analysis demonstrates an increase in cleaved PARP and cleaved caspase 3, without altering the protein levels of AURKA. Representative blots are from one of three independent experiments with similar results. The relative density of bands are shown under the immunoblot after normalization to the levels of actin. Quantification of Western blot data is included in Supplementary Figure S1 B, C and D. Black arrows indicate the molecular weight of proteins.
Knockdown of RPS6KB1 reduces survival of KRAS-mutant GI cancer cells
To validate the pro-survival role of RPS6KB1 in our GI cancer cell models, we knocked down RPS6KB1 using siRNA approach (ESO26, AGS, SNU-601, SNU-1, HCT116, SW480, SW620). Our data indicated that RPS6KB1 knockdown increased the protein level of pro-apoptotic markers such as cleaved caspase 3 and cleaved PARP (Figure 3C). The CellTiter-Glo Assay indicated that RPS6KB1 knockdown suppressed cancer cell viability (Figure S4C), consistent with our results showing alisertib-or AURKA knockdown-mediated inhibition of RPS6KB1 phosphorylation. Together, these data suggest that RPS6KB1 is a critical pro-survival signaling molecule downstream of AURKA in KRAS-driven GI cancer cells.
AURKA directly interacts with and phosphorylates RPS6KB1 at T389
Recent studies have shown that aberrant overexpression of AURKA in cancer cells is associated with the gain of oncogenic function due to its kinase activity by which the aberrant overexpressed protein directly binds to and phosphorylates several proteins such as HDM2, IKB, p73, and GSK3β 17, 27, 29, 30. Therefore, our next step was to determine if RPS6KB1 is a novel direct substrate for AURKA activity in cancer cells. Using immunofluorescence and confocal microscopy analysis, we observed that AURKA co-localized with RPS6KB1 in SW480 and AGS cells (Figure 4A). To verify if there is an interaction between AURKA and RPS6KB1, we performed protein immunoprecipitation and Western blot analysis of endogenous proteins. The results indicated that AURKA interacts and co-exists in the same protein complex with RPS6KB1. Inhibition of AURKA using alisertib abolished this interaction, suggesting that the binding of AURKA to RPS6KB1 is dependent on AURKA activity (Figure 4B). Additionally, using proximity ligation assay analysis, we observed a positive ligation between AURKA and RPS6KB1 (visualized as red dots), whereas alisertib treatment blocked this interaction in SW480 and AGS cells (Figure 4C). To further validate that inhibition of AURKA can effectively abrogate phosphorylation of RPS6KB1, we performed immunoprecipitation analysis by pulling down the endogenous total serine and threonine phosphorylated proteins in SW480 and AGS cells, followed by Western blot analysis. The results revealed that alisertib treatment completely abrogated the serine and threonine phosphorylation of RPS6KB1, as compared with the control group (Figure 4D). Next, we utilized an in vitro recombinant protein kinase activity assay which demonstrated that AURKA can directly phosphorylate RPS6KB1 at T389, with concentrations as low as 0.05 ng/μl (Figure 4E). This phosphorylation was not observed in our control experiment using glutathione peroxidase 7 (GPX7) recombinant protein. To further corroborate the role of kinase activity of AURKA, we repeated the same experiment and added alisertib (1–500 nM). Indeed, the ability of AURKA to phosphorylate RPS6KB1 was inhibited upon adding alisertib in doses as low as 5 nM (Figure 4F). We confirmed that alisertib was effective in suppressing p-AURKA (T288), a measure of its kinase activity (Figure 4G). Following in vitro kinase assay, we pulled down products of the recombinant protein kinase activity experiment by using antibodies against RPS6KB1. Western blot analysis indicated that AURKA can directly bind to RPS6KB1 (Figure 4H). Interestingly, alisertib treatment reduced the amount of AURKA binding to RPS6KB1 (Figure 4H). Collectively, these data indicated that AURKA can directly interact with and phosphorylate RPS6KB1, uncovering a novel mechanism for activation of RPS6KB1.
