

Abstract
This review focuses on the pathways that regulate lysosome biogenesis and that are implicated in numerous degenerative storage diseases, including lysosomal storage disorders and late-onset neurodegenerative diseases. Lysosomal proteins are synthesized in the endoplasmic reticulum and trafficked to the endolysosomal system through the secretory route. Several receptors have been characterized that execute post-Golgi trafficking of lysosomal proteins. Some of them recognize their cargo proteins based on specific amino acid signatures, others based on a particular glycan modification that is exclusively found on lysosomal proteins. Nearly all receptors serving lysosome biogenesis are under the transcriptional control of transcription factor EB (TFEB), a master regulator of the lysosomal system. TFEB coordinates the expression of lysosomal hydrolases, lysosomal membrane proteins, and autophagy proteins in response to pathways sensing lysosomal stress and the nutritional conditions of the cell among other stimuli. TFEB is primed for activation in lysosomal storage disorders but surprisingly its function is impaired in some late-onset neurodegenerative storage diseases like Alzheimer’s and Parkinson’s, due to specific detrimental interactions that limit TFEB expression or activation. Thus, disrupted TFEB function presumably plays a role in the pathogenesis of these diseases. Multiple studies in animal models of degenerative storage diseases have shown that exogenous expression of TFEB and pharmacological activation of endogenous TFEB attenuate disease phenotypes. These results highlight TFEB-mediated enhancement of lysosomal biogenesis and function as a candidate strategy to counteract the progression of these diseases.
Keywords: lysosomal biogenesis, sorting receptors, autophagy, lysosomal storage disorders, TFEB, neurodegenerative disease
Graphical abstract
This review discusses how the transcription factor EB (TFEB) is implicated in lysosomal biogenesis and degenerative storage diseases. TFEB coordinates the expression of proteins participating in the autophagy-lysosome pathway, including lysosomal enzymes and their transporters. TFEB is primed for activation in lysosomal storage disorders due to lysosomal stress, but TFEB function is impaired in some late-onset neurodegenerative storage diseases due to specific detrimental interactions that limit TFEB expression or activation. Increased expression or activation of TFEB results in the enhancement of the autophagy-lysosome pathway and ameliorates disease phenotypes in models of lysosomal storage disorders and other degenerative storage diseases.
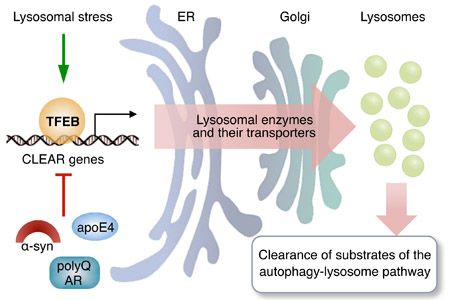
Introduction
Antonie Van Leeuwenhoek’s description of plant vacuoles as empty sacs filling up to 90% of the plant cell dates back to 1676, preceding by ~300 years Christian De Duve’s discovery of lysosomes as lytic and degradative organelles (De Duve et al., 1955). It is now appreciated that the lysosome hosts more than 60 soluble lysosomal hydrolases and accessory proteins and over 120 lysosomal membrane proteins and transitory residents (Braulke and Bonifacino, 2009). The characterization of the function of these proteins has caused a paradigm shift in study and description of the lysosome, which is now understood as a key cellular metabolic hub with multiple roles in processes as different as nutrient sensing, gene regulation, secretion, plasma membrane repair, metal ion homeostasis, cholesterol transport, and immune response. Interestingly, as many of these processes are similar in different phyla, it is not surprising that many of their underlying factors have been conserved over the course of evolution. Mutations in genes encoding lysosomal enzymes or proteins participating in their maturation and trafficking can lead to lysosomal storage disorders, of which more than 50 genetically distinct types have been classified to date. While individually rare, lysosomal storage disorders as a group have a relatively high incidence in the general population—more than 1:5000 live births is affected by one of the lysosomal storage disorders (Boustany, 2013). The clinical manifestations of lysosomal storage disorders vary widely, even though neurological symptoms and progressive neurodegeneration are common features. At the subcellular level, lysosomal storage disorders are characterized by the aberrant intralysosomal storage of metabolites that cannot be properly eliminated due to defects in one or multiple catabolic pathways (Walkley, 2009). In this Review, we will focus on lysosomal biogenesis from the point of view of the regulatory pathways and signals that contribute to the synthesis and trafficking of lysosomal proteins. As illustrated by numerous examples, the characterization of these pathways is providing powerful instruments to better understand—and potentially counteract—various human diseases that are rooted in, or worsened by, impaired lysosomal biogenesis or function.
Transcriptional control of lysosomal pathways
Under microscopic examination, lysosomal storage has the appearance of an expansion of the lysosomal compartment. This expansion could be interpreted not only as the consequence of the impaired capacity of the cell to eliminate its catabolic substrates but also as the result of an increased lysosomal biogenesis. Early findings indicated an increased activity of several lysosomal enzymes in multiple lysosomal storage disorders; these findings supported the notion that the cell could increase lysosomal biogenesis as a compensatory mechanism to counteract lysosomal stress (Karageorgos et al., 1997). Additional observations indicated that lysosomal storage stress and the subsequent lysosomal biogenesis could be recapitulated experimentally by providing sucrose to cultured cells. In some cultured cell lines, sucrose is indeed internalized into the lysosomes but cannot be eliminated due to the absence of the invertase enzyme. The progressive intralysosomal accumulation of sucrose results in the formation of enlarged lysosomes (“sucrosomes”) that offer an experimental model of lysosomal storage (Cohn and Ehrenreich, 1969; Karageorgos et al., 1997).
The hypothesis that the activity of the lysosomal system is coordinated as an adaptive response to the cell’s needs was demonstrated by the identification of transcription factor EB (TFEB) as a master transcriptional regulator of lysosomal biogenesis and function (Sardiello et al., 2009). Many genes contributing to the functioning of the lysosomal system have, in their promoter regions, one or multiple binding sites for TFEB (named as ‘coordinated lysosomal expression and regulation’ or CLEAR motif); genes with CLEAR sequences in their promoters are induced by TFEB activation or increased expression (Sardiello et al., 2009). The sucrosome experimental model led to the identification of TFEB cytosol-to-nucleus translocation as the mode of activation of TFEB. It was indeed noticed that TFEB is localized in the cytosol in the majority of cells cultured in normal conditions; however, supplementation of sucrose neatly promoted nuclear translocation of TFEB, consequently increasing both the binding of TFEB to the promoters of genes of the lysosomal system and the expression levels of these genes. Similarly, cells from models of lysosomal storage disorders have increased TFEB nuclear translocation (Sardiello et al., 2009).
These observations triggered a series of studies that documented a number of kinases and phosphatases able to modulate TFEB subcellular distribution by direct modification of several TFEB amino acid residues. Among them, the atypical serine/threonine kinase mechanistic target of rapamycin complex 1 (mTORC1) phosphorylates TFEB on Ser211, which triggers TFEB cytosolic sequestration by 14–3-3 proteins (Martina et al., 2012; Roczniak-Ferguson et al., 2012); mTORC1 also phosphorylates TFEB on Ser142 (Settembre et al., 2012) and Ser122 (Vega-Rubin-de-Celis et al., 2017); the serine/threonine kinase mitogen-activated protein kinase 1 (MAPK1 or ERK) phosphorylates TFEB on Ser142 (Settembre et al., 2011); the serine/threonine kinase protein kinase B (PKB or Akt) phosphorylates TFEB on Ser467 (Palmieri et al., 2017a; Palmieri et al., 2017b); the serine/threonine kinase glycogen synthase kinase 3β (GSK3β) phosphorylates TFEB on Ser134 and Ser138 (Li et al., 2016); and the serine/threonine kinase mitogen-activated protein kinase kinase kinase kinase 3 (MAP4K3) phosphorylates TFEB on Ser3 (Hsu et al., 2018) (Figure 1). These phosphorylation events all limit TFEB nuclear translocation and indeed, chemical inhibition of any of these kinases promotes TFEB activation. In response to various cellular stimuli, TFEB is dephosphorylated and activated by the phosphatases calcineurin (Medina et al., 2015) and protein phosphatase 2 (PP2A) (Chen et al., 2017). These regulatory sites are part of evolutionarily constrained regions in the protein sequence of TFEB (Chang et al., 2018).
Figure 1.
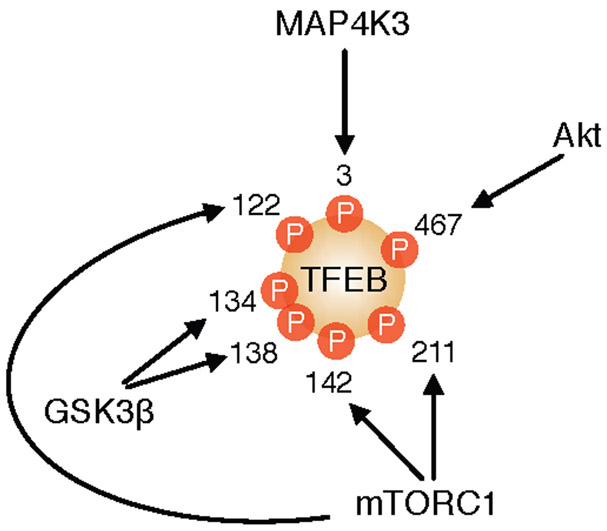
Phosphorylation of TFEB by serine/threonine protein kinases. mTORC1, Akt, GSK3β and MAP4K3 phosphorylate TFEB at various serine residues. The positions of the phosphorylated residues refer to the human TFEB protein.
Active TFEB promotes the transcription of genes of the autophagy-lysosome pathway and globally regulates phenomena such as lysosomal biogenesis (Sardiello et al., 2009), autophagy (Settembre et al., 2011), lysosomal proteostasis (Song et al., 2013), lysosomal positioning (Willett et al., 2017) and lysosomal exocytosis (Medina et al., 2011). More recently, cross-talks with pathways underlying ER stress (Martina et al., 2016), mitochondrial biogenesis (Mansueto et al., 2017), and peroxisomal biogenesis (Eun et al., 2018) have been described. The TFEB homologs TFE3 and MITF, which belong to the same subfamily of MiT/TFE transcription factors, also contribute to the transcriptional regulation of the autophagy-lysosome pathway (Martina et al., 2014) and are similarly regulated by mTORC1 (Martina and Puertollano, 2013; Roczniak-Ferguson et al., 2012) and Akt (Palmieri et al., 2017a). As detailed in the following paragraphs, TFEB coordinates the expression of various lysosomal proteins with that of their transporters, and is part of the pathogenic cascade in several neurodegenerative storage diseases.
Lysosomal sorting signals and sorting receptors
Soluble and membrane-bound vacuolar/lysosomal proteins have sorting signals that are recognized by sorting receptors. These receptors also contain signals, which allow them to be recognized by the trafficking machinery (clathrins and adaptor proteins) for their proper delivery to the endolysosomal system through various trafficking routes.
