

Abstract
Toxoplasma gondii is an intracellular parasite that causes disseminated infections that can produce neurological damage in fetuses and immunocompromised individuals. Microneme protein 2 (MIC2), a member of the thrombospondin-related anonymous protein (TRAP) family, is a secreted protein important for T. gondii motility, host cell attachment, invasion, and egress. MIC2 contains six thrombospondin type I repeats (TSRs) that are modified by C-mannose and O-fucose in Plasmodium spp. and mammals. Here, using MS analysis, we found that the four TSRs in T. gondii MIC2 with protein O-fucosyltransferase 2 (POFUT2) acceptor sites are modified by a dHexHex disaccharide, whereas Trp residues within three TSRs are also modified with C-mannose. Disruption of genes encoding either POFUT2 or the putative GDP–fucose transporter (NST2) resulted in loss of MIC2 O-fucosylation, as detected by an antibody against the GlcFuc disaccharide, and in markedly reduced cellular levels of MIC2. Furthermore, in 10–15% of the Δpofut2 or Δnst2 vacuoles, MIC2 accumulated earlier in the secretory pathway rather than localizing to micronemes. Dissemination of tachyzoites in human foreskin fibroblasts was reduced for these knockouts, which both exhibited defects in attachment to and invasion of host cells comparable with the Δmic2 phenotype. These results, indicating that O-fucosylation of TSRs is required for efficient processing of MIC2 and for normal parasite invasion, are consistent with the recent demonstration that Plasmodium falciparum Δpofut2 strain has decreased virulence and also support a conserved role for this glycosylation pathway in quality control of TSR-containing proteins in eukaryotes.
Keywords: invasion, Toxoplasma gondii, fucosyltransferase, glycosylation, glycoprotein secretion, apicomplexa parasite, MIC2, O-fucosylation, protein glycosylation, thrombospondin-like repeat
Introduction
Toxoplasma gondii is a eukaryotic parasite that belongs to the Apicomplexa phylum, together with other medically relevant parasites, such as Plasmodium spp. and Cryptosporidium parvum. T. gondii, which has the ability to invade all warm-blooded animals, causes transient, disseminated infections that can lead to neurological damage in immunocompromised individuals and developmental defects in fetuses (1, 2). T. gondii replicates asexually in all of its hosts, with the exception of felids (cats), where the parasite also undergoes a sexual cycle, which concludes in production and shedding of infectious oocysts in the feces (3).
Because T. gondii is an obligate, intracellular pathogen, its virulence depends on the ability to invade host cells. The parasite has specialized secretory organelles, which localize at the apical end and whose content is sequentially secreted during the invasion process (4). Secretion of microneme proteins depends upon elevation of parasite calcium levels and is the first event upon parasite attachment (4, 5).
Microneme protein 2 (MIC2)4 is a member of the thrombospondin-related anonymous protein (TRAP) family (6). MIC2 is a type 1 membrane protein composed of an A/I von Willebrand domain, six thrombospondin-like repeats, and a short cytoplasmic tail (7). The A/I domain has been shown to be involved in recognition of host cells, possibly through binding to ICAM-1, heparin, and sulfated glycosaminoglycans (8–10). MIC2 forms a hexameric complex with the MIC2-associated protein (M2AP) in the endoplasmic reticulum; together, they traffic to the micronemes and, upon secretion, to the apical cell surface (8, 11). As host cell penetration concludes, the MIC2/M2AP complex relocates to the posterior end of the parasite and is released into the environment by cleavage of the C-terminal transmembrane helix that is catalyzed by the MPP1 protease (7, 12).
Although knockout of the mic2 gene strongly affects motility and invasion, MIC2 is not essential for parasite viability. Tachyzoites lacking this protein still glide, attach to, and invade fibroblasts, albeit inefficiently (13). Numerous studies have shown that correct trafficking of the M2AP/MIC2 complex to the micronemes is important for its function. Knockout of m2ap results in inefficient localization of MIC2 to the micronemes and defects in the parasite's ability to glide and invade (14). Cleavage-resistant mutations of m2ap, which affect protein maturation and consequently localization of the M2AP/MIC2 complex, also result in inefficient gliding and attachment (15, 16).
Thrombospondin-like repeats, which are each about 60 amino acids long, are present in many extracellular proteins. TSRs have a conserved structure characterized by a tryptophan ladder composed of stacked Trp and arginine residues and a set of conserved cysteines that are involved in disulfide bridges (17). In metazoans, TSRs are glycosylated via two pathways: O-fucosylation by action of the protein O-fucosyltransferase 2 (POFUT2) and C-mannosylation by the C-mannosyltransferase DPY-19 (18, 19). C-Man is transferred via an unusual carbon–carbon bond to the Trp on a WXXWXXC1 sequence, where C1 is the first conserved cysteine in the TSR (20). Plasmodium falciparum and T. gondii each encode for a DPY-19 orthologue that can transfer C-Man to the Trp residues in the conserved motif, both in vitro and in vivo (21).
POFUT2 transfers α-fucose to the hydroxyl group on Ser/Thr residues in the consensus sequence C1X2–3(S/T)C2X2G, where C1 and C2 are the first two conserved cysteines (22). In metazoans, fucose is transferred on already folded TSR domains (23) and can be elongated by a β1,3-glucose by action of the glucosyltransferase B3GLCT (24). Mass spectrometry analyses of P. falciparum TRAP and circumsporozoite protein indicate that a hexose (Hex) residue elongates the fucose on Apicomplexa TSRs; this sugar may be glucose, but it has not yet been unequivocally identified (21, 25).
TSRs glycosylation is carried out in the endoplasmic reticulum (22, 26), and in studies performed in mammalian cell and Caenorhabditis elegans, defects in both C-mannosylation and O-fucosylation have been shown to affect protein stabilization and secretion, albeit with protein-to-protein variability (19, 23, 27–31). A knockout of pofut2 is embryonically lethal in mice (31). The human recessive disorder Peter Plus syndrome is caused by mutations in the B3GLCT glycosyltransferase (23). Recently, a knockout of P. falciparum pofut2 showed that O-fucosylation of TSRs is required for efficient mosquito infection by ookinetes and for hepatocyte invasion by sporozoites (32). These phenotypes are due to defective folding/stabilization of the parasite TSR-containing proteins, such as TRAP and circumsporozoite protein, consistent with the observations made in metazoans and a potential role for O-fucosylation in efficient folding and/or stabilization of TSRs (23, 32).
In this paper, we use MS to show that four of six TSRs of MIC2 are modified by O-fucose and that in addition to TSR5 (as shown by Hoppe et al. (25)), two other TSRs are modified with C-Man. We also identify the putative POFUT2 and GDP–fucose transporter (NST2) and show that knockout of either pofut2 or nst2 results in loss of O-fucosylation of MIC2, defects in the protein stability and localization, and a decrease in the ability of parasites to invade host cells.
Results
Four TSRs of MIC2 are modified by a dHex with or without Hex
Glycosylation on tachyzoite secreted proteins was analyzed as described (25). Briefly, secretion was induced with 1% ethanol (7), and the peptides resulting from reduction, alkylation, and trypsin digestion of the secreted protein fraction were analyzed by nano-LC-MS/MS using a Thermo Q Exactive quadrupole Orbitrap MS with low-energy CID. Following this discovery run, selected peptide species containing the O-fucosylation motif were further analyzed by higher-collision energy dissociation (HCD) fragmentation and by electron transfer dissociation (ETD), which allows fragmentation of the peptide backbone while preserving post-translational modifications (33).
The initial analysis identified numerous peptides from MIC2 that cover about 66% of the secreted sequence (7) (Fig. S1). In silico mining of the T. gondii predicted proteome indicated the possibility for detection of three additional TSR-containing proteins encoding the POFUT2 consensus motif: MIC14, TGGT1_209060, and TGGT1_223480 (Table S1). However, we did not observe any peptides from these three T. gondii proteins.
MIC2 has six contiguous TSRs. All six contain C-mannosylation sites, whereas only four (TSR1, -3, -4, and -5) have the consensus O-fucosylation motif (C1X2–3(S/T)C2X2G) (Fig. 1A). The targeted HCD and ETD experiments allowed localization of a dHexHex disaccharide on the predicted POFUT2 acceptor residues on MIC2 TSR1 and TSR4 and a dHex with or without Hex on TSR5 (25). TSR3 is also modified with a dHexHex, although the modification site could not be assigned (Table 1, Figs. 1–3, and Fig. S3). TSR6 lacks a consensus site for POFUT2 and is not modified by C-Man; TSR2 lacks an O-fucosylation consensus site and was not sampled. TSR3 and TSR4, in addition to the previously described TSR5 (25), are modified with C-Man. Last, whereas MIC2 contains four potential N-glycosylation sites, we observed peptides containing unmodified Asn at residues 463, 470, and 680, suggesting that these N-glycan sites are unoccupied (Fig. S1). The fourth potential N-glycosylation site was not observed.
Figure 1.