Figure 4. AURKA directly binds and phosphorylates RPS6KB1 at Thr-389.

A, Dual immunofluorescence showed co-localization (yellow) of AURKA (green) and RPS6KB1 (red) in SW480 and AGS cells. Z-stacking was displayed on top of each panel for X axis and on the right side of each panel for Y axis. Representative image is from one of three independent experiments. B, Immunoprecipitation (IP) analysis, following the treatment with or without alisertib for 3 days, indicates that AURKA directly binds to and phosphorylates RPS6KB1 (T389) protein. C, Proximity ligation assay (PLA) analysis indicates that AURKA co-localizes and interacts with RPS6KB1 in control groups (red dots, top row). Co-localization of AURKA and RPS6KB1 was reduced in alisertib treatment groups (bottom row). D, Western blot analysis of immunoprecipitation assay products, following pull-down of serine and threonine phosphorylated proteins, showed alisertib treatment downregulated phosphorylation of RPS6KB1 at threonine 389 in SW480 and AGS cells. E, Western blot analysis of in vitro kinase assay products by using increasing-concentrations of recombinant AURKA protein to react with 0.2 pg/pl recombinant RPS6KB1 proteins, or Glutathione peroxidase 7 protein (GPX7) (as a negative control). The data showed increased binding of AURKA with p-RPS6KB1 (T389). F, Western blot analysis of in vitro kinase assay products following increasing concentrations of alisertib. G, In vitro kinase assay for AURKA and RPS6KB1 with or without alisertib (0.5 μM) was performed followed by Western blotting for p-AURKA (T288), AURKA, p-RPS6KB1 (T389), and RPS6KB1. H, Western blot analysis following immunoprecipitation of the in vitro kinase assay products. The data indicate that AURKA directly binds to RPS6KB1.
AURKA is an important downstream effector of mutant KRAS that regulates RPS6KB1
To determine if AURKA-mediated phosphorylation is phenomenon equally present in KRAS-mutant and wild-type cells, we investigated several KRAS wild-type cell models (MKN45, STKM2, and RKO), following stable lentiviral-based expression of mutant KRAS (G12D). As expected, we observed an increase in protein expression levels of p-MEK (Ser217/221) and p-MTOR (Ser2488) in all three cell lines (Figure 5A). Stable expression of mutant KRAS led to an increase in protein expression levels of AURKA and p-RPS6KB1 (T389) in all three cell models (MKN45, STKM2, and RKO) (Figure 5A). Using lentivirus-mediated shRNA knockdown in cell lines with endogenous mutant (SW480, SNU-1) or amplified (ESO26) KRAS, we detected downregulation of AURKA and p-RPS6KB1 (T389) (Figure 5B). Surprisingly, knockdown of endogenous wild-type KRAS (STKM2 and RKO) did not alter the expression of AURKA or p-RPS6KB1 (T389) (Figure 5C). To uncover if AURKA phosphorylates RPS6KB1 only in mutant KRAS cells, we inhibited AURKA by alisertib treatment or genetic knockdown in STKM2 and RKO cells that were engineered to stably express mutant-KRAS, as compared to control. Consistent with our previous data, inhibition or knockdown of AURKA abolished RPS6KB1 phosphorylation that was induced by mutant KRAS-G12D in these cells (Figure 5D, E). Next, we tested whether AURKA knockdown decreases RPS6KB1 phosphorylation in endogenous mutant or wild-type KRAS cells. Western blot data demonstrated that AURKA knockdown in mutant KRAS SUN1 (KRAS-G12D) cells dramatically decreased RPS6KB1 phosphorylation (Supplementary Figure S5), whereas AURKA knockdown in wild-type KRAS cells (MKN45 or STKM2) had minimal effects on decreasing RPS6KB1 phosphorylation levels (Supplementary Figure S5). Collectively, these data demonstrate that mutant KRAS, not wild-type, and AURKA are important for phosphorylation of RPS6KB1, suggesting that the AURKA-RPS6KB1 axis is an active druggable downstream effector pathway in mutant KRAS cancer cells.