The sorting signals of soluble proteins can be either folded polypeptide sequences displayed on the protein surface or specific glycan modifications as detailed below. In plants, a sequence-specific vacuolar sorting signal (ssVSS) present at the protein N-terminus consists of the conserved sequence Asn-Pro-Ile-Arg (NPIR motif) (Holwerda et al., 1992; Koide et al., 1997; Matsuoka et al., 1990; Matsuoka et al., 1995). In the yeast Saccharomyces cerevisiae, an N-terminal QXXΦ motif (where X is any amino acid and Φ is a bulky hydrophobic residue) is recognized by vacuolar protein sorting 10 protein (Vps10p), a type I membrane-spanning sorting receptor (Valls et al., 1990). However, alternative signals might be present in the yeast as not all vacuolar proteins contain the QXXΦ motif (Westphal et al., 1996).
Compared to lower phyla/kingdoms, vertebrates have fewer well-characterized examples of sorting receptors with specificity to determined peptide sequences. Sortilin and LIMP-2 are sorting receptors for lysosomal soluble proteins that are both known to interact with their soluble cargo using short stretches of amino acids, though a consensus motif is yet to be discovered. As discussed below, other sorting receptors present in vertebrates interact with their cargo proteins through specific glycan modifications. Regardless of their mode of action, most mammalian receptors share similar structural features, including a single transmembrane domain, a large luminal domain for interaction with their cargo, and a C-terminus containing signals necessary for their own trafficking.
Sortilin.
A quintessential example of sorting receptors is sortilin (SORT1). Sortilin belongs to the Vps10p domain receptor family, which in mammals includes other four members (SorCS1, SorCS2, SorCS3 and SorLA) (Hermey et al., 1999; Jacobsen et al., 1996; Rezgaoui et al., 2001). About 90% of the total pool of sortilin receptors is found at the Golgi system and Golgi-derived vesicles, whereas only a minor fraction is expressed at the cell surface, suggesting Golgi-endosome sorting. Sortilin and some of its homologs contain the classic tyrosine- and dileucine-based consensus motifs at their C-terminus, which guide intracellular sorting and internalization via interactions with cytosolic adaptors such as AP-1 and GGA2 (discussed below). Sortilin has been shown to be involved in the lysosomal sorting of GM2 ganglioside activator, prosaposin (the precursor of sphingolipid activator proteins) (Hassan et al., 2004; Lefrancois et al., 2003; Zeng et al., 2009), acid sphingomyelinase (Ni and Morales, 2006), and cathepsins D and H (Canuel et al., 2008), demonstrating its involvement in lysosomal biogenesis pathways. In experimental sucrosome models, expression of SORT1 was upregulated along with that of the genes coding for several sortilin cargo proteins; direct overexpression of TFEB also increased the expression of SORT1 and related cargo proteins (Sardiello et al., 2009; Song et al., 2013).
Unexpectedly, sortilin-deficient mice do not show any obvious clinical signs of lysosomal pathology, suggesting compensation by alternative mechanisms of sorting (Zeng et al., 2009). This could be explained by redundancies between sortilin and other receptors of the same protein family, of which SorLA is one of the most important contenders for a role in lysosomal cargo transportation. While no human disease has been associated with loss-of-function mutations in sortilin protein, a non-coding single-nucleotide polymorphism in an enhancer controlling SORT1 expression has been causally associated with plasma low-density lipoprotein cholesterol and myocardial infarction (OMIM: 613589) (Musunuru et al., 2010). Molecular studies have found that this polymorphism creates a C/EBP (CCAAT/enhancer binding protein) transcription factor binding site that increases SORT1 expression in the liver, finally resulting in impaired homeostasis of total plasma cholesterol and low-density lipoprotein cholesterol (Musunuru et al., 2010). These data illustrate a possible function of sortilin in the control of hepatic lipoprotein metabolism that goes beyond its role as a sorting receptor for lysosomal proteins (Carlo et al., 2014).
Interestingly, Vps10p sorting receptors are substrates for presenilin-dependent γ-secretase cleavage (Nyborg et al., 2006). As both SorLA and presenilin have been implicated in the pathogenesis of Alzheimer’s disease (Andersen et al., 2005; Wolfe et al., 1999), these findings indicate the existence of connections between Alzheimer’s disease pathogenesis and lysosomal biogenesis, a theme that will be explored further in this Review.
LIMP-2.
Lysosomal integral membrane protein 2 (LIMP-2) is another mammalian lysosomal enzyme sorting receptor that interacts with its cargo based on proteinaceous signals (Blanz et al., 2015). LIMP-2 serves as a receptor for the transport of β-glucocerebrosidase (GCase) from the endoplasmic reticulum throughout endolysosomal compartments (Reczek et al., 2007). Loss-of-function mutations in the GCase gene, glucosylceramidase beta (GBA), cause Gaucher disease (GD), the most prevalent lysosomal storage disorder. The binding between LIMP-2 and GCase depends on the helical arrangement and the amphipathic nature of critical residues (152 to 167) in the coiled-coil domain of LIMP-2 (Blanz et al., 2010). The binding between LIMP-2 and GCase also depends on pH, with a single histidine residue of LIMP-2 (His171) functioning as a pH sensor (Zachos et al., 2012). The neutral environment of the ER lumen allows association of GCase with LIMP-2, whereas the acidic pH in endolysosomes triggers their dissociation (Reczek et al., 2007). Similar to sortilin, LIMP-2 is targeted to endolysosomes using a dileucine motif contained in its C-terminal region (Sandoval et al., 1994). The levels of both LIMP-2 and GCase are regulated by TFEB (Sardiello et al., 2009; Song et al., 2013). Recent structural studies have shown that LIMP-2 is also modified by the post-translational addition of mannose 6-phosphate (M6P) (Zhao et al., 2014). By virtue of this modification, LIMP-2 interacts with mannose-6-phosphate receptors (M6PR, discussed below), suggesting the presence of a ternary complex between LIMP-2, GCase, and M6PR (Zhao et al., 2014).
Deficiency of LIMP-2 in mice leads to a severe reduction of GCase activity in various tissues, with the majority of the enzyme being secreted to the extracellular compartment (Reczek et al., 2007). Providing exogenous LIMP-2 to LIMP-2-deficient fibroblasts reconstituted both GCase activity and trafficking (Reczek et al., 2007). Mutations in SCARB2 (the gene that encodes LIMP-2) cause a rare form of progressive myoclonus epilepsy (PME) often associated with action myoclonus-renal failure syndrome (AMRF; OMIM: 254900). PME and AMRF are part of the phenotypic spectrum displayed by GD patients (Berkovic et al., 2008; Dibbens et al., 2011; Hopfner et al., 2011; Velayati et al., 2011). Similar to most pathogenic mutations affecting lysosomal proteins, there is not a clear genotype-phenotype correlation between mutations in SCARB2 and the associated clinical manifestations. In particular, it is not clear why some patients develop PME or renal failure, whereas others show mild hearing impairment or neuropathy. LIMP-2-deficient mice show signs of ureteropelvic junction obstruction, deafness, and peripheral neuropathy, and cell lines derived from these mice have clear signs of GCase missorting and secretion (Gamp et al., 2003). The functional relationship between LIMP-2 and GCase is further underlined by the finding that SCARB2 can act as a modifier of Gaucher disease (Velayati et al., 2011). In an examined family, two siblings were diagnosed with GD, with the additional diagnosis of myoclonic epilepsy only for one of the two. Sequencing of SCARB2 genomic and cDNA sequences identified a heterozygous, maternally inherited novel mutation in the sibling with GD and epilepsy that was responsible for diminished GCase lysosomal transport and activity (Velayati et al., 2011). This suggests the potential usefulness of investigating the effects of modifiers such as protein transporters or activators in cases in which there is discordance between genotype and phenotype.
Additional consequences of reduced GCase activity in LIMP-2 deficient mice are lipid storage and disturbed autophagic-lysosomal function (Rothaug et al., 2014). This results in α-synuclein accumulation, which in turn causes neurotoxicity in dopaminergic neurons. Reintroducing LIMP-2 by heterologous overexpression accelerated clearance of α-synuclein, indicating modulation of GCase activity as a potential strategy for treating synucleopathies (Rothaug et al., 2014).
Transport mediated by the mannose-6-phosphate receptors
The trafficking of many lysosomal soluble proteins requires the intervention of a set of highly specialized protein activities along the trafficking route, including specific post-translational modifications, recognition of unique tags, and cargo packaging and delivery. Once synthesized and N-glycosylated in the endoplasmic reticulum, lysosomal soluble proteins are transported to the Golgi complex where most of them are modified to expose M6P residues. The M6P-tagged proteins are consequently recognized by the M6PR and delivered from the trans-Golgi network to the endolysosomal system. The importance of these processes for the correct biogenesis and function of lysosomes is underlined by the existence of various human diseases caused by mutations in the genes that are involved in these trafficking steps.
M6P modification.
The series of posttranslational modifications that ensure the correct sorting of most lysosomal soluble proteins to the lysosome begin at early stages of their synthesis. Native lysosomal proteins are co-translationally glycosylated by the oligosaccharyltransferase (OST) onto selected asparagine residues belonging to the consensus sequence Asn-X-Ser/Thr (Kornfeld and Kornfeld, 1985). These N-linked oligosaccharides are further modified at the Golgi compartment by the addition of carbohydrate groups of higher complexity. The modification of high-mannose type oligosaccharides to M6P is catalyzed by the cooperative function of two enzymes: the UDP-N-acetylglucosamine 1-phosphotransferase (GlcNAc-1-phosphotransferase) and the N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase, also known as the uncovering enzyme (UCE) (Do et al., 2002). This last modification is essential for the sorting of lysosomal soluble proteins from the Golgi compartment to the endolysosomal system.
The GlcNAc-1-phosphotransferase catalyzes the addition of GlcNAc-P-mannose diester onto the high-mannose type oligosaccharides. It is a transmembrane protein composed of three subunits (α, β, γ) which appear twice, forming the hexamer α2β2γ2. The γ subunit is encoded by the GNPTG gene, whereas the α and β subunits are encoded as a single precursor by the GNPTAB gene. The release of the individual α and β subunits requires proteolytic processing between Lys928 and Asp929 by the site-1 protease (Marschner et al., 2011). The α subunit carries both the catalytic and the protein binding domains, but alone it is unable to reconstitute the GlcNAc-1-phosphotransferase activity. In fact, the β and γ subunits are required as they appear to be important for the structural maintenance of this multimeric complex (Kudo and Canfield, 2006) and for the regulation of its catalytic activity (Qian et al., 2010), respectively.
The recognition of the high mannose lysosomal proteins by the GlcNAc-1-phosphotransferase requires a tridimensional determinant composed of amino acid residues that are far from each other in the folded protein. Studies involving chimeric products of the lysosomal protease cathepsin D and its non-lysosomal homolog pepsinogen, together with mutational studies of cathepsin L, identified a group of lysine residues organized in a precise tridimensional configuration as a crucial determinant for the selective recognition of these proteins as destined to the lysosome (Baranski et al., 1990; Cuozzo and Sahagian, 1994).