MIC2 TSR1, -3, -4, and -5 are modified by dHexHex and TSR3, -4, and -5 are also C-mannosylated. A, amino acid sequences of the six TSR domains of MIC2. Cys resides are labeled in green, the O-fucosylation acceptor Ser/Thr is marked in red. C-Mannosylation sites are shown in blue. B, ETD of a semi-tryptic peptide from TSR1. The presence of the three product ions z9, z10, and y10 containing dHexHex identifies Ser-285, the predicted POFUT2 acceptor, as the glycosylated residue. C, HCD fragmentation of a TSR3 semi-tryptic peptide is consistent with modification by dHexHex disaccharide plus two C-mannoses. Collision energy of 10 eV (top) shows ion species corresponding to the precursor minus a Hex, a dHexHex, or minus dHexHex and 120.04 Da (broken green circle). Increasing the collision energy to 15 eV (bottom) allows detection of the precursor minus Hex, dHexHex, and two Hex −120.04 Da fragments. D, ion trap ETD of a semi-tryptic peptide from TSR4 identified the z7 product ion carrying both Hex and dHex, indicating Ser-485, the predicted POFUT2 acceptor site, as the residue modified by a dHexHex disaccharide. E, 10-eV HCD fragmentation of the Hex2dHex glycoform, m/z 1120.7058 (top), and the Hex3dHex ion species, m/z 1161.2106 (bottom), is shown. The most abundant ion, corresponding to the precursor minus the O-linked sugar, was the same for both species, consistent with the difference between the glycoforms being the Hex elongating the O-fucose and not one of the C-Man. Red triangle, fucose; green circle, mannose; broken green circle, C-Man cross-ring fragment; white circle, Hex; *, cysteine carbamidomethylation.
Table 1.
Peptides and glycopeptides from the TSRs of MIC2 detected in this study
| TSR | Amino acid sequence | Modification | m/z | z |
|---|---|---|---|---|
| 1 | SVSCDGSQIR | HexdHex + 1 CAMa | 737.3218 | 2 |
| 1 | SVSCDGSQIR | 1 CAM | 583.2670 | 2 |
| 3 | ITIPPTPPEWAAWSDCTVTCGGGNR | Hex3dHex + 2 CAM | 1125.8290 | 3 |
| 4 | QEEACNTSPCPINATCGQFEEWSTCSVSCGGGLK | Hex2dHex + 5 CAM | 1431.2498 | 3 |
| 4 | SVSCGGGLK | HexdHex + 1 CAM | 586.7708 | 2 |
| 5 | TETVTCNPQACPVDERPGEWAEWGECSVTCGDGVR | Hex3dHex + 4 CAM | 1161.2313 | 4 |
| 5 | TETVTCNPQACPVDERPGEWAEWGECSVTCGDGVR | Hex2dHex + 4 CAM | 1120.7179 | 4 |
| 5 | ACPVDERPGEWAEWGECSVTCGDGVR | Hex2dHex + 3 CAM | 1150.4752 | 3 |
| 5 | PGEWAEWGECSVTCGDGVR | Hex3dHex + 2 CAM | 928.7054 | 3 |
| 5 | PGEWAEWGECSVTCGDGVR | Hex2dHex + 2 CAM | 1311.5304b | 2 |
| 5 | SVTCGDGVR | HexdHex + 1 CAM | 629.7768 | 2 |
| 5 | SVTCGDGVR | dHex + 1 CAM | 548.7500 | 2 |
| 6 | CTYVWSDWNK | 1CAM | 679.7951 | 2 |
a CAM, cysteine carbamidomethylation.
b Described previously by Hoppe et al. (25).
Figure 2.

HCD MS/MS of an O-fucosylated and C-mannosylated glycopeptide from TSR3 of MIC2. An HCD MS/MS spectrum was recorded with collision energy of 25 eV. Detection of the singly charged b11 and doubly charged y13, y13-NH3 ions allows positioning of C-Man residues to the first and second Trp residues of the WXXWXXC motif, respectively. Other ions of interest are the doubly charged y19 (two Hex residues) and y22 (two intact Hex(s) or one intact Hex plus one Hex −120.04 Da fragment). Red triangle, fucose; green circle, mannose; white circle, Hex; *, cysteine carbamidomethylation.
Figure 3.

HCD MS/MS of an O-fucosylated and C-mannosylated glycopeptide from TSR4 of MIC2. HCD MS/MS spectrum was obtained with a collision energy of 20 eV. Detection of the Hex in the y-series ions only from y13 onward, both on singly and doubly charged species, confirms positioning of the Hex on the Trp in the WXXC motif, consistent with C-mannosylation. The y7 + dHexHex ion supports the presence of the disaccharide on Ser-485, the predicted POFUT2 site. Red triangle, fucose; green circle, mannose; white circle, Hex; *, cysteine carbamidomethylation.
A TSR1 semi-tryptic peptide, 282SVSCDGSQIR294, modified with a dHex and a Hex, was observed as the [M + 2H]2+ ion at m/z 737.3218 (Table 1). ETD fragmentation on an LTQ-Orbitrap resulted in three product ions that carried both Hex and dHex: z9, z10, and y10 (Fig. 1B). No other dHex- and/or Hex-containing product ions were observed. These observations allow assignment of Ser-285, the predicted POFUT2 acceptor site, as the glycosylated residue and indicate that the two sugars compose a disaccharide modifying this serine (22). It should be noted that this analytical technique cannot define the hexose monosaccharide substituting the dHex. The same peptide was also observed as a [M + 2H]2+ ion without any sugar modification at m/z 583.2670 (Table 1). Comparison of the extracted ion chromatograms for the two species suggests that the glycosylated version is the more abundant of the two (Fig. S2A).
For the MIC2 TSR3, we observed the semi-tryptic peptide 399ITIPPTPPEWAAWSDCTVTCGGGNR425 modified with a dHex and 3 Hex as a [M + 3H]3+ species at m/z 1125.8290 (Table 1). HCD fragmentation using a low collision energy setting (10 eV) resulted in the detection of triply charged product ions corresponding to loss of Hex from the precursor (m/z 1071.8176), loss of dHexHex (m/z 1023.1157), and subsequent loss of 120.04 Da, resulting from a cross-ring cleavage characteristic of C-mannose (m/z 983.1221) (Fig. 1C) (20). Increasing the collision energy to 15 eV resulted in the detection of the ion species observed at 10 eV plus the precursor minus Hex, dHexHex, and two Hex fragments originating from cross-ring cleavages (m/z 943.0975). These results are consistent with peptide modification with two C-Man residues and a dHexHex disaccharide on the POFUT2 site. Additionally, loss of the Hex and then the dHex is consistent with the dHex as the monosaccharide that is attached to the protein. We also observed a lower-abundance ion corresponding to loss of dHex from the precursor (m/z 1077.1379; Fig. 1C). This ion could indicate either a minor glycoform where the Hex is the monosaccharide attached to the protein or a rearrangement of the glycan during the MS analysis (34). Because rearrangement should be observed for other O-fucosylated TSRs, we analyzed tryptic peptides from human thrombospondin 1 (TSP1). HCD MS/MS of a semi-tryptic peptide from the TSR2 of TSP1, using a collision energy of 15 eV, resulted in a product ion ([M + 2H]2+ m/z 663.8168) corresponding to the precursor after loss of a dHex (Fig. S3). This observation is consistent with the M-dHex ion resulting from rearrangement of the dHexHex. Confirmation of the presence of Hex residues on each of the two Trp in the WXXWXXC motif was obtained by HCD fragmentation using higher collision energies (20 and 25 eV), which resulted in almost complete elimination of the precursor ion (Fig. 2). In particular, detection of the doubly charged y13 and y13-NH3 ions allowed positioning of one C-Man at the second Trp of the WXXWXXC motif, whereas the singly charged b11 was diagnostic of the presence of another C-Man on the first Trp of the motif. These assignments were supported by the detection of the doubly charged ions y19 (two intact Hex residues, m/z 1223.0099) and y22 (two intact Hex(s), m/z 1370.5773), accompanied by product ions that arise via loss of water or 120.04 Da from y22 (m/z 1361.5885 and m/z 1310.5645, respectively) (Fig. 2). We observed neither the unmodified peptide nor additional glycoforms.
Two peptides were observed for the MIC2 TSR4: 457QEEACNTSPCPINATCGQFEEWSTCSVSCGGGLK492 as a [M + 3H]3+ species, modified with dHex and 2 Hex, at m/z 1431.2498 and a smaller semi-tryptic peptide, 482SVSCGGGLK492 as an [M + 2H]2+ species, modified with a dHexHex, at m/z 586.7708 (Table 1). ETD analysis of the smaller semi-tryptic peptide (m/z 586.7708) showed the presence of the z7 product ion carrying both Hex and dHex. This observation allowed assignment of Ser-485, the predicted POFUT2 acceptor site, as the residue modified by dHexHex (Fig. 1D). Additionally, HCD fragmentation using a low-collision energy setting (10 eV) of the [M + 3H]3+ species showed ions corresponding to the precursor after loss of an intact Hex (m/z 1377.2067), a Hex and dHex (m/z 1328.5223), or a dHexHex and a 120.04 Da from a Hex cross-ring cleavage (m/z 1288.5155) (Fig. S4). These results are consistent with modification of the Trp on the WXXC motif with one Hex and a dHexHex disaccharide modifying the O-fucosylation site. HCD fragmentation using higher collision energy (20 eV) allowed confirmation of Trp modification by a Hex upon detection of the sugar in the y-series ions only from y13 onward, both as singly and doubly charged species (Fig. 3). HCD fragmentation also identified a y7 + dHexHex ion, consistent with the presence of the disaccharide on Ser-485. Neither additional glycoform nor the unmodified peptide were observed.