Figure 5. AURKA is a downstream effector of mutant KRAS in GI cancer cells.

A, KRAS WT cancer cells (MNK45, STKM2, RKO) were infected with control lentivirus or lentiviral particles expressing KRAS-G12D. Western blot analysis data showed overexpression of KRAS-G12D significantly increased the protein levels of AURKA, p-RPS6KB1 (T389), p-MEK, and p-MTOR. B, KRAS-driven cancer cells (SW480, SNU-1, ESO26) were infected with control lentivirus or lentivirus-mediated expression of shRNA specific to KRAS, Western blot analysis data showed knockdown of KRAS decreased protein expression of AURKA, p-RPS6KB1 (T389), and p-MEK. C, KRAS WT cancer cells (STKM2, RKO) were infected as described in B. Western blot analysis data showed knockdown of KRAS decreased protein expression of p-MEK, without altering expression of AURKA and p-RPS6KB1 (T389). D-E, STKM2 and RKO cells were infected as described in A, then treated with alisertib for 3 days (D) or cells were transfected siAURKA for 48 hrs (E). Western blot analysis data showed downregulation of p-RPS6KB1 (T389). Representative blot is from one of two independent experiments. The relative density of bands are shown under immunoblot after normalization to the levels of actin). Quantification of Western blot data is included in Supplementary Figure S1 E to I. Black arrows indicate the molecular weight of proteins.
Targeting AURKA by alisertib demonstrates therapeutic efficacy in tumor xenograft models of KRAS-driven GI cancers
To confirm that AURKA is a potential therapeutic target in luminal GI cancers with mutant KRAS, we used four tumor xenograft mouse models (SNU-601, SNU-1, HCT116, and SW480). Treatment with alisertib was initiated after tumors reached 150 – 200 mm3 and continued for 28 days. The results demonstrated a significant reduction in tumor volumes in all tested models (p < 0.001, Figure 6A, B), as compared to control group. Our next step was to validate the in vitro findings in the in vivo models. However, all tumors almost vanished at the end of 28 days of treatment. Therefore, we performed an additional experiment using tumor xenografts and limited treatment with alisertib for one week in order to detect the molecular effects of treatment in vivo. Western blot analysis of harvested tumors demonstrated that inhibition of AURKA by alisertib led to decreased expression of p-RPS6KB1 (T389) in tumor xenografts, confirming the in vitro data (Figure 6C). Immunohistochemical staining data showed that alisertib-induced tumor regression was associated with decreased proliferation and increased apoptosis as shown by Ki-67 and cleaved caspase 3 staining in SW480 and SNU-601 xenografts (Figure 6D, E). Taken together, our results indicate that AURKA-RPS6KB1 is a druggable signaling axis downstream of mutant KRAS luminal GI cancers. These findings could have a broader therapeutic potential for treating KRAS-driven malignancies, including those resistant to currently available therapeutic options.
Figure 6. Alisertib effectively induces regression of tumor xenografts.

A, B, SNU-601, SNU-1, HCT116, and SW480 subcutaneous xenograft tumors (n=8, 16 tumors) (150-200 mm3 in size) were treated with alisertib (40 mg/kg, 5 times a week) for 4 weeks, measuring tumor size every 3 days, error bars indicate standard deviation (SD), *p < 0.05; **p < 0.01; ***p < 0.001. Data indicated that alisertib has significant anti-tumor activity. C, SNU-601 and SW480 subcutaneous xenograft tumors were treated with alisertib (40 mg/kg/day) for 7 days. Tumor tissue lysates were subjected to Western blot analysis of the indicated proteins. Data showed the significant decrease in expression of p-RPS6KB1 (T389). D, After 7 days of treatment, tumors were resected and immunohistochemical staining of cleaved caspase-3 (apoptosis) and Ki-67 (proliferation) was performed. E, Quantification data for cleaved caspase-3 and Ki-67 levels, error bars indicate standard deviation (SD), *p < 0.05; **p < 0.01. Black arrows indicate the molecular weight of p-AURKA or AURKA at 48 KD.