The UCE catalyzes the uncovering of the M6P signal by removing the GlcNAc from the GlcNAc-P-mannose diester. The UCE is a tetrameric transmembrane protein, coded as a precursor by the NAGPA gene. To be active, the UCE precursor needs the removal of a propeptide of 24 amino acids catalyzed by furin in the trans-Golgi network (Do et al., 2002).
M6P receptors.
Once tagged with M6P residues, lysosomal soluble proteins are diverted from the secretory pathway and rerouted to the lysosomes by the action of the M6PRs, which bind the cargo at pH 6.7 in the trans-Golgi network and release it in the endosomes at pH 6. There are two types of M6PRs: cation-dependent (CD-MPR, encoded by the M6PR gene) and cation-independent (CI-MPR, encoded by the IGF2R gene). The CI-MPR is also known as MPR300 due to its molecular weight of 300 kDa, or insulin-like growth factor II receptor (IGFIIR), due to its ability to bind and transport the IGF-II hormone. The extracytoplasmic domain of the CI-MPR is composed of 15 repeated homology segments of about 147 amino acids each. Studies have shown that this domain contains two binding sites for the M6P, one in domain 3 and one in domain 9. Mutagenesis of Arg435 in domain 3 and Arg1334 in domain 9 reduced dramatically the ligand binding activity, confirming the ability of these two domains to bind the M6P. The CI-MPR also contains a binding site for the IGF-II (Westlund et al., 1991) and a different one for a Man6P-GlcNAc phosphodiester (Marron-Terada et al., 2000). The CD-MPR is also known as MPR46 due to its molecular weight of 46 kDa. The extracytoplasmic side of CD-MPR contains one single domain which is homologous to the 15 repetitive domains presents in the CI-MPR (Tong and Kornfeld, 1989). The CD-MPR works either as a homodimer or as a tetramer, and since each monomer contains only one binding site for the M6P, the CD-MPR protein complex is able to bind two or more M6P tags at the same time (Dahms and Kornfeld, 1989).
Although the two M6PRs are related and share similar domains, studies in fibroblasts deficient for CI-MPR, CD-MPR, or both, revealed a different affinity for different M6P-tagged substrates, indicating that the function of the two receptors is mostly complementary and only partially redundant (Munier-Lehmann et al., 1996a; Munier-Lehmann et al., 1996b). In vivo disruption of CD-MPR or CI-MPR caused a moderately impaired trafficking of lysosomal hydrolases to the lysosomes, although mutant mice lacking the CI-MPR die prenatally due to the accumulation of IGF-II (Sohar et al., 1998).
The anterograde transport of the M6PRs is mediated by the Golgi-localized, gamma-ear-containing, ARF-binding proteins (GGAs). The GGAa recognize acidic-cluster-dileucine signals that are present at the cytosolic C-terminus of the M6PRs, and dominant-negative mutations of the GGAs retain both CI-MRP and CD-MPR proteins in the trans-Golgi network (Puertollano et al., 2001). Therefore, GGA function is crucial for the correct sorting of lysosomal soluble proteins. Once these proteins are released in the lysosomes, the M6PRs are recycled back to the trans-Golgi network by the combined action of the retromer and the Rab9/TIP47 protein complex (Seaman et al., 1998). The cytoplasmic tail of CD-M6PR contains a Phe in position 18 and a Trp in position 19 that interact with TIP47 and are required for the correct retrieval of the CD-M6PR (Schweizer et al., 1997). Retrieval of CI-M6PR to the trans-Golgi network is mediated by the interaction of the retromer, in concert with the clathrin adaptor AP-1, with the Trp/Phe-Leu-Met/Val tri-peptide motif that is present in the cytoplasmic tail of the receptor (Seaman, 2007).
Importantly, not all the lysosomal proteins that are correctly tagged with M6P are delivered to the lysosome. Some escape the binding with the M6PRs and are exocytosed in the extracellular compartment. Since the CI-MPR is localized not only at the trans-Golgi network and endosomes but also at the plasma membrane, the escaped proteins can be retrieved by CI-MPR and rerouted back to the endosomal system and, hence, to the lysosomal compartment. This mechanism represents the foundation of the concept of enzyme replacement therapy (ERT). Missing lysosomal enzymes can be provided exogenously, upon modification with M6P tags before administration so to be rescued from the extracellular compartment via CI-MPR interaction. ERT is being used successfully for the treatment of some lysosomal storage disorders, including Fabry disease, Pompe disease and CLN2 disease (Schiffmann et al., 2000; Schulz et al., 2018; Thurberg et al., 2006).
From a cell biology perspective, it is interesting that both M6PRs, as well as the GlcNAc-1-phosphotransferase and the uncovering enzyme, are encoded by genes that are part of the CLEAR network. Like SORT1 and SCARB2, the M6PR, IGF2R, NAGPA and GNPTG genes indeed contain single or multiple binding sites for TFEB and are responsive to TFEB activation or overexpression (Palmieri et al., 2011; Sardiello et al., 2009; Song et al., 2013). This suggests that the expression of lysosomal proteins needs to be coordinated with that of their modifying proteins and sorting receptors in order to allow efficient biogenesis of lysosomes and, presumably, appropriate responses to external and internal stimuli (Figure 2).
Figure 2.

Regulation of the synthesis and trafficking of lysosomal enzymes. TFEB modulates the expression of various lysosomal enzymes and of their transporters: LIMP-2 (SCARB2), sortilin (SORT1), and the mannose-6-phosphate (M6P) receptors (M6PR and IGF2R). TFEB also modulates the expression of a subunit of GlcNac-1-phosphotransferase (GNPTG) and of the uncovering enzyme (NAGPA), the Golgi-residing enzymes that generate the mannose-6-phosphate tags on most lysosomal enzymes.
M6P and human disease.
Mutations in any of the components involved in M6P-based trafficking of lysosomal enzymes may result in devastating disorders. Mucolipidosis II (ML II) and Mucolipidosis III (ML III) are caused by mutations in the genes encoding the GlcNAc-1-phosphotransferase: ML II α/β (OMIM: 252500) are caused by mutations in the GNPTAB gene, whereas ML III α/β (OMIM: 252600) are caused by mutations in the GNPTG gene. ML II, also known as I-cell disease, is characterized by a complete loss of the GlcNAc-1-phosphotransferase activity and the presence of large intracellular inclusion bodies. Clinically, the skeletal system is severely affected, the development is delayed, and death occurs prematurely, usually within the first decade of life. ML III is much milder and characterized by a typical Hurler-like dysmorphism, with slower progression, short stature, impaired mobility of the joints and mental retardation in half of the affected patients. Death occurs late in life, with some patients living until the eighth decade (Kollmann et al., 2010). Conversely, loss of UCE enzyme activity is well tolerated and to date there are no reported pathologies that are associated with mutations in the NAGPA gene. This is probably due to the ability of the CI-MPR to bind and transport the Man6P-GlcNAc phosphodiesters, although with lower efficiency (Chavez et al., 2007).
Importantly, ML II presents with a severely impaired—but not completely abolished—trafficking of lysosomal enzymes to the lysosome. Since the GlcNAc-1-phosphotransferase activity is completely absent, one would expect to observe nearly complete loss of enzymes in the lysosomes. Instead, studies involving ML II patient-derived cell lines have shown that many lysosomal enzymes are able to reach the lysosomes despite the lack of M6P tags, suggesting that alternative routes such as those based on sortilin and LIMP-2 could at least partially take over in the sorting of the enzymes (Staudt et al., 2016; Tsuji et al., 1988).
Sorting of membrane proteins to the endolysosomal system
The mammalian lysosome contains several dozens of lysosomal membrane proteins (LMPs), and many others likely remain to be identified (Lubke et al., 2009). LMPs are involved in diverse functions, ranging from the acidification of the lysosomal lumen to the regulation of membrane fusion and fission events, import of sorting machineries, and recycling of lysosomal degradation products. LMPs and transmembrane transporters that are involved in the sorting of soluble lysosomal proteins are mostly known to utilize tyrosine-based (NPXY or YXXΦ) or dileucine-based ([DE]XXXL[LI] or DXXLL) signals for their targeting to endolysosomes through clathrin-coated vesicles—APs (adaptor proteins) or GGAs (Golgi-localising, Gamma-adaptin ear domain homology, ARF-binding proteins) (Hermey et al., 2003; Hunziker and Fumey, 1994; Johnson and Kornfeld, 1992; Nielsen et al., 2007; Nielsen et al., 2008; Nielsen et al., 2001). Examples of ‘conventional’ tyrosine- and dileucine-based signal motifs that drive protein sorting in endosome-derived organelles are found in the cytosolic termini of LAMP1 (Guarnieri et al., 1993), tyrosinase (Simmen et al., 1999) and endolyn (Ihrke et al., 2000) among others (Bonifacino and Traub, 2003). However, alternatives to conventional signal motifs and trafficking carriers are now emerging which have likely evolved because of the different topologies of LMPs. For example, the polytopic LAPTM5 (lysosomal-associated transmembrane protein 5) contains three modified tyrosine motifs (L/PPxY) within its C-terminus that promote protein recruitment and transport to lysosomes (Pak et al., 2006). Similarly, the LAPTM5 protein homolog LAPTM4α (lysosomal-associated transmembrane protein 4 alpha) necessitates of two tandem tyrosine-based motifs in its C-terminus for efficient localization to lysosomes (Hogue et al., 2002).
A different example is TMEM106B, a protein associated with frontotemporal lobar degeneration (Lang et al., 2012) which contains an extended dileucine-based signal (EXXXXXLI) for lysosomal sorting (Busch et al., 2016). Similarly, the Batten disease protein CLN3 has an EEEX(8)LI signal in its second cytosolic loop that is necessary for efficient lysosomal targeting (Kyttala et al., 2004; Storch et al., 2004).
Very different from the tyrosine- or dileucine-based signals is the case of MLN64 (metastatic lymph node 64), an integral endolysosomal membrane protein involved in cholesterol transport (Alpy et al., 2001; Zhang et al., 2002) whose sorting depends on a KSASNP motif. This motif is present in the C-terminal START domain of the protein and mediates binding with 14–3-3 proteins. Alanine substitution of key amino acids in this motif results in delayed transport of MLN64 to endosomes (Liapis et al., 2012).
Sorting motifs in LMPs do not only come in different flavors of amino acid stretches but may also be based on specific post-translational modifications. For example, prenylation of a C-terminal CAAX box is known to be an additional guide for endolysosomal transport of CLN3 protein (Storch et al., 2007). Similarly, palmitoylation of the C-terminal tail of mucolipin (MCOLN1/TRPML1) promotes its internalization from the plasma membrane (Vergarajauregui and Puertollano, 2006).