Last, multiple peptides containing the C-mannosylation and O-fucosylation sites were observed for MIC2 TSR5. Although tryptic cleavage rarely occurs when proline follows arginine, the fully tryptic peptide 533PGEWAEWGECSVTCGDGVR553 modified by dHexHex2 was observed as a [M + 2H]2+ at m/z 1311.5304 and the semi-tryptic peptide with one missed cleavage 526ACPVDERPGEWAEWGECSVTCGDGVR553 also carrying dHexHex2 as [M + 3H]3+ at m/z 1150.4752 (Table 1). HCD MS/MS for the glycopeptide at m/z 1311.5304 has been shown previously (25); it demonstrated that both Trp residues in the WXXWXXC motif are modified with Hex. The previously published HCD analysis also detected a y7 + dHex ion, consistent with the presence of deoxyhexose on Thr-546 (25). In this study, we also observed the fully tryptic peptide 533PGEWAEWGECSVTCGDGVR553 as [M + 3H]3+ m/z 928.7054, consistent with its modification by dHex and three Hex residues. Additionally, we observed the tryptic peptide (one missed cleavage) 517TETVTCNPQACPVDERPGEWAEWGECSVTCGDGVR553 modified with dHexHex2 ([M + 4H]4+ m/z 1120.7179) and with dHexHex3 ([M + 4H]4+ m/z 1161.2313). HCD fragmentation using low collision energy (10 eV) was performed on both species. The identified fragment ions are consistent with the difference between these two glycoforms being a Hex on the O-fucosylation site because the m/z value measured for the most abundant ion, corresponding to the precursor minus the O-linked sugar, was the same within 10 ppm: m/z 1084.2029 and 1084.1923, respectively (Fig. 1E). Comparison from the extracted ion chromatograms for m/z 1120.7179 and 1161.2313 suggests that these two glycoforms have similar abundances (Fig. S2B). Finally, a semi-tryptic peptide containing the TSR5 O-fucosylation site, 543SVTCGDGVR553, was also observed in two different glycoforms: modified with only a dHex ([M + 2H]2+ m/z 548.7500) and with dHexHex ([M + 2H]2+ m/z 629.7768).
In conclusion, all four MIC2 TSRs with POFUT2 acceptor sites are modified by a dHexHex disaccharide, consistent with FucGlc, and TSR5 can also be modified by only a dHex. Furthermore, Trp residues on TSR3 and -4, in addition to the already described TSR5 (25), are modified with C-mannose.
Identification and gene disruption of T. gondii orthologues of host POFUT2 and GDP–fucose transporter
The T. gondii genome encodes for a putative POFUT2 (TGGT1_273550), which belongs to the CAZy GT68 family of glycosyltransferases that includes Plasmodium and human POFUT2 (Fig. S5) (35). Three conserved motifs, which are shared with the members of the α1,2-, α1,6-, and O-FTs superfamily (black boxes), can be identified, including Motif I that forms part of the substrate binding site (36). A GT68-specific region (amino acids 229–357) includes the catalytic glutamic acid and some of the residues involved in TSR recognition (gray boxes in Fig. S4) (36, 37). T. gondii POFUT2 also has a hydrophobic N-terminal sequence (amino acids 1–180), which is absent from the other POFUT2s identified to date (Fig. S5). Using CRISPR/Cas9 gene editing (38) in the RH ΔKu80 strain, which is deficient in the nonhomologous end-joining DNA repair system (39), we added a sequence encoding a C-terminal 3xMYC tag to the endogenous pofut2 locus. Specifically, Cas9 was directed to excise the pofut2 gene 20 bp upstream of the stop codon, and an oligonucleotide, flanked by about 50 bp of homology sequence to the break site, was provided as template for the homologous recombination. (Fig. 4A and Table S2). Insertion of the 3xMYC tag at the correct locus was confirmed by PCR from genomic DNA extracted at 72 h post-electroporation. A reaction product was observed only when cells were electroplated with the CRISPR/Cas9 construct directing to the pofut2 locus and the 3xMYC recombination sequence (Fig. 4B and Table S2). The parasites were analyzed by immunofluorescence at 24 h post-electroporation to look for MYC-positive vacuoles. T. gondii POFUT2–3xMYC has a similar distribution to P30HDEL-YFP (40) (Fig. 4C), showing that POFUT2 is expressed in tachyzoites and, for the most part, localizes to the endoplasmic reticulum, as expected (22).
Figure 4.

Localization of POFUT2 and generation of Δpofut2 and Δnst2 mutants. A, schematic representation of the strategy used to in situ tag pofut2 and the regions recognized by the primers used in B. B, PCR to show integration of the 3xMYC tag in the correct locus (P35 + P40). A control reaction amplifying a fragment of the pofut2 ORF was also performed (P37 + P38). C, IFA of RH ΔKu80 tachyzoites expressing pofut2 tagged at the C terminus with a 3xMYC tag and an ER marker (p30HDEL-YFP) shows partial co-localization. Scale bar, 2 μm. D, schematic representation of the strategy used to generate the pofut2 and nst2 gene disruptions. Cas9 was directed to excise each gene in the first and last exon. An mGFP-expressing cassette was inserted in each locus. The primers used in E and F are marked by arrows. E and F, genomic DNA was extracted after a first enrichment for mGFP-positive cells (mixed population) and after the final cloning step (KO). PCR analyses show substitution of the WT ORF with the mGFP cassette in both strains. Mixed pop, mixed population. All primer sequences can be found in Table S2. G, Δpofut2 parasites were complemented by expression of T. gondii POFUT2 fused to a 3xMYC C-terminal tag. MYC staining by IFA is consistent with ER localization. Scale bar, 2 μm.
Transport of the activated sugar GDP–Fuc from the cytosol to the lumen of the ER is required for POFUT2 activity. A BLASTP search, using the human Golgi GDP–Fuc transporter as template, identified a single putative GDP–sugar transporter, TGGT1_267730 or NST2. There are 10 predicted transmembrane helices (41) and conserved nucleotide and sugar binding motifs (black dashed boxes) at the C terminus (42) (Fig. S6).
Using CRISPR/Cas9 in the RH ΔKu80 strain, the coding regions of T. gondii pofut2 and nst2 were disrupted. Briefly, Cas9 was directed to excise each gene in the first and last exon. A cassette expressing mGFP, under the gra1 promoter (43) and flanked by about 30 bp of homology sequence to each break site, was provided as template for the homologous recombination (Fig. 4D and Table S2). After selection for mGFP-positive cells, clones were obtained by limiting dilution. Integration at the correct locus was verified by diagnostic PCR for both Δpofut2 (Fig. 4E) and Δnst2 (Fig. 4F) clones (see Table S2 for primer sequences). In both cases, PCR with locus-specific primers (Table S2) resulted in a lower-molecular weight band upon integration of the mGFP cassette. Additionally, a reaction product using a mGFP-specific reverse primer was observed only in the Δpofut2 and Δnst2 cell lines and not in the parental strain (Fig. 4, E and F).
Additionally, Δpofut2 was complemented by electroporation of a construct expressing T. gondii pofut2 cDNA fused to a C-terminal 3xMYC tag under the control of tubulin 5′-UTRs (ptubPOFUT2–3xMYC/sagCAT). Selection with chloramphenicol was used to isolate the parasites expressing POFUT2–3xMYC and immunofluorescence analysis showed that the resulting population is positive for anti-MYC, and the observed staining pattern is consistent with ER localization (Fig. 4G).
MIC2 is not O-fucosylated in Δpofut2 and Δnst2
To determine the glycosylation status of the mutants, we used an antibody against the disaccharide FucGlc. To generate the antibody, chickens were immunized with Glc-β-1,3-Fuc-α-KLH (16) as antigen (Fig. 5A and supporting information), and the disaccharide-specific polyclonal IgY were purified from the antibody fraction via Glc-β-1,3-Fuc-α-agarose beads. To analyze the anti-GlcFuc IgY specificity, the disaccharide and the two monosaccharides (Fuc and Glc) were conjugated to BSA. As there is no evidence of O-Glc directly linked to proteins in T. gondii, we used only BSA-GlcFuc and BSA-Fuc for ELISA and Western blotting assessment of antibody specificity. Antibody titration by ELISA identified 1 μg/ml as the concentration with the best signal-to-noise ratio (Fig. 5B and Fig. S7). Western blot analysis also confirmed the antibody specificity for BSA-GlcFuc, although a minor reactivity to BSA-Fuc was also observed (Fig. 5C).
Figure 5.