Aberrant overexpression of AURKA in gastrointestinal cancers
Using immunohistochemistry on tissue microarrays, we evaluated the protein expression of AURKA in human normal, adenoma, and cancer colonic tissues. IHC data showed significantly stronger expression of AURKA in human adenomas (P=0.0003) and cancer tissues (p=0.0001) than in human normal colonic tissues (Figure 7A, B). We did not detect significant differences in the pattern of AURKA expression in MSS versus MSI colon cancers. However, there was a trend suggesting associations between AURKA expression levels with tumor stages (p=0.19) and KRAS mutation (p=0.16), although not reaching statistical significance possibly due to small sample size. A summary of IHC results and molecular data of normal colon, colon adenoma and tumors is provided in Supplementary Table S1 and S3. Of note, these results are concordant with our earlier analyses of IHC that demonstrated strong diffuse cytosolic overexpression of AURKA in gastric and esophageal adenocarcinoma tissue samples 27, 31. Altogether, these findings suggest that AURKA gain of novel oncogenic functions is possibly related to its aberrant overexpression in cancer cells.
Figure 7. Immunohistochemistry analysis of AURKA on human tissue microarray.

A, Immunohistochemistry analysis of AURKA expression and localization in representative samples from tissue microarrays containing 189 paraffin-embedded colon tissue samples. B, The quantification data of cytoplasmic AURKA composite expression scores (CES) in tissue microarrays. Adenoma and cancer tissues showed significantly higher levels of cytoplasmic AURKA expression, as compared to normal tissues (p < 0.0001). The box extends from the 25th to 75th percentiles. The line in the middle of the box is plotted at the median. Whiskers are shown down to the minimum and up to the maximum value.
Discussion
In this study, we investigated the functional and mechanistic roles of AURKA and its targeting in KRAS-driven luminal gastrointestinal cancers. Mutations in KRAS are frequent early events in colorectal cancers that have been identified in approximately 30% of adenomas32. While gastric and esophageal adenocarcinomas have lower frequencies of mutant KRAS, amplifications and non-mutational activation of oncogenic KRAS signatures have been frequently described in these cancers4, 33. Mutant KRAS can activate a plethora of oncogenic signaling pathways that include PI3K/AKT, ERK1/2, MEK1/2, and mTOR that promote cancer cell survival leading to poor therapeutic outcomes34,35, 36. Based on preclinical and clinical studies, mutant KRAS poses a significant therapeutic challenge with significant resistance to first-line therapeutic agents 37. In addition, GI tumors with mutant KRAS are resistant to treatment with targeted agent therapies, such as EGFR inhibitors (cetuximab and panitumumab) 38, MEK inhibitor (CI-1040) 39, and BRAF inhibitor (vemurafenib) 40,41. The development of therapeutic approaches that can target oncogenic KRAS has been a challenging task with limited success to date. Therefore, identification of an innovative approach to target oncogenic KRAS critical effector signaling pathways can be a promising alternative treatment strategy.
We have shown that pharmacological inhibition of AURKA by alisertib or its knockdown can significantly reduce viability of luminal GI cancer cells with KRAS activation, in vitro and in vivo. A recent study has shown that AURKA is possibly an important KRAS target in lung cancer 42. Although some data suggest that AURKA can interact with RAS-MAPK signaling pathways in colorectal cancer 43, we did not observe consistent changes in ERK1/2 or MEK1/2, following pharmacologic inhibition of AURKA by alisertib or knockdown in luminal GI cancer models. These data suggest that the observed anti-cancer effects and biological outcomes possibly involve additional key pro-survival signaling pathways downstream of AURKA and KRAS in our cancer cell models.