Intriguingly, instead of evolving sorting motifs, several LMPs have leveraged their interactions with other LMPs as a piggyback mechanism of trafficking to the endolysosomal compartment. For example, ABCD4, a member of ATP-binding cassette transporter family, associates with LMBD1 (LMBR1 domain containing protein 1). LMBD1 is internalized in clathrin-coated vesicles by using a conventional tyrosine-based signal in its C-terminus, carrying ABCD4 in the process (Kawaguchi et al., 2016; Tseng et al., 2013). Similarly, synaptotagmin VII associates with the lysosomal tetraspanin CD63/LAMP3 upon palmitoylation of cysteine residues in its luminal domain. Synaptotagmin exploits this interaction to reach the lysosomal compartment together with CD63, which is sorted to lysosomes using tyrosine-based sorting motifs in its C-terminus (Flannery et al., 2010).
Lysosome-associated membrane protein 1 and 2 (LAMP1 and LAMP2) are some of the most abundant and vastly studied LMPs. Their packaging in clathrin-coated vesicles using tyrosine-based signals has long been known; however, studies based on LAMP1 and LAMP2 sorting and packaging in certain cell types devoid of clathrin vesicles have revealed unconventional clathrin-independent routes of trafficking (Karlsson and Carlsson, 1998). Using immunoelectron microscopy, LAMP1 was found to be present in carriers devoid of APs but rather containing the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein VAMP7 (vesicle associated membrane protein 7) and an accessory protein of the homotypic fusion and protein sorting (HOPS) complex, hVps41 (Pols et al., 2013). An additional study reported that CD63/LAMP3 is internalized via caveolae-mediated endocytosis independent of clathrin-coated vesicles (Pols and Klumperman, 2009). These examples indicate that the contribution of unconventional trafficking routes for lysosomal protein sorting may play a more general role than initially anticipated.
Although several genes encoding LMPs are well-characterized members of the CLEAR network and TFEB targets (Palmieri et al., 2011; Sardiello et al., 2009), the relationship between TFEB function and the machineries governing the transport of lysosomal membrane proteins remains unexplored.
Impaired TFEB signaling in neurodegenerative disease
In recent years, the lysosome has gained the spotlight in the study of the pathogenesis of late-onset neurodegenerative diseases (Sharma et al., 2018). Multiple studies have indeed identified very definite molecular events that lead to impairment of lysosome-dependent processes as an important component of neuronal stress in these diseases. Interestingly, many of these processes converge to impaired lysosome biogenesis and function via interference with TFEB levels or activity (Sardiello, 2016). Chief among the lysosomal functions that are impaired in neurodegenerative diseases is macroautophagy (henceforth referred to as ‘autophagy’). The lysosome is indeed the terminal degradative organelle for the breakdown of material that is fed to lysosomes through the autophagic complex—macromolecules, protein aggregates, damaged cellular components and exhausted organelles. Autophagy encompasses material sequestration by double-membrane structures (the ‘phagophore’) and multiple vesicle fusion events that culminate in lysosomal degradation of autophagic cargo. Defects in the assembly, fusion, or degradation of these structures are consistently observed not only in lysosomal storage disorders but also in the most widespread late-onset neurodegenerative diseases (Seranova et al., 2017; Sharma et al., 2018). Among the proposed mechanisms that lead to lysosomal-autophagic failure in neurodegenerative diseases, TFEB deregulation has gained increasing recognition owing to the accurate dissection of multiple molecular events that can interfere with normal TFEB function. The best-defined examples pertain to the identification of possible pathogenic mechanisms in Parkinson’s disease, Alzheimer’s disease, and spinobulbar muscular atrophy (Figure 3), as detailed below.
Figure 3.
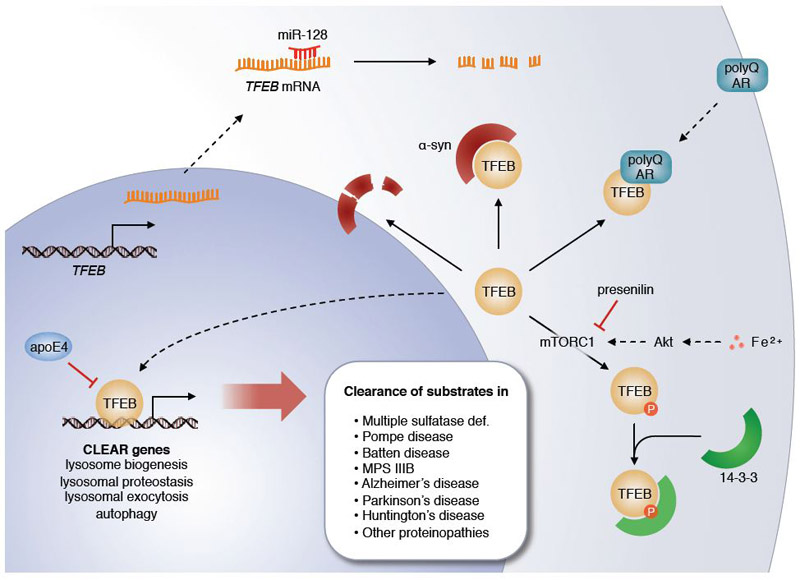
Interplay between TFEB and various molecular pathways in neurodegenerative disease. TFEB binding to CLEAR sites can be outcompeted by apoE4, a protein encoded by the Alzheimer’s disease risk factor APOE ε4 allele. TFEB mRNA levels can be decreased by miR-128, a microRNA that is upregulated in Alzheimer’s disease. Loss of functional presenilin in familial Alzheimer’s disease leads to increased phosphorylation of TFEB by mTORC1 and subsequent cytosolic sequestration by 14–3-3 proteins. Increased TFEB sequestration can also result from increased Akt/mTORC1 pathway activity downstream of iron accumulation. α-synuclein and polyQ-expanded androgen receptor (AR) also can sequester TFEB and decrease its action. Active TFEB promotes various lysosome-based clearance pathways that can counteract pathogenic storage of undegraded molecules in lysosomal storage disorders and proteinopathies.
Parkinson’s disease is characterized by selective loss of dopaminergic neurons in the substantia nigra which is preceded by an aberrant accumulation of α-synuclein in Lewy bodies (Dauer and Przedborski, 2003). In addition to various environmental causes (Greenamyre and Hastings, 2004; Pal et al., 2014), several genes of the lysosomal system have been characterized as risk factors for Parkinson’s disease, including GBA (Goker-Alpan et al., 2004; Tayebi et al., 2003), SMPD1 (Foo et al., 2013; Gan-Or et al., 2013), and others (Chang et al., 2017; Do et al., 2011; Robak et al., 2017; Shachar et al., 2011). These findings have introduced the concept that lysosomal storage disorders and Parkinson’s disease may share a common genetic mechanism, an idea corroborated by the observations that the lysosome is implicated in the clearance of α-synuclein aggregates (Cuervo et al., 2004) and that excessive α-synuclein disrupts neuronal lysosomal function (Mazzulli et al., 2011). In addition, lysosomal depletion has been described as a prominent cellular phenotype in Parkinson’s disease (Dehay et al., 2010). A straightforward link between α-synuclein accumulation and impairment of lysosomal function has been provided with the finding that α-synuclein behaves as a TFEB-sequestering molecule; thus, α-synuclein impedes TFEB nuclear translocation, thereby inhibiting lysosomal biogenesis (Decressac et al., 2013). The analysis of postmortem tissues from patients with Parkinson’s disease revealed depletion of nuclear TFEB and its accumulation with α-synuclein in Lewy bodies in dopaminergic neurons of the substantia nigra (Decressac et al., 2013). Iron accumulation could be an additional component of this pathogenic loop. Iron indeed accumulates in various neurodegenerative disorders and particularly in synucleinopathies (Snyder and Connor, 2009), where it contributes to α-synuclein aggregation and transmission by interfering with TFEB function via activation of the Akt/mTORC1 axis (Xiao et al., 2018).
Alzheimer’s disease is characterized by progressive hippocampal (and overall brain) atrophy. At the cellular level, accumulation of β-amyloid and hyperphosphorylated tau parallels the progressive disruption of the autophagy-lysosomal pathway (Nixon et al., 2005). Interestingly, studies have shown that the hippocampus of Alzheimer’s disease patients express higher levels of microRNA-128 (miR-128) (Lukiw, 2007), a microRNA that targets TFEB mRNA and thus lowers expression of TFEB and lysosomal genes (Sardiello et al., 2009). In blood cells from Alzheimer’s disease patients, increased expression of miR-128 correlated with decreased activities of lysosomal enzymes and a decreased ability of the cells to degrade β-amyloid, which was restored by suppression of miR-128 expression (Tiribuzi et al., 2014). A different link between Alzheimer pathology and TFEB function was observed with the finding that activation of TFEB is negatively impacted by the deficiency of presenilin, a cause of familial Alzheimer’s disease (Reddy et al., 2016). In the absence of presenilin, the levels of the mTORC1 amino acid sensor sestrin2 are reduced and mTORC1 is constitutively tethered to the lysosomal membrane, thereby increasing inhibitory phosphorylation of TFEB (Reddy et al., 2016). Increased phosphorylation of TFEB in Alzheimer’s is consistent with the observed progressive nuclear exclusion of TFEB in brain samples from Alzheimer’s disease patients (Wang et al., 2016b). A third, independent connection between Alzheimer pathology and TFEB function was reported with the observation that TFEB binding to the promoters of lysosomal genes can be outcompeted by apoE4 (Parcon et al., 2018). ApoE4 is the protein encoded by the APOE ɛ4 allele, the single greatest genetic risk factor for the development of Alzheimer’s disease (Strittmatter and Roses, 1995). In vitro, apoE4 could bind to DNA probes carrying the CLEAR motif with much greater affinity than apoE3, the protein product of APOE ɛ3 allele which is not a risk factor for Alzheimer’s disease; increased apoE4 binding to CLEAR probes was paralleled by decreased TFEB binding to the same probes, and expression of apoE4 diminished the expression of TFEB target genes (Parcon et al., 2018). Thus, several independent pathways seem to converge to disturbed TFEB-mediated lysosomal biogenesis as a potential common leitmotif in the pathogenesis of Alzheimer’s disease. More in vivo work is needed to clarify whether these pathways cross-talk and where they are singularly placed in the sequence of cellular events that culminate with neuronal cell death.
Spinobulbar muscular atrophy (SBMA) is a disease caused by the expansion of a polyglutamine tract (polyQ) in androgen receptor (AR) protein. Recent work has shown that normal AR interacts with TFEB and promotes TFEB expression, whereas polyQ AR interferes with TFEB activation and impairs neuronal autophagy, thereby contributing to disease pathology (Cortes et al., 2014).
Together, these examples highlight impaired function of TFEB as a potential contributor of neuronal degeneration and prompt the question of whether interventions aimed at increasing TFEB function would counteract disease progression.