MIC2 is not O-fucosylated in either Δpofut2 and Δnst2 strains. A, structure of Glc-β-1,3-Fuc-α-KLH antigen. B, ELISA of the purified anti-GlcFuc antibody (1 μg/ml) shows the specificity of the polyclonal IgY for BSA-GlcFuc versus BSA-Fuc. C, Western blot testing of purified anti-GlcFuc antibody confirms its specificity for BSA modified with the GlcFuc disaccharide. No reactivity is observed probing with the preimmune IgY fraction. D, Western blot analysis with anti-FucGlc antibody identifies a β-elimination–sensitive band in WT corresponding to MIC2. No reactivity is shown in all three knockouts analyzed. MIC2 and tubulin controls are also shown. E, Western blotting of tachyzoites cell lysate with monoclonal anti-MIC2 shows reduced levels of cellular MIC2 in both knockouts. F, MIC2 levels were normalized to tubulin, and the average of three biological repeats is shown. G, Western blot analysis shows that complementation with POFUT2 restores reactivity to the anti-GlcFuc antibody, consistent with restored O-fucosylation. Additionally, MIC2 levels are again comparable with WT. Student's t test was used to compare the samples, and significant differences are marked: *, 0.05 < p < 0.01; **, p = 0.001. Error bars, S.D.
When total cell lysates from the parental strain and the O-fucosylation mutants were analyzed by Western blotting using the antibody against GlcFuc, a β-elimination–sensitive band at an apparent molecular mass of about 110 kDa was identified in WT lysate, but not in the Δpofut2 and Δnst2 lysates (Fig. 5D). Additionally, the anti-GlcFuc antibody did not bind to Δmic2 lysate, confirming that the band observed in WT is indeed MIC2 and that MIC2 TSRs are not being O-fucosylated in the two O-fucosylation mutants (Fig. 5D). These results also indicate that the dHexHex identified by MS is indeed GlcFuc. Furthermore, complementation of Δpofut2 with the T. gondii enzyme restored MIC2 reactivity against the anti-GlcFuc (Fig. 5G).
Using the fucose-specific Aleuria aurantia lectin (AAL), we have previously described O-fucosylated proteins at the nuclear periphery of T. gondii (44). To assess the role of TgPOFUT2 in nuclear O-fucosylation, we analyzed the mutant parasites by AAL immunofluorescence. No effect on AAL binding was observed upon knockout of pofut2, consistent with nuclear O-fucosylation being mediated by a different glycosyltransferase (Fig. S8).
Loss of O-fucosylation resulted in a decrease of more than 50% in cellular MIC2 levels in both mutants, as determined by Western blot analysis (Fig. 5, E and F). Additionally, analysis of cell lysate from Δpofut2 + POFUT2 parasites indicates that complementation rescued the defect in MIC2 cellular levels (Fig. 5G). This result suggests that O-fucosylation may be important for protein folding or processing of MIC2, because nonfucosylated MIC2 is not being processed as efficiently as WT MIC2.
Absence of O-fucosylation results in defective MIC2 localization to the micronemes
Localization of MIC2 was analyzed by immunofluorescence microscopy. As shown in the left panel of Fig. 6A, 12–15% of vacuoles in both Δpofut2 and Δnst2 show an accumulation of MIC2 to the early/mid-secretory pathway (about 15% of vacuoles for Δpofut2 and 12% for Δnst2; Fig. 6B). The same abnormal accumulation was observed for M2AP (Fig. 6A, middle), but not for AMA4 (Fig. 6, A (right) and B), a protein with no TSRs and no reported strong association with MIC2. These observations suggest that we are observing an effect specific to O-fucosylated proteins and not to all micronemal proteins. In both the Δpofut2 and Δnst2 strains, transiently expressed GRASP55-mRFP, a cis-Golgi marker (40), partially co-localizes with the fraction of MIC2 that accumulates in the early/mid-secretory pathway (Fig. 7A). IFA also suggests that GRASP55 distribution is aberrant in these vacuoles, as it looks more diffused and unlike the observed cisternae-shaped structure observed in the parental strain (Fig. 7A). No co-localization was observed between MIC2 and the ER-resident protein P30HDEL-mCherry (Fig. 7B). Both the reduction of MIC2 levels and its defective localization are consistent with a role for O-fucosylation in protein folding and secretion, as has been described in humans and P. falciparum (23, 32).
Figure 6.

MIC2 is mislocalized in both Δpofut2 and Δnst2 strains. A, MIC2 and, consequently, M2AP localization is affected in the KO, with both proteins accumulating in the early secretory pathway in a fraction of the vacuoles (white arrows). This is not a general microneme defect, as AMA4 location is unaffected. The white squares indicate the areas magnified on the right. Merge, antibody staining (red), DAPI (blue), and mGFP (green) for the O-fucosylation mutants. B, quantification of the percentage of vacuoles with abnormal MIC2, M2AP, and AMA4 localization. The average of three biological repeats ± S.D. (error bars) is shown. Student's t test was used to compare the samples, and significant differences are marked: *, 0.05 < p < 0.01; **, 0.01 < p < 0.0001; ***, p < 0.0001. n.s., not significant.
Figure 7.

Accumulated MIC2 partially co-localizes with a cis-Golgi marker. Parasites were transiently electroporated with GRASP55-mRFP (A) or P30HDEL-mCherry (B) and analyzed by structured illumination microscopy. Mislocalized MIC2 partially co-localizes with the cis-Golgi marker (GRASP55), but not with an ER-resident protein (P30HDEL). Additionally, GRASP55 localization itself is aberrant (A, arrowheads). White arrows, accumulation of MIC2 in the early/mid-secretory pathway. White squares, areas magnified below. Scale bars, 2 μm.
Phenotypic analyses show a defect in attachment and invasion
The genome-wide CRISPR/Cas9 screen that was performed in T. gondii (45) identified both pofut2 and nst2 as nonessential genes (phenotypic scores of −0.34 and +1.23, respectively). The ability of the two O-fucosylation–deficient strains to disseminate was determined by assessing plaque formation on human foreskin fibroblast (HFF) monolayer. Here, we found that both Δpofut2 and Δnst2 displayed a 40% reduction in the number of plaques compared with the parental strain (Fig. 8, A and B), whereas no difference between mutants and WT was observed in the average area of the plaques (Fig. S9). These observations are usually indicative of a defect in the parasite's ability to either invade or egress from the host cells.
Figure 8.

Growth, attachment, and invasion are affected in Δpofut2 and Δnst2. A, representative images from the plaque assay. Scale bars, 2 mm. B, quantification of the number of plaques shows a 40% reduction plaque formation in the KOs compared with the parental strain. The average of four biological repeats ± S.D. (error bars) is shown. ***, p < 0.0005. C, representative images from the red/green invasion assays. Attached parasites look yellow (red + green), and invaded parasites are green. D, box plot for the red/green assay, comparing the invasion rate between parental lines and mutants (ratio of invaded to total parasites). E, box plot for the red/green assay, comparing the invasion rates between parental strain, Δpofut2, and the complemented parasites (Δpofut2 + POFUT2). Student's t test was used to compare the samples. and significant differences are marked: *, 0.05 < p < 0.01; **, p < 0.001; ***, p < 0.0005; ****, p < 0.00001. All p values are reported in Table S3. Scale bars, 5 μm.
Phenotypic assays were performed to analyze the knockouts' ability to attach, invade, and egress. Δpofut2 and Δnst2 were compared with the parental strain and the recently published Δmic2 (13), as the data presented here indicate that MIC2 is the most abundant O-fucosylated protein in tachyzoites and is incorrectly processed in both knockouts. Attachment and invasion were assessed via the previously described red/green assay (Fig. 8C) (14). Briefly, extracellular parasites were labeled with anti-SAG1, and after permeabilization, all parasites were labeled with anti-β-tubulin. As a result, attached parasites were stained with both antibodies (red + green) and invaded parasites only with anti-β-tubulin (green). A significant reduction in the total number of parasites that attached to or invaded the host cells (HFF) was observed in all three knockout cell lines, compared with the RH Δku80 strain (Fig. 8D). This phenotype was rescued when the Δpofut2 parasites were complemented with the endogenous enzyme (Δpofut2 + POFUT2) (Fig. 8E). Additionally, no statistically significant difference was observed between Δpofut2 or Δnst2 and Δmic2, suggesting that lack of O-fucosylation is sufficient to reproduce the attachment/invasion defect observed when MIC2 is not being synthesized (Table S3). Furthermore, there was no statistically significant difference between pofut2 and nst2 knockouts.
Egress was stimulated with the addition of the calcium ionophore A23178, as described under “Experimental procedures,” and intact, permeabilized, or completely egressed parasitophorous vacuoles (PVs) were quantified (Fig. 9A). As shown in Fig. 9B and Table 2, all three knockouts had a significantly larger proportion of PVs that were just permeabilized, compared with the RH Δku80 strain. However, the egress defect in Δmic2 was found to be significantly more severe than in either Δpofut2 or Δnst2 (Table 2). The increased egress defect in the Δmic2 strain may explain the markedly reduced infectivity in culture of the Δmic2 strain, reported by Gras et al. (13), versus the more modest reduction in the Δpofut2 and Δnst2 strains shown here.