We found that AURKA mediates phosphorylation and activation of RPS6KB1 in KRAS-driven cancer cells. An earlier study suggested that AURKA expression is associated with RPS6KB1 phosphorylation44. Therefore, we aimed to identify the missing mechanistic link between AURKA and RPS6KB1, a reported mammalian target of mTOR 45. Herein, we show that phosphorylation of RPS6KB1 was consistently and significantly downregulated following knockdown or inhibition of AURKA in KRAS-driven luminal GI cancer cell models, without notable changes in the levels of phospho-mTOR. Of note, an earlier study has shown that AURKA can directly activate EIF4E, a downstream target of mTOR, independent of mTOR 22. Using immunofluorescent staining, we showed co-localization of AURKA and RPS6KB1 in our cell models. In addition, co-immunoprecipitation, proximity ligation, and in vitro kinase assays indicated that AURKA co-localizes, binds, and directly phosphorylates RPS6KB1, supporting the notion that RPS6KB1 is a novel substrate for AURKA. We could not detect similar effects of AURKA on RPS6KB1 phosphorylation in KRAS wild-type cells. However, expression of exogenous mutant KRASG12D induced expression of AURKA and phosphorylation of RPS6KB1 in cancer cells with wild-type KRAS. Taken together, the data suggests RPS6KB1 as a novel substrate for AURKA in KRAS-driven GI cancer cells, possibly a unique phenomenon in KRAS-driven cancer cells that express high levels of endogenous AURKA.
Using IHC on tissue microarrays, we previously showed high levels of AURKA in gastric and esophageal adenocarcinoma cancer cells31. In this study, we observed similar findings in colon cancer tissue microarrays. A study by Tseng, et al revealed that AURKA and mutant RASv12 are overexpressed in bladder and colon cancers46. We detected a strong trend towards the association between AURKA and KRAS mutation, although not attaining statistical significance, possibly due to the relatively small sample size with this information in our colon cancer cohort. Studies in ovarian carcinoma and esophageal squamous cancers have suggested that AURKA overexpression occurs in late histological stages with poor clinical outcome47, 48. However, we detected aberrant overexpression of AURKA in premalignant colon adenomas, similar to a previous study, showing AURKA overexpression in pre-malignant lesions in gastric cancer mouse models27. Based on these data and earlier observations, the observed overexpression of AURKA in luminal gastrointestinal cancers could be an important early tumorigenic event.
Using in vitro and in vivo, our results demonstrated that inhibition of AURKA, using alisertib, in KRAS-driven cells inhibited cancer cell growth and significantly reduced tumor volumes. The use of alisertib in tumor xenograft models suppressed the levels of p-RPS6KB1, similar to in vitro findings. An earlier study has shown that RPS6KB1 activation can mediate resistance to selumetinib (MEK inhibitor) in colorectal cancer cells49. Our findings suggest that inhibition of AURKA may restore efficacy to selumetinib; however, given the complexity of signaling in cancer cells, additional investigations are needed. It is also important to note that AURKA can regulate a plethora of oncogenic pathways in cancer cells such as β-catenin30, HDM217, NF-κB27, EIF4E22, 50, and MYC51. Although these recent studies along with our results suggest that targeting AURKA could be a therapeutic strategy to target multiple signaling pathway, it is important to note that treatment with single agents often fail in clinical practice. Future studies are needed to test and optimize combination therapies that include AURKA inhibitor. To this end, we hope that our data will be useful when developing strategies for the treatment of KRAS mutant tumors.
In conclusion, our results identified a previously unknown mechanism by which AURKA can directly bind to and phosphorylate RPS6KB1 in KRAS-mutant cancer cells. Developing and testing therapeutic approaches that include targeting AURKA may be effective for treating KRAS-driven GI cancers.