Therapeutic effects of TFEB-mediated enhancement of the autophagy-lysosome pathway
Impairment of the autophagy-lysosome pathway and aberrant accumulation of non-degraded metabolites are shared focal points of most lysosomal storage disorders and several late-onset neurodegenerative diseases (Sharma et al., 2018). Enhancing autophagy and lysosomal function can, therefore, be pursued as a line of investigation to define possible treatments for these diseases. Several studies based on autophagic enhancement in pre-clinical models of neurodegenerative diseases have been conducted using the mTORC1 allosteric inhibitor, rapamycin, or analogue molecules (the rapalogs). The rationale was rooted in the notion that active mTORC1 inhibits autophagy by phosphorylating, among others, the autophagic initiating factor ULK1, leading to autophagy inhibition (Kim et al., 2011). Thus, inhibition of mTORC1 results in activation of autophagy. Results from various models of neurodegenerative disease, including Alzheimer’s, Parkinson’s and Huntington’s diseases and frontotemporal lobar degeneration have consistently shown improved pathology upon mTORC1 pharmacological inhibition with rapamycin/rapalogs (Berger et al., 2006; Caccamo et al., 2010; Ravikumar et al., 2004; Santini et al., 2009; Wang et al., 2012). Pharmacological inhibition of mTORC1 should also boost autophagy and lysosomal biogenesis by relieving mTORC1-mediated TFEB inhibition; unfortunately, however, TFEB is among the mTORC1 substrates that are insensitive to rapamycin and that therefore are not significantly activated by this pharmacological treatment (Roczniak-Ferguson et al., 2012; Settembre et al., 2012). Importantly, proof-of-principle studies using overexpression of exogenous TFEB have been conducted that demonstrate the therapeutic effects of TFEB-mediated enhancement of the autophagy-lysosome pathway in very different disease models. Thus, additional signaling pathways are being examined to leverage activation of endogenous TFEB as a therapeutic tool. The following paragraphs provide an updated summary of studies focused on genetic and pharmacological manipulation of TFEB to sustain an enhanced lysosomal function in various animal models of storage diseases (Figure 3).
Multiple sulfatase deficiency.
Multiple sulfatase deficiency (MSD) is a severe lysosomal storage disorder caused by defects in SUMF1, an ER-residing post-translational modifying factor of the sulfatases (Cosma et al., 2003; Dierks et al., 2003). Deficiency of functional SUMF1 leads to the simultaneous inactivity of all sulfatases, of which several reside at the lysosome and are themselves defective in various distinct lysosomal storage disorders (Diez-Roux and Ballabio, 2005). At the tissue level, a prominent MSD feature is the storage of undegraded glycosaminoglycans (GAGs). Systemic injection of an adeno-associated virus (AAV) carrying TFEB cDNA in a mouse model of MSD resulted in reductions of GAGs, macrophage infiltration (a marker of tissue inflammation) and cell death in the muscle and liver, indicating amelioration of tissue pathology (Medina et al., 2011). TFEB-induced clearance was mediated by enhanced lysosomal exocytosis, a process by which the lysosomal membrane fuses with the plasma membrane and the content of the lysosome is emptied outside of the cell; lysosomal exocytosis requires active TRPML1, a Ca2+ channel encoded by the MCOLN1 gene which is a direct target of TFEB (Medina et al., 2011).
Pompe disease.
Pompe disease is a lysosomal storage disorder caused by the deficiency of functional acid α-glucosidase, a lysosomal enzyme that breaks down glycogen to glucose in the lysosomal lumen. Deficiency of α-glucosidase function results in a severe myopathy characterized by the accumulation of glycogen and excessive autophagic buildup in muscle fibers (Raben et al., 2007). AAV-mediated TFEB overexpression in the muscle of a mouse model of Pompe disease reduced abnormal glycogen storage and alleviated the autophagic buildup (Spampanato et al., 2013). Similar to the MSD study, the clearance effect was mainly due to exocytosis of storage vesicles, which in this case included autophagosomes; suppressing autophagy indeed reduced the effects of TFEB overexpression (Spampanato et al., 2013).
Batten disease (neuronal ceroid lipofuscinosis).
Batten disease comprises a family of genetically distinct neurodegenerative diseases characterized, at the cellular level, by the intralysosomal storage of autofluorescent material (ceroid lipopigment) (Mole and Cotman, 2015). Juvenile Batten disease is caused by mutations in CLN3 gene, which encodes a multi-pass lysosomal membrane protein involved in lysosome homeostasis (Carcel-Trullols et al., 2015; Cotman and Staropoli, 2012). Pharmacological activation of endogenous TFEB via oral administration of trehalose reduced neuropathology and elongated the lifespan of a mouse model of juvenile Batten disease (Palmieri et al., 2017a). Trehalose activates TFEB through the inhibition of the activity of Akt, a negative regulator of TFEB; trehalose-treated animals had indeed increased nuclear translocation of TFEB in neurons and an enhanced expression of genes of the autophagy-lysosome pathway (Palmieri et al., 2017a). Trehalose treatment diminished the storage of ceroid lipopigment in brain cells and reduced neuroinflammation and neurodegeneration, while improving a sensory phenotype (Palmieri et al., 2017a). Since both the primary function of CLN3 protein and the link between CLN3 deficiency and the storage of ceroid lipopigment are unknown, it remains to be established whether TFEB activation reduces lysosomal storage in CLN3-deficient neurons by preserving lysosomal homeostasis or rather by enhancing lysosomal pathways that can compensate for the loss of CLN3 function. It is also possible that TFEB-induced lysosomal exocytosis contributes to reducing the cellular load of ceroid lipopigment.
Sanfilippo syndrome type B (mucopolysaccharidosis IIIB).
Sanfilippo syndrome is caused by the deficiency of one of four enzymes involved in the degradation of heparan sulfate (a mucopolysaccharide, MPS). Similar to several other lysosomal storage disorders, Sanfilippo syndrome is characterized by progressive neurodegeneration, neuroinflammation, and vision loss among other symptoms (Ashworth et al., 2006; Tse et al., 2015; Valstar et al., 2008). Sanfilippo syndrome type B (MPS IIIB) is caused by mutations in the NAGLU gene, which encodes the heparan sulfate-degrading lysosomal enzyme α-N-acetylglucosaminidase. At the cellular level, NAGLU deficiency causes abnormal accumulation of heparan sulfate in autophagic vacuoles (Li et al., 1999). TFEB activation in MPS IIIB mice via oral administration of trehalose prevented retinal degeneration, attenuated vision loss, and reduced both neuroinflammation and the load of autophagic vacuoles in brain cells (Lotfi et al., 2018). Trehalose treatment also elongated the lifespan of MPS IIIB mice (Lotfi et al., 2018). Since no other enzymes similar to α-N-acetylglucosaminidase are known that could compensate for its loss, it is likely that the clearance effect obtained upon TFEB activation was mainly mediated by lysosomal exocytosis, as seen for exogenous TFEB expression in other models of lysosomal enzyme deficiencies. This study demonstrated the therapeutic potential of drug-induced enhancement of the autophagy-lysosome pathway in a mouse model of lysosomal enzyme deficiency (Lotfi et al., 2018).
Other degenerative diseases with toxic protein aggregates.
The finding that TFEB overexpression could induce the degradation of polyQ-expanded huntingtin (htt) (Sardiello et al., 2009), the protein that forms pathogenic aggregates in Huntington’s disease (MacDonald et al., 1993), set the stage for testing TFEB as a therapeutic tool in neurodegenerative diseases with a component of autophagic substrate accumulation. TFEB is a transcriptional target of PGC1α, and overexpression of PGC1α increases the expression of TFEB (Tsunemi et al., 2012). In a mouse model of Huntington’s disease, overexpression of PGC1α eliminated htt protein aggregation and ameliorated neuropathology through TFEB-dependent enhancement of htt autophagic degradation (Tsunemi et al., 2012). Comparable results were obtained with a more direct approach, namely, injection of AAVs carrying TFEB cDNA into the striatum of Huntington’s disease mice (Vodicka et al., 2016). Similarly, injection of TFEB AAVs in the brain of a rat model of Parkinson’s disease decreased both α-synuclein accumulation and loss of nigral dopamine neurons, demonstrating a therapeutic effect (Decressac et al., 2013). Likewise, AAV-mediated expression of TFEB in the brain of the rTg4510 mouse model of tauopathy (a model of Alzheimer’s disease) reduced neurofibrillary tangle pathology and neurodegeneration, resulting in the rescue of synaptic and behavioral defects observed in these mice (Polito et al., 2014). Neuroprotective effects were also achieved by crossing a P301S model of tauopathy with a transgenic mouse line expressing TFEB in the adult brain (Wang et al., 2016a). Among the observed effects mediated by TFEB overexpression was the restored expression of spinophilin, a protein that regulates the formation and function of dendritic spines (Feng et al., 2000; Wang et al., 2016a). Finally, AAV-mediated expression of TFEB either in the astrocytes or neurons of APP/PS1 mice (a different model of Alzheimer’s disease displaying amyloid plaque pathogenesis) reduced β-amyloid levels and amyloid plaque load (Xiao et al., 2014; Xiao et al., 2015).
The brain is not the only organ that is affected by proteinopathies and that therefore could benefit from the enhancement of the autophagy-lysosome pathway. Alpha-1-antitrypsin (AAT) deficiency can present with the presence of AAT toxic aggregates resulting from mutations that impair normal AAT protein folding (Wu et al., 1994). TFEB viral delivery to the liver of a mouse model of AAT deficiency decreased the protein toxic aggregates and ameliorated liver fibrosis and cell death (Pastore et al., 2013). Similar therapeutic effects were observed when TFEB was delivered to the lung of mice with pathogenic aggregates of misfolded antitrypsin (Hidvegi et al., 2015) or in a Zebrafish model of amyloidogenic light chain-mediated cardiotoxicity (Guan et al., 2014).
Additional studies in cellular models of disease caused by abnormal protein accumulation—including SBMA (Cortes et al., 2014), spinocerebellar ataxia type 1 (Bouche et al., 2016) and CLN2 disease (a Batten disease subtype) (Song et al., 2014)—demonstrated the potential therapeutic effects of increased TFEB function, thereby expanding the spectrum of diseases that are potentially amenable to TFEB therapy.
Conclusions
Although a complex and multi-layered process, lysosomal biogenesis can be dissected into a relatively small number of streamlined events that are amenable to genetic and biochemical analysis. Somehow unexpectedly, the characterization of these pathways is revealing that highly complex organisms such as mammals are quite resistant to pathogenic mutations that would be predicted to be catastrophic based on the established cellular function of the affected components. Instead, the emerging picture is that of phenotypic manifestations that are much milder than anticipated—if observed at all. Degenerative storage diseases that are rooted in causes different from mutations in the genes participating in lysosomal biogenesis or function typically have a late onset, indicating that the system as a whole has been able to heal itself for a protracted period of time before being overwhelmed by some still undefined age-related component. Functional redundancy amongst various effectors of lysosomal biogenesis pathways could help explain the observed organism’s resilience to defects in the autophagy-lysosome system. The CLEAR network and its chief regulator TFEB could also contribute to this resilience. Indeed, experimental activation of the CLEAR network in very different models of degenerative storage diseases has consistently resulted in phenotypic improvements in the affected organ(s) and even elongation of lifespan in the two studies that included lifelong analyses. It is significant that many of these diseases do not currently have any therapeutic options other than palliative care. Translational studies that leverage the improved understanding of pathogenic mechanisms and regulatory pathways related to the autophagy-lysosome system are therefore urgently needed to test lysosomal enhancement as a therapeutic tool in degenerative storage diseases.