Figure 9.

Egress defect in Δpofut2 and Δnst2 is not as severe as in the Δmic2. A, representative images showing the different phenotypes observed in the assay. Extracellular parasites were labeled with anti-SAG1 (in red). After permeabilization, all parasites were labeled with anti-β-tubulin (in green). As a result, parasites in intact vacuoles are green, whereas parasites in permeabilized vacuoles or that have egressed are labeled in green and red. B, quantification of the egress phenotypes. Quantifications are given as the average of at least three biological repeats ± S.D. (error bars). Student's t test was used to compare the samples, and all p values are reported in Table 2. Scale bars, 5 μm.
Table 2.
Comparison of egress phenotypes between WT and knockout cell lines analyzed in this study
Student's t test was used to calculate the p values. n.s., not significant.
| Phenotype |
p value |
|||||
|---|---|---|---|---|---|---|
| Δku80 versus Δpofut2 | Δku80 versus Δnst2 | Δku80 versus Δmic2 | Δpofut2 versus Δnst2 | Δpofut2 versus Δmic2 | Δnst2 versus Δmic2 | |
| Egressed | 0.04 | 0.0007 | 8.1 × 10−15 | n.s. (0.18) | 2.3 × 10−10 | 1.8 × 10−8 |
| Permeabilized | 0.007 | 0.0001 | 1.8 × 10−15 | n.s. (0.13) | 9.5 × 10−11 | 8.8 × 10−8 |
| Intact PSV | n.s. (0.92) | n.s. (0.88) | n.s. (0.62) | n.s. (0.95) | n.s. (0.56) | n.s. (0.53) |
Discussion
MIC2 is a well-characterized T. gondii virulence factor. This study adds to our understanding of MIC2 structure and function by characterizing the glycosylation on its TSRs and describing the importance of O-fucosylation on MIC2 processing and, consequently, the parasite's ability to invade.
Mass spectrometry analysis, using both HCD and ETD, showed that, in MIC2, all four TSRs containing the O-fucosylation motif (TSR1, -3, -4, and -5) are modified by O-FucGlc. Two other TSRs (TSR3 and -4), in addition to TSR5 (25), are also modified with C-Man. TSR5 was observed in vivo in two glycoforms, indicating heterogeneity at the O-fucosylation site, which is modified with either Fuc or FucGlc. Although ETD-based fragmentation is a very effective technique to characterize glycan modifications and identify the glycosylated amino acids, it is not able to distinguish between isomeric sugars (e.g. glucose versus galactose). The Western blotting evidence that indicates the disaccharide is FucGlc is the binding by an antibody generated here against the Glcβ1,3Fuc disaccharide. Consistent with this observation, a putative B3GLCT, the enzyme that adds Glc to elongate the Fuc on TSRs, is also present in the T. gondii genome (TGGT1_239752).
MIC2 was the only one of four predicted POFUT2 substrates that was identified in our MS analysis of secreted proteins; this result is consistent with the previous report (25). Whereas it is possible that MIC2 is the only tachyzoite protein with TSRs modified by O-Fuc and C-Man, it is more likely that the failure to detect other targets was due to either their low abundance or lack of secretion. TGGT1_223480, for example, is predicted to have higher expression in unsporulated oocysts (47) and could be the protein responsible for the previously observed staining of oocyst walls by the fucose-specific AAL (44), provided that O-Fuc is not capped in this life stage.
Eukaryotic POFUT2s have been described as soluble ER lumen proteins (22), and immunofluorescence after in situ tagging of T. gondii POFUT2 showed a pattern similar, although not identical, to the one observed for the lumenal ER marker. Furthermore, when an ectopic copy of T. gondii POFUT2, with a 3xMYC tag, was used to complement Δpofut2 parasites, this construct also showed ER localization. Taken together, these results suggest that the ER is the primary subcellular localization for POFUT2 in the parasite. However, in silico analyses suggested a different topology, as four putative transmembrane domains are predicted in the hydrophobic N-terminal region of T. gondii POFUT2, and additional experiments will be required to verify this prediction. Attempts at recombinantly expressing POFUT2 either plus or minus the N-terminal domain or with only the fourth transmembrane domain in CHO cells, as previously done for DPY19 (25), did not result in active protein (data not shown). Even in the absence of in vitro activity data, loss of O-fucosylation on MIC2 in the Δpofut2 strongly supports the model that this enzyme is the O-fucosyltransferase that modifies TSRs in the parasite. Additionally, a CRISPR/Cas9 screen directed to the glycosylation machinery of the parasite was published during the revision of this paper (48). Disruption of pofut2 in Gas-Pascual et al. (48) resulted in loss of the dHexHex disaccharide from the T. gondii β-eliminated glycan fraction, consistent with a role of this enzyme in the synthesis of O-FucGlc (48).
T. gondii is predicted to synthesize both GDP–Man and GDP–Fuc, but only one putative GDP–sugar transporter, NST2, can be identified in its genome. Consistent with this observation, the C-mannosyltransferase DPY-19 (25) utilizes Dol-P-Man as a sugar donor (26), and no putative Golgi mannosyl- or fucosyltransferases can be identified. Additionally, no putative POFUT1, the enzyme responsible for O-fucosylation of endothelial growth factor–like domains (49), is present in the parasite genome, even though T. gondii is predicted to encode for multiple EGF domain–containing proteins (50). The lack of O-fucosylation in Δnst2 parasites and the similar phenotype between Δpofut2 and Δnst2 mutants support the identification of NST2 as a T. gondii GDP–Fuc transporter and suggest that in the parasite secretory system, GDP–Fuc is primarily used for O-fucosylation by POFUT2. Identification of the GDP–Fuc transporter involved in TSR and EGF O-fucosylation has proven more difficult in metazoans, with various studies suggesting that the players involved in GDP–Fuc transport into the ER are not as well-conserved as the O-fucosyltransferases themselves (51–53).
Studies on mammalian POFUT2 indicate that this enzyme recognizes already folded TSRs, and defects in its activity affect the folding dynamics or the stabilization of TSR-containing proteins (37, 54). Our observations in T. gondii are consistent with this model. Although we detect nonfucosylated MIC2 that is correctly trafficked (and presumably folded), we also observe in a subset of vacuoles a clear accumulation of likely misfolded MIC2 in the early/mid-secretory pathway. Aberrant GRASP55 staining, which is also observed in this subpopulation of vacuoles, suggests that accumulation of misfolded MIC2 causes a disruption in the Golgi structure. Our results also suggest that the M2AP/MIC2 complex is not affected in either Δpofut2 or Δnst2, as we observed mislocalization of MIC2 and M2AP to the early/mid-secretory pathway in a statistically comparable number of vacuoles. This is unlike the Δmic2, where M2AP is redirected to the constitutive secretion pathway (13). Previous work showed that M2AP binds MIC2 TSR6 through its galectin-like domain, and structural studies indicated that the recognition is due to protein–protein, not carbohydrate–protein, interactions (16). Whereas MIC2 TSR6 contains a C-mannosylation motif, we observed it exclusively in its unglycosylated form, supporting the protein–protein interaction model.
Comparison between the two O-fucosylation–deficient strains generated in this study and the previously described Δmic2 (13) indicates that glycosylation of TSRs has the strongest effects on attachment and invasion (Fig. 10). This result suggests a role for MIC2 TSRs in the attachment and/or invasion processes, consistent with the known role of these domains in cell–cell interaction (17). These results are in agreement with the recently described pofut2 knockout in P. falciparum, which also resulted in reduced invasion efficiency by sporozoites (32). It has been shown previously that disruption of TgGAMA, a GPI-anchored microneme protein, resulted in a defective parasite attachment, causing a delayed invasion phenotype while not affecting parasite plaquing ability (55). This is similar to the phenotype described here upon disruption of TSRs O-fucosylation, suggesting that the defect we observe in both pofut2 and nst2 might be at the level of parasite attachment. Further experiments will be necessary to verify this hypothesis.
Figure 10.
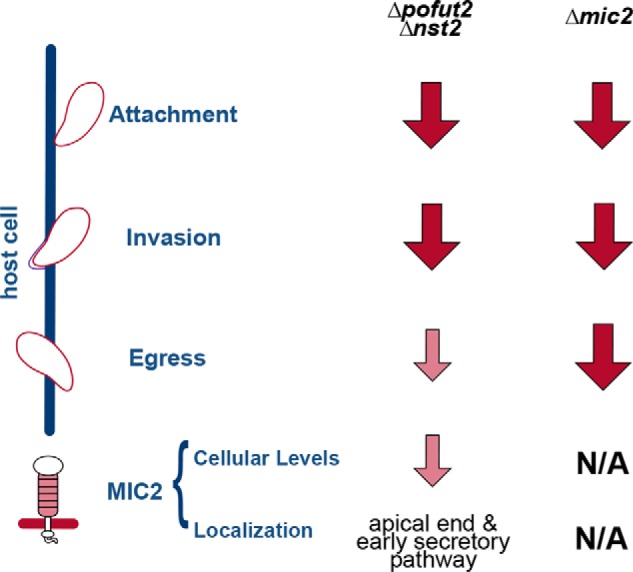
Comparison of mic2 and O-fucosylation KOs. Shown is a schematic representation of differences and similarities between Δmic2 and Δpofut2 and Δnst2. Disruption of O-fucosylation is sufficient to reproduce the attachment and invasion defects observed when MIC2 is not being synthesized. Conversely, the parasite ability to egress is much more compromised in the Δmic2 versus Δpofut2 and Δnst2. Dark red arrows indicate a stronger defect, whereas small light red arrows indicate a milder phenotype. N/A, not applicable.