Supplementary Material
Acknowledgment
We would like to thank the members of the Flow Cytometry Shared Resources and Bioinformatics and Biostatistics at Sylvester Comprehensive Cancer Center at the University of Miami Miller School of Medicine, for their technical support.
Grant Support: This study was supported by grants from the U.S. National Institutes of Health (R01CA131225), the U.S. Department of Veterans Affairs (1IK6BX003787 and I01BX001179), and University of Miami, Sylvester Comprehensive Cancer Center (SCCC) intramural funds. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, Department of Veterans Affairs, or University of Miami.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have declared no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takashima A, Faller DV. Targeting the RAS oncogene. Expert Opin Ther Targets 2013;17:507–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrington AK, Heinrich EL, Lee W, et al. Prognostic and predictive roles of KRAS mutation in colorectal cancer. Int J Mol Sci 2012;13:12153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao W, Chan TL, Chu KM, et al. Mutations of BRAF and KRAS in gastric cancer and their association with microsatellite instability. Int J Cancer 2004;108:167–9. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong GS, Zhou J, Liu JB, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prenen H, Tejpar S, Van Cutsem E. New strategies for treatment of KRAS mutant metastatic colorectal cancer. Clin Cancer Res 2010;16:2921–6. [DOI] [PubMed] [Google Scholar]
- 7.Siddiqui AD, Piperdi B. KRAS mutation in colon cancer: a marker of resistance to EGFR-I therapy. Ann Surg Oncol 2010;17:1168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis 2015;2:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol 2011;43:47–59. [DOI] [PubMed] [Google Scholar]
- 10.Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 1999;253:100–9. [DOI] [PubMed] [Google Scholar]
- 11.Tavares MR, Pavan IC, Amaral CL, et al. The S6K protein family in health and disease. Life Sci 2015;131:1–10. [DOI] [PubMed] [Google Scholar]
- 12.Xiao L, Wang YC, Li WS, et al. The role of mTOR and phospho-RPS6KB1 in pathogenesis and progression of gastric carcinomas: an immunohistochemical study on tissue microarray. J Exp Clin Cancer Res 2009;28:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katsha A, Belkhiri A, Goff L, et al. Aurora kinase A in gastrointestinal cancers: time to target. Mol Cancer 2015;14:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 1998;17:3052–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang SB, Zhou XB, Zhu HX, et al. Amplification and overexpression of Aurora-A in esophageal squamous cell carcinoma. Oncol Rep 2007;17:1083–8. [PubMed] [Google Scholar]
- 16.Koh HM, Jang BG, Hyun CL, et al. Aurora Kinase A is a Prognostic Marker in Colorectal Adenocarcinoma. J Pathol Transl Med 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sehdev V, Katsha A, Arras J, et al. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clin Cancer Res 2014;20:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vilgelm AE, Pawlikowski JS, Liu Y, et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 2015;75:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng F, Yue C, Li G, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun 2016;7:10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnepp RW, Khurana P, Attiyeh EF, et al. A LIN28B-RAN-AURKA Signaling Network Promotes Neuroblastoma Tumorigenesis. Cancer Cell 2015;28:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S, He S, Zhou X, et al. Suppression of Aurora-A oncogenic potential by c-Myc downregulation. Exp Mol Med 2010;42:759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katsha A, Wang L, Arras J, et al. Activation of EIF4E by Aurora kinase A depicts a novel druggable axis in everolimus resistant cancer cells. Clin Cancer Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sehdev V, Katsha A, Ecsedy J, et al. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer 2013;119:904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis SL, Robertson KM, Pitts TM, et al. Combined inhibition of MEK and Aurora A kinase in KRAS/PIK3CA double-mutant colorectal cancer models. Front Pharmacol 2015;6:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DuBois SG, Marachelian A, Fox E, et al. Phase I Study of the Aurora A Kinase Inhibitor Alisertib in Combination With Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma: A NANT (New Approaches to Neuroblastoma Therapy) Trial. J Clin Oncol 2016;34:1368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol 2014;32:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsha A, Soutto M, Sehdev V, et al. Aurora kinase A promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology 2013;145:1312–22 e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Arras J, Katsha A, et al. Cisplatin-resistant cancer cells are sensitive to Aurora kinase A inhibition by alisertib. Mol Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katayama H, Wang J, Treekitkarnmongkol W, et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell 2012;21:196–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene 2009;28:866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dar AA, Zaika A, Piazuelo MB, et al. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer 2008;112:1688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zauber P, Marotta S, Sabbath-Solitare M. KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int J Mol Epidemiol Genet 2013;4:1–10. [PMC free article] [PubMed] [Google Scholar]
- 33.Peng D, Guo Y, Chen H, et al. Integrated molecular analysis reveals complex interactions between genomic and epigenomic alterations in esophageal adenocarcinomas. Sci Rep 2017;7:40729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coulson R Molecular Profiling in Resectable Colorectal Liver Metastases: The Role of KRAS Mutation Status in Assessing Prognosis in the Preoperative Setting. J Adv Pract Oncol 2015;6:470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuracha MR, Thomas P, Loggie BW, et al. Bilateral blockade of MEK- and PI3K-mediated pathways downstream of mutant KRAS as a treatment approach for peritoneal mucinous malignancies. PLoS One 2017;12:e0179510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCormick F KRAS as a Therapeutic Target. Clin Cancer Res 2015;21:1797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russo M, Di Nicolantonio F, Bardelli A. Climbing RAS, the everest of oncogenes. Cancer Discov 2014;4:19–21. [DOI] [PubMed] [Google Scholar]
- 38.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091–6. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Van Becelaere K, Jiang P, et al. A role for K-ras in conferring resistance to the MEK inhibitor, CI- 1040. Neoplasia 2005;7:336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sclafani F, Gullo G, Sheahan K, et al. BRAF mutations in melanoma and colorectal cancer: a single oncogenic mutation with different tumour phenotypes and clinical implications. Crit Rev Oncol Hematol 2013;87:55–68. [DOI] [PubMed] [Google Scholar]
- 41.Danysh BP, Rieger EY, Sinha DK, et al. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget 2016;7:30907–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dos Santos EO, Carneiro-Lobo TC, Aoki MN, et al. Aurora kinase targeting in lung cancer reduces KRAS- induced transformation. Mol Cancer 2016;15:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobsen A, Bosch LJW, Martens-de Kemp SR, et al. Aurora kinase A (AURKA) interaction with Wnt and Ras-MAPK signalling pathways in colorectal cancer. Sci Rep 2018;8:7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou Z, Yuan Z, Zhang Q, et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012;8:1798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amaral CL, Freitas LB, Tamura RE, et al. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016;16:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tseng YS, Lee JC, Huang CY, et al. Aurora-A overexpression enhances cell-aggregation of Ha-ras transformants through the MEK/ERK signaling pathway. BMC Cancer 2009;9:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mignogna C, Staropoli N, Botta C, et al. Aurora Kinase A expression predicts platinum-resistance and adverse outcome in high-grade serous ovarian carcinoma patients. J Ovarian Res 2016;9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamotsu K, Okumura H, Uchikado Y, et al. Correlation of Aurora-A expression with the effect of chemoradiation therapy on esophageal squamous cell carcinoma. BMC Cancer 2015;15:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grasso S, Tristante E, Saceda M, et al. Resistance to Selumetinib (AZD6244) in colorectal cancer cell lines is mediated by RPS6KB1 and RPS6 activation. Neoplasia 2014;16:845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Arras J, Katsha A, et al. Cisplatin-resistant cancer cells are sensitive to Aurora kinase A inhibition by alisertib. Mol Oncol 2017;11:981–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silva A, Wang J, Lomahan S, et al. Aurora kinase A is a possible target of OSU03012 to destabilize MYC family proteins. Oncol Rep 2014;32:901–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.