Acknowledgments
Work in the M.S. laboratory is supported by research grant NS079618 from NIH/NINDS and grants from the Beyond Batten Disease Foundation, James Family/Fighting for Maya Foundation, and Charlotte & Gwenyth Gray Foundation.
Abbreviations
- TFEB
transcription factor EB
- mTORC1
mechanistic/mammalian target of rapamycin complex 1
- CLEAR
Coordinated Lysosomal Expression and Regulation
- PKB
protein kinase B
- GSK3β
glycogen synthase kinase 3β
- MAP4K3
mitogen-activated protein kinase kinase kinase kinase 3
- PP2A
protein phosphatase 2
- MITF
microphtalmia-associated transcription factor
- ssVSS
vacuolar sorting signal
- LIMP-2
lysosome membrane protein 2
- SORT1
sortilin 1
- SAPs
sphingolipid activator proteins
- GBA
glucosylceramidase beta
- GD
Gaucher disease
- GCase
glucocerebrosidase
- AP-1
adaptor protein-1
- M6P
mannose 6-phosphate
- M6PR
mannose-6-phosphate receptors
- AMRF
action myoclonus-renal failure syndrome
- PME
progressive myoclonus epilepsy
- GNPTG
N-Acetylglucosamine-1-Phosphotransferase Subunit Gamma
- OST
oligosaccharyl transferase
- IGFIIR
insulin-like growth factor II receptor
- ERT
enzyme replacement therapy
- GNPTAB
N-Acetylglucosamine-1-Phosphate Transferase Alpha And Beta Subunits
- ML III
mucolipidosis III
- LMPs
lysosomal membrane proteins
- NAGPA
N-Acetylglucosamine-1-Phosphodiester Alpha-N-Acetylglucosaminidas
- UCE
uncovering enzyme
- LAPTM5
lysosomal-associated transmembrane protein 5
- LAPTM4α
lysosomal-associated transmembrane protein 4
- MLN64
metastatic lymph node 64
- LMBD1
LMBR1 domain containing protein 1
- LAMP1
Lysosome-associated membrane protein 1
- LAMP2
Lysosome-associated membrane protein 2
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- VAMP7
vesicle associated membrane protein 7
- HOPS
homotypic fusion and protein sorting
- SBMA
spinobulbar muscular atrophy
- polyQ
polyglutamine tract
- AR
androgen receptor
- MSD
multiple sulfatase deficiency
- GAGs
glycosaminoglycans
- AAV
adeno-associated virus
- MPS IIIB
mucopolysaccharidosis IIIB
- NAGLU
α-N-acetylglucosaminidase
- htt
huntingtin
- AAT
alpha-1-antitrypsin
- SBMA
spinocerebellar ataxia type 1
- SUMF1
sulfatase modifying factor 1
- TRPML1
TRP channel subfamily 1
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1
- apoE
apolipoprotein E
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Alpy F, Stoeckel ME, Dierich A, Escola JM, Wendling C, Chenard MP, Vanier MT, Gruenberg J, Tomasetto C, and Rio MC. 2001. The steroidogenic acute regulatory protein homolog MLN64, a late endosomal cholesterol-binding protein. J Biol Chem. 276:4261–4269. [DOI] [PubMed] [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, and Willnow TE. 2005. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 102:13461–13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth JL, Biswas S, Wraith E, and Lloyd IC. 2006. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 51:1–17. [DOI] [PubMed] [Google Scholar]
- Baranski TJ, Faust PL, and Kornfeld S. 1990. Generation of a lysosomal enzyme targeting signal in the secretory protein pepsinogen. Cell. 63:281–291. [DOI] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O’Kane CJ, and Rubinsztein DC. 2006. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 15:433–442. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, Lullmann-Rauch R, Blanz J, Zhang KW, Stankovich J, Kalnins RM, Dowling JP, Andermann E, Andermann F, Faldini E, D’Hooge R, Vadlamudi L, Macdonell RA, Hodgson BL, Bayly MA, Savige J, Mulley JC, Smyth GK, Power DA, Saftig P, and Bahlo M. 2008. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 82:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanz J, Groth J, Zachos C, Wehling C, Saftig P, and Schwake M. 2010. Disease-causing mutations within the lysosomal integral membrane protein type 2 (LIMP-2) reveal the nature of binding to its ligand beta-glucocerebrosidase. Hum Mol Genet. 19:563–572. [DOI] [PubMed] [Google Scholar]
- Blanz J, Zunke F, Markmann S, Damme M, Braulke T, Saftig P, and Schwake M. 2015. Mannose 6-phosphate-independent Lysosomal Sorting of LIMP-2. Traffic. 16:1127–1136. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, and Traub LM. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 72:395–447. [DOI] [PubMed] [Google Scholar]
- Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, and Botas J. 2016. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 12:484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boustany RM 2013. Lysosomal storage diseases--the horizon expands. Nat Rev Neurol. 9:583–598. [DOI] [PubMed] [Google Scholar]
- Braulke T, and Bonifacino JS. 2009. Sorting of lysosomal proteins. Biochim Biophys Acta. 1793:605–614. [DOI] [PubMed] [Google Scholar]
- Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, and Chen-Plotkin AS. 2016. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet. 25:2681–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, and Oddo S. 2010. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 285:13107–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canuel M, Korkidakis A, Konnyu K, and Morales CR. 2008. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem Biophys Res Commun. 373:292–297. [DOI] [PubMed] [Google Scholar]
- Carcel-Trullols J, Kovacs AD, and Pearce DA. 2015. Cell biology of the NCL proteins: What they do and don’t do. Biochim Biophys Acta. 1852:2242–2255. [DOI] [PubMed] [Google Scholar]
- Carlo AS, Nykjaer A, and Willnow TE. 2014. Sorting receptor sortilin-a culprit in cardiovascular and neurological diseases. J Mol Med (Berl). 92:905–911. [DOI] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, C. International Parkinson’s Disease Genomics, T. andMe Research, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, and Graham RR. 2017. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 49:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Guo J, di Ronza A, and Sardiello M. 2018. Aminode: Identification of Evolutionary Constraints in the Human Proteome. Sci Rep. 8:1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez CA, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, and Dahms NM. 2007. Domain 5 of the cation-independent mannose 6-phosphate receptor preferentially binds phosphodiesters (mannose 6-phosphate N-acetylglucosamine ester). Biochemistry. 46:12604–12617. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang K, Long A, Jia L, Zhang Y, Deng H, Li Y, Han J, and Wang Y. 2017. Fasting-induced hormonal regulation of lysosomal function. Cell Res. 27:748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn ZA, and Ehrenreich BA. 1969. The uptake, storage, and intracellular hydrolysis of carbohydrates by macrophages. J Exp Med. 129:201–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, Garden GA, and La Spada AR. 2014. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci. 17:1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, and Ballabio A. 2003. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 113:445–456. [DOI] [PubMed] [Google Scholar]
- Cotman SL, and Staropoli JF. 2012. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin Lipidol. 7:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, and Sulzer D. 2004. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 305:1292–1295. [DOI] [PubMed] [Google Scholar]
- Cuozzo JW, and Sahagian GG. 1994. Lysine is a common determinant for mannose phosphorylation of lysosomal proteins. J Biol Chem. 269:14490–14496. [PubMed] [Google Scholar]
- Dahms NM, and Kornfeld S. 1989. The cation-dependent mannose 6-phosphate receptor. Structural requirements for mannose 6-phosphate binding and oligomerization. J Biol Chem. 264:11458–11467. [PubMed] [Google Scholar]
- Dauer W, and Przedborski S. 2003. Parkinson’s disease: mechanisms and models. Neuron. 39:889–909. [DOI] [PubMed] [Google Scholar]
- De Duve C, Pressman BC, Gianetto R, Wattiaux R, and Appelmans F. 1955. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. The Biochemical journal. 60:604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, and Bjorklund A. 2013. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 110:E1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, and Vila M. 2010. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 30:12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbens LM, Karakis I, Bayly MA, Costello DJ, Cole AJ, and Berkovic SF. 2011. Mutation of SCARB2 in a patient with progressive myoclonus epilepsy and demyelinating peripheral neuropathy. Arch Neurol. 68:812–813. [DOI] [PubMed] [Google Scholar]
- Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, and von Figura K. 2003. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 113:435–444. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, and Ballabio A. 2005. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 6:355–379. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, Wojcicki A, and Eriksson N. 2011. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 7:e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Lee WS, Ghosh P, Hollowell T, Canfield W, and Kornfeld S. 2002. Human mannose 6-phosphate-uncovering enzyme is synthesized as a proenzyme that is activated by the endoprotease furin. J Biol Chem. 277:29737–29744. [DOI] [PubMed] [Google Scholar]
- Eun SY, Lee JN, Nam IK, Liu ZQ, So HS, Choe SK, and Park R. 2018. PEX5 regulates autophagy via the mTORC1-TFEB axis during starvation. Exp Mol Med. 50:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, and Greengard P. 2000. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 97:9287–9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery AR, Czibener C, and Andrews NW. 2010. Palmitoylation-dependent association with CD63 targets the Ca2+ sensor synaptotagmin VII to lysosomes. J Cell Biol. 191:599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL, Prakash KM, Tan LC, and Tan EK. 2013. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol Aging. 34:2890 e2813–2895. [DOI] [PubMed] [Google Scholar]
- Gamp AC, Tanaka Y, Lullmann-Rauch R, Wittke D, D’Hooge R, De Deyn PP, Moser T, Maier H, Hartmann D, Reiss K, Illert AL, von Figura K, and Saftig P. 2003. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet. 12:631–646. [PubMed] [Google Scholar]
- Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, Gana-Weisz M, Raymond D, Rozenkrantz L, Deik A, Gurevich T, Gross SJ, Schreiber-Agus N, Giladi N, Bressman SB, and Orr-Urtreger A. 2013. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology. 80:1606–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, and Sidransky E. 2004. Parkinsonism among Gaucher disease carriers. J Med Genet. 41:937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, and Hastings TG. 2004. Biomedicine. Parkinson’s--divergent causes, convergent mechanisms. Science. 304:1120–1122. [DOI] [PubMed] [Google Scholar]
- Guan J, Mishra S, Qiu Y, Shi J, Trudeau K, Las G, Liesa M, Shirihai OS, Connors LH, Seldin DC, Falk RH, MacRae CA, and Liao R. 2014. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med. 6:1493–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri FG, Arterburn LM, Penno MB, Cha Y, and August JT. 1993. The motif Tyr-X-X-hydrophobic residue mediates lysosomal membrane targeting of lysosome-associated membrane protein 1. J Biol Chem. 268:1941–1946. [PubMed] [Google Scholar]
- Hassan AJ, Zeng J, Ni X, and Morales CR. 2004. The trafficking of prosaposin (SGP-1) and GM2AP to the lysosomes of TM4 Sertoli cells is mediated by sortilin and monomeric adaptor proteins. Mol Reprod Dev. 68:476–483. [DOI] [PubMed] [Google Scholar]
- Hermey G, Keat SJ, Madsen P, Jacobsen C, Petersen CM, and Gliemann J. 2003. Characterization of sorCS1, an alternatively spliced receptor with completely different cytoplasmic domains that mediate different trafficking in cells. J Biol Chem. 278:7390–7396. [DOI] [PubMed] [Google Scholar]