It should be noted that an independent study by Khurana et al. (56) also identified pofut2 as the enzyme responsible for TSRs O-fucosylation in T. gondii; however, in their hands, its knockout did not result in any detectable invasion phenotype or defect in the parasite plaquing ability. Consistently, in their report, MIC2 levels were close to those of the parental strain (54). The hypothesis of an off-target mutation being responsible for the phenotype reported here is not supported by the complementation data, as adding back of T. gondii POFUT2 rescued the attachment/invasion phenotype of Δpofut2 parasites and restored cellular MIC2 levels. Our strategy differed from that used by Khurana et al. (56) to generate the pofut2 knockout. Additionally, we used different assay conditions to assess parasite invasion. It is possible that a combination of these experimental divergences might be responsible for the contrasting phenotypes observed.
This work complements the recent studies that have looked at the role of fucosylation in T. gondii biology. Fucose has been described previously in this parasite as part of two uncommon nucleocytosolic glycosylation pathways: modification of the E3-ubiquitin ligase adaptor Skp1 by a fucose-containing pentasaccharide (57, 58) and an O-fucosylation pathway that modifies Ser/Thr on proteins that accumulate at the nuclear periphery (44). In this study, we add a role for fucose in folding or stabilization of TSRs in T. gondii, a function conserved from Plasmodium to metazoans (23, 31, 32).
Experimental procedures
In silico analyses
Human POFUT2 (UniProt number Q9Y2G5-3) and GDP-fucose transporter (UniProt number Q96A29-1) protein sequences were used as templates to mine the T. gondii genome (http://toxodb.org/toxo)5 (47). The hits from these searches were confirmed by using the hits themselves as templates for searches on the NCBI BLASTP engine. Alignments were performed using Clustal Omega (59) and were colored by percentage identity and edited using Jalview (60).
To identify the putative POFUT2 protein acceptors, a protein motif search was performed on ToxoDB for C1X2–3(S/T)C2X2G (47). The resulting hits were manually verified to confirm that the proteins had a predicted signal peptide and the motif was indeed part of a TSR. The identified proteins are listed in Table S1.
T. gondii culture and manipulation
Cell culture of T. gondii tachyzoites was performed as described previously (39, 61). The Δmic2 cell line was a kind gift of Dr. Markus Meissner (Ludwig-Maximilians-University, Munich, Germany) (13). RH Δku80 was used as parental strain for all T. gondii transgenic cell lines generated in this study (39). To generate the transgenic cell lines, the required amount of DNA (see below) was electroporated by adapting the protocol described previously (45). Briefly, tachyzoites were washed in HHE buffer and resuspended in Cytomix to be 1.6 × 107/ml. Purified DNA that had been previously ethanol-precipitated was redissolved in Cytomix (supplemented with GSH, CaCl2, and ATP) and combined with 106 tachyzoites in a 2-mm gap BTX cuvette (BTX Harvard Apparatus). Electroporation was performed using the program X-014 on an Amaxa® Nucleofactor® II electroporator (Lonza).
CRISPR/Cas9-mediated gene disruption
The approach to generate the pofut2 (TGGT1_273550) and nst2 (TGGT1_267730) knockouts is described in Fig. 4D. The protospacers to direct Cas9 were cloned using oligonucleotides P24-25 (pofut2_Nterm), P26-27 (pofut2_Cterm), P28-29 (nst2_Nterm), and P30-31 (nst2_Cterm) (Table S2). Oligonucleotides were annealed and phosphorylated, and the resulting dsDNA was cloned in the BsaI-digested pU6Universal plasmid (38) to generate the following plasmids: pU6_P2N, pU6_P2C, pU6_N2N, and pU6_N2C. To replace the gene of interest, the mGFP-expressing cassette from pDHFR-Luc-mGFP (43) (gra1 5′-UTRs-mGFP-gra2 3′-UTRs) was amplified by PCR using primers containing about 30–40 bp of homology sequence to the double-stranded break sites (P20-21 and P22-23, Table S2) and cloned into pCR2.1TOPO (Invitrogen), generating pP2KO_mGFP and pN2KO_mGFP. The cassettes also contained blunt restriction sites (AfeI/BsrBI for pofut2 and StuI/SnaBI for nst2) to allow their isolation from plasmid DNA, without adding or removing bases to the homology sequences. DNA was purified from pP2KO_mGFP and pN2KO_mGFP, and the cassettes were isolated by restriction digestion and gel purification. About 20 μg each of pU6 guide RNAs and at least a 5-fold molar excess of purified recombination cassette were used for each electroporation. Cells were recovered in 15-cm culture dishes (Corning), and mGFP-positive plaques were isolated and transferred to 96-well plates for cloning by limiting dilution. Two to three rounds of cloning were required to isolate clonal populations of mGFP-positive parasites. DNA was extracted before and after cloning using DNAzol (Invitrogen) according to the manufacturer's instructions. Insertion of the cassette in the correct locus was verified by PCR using primers P34, P36, and P39 (pofut2) or P32, P33, P34, and P41 (nst2) (Table S2).
To generate the complementation construct, total RNA was extracted from RH using TriReagent (Life Technologies, Inc.) according to the manufacturer's instructions and used for cDNA synthesis using Moloney murine leukemia virus retrotranscriptase and Oligo(dT)23VN primers (New England Biolabs). The POFUT2 ORF was amplified from the cDNA using primers P42 and P43 and cloned into the final vector using AvrII and BglII to obtain ptubPOFUT2–3xMYC/sagCAT. The original vector was a kind gift of Marc-Jan Gubbels (Boston College). About 20 μg of the plasmid were linearized with NruI and electroporated in T. gondii Δpofut2. Parasites expressing POFUT2–3xMYC were selected with chloramphenicol.
In situ tagging of POFUT2
This approach is shown in Fig. 4A. Briefly, pU6_P2C was used to generate a Cas9-mediated double-stranded break near the stop codon of the pofut2 coding sequence. A 258-bp recombination sequence (O1; Table S2) was designed to include a 3xMYC tag, flanked at both sides by about 50 bp of homology sequence and blunt restriction enzymes (MscI/PvuII) to allow release from the plasmid (pUC57-P2tRO), and sent for synthesis (GenScript). Additionally, a synonymous point mutation was introduced in O1 to remove the Cas9 PAM site after recombination (mutated base in lowercase in Table S2) (38). About 20 μg of pU6_P2C and a 25-fold molar excess of the O1 sequence were electroporated as described above in the presence or absence of 10 μg of ptubP30HDEL/sagCAT as an endoplasmic reticulum marker (40). Parasites were then allowed to infect HFF monolayers on coverslips in 24-well plates for up to 72 h. Cells were fixed for immunofluorescence analysis at 24 h post-electroporation. DNA was extracted 72 h post-electroporation and used to verify insertion of the 3xMYC tag at the correct locus by PCR (primers P35, P37, P38, and P40).
Immunofluorescence analysis
Unless otherwise specified, intracellular tachyzoites were fixed in 4% paraformaldehyde (PFA) in phosphate buffer (PB) for 20 min at room temperature (RT). Permeabilization was performed in 0.25% Triton X-100 in PBS for 15 min at RT, and it was followed by blocking in 3% BSA in PBS for 1 h, at RT. ptubGRASP55-mRPF/sagCAT (40) and ptubP30HDEL-mCherry/sagCAT were a kind gifts of Marc-Jan Gubbels. Electroporation was performed as described above, using 20 μg of plasmid, and cells were fixed 24 h post-electroporation. Primary antibodies were used at the following concentrations: mAb anti-MIC2 6D10, 1:500; rabbit anti-MIC2, 1:500; rabbit anti-AMA4, 1:1000; mouse anti-c-Myc, 9E10 (Developmental Studies Hybridoma Bank), 4 μg/ml; and rabbit anti-M2AP, 1:500. Antibodies to MIC2, M2AP were kind gifts of Dr. Vern Carruthers (University of Michigan), whereas the antibody against AMA4 was a kind gift of Maryse Lebrun (UMR 5235 CNRS, Université de Montpellier). Goat Alexa Fluor–conjugated anti-mouse and anti-rabbit antibodies (Molecular Probes) were used at a 1:800 dilution. AAL was purchased from Vector Laboratories and conjugated to Alexa Fluor 594 succinimidyl esters (Molecular Probes) following the manufacturer's instructions. The final conjugate (AAL–Alexa Fluor 594) was used at 1:250. Cells were labeled with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 10 min at RT. Coverslips were mounted on glass slides using Vectashield (Vector Laboratories) as mounting medium and examined by deconvolution microscopy on a Zeiss AXIO inverted microscope with a Colibri LED. Images were collected at 0.2-μm optical sections with a Hamamutsu Orca-R2 camera and deconvolved using ZEN software (Zeiss). Images were further processed, and single optical sections were selected with Fiji (62). When counting the number of vacuoles with mislocalized MIC2 or M2AP, cells were observed at a total magnification of ×1000, and 100 vacuoles/biological repeat were counted. The average ± S.D. of three independent repeats is shown. A two-tailed, homoscedastic Student's t test was used to calculate the p values. Superresolution microscopy was performed on a Zeiss ELYRA microscope. Images were acquired with a ×63/1.4 numerical aperture oil immersion objective, 0.089-μm z sections at RT, and processed for structured illumination using ZEN software. Single optical sections were selected using Fiji (62).
Affinity purification of polyclonal chicken antibody
Glc-β-1,3-Fuc-α-KLH (16) (see supporting information) was sent to Genway Biotech Inc. to generate a chicken antibody using standard protocols. Crude chicken IgY (50 ml at 10 mg/ml) were centrifuged at 5000 × g for 5 min. The debris was removed, and the clear IgY was incubated with agitation overnight with the disaccharide agarose beads (see supporting information). The mixture was loaded on a column, and the flow-through fraction was collected. The column was washed with PBS buffer and eluted with 0.1 m glycine buffer, pH 2. The flow-through fraction was reincubated with the agarose beads, and elutions were collected twice more to ensure maximum yield of the GlcFuc antibody. The elutions were immediately neutralized with 1 m Tris-HCl, pH 9.0, pooled, and concentrated to 1 mg/ml using Amicon ultracentrifugal filters (EMD Millipore) (3000 molecular weight cut-off) (63).
Generation of BSA neoglycoproteins
The disaccharide (GlcFuc) or the monosaccharide (Fuc) with an azide linker group was reacted with the NHS-linker (12) (see supporting information) and BSA to generate the BSA neoglycoproteins, using the same method for generating KLH-GlcFuc (see supporting information, synthesis of compound 16). The products were purified by dialysis in distilled water for 24 h twice. The resulting BSA-GlcFuc and BSA-Fuc were concentrated to 1 mg/ml using Amicon Ultra centrifugal filters (3000 molecular weight cut-off).
Testing of polyclonal anti-GlcFuc IgY
For the ELISAs, BSA-GlcFuc, BSA-Fuc, and BSA were diluted at different concentrations in PBS supplemented with 1% BSA and 0.1% NaN3. Samples were incubated on an ELISA plate at 4 °C overnight. Following blocking in PBST (PBS, pH 7.4, 0.05% Tween 20) with 10% BSA at RT for 1 h, the wells were incubated with 1 μg/ml purified anti-GlcFuc antibody in PBST + 10% BSA for 2 h. The secondary antibody (rabbit anti-chicken IgY, HRP-conjugated) was diluted 1:100,000 in blocking buffer and incubated for 2 h at RT. Signal was developed by using o-phenylenediamine dihydrochloride reagent and read at the wavelengths of 492 and 620 nm. For Western blotting, 0.5 μg of protein/lane was loaded on an 8% SDS-polyacrylamide gel with 4% stacking, transferred onto nitrocellulose membranes, and blocked in blocking buffer (percentage milk in PBST) for 1 h at RT. Anti-GlcFuc antibody was diluted in blocking buffer to 1 μg/ml and incubated for 1 h at RT. The secondary antibody (rabbit anti-chicken IgY, HRP-conjugated) was diluted 1:5000 in blocking buffer and incubated for 1 h at RT. Blots were developed using SuperSignal West Pico (Pierce).
Western blotting
For total cell lysate, extracellular tachyzoites were harvested by centrifugation, washed twice in PBS, and lysed in 1× reducing SDS-PAGE loading buffer with an additional 0.1 m DTT. Lysates were heated for 10 min at 96 °C, and about 5 × 106 cell eq/lane were loaded on 8–16% TGX gels (Life Technologies). PAGE-separated proteins were blotted on polyvinylidene difluoride, and the membranes were then blocked in 50 mm Tris-HCl, 0.15 m NaCl, 0.25% BSA, 0.05% Nonidet P-40, pH 7.4 (64). β-Elimination on blot was performed as described previously (65). Briefly, after blotting, the membrane was washed in PBS and incubated with rotation for 16 h in 55 mm NaOH at 40 °C. After being rinsed in water, the membrane was blocked as described above. Both primary and secondary antibodies were diluted in blocking buffer as follows: mouse mAb anti-MIC2 6D10 (1:5000), mouse mAb anti-α-tubulin (1:800) (Developmental Studies Hybridoma Bank), chicken anti-GlcFuc (1.1 μg/ml), anti-mouse HRP-conjugated (Bio-Rad) (1:1000), and anti-chicken HRP (1:5000). Blots were developed by chemiluminescence (SuperSignal West Pico PLUS) using an ImageQuant LAS4000 imager (GE Healthcare). Quantification was performed using the ImageQuant TL software. The average of three biological repeats ± S.D. is shown. A two-tailed, heteroscedastic Student's t test was used to calculate the p values (t.test function in Excel).
Glycopeptide analysis by nUPLC-MS/MS (HCD)
Secreted proteins were isolated and prepared for nano-ultra-performance liquid chromatography tandem mass spectrometry (nUPLC-MS/MS) analysis on a Thermo Q Exactive quadrupole Orbitrap MS as described previously (7, 25). Briefly, 4 × 1010 tachyzoites were isolated and resuspended in Dulbecco's modified Eagle's medium with 1% ethanol. After removal of parasites by centrifugation, the supernatant was filtered and concentrated. About 110 μg of total protein was precipitated in 0.1 m ammonium acetate in MeOH for at least 16 h at −20 °C. The sample was washed, dried by speed vacuum (SpeedVac Plus, Savant), and resuspended in 50 mm ammonium bicarbonate, pH 8. The proteins were then reduced, alkylated with iodoacetamide, and digested with trypsin, and the resulting peptides were desalted on a silica C18 column (Nest Group). After speed vacuum drying, the sample was reconstituted in 99% water, 1% acetonitrile, 0.1% formic acid. For the initial discovery method, a 2-μl aliquot was analyzed as described previously (25). Precursor ions assigned from the Mascot search as peptides containing the O-fucosylation peptide motif were analyzed by targeted nUPLC-MS/MS (HCD) in parallel reaction monitoring (PRM) mode with an inclusion list containing both the m/z value and retention times of the ions of interest. The PRM experiment was set up as follows. A full mass scan was acquired with 70,000 resolution at m/z 200, automatic gain control (AGC) target 1 × 106, 100-ms maximum injection time, 1 microscan/spectrum over the range m/z 370–2000. The selected precursor masses were calibrated with background ions at m/z 371.1012, 391.2843, 429.0887, and 445.1200 as the lock masses. The full mass range scan event was followed by seven PRM scan events, each with a different normalized collision energy. The PRM scan events were recorded with 17,500 resolution at m/z 200, AGC target 1 × 106, 150-ms maximum injection time, 2 microscans/spectrum, and their first mass was fit to m/z 100. The isolation window was set to 2 m/z units (Th) with an isolation offset of 0.4. An inclusion list with the m/z and retention times of the ions of interest was loaded, and the inclusion list feature was set to “on.” Ion dissociation/activation was achieved by quadrupole/beam-type collisionally activated dissociation or HCD. For each of the seven PRM scan events, a different normalized collision energy was used. The normalized collision energy was set from 10 to 40 eV by increments of 5 eV.
Glycopeptide analyses by nUPLC-MS/MS (ETD)
Precursor ions of most interest that had been identified in the initial HCD experiment were used for a targeted nUPLC-MS/MS ETD experiment. An inclusion list of m/z with retention times was used in the ETD-targeted experiment. Five-μl aliquots of the sample were injected into a nanoAcquity-UPLC (Waters) equipped with reversed-phase columns (trap column, 5-μm Symmetry C18, 180 μm × 20 mm; analytical column, 1.7-μm BEH130 C18, 150 μm × 100 mm (both from Waters)) and separated as described before (25). The nano-UPLC was connected online to an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific) equipped with a Triversa NanoMate (Advion) electrospray ionization source. The mass spectrometer was operated in the positive-ion mode. The MS and tandem MS data were acquired in automatic data-dependent “top 3” mode. The molecular ions were detected in the Orbitrap mass analyzer at a resolution of 30,000 at m/z 400, over the full scan range m/z 300–2000, 1 microscan/spectrum, maximum injection time (ion accumulation time) of 500 ms with a target AGC of 500,000 ion population. The selected precursor masses were calibrated with background ions at m/z 371.1012, 391.2842, and 445.1200 as the lock masses. The top three most abundant ions with charge state higher than 2 in the survey MS scan were selected, using a 2-Da isolation window, and were fragmented by ETD in the linear ion trap (LTQ) mass analyzer. The fluoranthene radical anions were accumulated for a maximum time of 125 ms and AGC of 500,000 prior to the ETD reaction. The ETD activation time was 125 ms, with supplemental activation of 15 V. For m/z 737.3218 (2+), the fragments were detected in the Orbitrap mass analyzer with 2 microscans/spectrum, maximum injection time of 700 ms with AGC of 200,000 ion population. For m/z 586.7708 (2+), the fragments were detected in the LTQ mass analyzer with 3 microscans/spectrum, maximum injection time of 150 ms with AGC of 10,000. All MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD010622 (46).