- Hermey G, Riedel IB, Hampe W, Schaller HC, and Hermans-Borgmeyer I. 1999. Identification and characterization of SorCS, a third member of a novel receptor family. Biochem Biophys Res Commun. 266:347–351. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Stolz DB, Alcorn JF, Yousem SA, Wang J, Leme AS, Houghton AM, Hale P, Ewing M, Cai H, Garchar EA, Pastore N, Annunziata P, Kaminski N, Pilewski J, Shapiro SD, Pak SC, Silverman GA, Brunetti-Pierri N, and Perlmutter DH. 2015. Enhancing Autophagy with Drugs or Lung-directed Gene Therapy Reverses the Pathological Effects of Respiratory Epithelial Cell Proteinopathy. J Biol Chem. 290:29742–29757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue DL, Nash C, Ling V, and Hobman TC. 2002. Lysosome-associated protein transmembrane 4 alpha (LAPTM4 alpha) requires two tandemly arranged tyrosine-based signals for sorting to lysosomes. Biochem J. 365:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwerda BC, Padgett HS, and Rogers JC. 1992. Proaleurain vacuolar targeting is mediated by short contiguous peptide interactions. Plant Cell. 4:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner F, Schormair B, Knauf F, Berthele A, Tolle TR, Baron R, Maier C, Treede RD, Binder A, Sommer C, Maihofner C, Kunz W, Zimprich F, Heemann U, Pfeufer A, Nabauer M, Kaab S, Nowak B, Gieger C, Lichtner P, Trenkwalder C, Oexle K, and Winkelmann J. 2011. Novel SCARB2 mutation in action myoclonus-renal failure syndrome and evaluation of SCARB2 mutations in isolated AMRF features. BMC Neurol. 11:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Lee EX, Gordon KL, Paz EA, Shen WC, Ohnishi K, Meisenhelder J, Hunter T, and La Spada AR. 2018. MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat Commun. 9:942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, and Fumey C. 1994. A di-leucine motif mediates endocytosis and basolateral sorting of macrophage IgG Fc receptors in MDCK cells. Embo J. 13:2963–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrke G, Gray SR, and Luzio JP. 2000. Endolyn is a mucin-like type I membrane protein targeted to lysosomes by its cytoplasmic tail. Biochem J. 345 Pt 2:287–296. [PMC free article] [PubMed] [Google Scholar]
- Jacobsen L, Madsen P, Moestrup SK, Lund AH, Tommerup N, Nykjaer A, Sottrup-Jensen L, Gliemann J, and Petersen CM. 1996. Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem. 271:31379–31383. [DOI] [PubMed] [Google Scholar]
- Johnson KF, and Kornfeld S. 1992. A His-Leu-Leu sequence near the carboxyl terminus of the cytoplasmic domain of the cation-dependent mannose 6-phosphate receptor is necessary for the lysosomal enzyme sorting function. J Biol Chem. 267:17110–17115. [PubMed] [Google Scholar]
- Karageorgos LE, Isaac EL, Brooks DA, Ravenscroft EM, Davey R, Hopwood JJ, and Meikle PJ. 1997. Lysosomal biogenesis in lysosomal storage disorders. Exp Cell Res. 234:85–97. [DOI] [PubMed] [Google Scholar]
- Karlsson K, and Carlsson SR. 1998. Sorting of lysosomal membrane glycoproteins lamp-1 and lamp-2 into vesicles distinct from mannose 6-phosphate receptor/gamma-adaptin vesicles at the trans-Golgi network. J Biol Chem. 273:18966–18973. [DOI] [PubMed] [Google Scholar]
- Kawaguchi K, Okamoto T, Morita M, and Imanaka T. 2016. Translocation of the ABC transporter ABCD4 from the endoplasmic reticulum to lysosomes requires the escort protein LMBD1. Scientific reports. 6:30183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide Y, Hirano H, Matsuoka K, and Nakamura K. 1997. The N-terminal propeptide of the precursor to sporamin acts as a vacuole-targeting signal even at the C terminus of the mature part in tobacco cells. Plant Physiol. 114:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann K, Pohl S, Marschner K, Encarnacao M, Sakwa I, Tiede S, Poorthuis BJ, Lubke T, Muller-Loennies S, Storch S, and Braulke T. 2010. Mannose phosphorylation in health and disease. Eur J Cell Biol. 89:117–123. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, and Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 54:631–664. [DOI] [PubMed] [Google Scholar]
- Kudo M, and Canfield WM. 2006. Structural requirements for efficient processing and activation of recombinant human UDP-N-acetylglucosamine:lysosomal-enzyme-N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 281:11761–11768. [DOI] [PubMed] [Google Scholar]
- Kyttala A, Ihrke G, Vesa J, Schell MJ, and Luzio JP. 2004. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol Biol Cell. 15:1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, and Haass C. 2012. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 287:19355–19365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois S, Zeng J, Hassan AJ, Canuel M, and Morales CR. 2003. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. Embo J. 22:6430–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, and Neufeld EF. 1999. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc Natl Acad Sci U S A. 96:14505–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu M, Ding X, Yan C, Song Z, Chen L, Huang X, Wang X, Jian Y, Tang G, Tang C, Di Y, Mu S, Liu X, Liu K, Li T, Wang Y, Miao L, Guo W, Hao X, and Yang C. 2016. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 18:1065–1077. [DOI] [PubMed] [Google Scholar]
- Liapis A, Chen FW, Davies JP, Wang R, and Ioannou YA. 2012. MLN64 transport to the late endosome is regulated by binding to 14–3-3 via a non-canonical binding site. PLoS One. 7:e34424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi P, Tse DY, di Ronza A, Seymour ML, Martano G, Cooper JD, Pereira FA, Passafaro M, Wu SM, and Sardiello M. 2018. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency Autophagy. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubke T, Lobel P, and Sleat DE. 2009. Proteomics of the lysosome. Biochim Biophys Acta. 1793:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ 2007. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 18:297–300. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, and Groot N. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 72:971–983. [DOI] [PubMed] [Google Scholar]
- Mansueto G, Armani A, Viscomi C, D’Orsi L, De Cegli R, Polishchuk EV, Lamperti C, Di Meo I, Romanello V, Marchet S, Saha PK, Zong H, Blaauw B, Solagna F, Tezze C, Grumati P, Bonaldo P, Pessin JE, Zeviani M, Sandri M, and Ballabio A. 2017. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 25:182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marron-Terada PG, Hancock MK, Haskins DJ, and Dahms NM. 2000. Recognition of Dictyostelium discoideum lysosomal enzymes is conferred by the amino-terminal carbohydrate binding site of the insulin-like growth factor II/mannose 6-phosphate receptor. Biochemistry. 39:2243–2253. [DOI] [PubMed] [Google Scholar]
- Marschner K, Kollmann K, Schweizer M, Braulke T, and Pohl S. 2011. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 333:87–90. [DOI] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, and Puertollano R. 2012. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, and Puertollano R. 2016. TFEB and TFE3 are novel components of the integrated stress response. Embo J. 35:479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, and Puertollano R. 2014. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 7:ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, and Puertollano R. 2013. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 200:475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Matsumoto S, Hattori T, Machida Y, and Nakamura K. 1990. Vacuolar targeting and posttranslational processing of the precursor to the sweet potato tuberous root storage protein in heterologous plant cells. J Biol Chem. 265:19750–19757. [PubMed] [Google Scholar]
- Matsuoka K, Watanabe N, and Nakamura K. 1995. O-glycosylation of a precursor to a sweet potato vacuolar protein, sporamin, expressed in tobacco cells. Plant J. 8:877–889. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, and Krainc D. 2011. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, and Ballabio A. 2015. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 17:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, and Ballabio A. 2011. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 21:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole SE, and Cotman SL. 2015. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta. 1852:2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier-Lehmann H, Mauxion F, Bauer U, Lobel P, and Hoflack B. 1996a. Re-expression of the mannose 6-phosphate receptors in receptor-deficient fibroblasts. Complementary function of the two mannose 6-phosphate receptors in lysosomal enzyme targeting. J Biol Chem. 271:15166–15174. [DOI] [PubMed] [Google Scholar]
- Munier-Lehmann H, Mauxion F, Bauer U, Lobel P, and Hoflack B. 1996b. Re-expression of the mannose 6-phosphate receptors in receptor-deficient fibroblasts. Complementary function of the two mannose 6-phosphate receptors in lysosomal enzyme targeting. The Journal of biological chemistry. 271:15166–15174. [DOI] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, and Rader DJ. 2010. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 466:714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, and Morales CR. 2006. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic. 7:889–902. [DOI] [PubMed] [Google Scholar]
- Nielsen MS, Gustafsen C, Madsen P, Nyengaard JR, Hermey G, Bakke O, Mari M, Schu P, Pohlmann R, Dennes A, and Petersen CM. 2007. Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA Mol Cell Biol. 27:6842–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Keat SJ, Hamati JW, Madsen P, Gutzmann JJ, Engelsberg A, Pedersen KM, Gustafsen C, Nykjaer A, Gliemann J, Hermans-Borgmeyer I, Kuhl D, Petersen CM, and Hermey G. 2008. Different motifs regulate trafficking of SorCS1 isoforms. Traffic. 9:980–994. [DOI] [PubMed] [Google Scholar]
- Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, Pohlmann R, and Petersen CM. 2001. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. Embo J. 20:2180–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, and Cuervo AM. 2005. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 64:113–122. [DOI] [PubMed] [Google Scholar]
- Nyborg AC, Ladd TB, Zwizinski CW, Lah JJ, and Golde TE. 2006. Sortilin, SorCS1b, and SorLA Vps10p sorting receptors, are novel gamma-secretase substrates. Mol Neurodegener. 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak Y, Glowacka WK, Bruce MC, Pham N, and Rotin D. 2006. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. J Cell Biol. 175:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Monroe TO, Palmieri M, Sardiello M, and Rodney GG. 2014. Rotenone induces neurotoxicity through Rac1-dependent activation of NADPH oxidase in SHSY-5Y cells. FEBS Lett. 588:472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, and Ballabio A. 2011. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 20:3852–3866. [DOI] [PubMed] [Google Scholar]
- Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, and Sardiello M. 2017a. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 8:14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Pal R, and Sardiello M. 2017b. AKT modulates the autophagy-lysosome pathway via TFEB. Cell Cycle. 16:1237–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ, Barger SW, Mrak RE, and Griffin WST. 2018. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 14:230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, Vetrini F, Palmer D, Ng P, Polishchuk E, Iacobacci S, Polishchuk R, Teckman J, Ballabio A, and Brunetti-Pierri N. 2013. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med. 5:397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, Sardiello M, Ballabio A, and Zheng H. 2014. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 6:1142–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols MS, and Klumperman J. 2009. Trafficking and function of the tetraspanin CD63. Exp Cell Res. 315:1584–1592. [DOI] [PubMed] [Google Scholar]
- Pols MS, van Meel E, Oorschot V, ten Brink C, Fukuda M, Swetha MG, Mayor S, and Klumperman J. 2013. hVps41 and VAMP7 function in direct TGN to late endosome transport of lysosomal membrane proteins. Nature communications. 4:1361. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, and Bonifacino JS. 2001. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 292:1712–1716. [DOI] [PubMed] [Google Scholar]
- Qian Y, Lee I, Lee WS, Qian M, Kudo M, Canfield WM, Lobel P, and Kornfeld S. 2010. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 285:3360–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Roberts A, and Plotz PH. 2007. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 26:45–48. [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, and Rubinsztein DC. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Reczek D, Schwake M, Schroder J, Hughes H, Blanz J, Jin X, Brondyk W, Van Patten S, Edmunds T, and Saftig P. 2007. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 131:770–783. [DOI] [PubMed] [Google Scholar]
- Reddy K, Cusack CL, Nnah IC, Khayati K, Saqcena C, Huynh TB, Noggle SA, Ballabio A, and Dobrowolski R. 2016. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 14:2166–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezgaoui M, Hermey G, Riedel IB, Hampe W, Schaller HC, and Hermans-Borgmeyer I. 2001. Identification of SorCS2, a novel member of the VPS10 domain containing receptor family, prominently expressed in the developing mouse brain. Mech Dev. 100:335–338. [DOI] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, C. International Parkinson’s Disease Genomics, Heutink P, and Shulman JM. 2017. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 140:3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, and Ferguson SM. 2012. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 5:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, Kallemeijn WW, Gaspar P, Aerts JM, Glatzel M, Saftig P, Krainc D, Schwake M, and Blanz J. 2014. LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc Natl Acad Sci U S A. 111:15573–15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval IV, Arredondo JJ, Alcalde J, Gonzalez Noriega A, Vandekerckhove J, Jimenez MA, and Rico M. 1994. The residues Leu(Ile)475-Ile(Leu, Val, Ala)476, contained in the extended carboxyl cytoplasmic tail, are critical for targeting of the resident lysosomal membrane protein LIMP II to lysosomes. J Biol Chem. 269:6622–6631. [PubMed] [Google Scholar]
- Santini E, Heiman M, Greengard P, Valjent E, and Fisone G. 2009. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci Signal. 2:ra36. [DOI] [PubMed] [Google Scholar]
- Sardiello M 2016. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci. 1371:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, and Ballabio A. 2009. A gene network regulating lysosomal biogenesis and function. Science. 325:473–477. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, and Brady RO. 2000. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proceedings of the National Academy of Sciences of the United States of America. 97:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschutter A, and Group CLNS. 2018. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. 378:1898–1907. [DOI] [PubMed] [Google Scholar]
- Schweizer A, Kornfeld S, and Rohrer J. 1997. Proper sorting of the cation-dependent mannose 6-phosphate receptor in endosomes depends on a pair of aromatic amino acids in its cytoplasmic tail. Proceedings of the National Academy of Sciences of the United States of America. 94:14471–14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN 2007. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. Journal of cell science. 120:2378–2389. [DOI] [PubMed] [Google Scholar]
- Seaman MN, McCaffery JM, and Emr SD. 1998. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol. 142:665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI, and Sarkar S. 2017. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61:733–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, and Ballabio A. 2011. TFEB links autophagy to lysosomal biogenesis. Science. 332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, and Ballabio A. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 31:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, and Futerman AH. 2011. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 26:1593–1604. [DOI] [PubMed] [Google Scholar]
- Sharma J, di Ronza A, Lotfi P, and Sardiello M. 2018. Lysosomes and Brain Health. Annu Rev Neurosci. [DOI] [PubMed] [Google Scholar]
- Simmen T, Schmidt A, Hunziker W, and Beermann F. 1999. The tyrosinase tail mediates sorting to the lysosomal compartment in MDCK cells via a di-leucine and a tyrosine-based signal. J Cell Sci. 112 (Pt 1):45–53. [DOI] [PubMed] [Google Scholar]
- Snyder AM, and Connor JR. 2009. Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta. 1790:606–614. [DOI] [PubMed] [Google Scholar]
- Sohar I, Sleat D, Gong Liu C, Ludwig T, and Lobel P. 1998. Mouse mutants lacking the cation-independent mannose 6-phosphate/insulin-like growth factor II receptor are impaired in lysosomal enzyme transport: comparison of cation-independent and cation-dependent mannose 6-phosphate receptor-deficient mice. The Biochemical journal. 330 (Pt 2):903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Wang F, Lotfi P, Sardiello M, and Segatori L. 2014. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J Biol Chem. 289:10211–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Wang F, Savini M, Ake A, di Ronza A, Sardiello M, and Segatori L. 2013. TFEB regulates lysosomal proteostasis. Hum Mol Genet. 22:1994–2009. [DOI] [PubMed] [Google Scholar]
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G, Ballabio A, and Raben N. 2013. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 5:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt C, Puissant E, and Boonen M. 2016. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int J Mol Sci. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch S, Pohl S, and Braulke T. 2004. A dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of the batten disease-related CLN3 protein are required for efficient lysosomal targeting. J Biol Chem. 279:53625–53634. [DOI] [PubMed] [Google Scholar]
- Storch S, Pohl S, Quitsch A, Falley K, and Braulke T. 2007. C-terminal prenylation of the CLN3 membrane glycoprotein is required for efficient endosomal sorting to lysosomes. Traffic. 8:431–444. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, and Roses AD. 1995. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci U S A. 92:4725–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, Rosenbaum H, Schiffmann R, Bembi B, and Sidransky E. 2003. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. 79:104–109. [DOI] [PubMed] [Google Scholar]
- Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, and O’Callaghan M. 2006. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 86:1208–1220. [DOI] [PubMed] [Google Scholar]
- Tiribuzi R, Crispoltoni L, Porcellati S, Di Lullo M, Florenzano F, Pirro M, Bagaglia F, Kawarai T, Zampolini M, and Orlacchio A. 2014. miR128 up-regulation correlates with impaired amyloid beta(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol Aging. 35:345–356. [DOI] [PubMed] [Google Scholar]
- Tong PY, and Kornfeld S. 1989. Ligand interactions of the cation-dependent mannose 6-phosphate receptor. Comparison with the cation-independent mannose 6-phosphate receptor. J Biol Chem. 264:7970–7975. [PubMed] [Google Scholar]
- Tse DY, Lotfi P, Simons DL, Sardiello M, and Wu SM. 2015. Electrophysiological and Histological Characterization of Rod-Cone Retinal Degeneration and Microglia Activation in a Mouse Model of Mucopolysaccharidosis Type IIIB. Sci Rep. 5:17143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng LT, Lin CL, Tzen KY, Chang SC, and Chang MF. 2013. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J Biol Chem. 288:32424–32432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji A, Omura K, and Suzuki Y. 1988. I-cell disease: evidence for a mannose 6-phosphate independent pathway for translocation of lysosomal enzymes in lymphoblastoid cells. Clin Chim Acta. 176:115–121. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, and La Spada AR. 2012. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 4:142ra197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls LA, Winther JR, and Stevens TH. 1990. Yeast carboxypeptidase Y vacuolar targeting signal is defined by four propeptide amino acids. J Cell Biol. 111:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, and Wijburg FA. 2008. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 31:240–252. [DOI] [PubMed] [Google Scholar]
- Vega-Rubin-de-Celis S, Pena-Llopis S, Konda M, and Brugarolas J. 2017. Multistep regulation of TFEB by MTORC1. Autophagy. 13:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayati A, DePaolo J, Gupta N, Choi JH, Moaven N, Westbroek W, Goker-Alpan O, Goldin E, Stubblefield BK, Kolodny E, Tayebi N, and Sidransky E. 2011. A mutation in SCARB2 is a modifier in Gaucher disease. Hum Mutat. 32:1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S, and Puertollano R. 2006. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic. 7:337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodicka P, Chase K, Iuliano M, Tousley A, Valentine DT, Sapp E, Kegel-Gleason KB, Sena-Esteves M, Aronin N, and DiFiglia M. 2016. Autophagy Activation by Transcription Factor EB (TFEB) in Striatum of HDQ175/Q7 Mice. J Huntingtons Dis. 5:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley SU 2009. Pathogenic cascades in lysosomal disease-Why so complex? J Inherit Metab Dis. 32:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang R, Carrera I, Xu S, and Lakshmana MK. 2016a. TFEB Overexpression in the P301S Model of Tauopathy Mitigates Increased PHF1 Levels and Lipofuscin Puncta and Rescues Memory Deficits. eNeuro. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang R, Xu S, and Lakshmana MK. 2016b. Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer’s and Amyotrophic Lateral Sclerosis Brains. Neurosci J. 2016:4732837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, and Shen CK. 2012. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 109:15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlund B, Dahms NM, and Kornfeld S. 1991. The bovine mannose 6-phosphate/insulin-like growth factor II receptor. Localization of mannose 6-phosphate binding sites to domains 1–3 and 7–11 of the extracytoplasmic region. J Biol Chem. 266:23233–23239. [PubMed] [Google Scholar]
- Westphal V, Marcusson EG, Winther JR, Emr SD, and van den Hazel HB. 1996. Multiple pathways for vacuolar sorting of yeast proteinase A. J Biol Chem. 271:11865–11870. [DOI] [PubMed] [Google Scholar]
- Willett R, Martina JA, Zewe JP, Wills R, Hammond GRV, and Puertollano R. 2017. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat Commun. 8:1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, and Selkoe DJ. 1999. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 398:513–517. [DOI] [PubMed] [Google Scholar]
- Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, and Perlmutter DH. 1994. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 91:9014–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, and Lee JM. 2014. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 34:9607–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL, Czerniewski L, Ballabio A, Cirrito JR, Diwan A, and Lee JM. 2015. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci. 35:12137–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Chen X, Huang S, Li G, Mo M, Zhang L, Chen C, Guo W, Zhou M, Wu Z, Cen L, Long S, Li S, Yang X, Qu S, Pei Z, and Xu P. 2018. Iron promotes alpha-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J Neurochem. 145:34–50. [DOI] [PubMed] [Google Scholar]
- Zachos C, Blanz J, Saftig P, and Schwake M. 2012. A critical histidine residue within LIMP-2 mediates pH sensitive binding to its ligand beta-glucocerebrosidase. Traffic. 13:1113–1123. [DOI] [PubMed] [Google Scholar]
- Zeng J, Racicott J, and Morales CR. 2009. The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp Cell Res. 315:3112–3124. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu P, Dwyer NK, Christenson LK, Fujimoto T, Martinez F, Comly M, Hanover JA, Blanchette-Mackie EJ, and Strauss JF 3rd. 2002. MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J Biol Chem. 277:33300–33310. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ren J, Padilla-Parra S, Fry EE, and Stuart DI. 2014. Lysosome sorting of beta-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat Commun. 5:4321. [DOI] [PMC free article] [PubMed] [Google Scholar]