Plaque assay
Host cell monolayers (HFF) on 6-well plates were infected with 250 parasites/well of either parental or knockout strains. Parasites were allowed to grow at 37 °C, 5% CO2 for 8 days. After washing in PBS, cells were fixed in ice-cold methanol at −20 °C for at least 20 min and stained with Giemsa for 10 min. After the cells were washed with double-distilled H2O and air-dried, plates were imaged on an ImageQuant LAS400 system. Plaque areas were determined using the ImageQuant TL software. Plaque numbers shown are the average of four biological repeats ± S.D. A two-tailed, homoscedastic Student's t test was used to calculate the p values (t.test function in Excel).
Attachment/invasion assay
This protocol was adapted from Huynh et al. (14). Briefly, HFF monolayers on a coverslip (24-well plate) were infected with 5 × 106 tachyzoites/well from either parental or knockout strains. The plate was centrifuged for 5 min at 200 × g and incubated for 30 min at 37 °C. After washing with PBS to remove unattached parasites, the coverslips were fixed in 4% PFA in PB for 20 min on ice and blocked in 3% BSA/PBS for 1 h at RT. Extracellular (attached) parasites were labeled with mAb anti-SAG1 T4 1E5 1:300 (BEI Resources). Cells were then permeabilized (10 min, 0.25% Triton X-100/PBS) and blocked once more (3% BSA/PBS, 30 min at RT). All parasites (attached and invaded) were labeled with rabbit anti-β-tubulin 1:1000 (this step was required as the parental strain does not express mGFP like the knockout strains analyzed in this study). Anti-mouse Alexa Fluor 594 and anti-rabbit Alexa Fluor 488 were used as secondary antibodies. Coverslips were mounted on glass slides using Prolong Diamond with DAPI (Molecular Probes) and examined on a Zeiss AXIO inverted microscope. Six randomly selected fields were imaged with a total magnification of ×400. Attached (red + green) and invaded (green) parasites were counted, and the invasion rate was calculated. This is the ratio of invaded to total parasites. The data from four or five biological repeats are shown in Fig. 8D and from two biological repeats for Fig. 8E. A two-tailed, heteroscedastic Student's t test was used to calculate the p values (t.test function in Excel).
Egress assay
This protocol was adapted from Refs. 13 and 45. Briefly, HFF monolayers on coverslip (24-well plate) were infected with 1 × 105 tachyzoites/well from either parental or knockout strains, and parasites were allowed to grow for 24–28 h. To induce egress, cells were incubated for 5 min at 37 °C with 2 μm A23178 (Sigma) in prewarmed Dulbecco's modified Eagle's medium (no FBS). Coverslips were immediately fixed in 4% PFA in PB for 20 min on ice and blocked in 3% BSA/PBS for 1 h at RT. Labeling and imaging were performed as described for the invasion/attachment assay with the difference that 8–10 fields were scored in each biological repeat. The average of three independent experiments ± S.D. is shown. A two-tailed, heteroscedastic Student's t test was used to calculate the p values (t.test function in Excel).
Author contributions
J. S. and G. B. conceived the study and designed the experiments. D. R. L. performed the mass spectrometry analysis; C. M. H. isolated the secreted proteins; Y. Z., L. K. M., and M. J. S. produced and characterized the antibody. G. B. performed all remaining experiments. G. B., J. S., D. R. L., C. A.-N., F. H. R., and C. E. C. performed the data analysis, and all authors contributed to the writing of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. Maryse Lebrun for the AMA4 antibody and Prof. Markus Meissner, Prof. Vern Carruthers, and Prof. Marc Jan Gubbels for reagents and helpful discussions. We thank Bret Judson and the Boston College Imaging Core for infrastructure and support (National Science Foundation Grant 1626072).
This work was supported in part by National Institutes of Health Grants R01 AI110638 (to J. S.) and P41 GM104603 (to C. E. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting Experimental procedures and Tables S1–S3 and Figs. S1–S9.
All MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD010622.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MIC2
- microneme protein 2
- TSR
- thrombospondin repeat
- POFUT2
- protein O-fucosyltransferase 2
- NST2
- nucleotide sugar transporter 2
- M2AP
- MIC2-associated protein
- mGFP
- modified green fluorescent protein
- mRFP
- modified red fluorescent protein
- AAL
- Aleuria aurantia lectin
- KLH
- keyhole limpet hemocyanin
- Hex
- hexose
- dHex
- deoxyhexose
- Fuc
- fucose
- Man
- mannose
- HCD
- higher-collision energy dissociation
- ETD
- electron transfer dissociation
- PV
- parasitophorous vacuole
- HFF
- human foreskin fibroblast(s)
- IFA
- immunofluorescence analysis
- nUPLC
- nano-ultra-performance liquid chromatography
- TRAP
- thrombospondin-related anonymous protein
- ER
- endoplasmic reticulum
- PFA
- paraformaldehyde
- PB
- phosphate buffer
- RT
- room temperature
- DAPI
- 4′,6-diamidino-2-phenylindole
- HRP
- horseradish peroxidase
- PRM
- parallel reaction monitoring
- AGC
- automatic gain control
- LTQ
- linear trap quadrupole.
References
- 1. Torgerson P. R., and Mastroiacovo P. (2013) The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508 10.2471/BLT.12.111732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torrey E. F., and Yolken R. H. (2013) Toxoplasma oocysts as a public health problem. Trends Parasitol. 29, 380–384 10.1016/j.pt.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 3. Ferguson D. J. P. (2004) Use of molecular and ultrastructural markers to evaluate stage conversion of Toxoplasma gondii in both the intermediate and definitive host. Int. J. Parasitol. 34, 347–360 10.1016/j.ijpara.2003.11.024 [DOI] [PubMed] [Google Scholar]
- 4. Carruthers V. B., Giddings O. K., and Sibley L. D. (1999) Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell Microbiol. 1, 225–235 10.1046/j.1462-5822.1999.00023.x [DOI] [PubMed] [Google Scholar]
- 5. Carruthers V. B., and Sibley L. D. (1999) Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol. Microbiol. 31, 421–428 10.1046/j.1365-2958.1999.01174.x [DOI] [PubMed] [Google Scholar]
- 6. Kappe S., Bruderer T., Gantt S., Fujioka H., Nussenzweig V., and Ménard R. (1999) Conservation of a gliding motility and cell invasion machinery in Apicomplexan parasites. J. Cell Biol. 147, 937–944 10.1083/jcb.147.5.937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou X. W., Blackman M. J., Howell S. A., and Carruthers V. B. (2004) Proteomic analysis of cleavage events reveals a dynamic two-step mechanism for proteolysis of a key parasite adhesive complex. Mol. Cell. Proteomics 3, 565–576 10.1074/mcp.M300123-MCP200 [DOI] [PubMed] [Google Scholar]
- 8. Harper J. M., Hoff E. F., and Carruthers V. B. (2004) Multimerization of the Toxoplasma gondii MIC2 integrin-like A-domain is required for binding to heparin and human cells. Mol. Biochem. Parasitol. 134, 201–212 10.1016/j.molbiopara.2003.12.001 [DOI] [PubMed] [Google Scholar]
- 9. Carruthers V. B., Håkansson S., Giddings O. K., and Sibley L. D. (2000) Toxoplasma gondii uses sulfated proteoglycans for substrate and host cell attachment. Infect. Immun. 68, 4005–4011 10.1128/IAI.68.7.4005-4011.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barragan A., Brossier F., and Sibley L. D. (2005) Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell. Microbiol. 7, 561–568 10.1111/j.1462-5822.2005.00486.x [DOI] [PubMed] [Google Scholar]
- 11. Rabenau K. E., Sohrabi A., Tripathy A., Reitter C., Ajioka J. W., Tomley F. M., and Carruthers V. B. (2001) TgM2AP participates in Toxoplasma gondii invasion of host cells and is tightly associated with the adhesive protein TgMIC2. Mol Microbiol. 41, 537–547 10.1046/j.1365-2958.2001.02513.x [DOI] [PubMed] [Google Scholar]
- 12. Carruthers V. B., Sherman G. D., and Sibley L. D. (2000) The Toxoplasma adhesive protein MIC2 is proteolytically processed at multiple sites by two parasite-derived proteases. J. Biol. Chem. 275, 14346–14353 10.1074/jbc.275.19.14346 [DOI] [PubMed] [Google Scholar]
- 13. Gras S., Jackson A., Woods S., Pall G., Whitelaw J., Leung J. M., Ward G. E., Roberts C. W., and Meissner M. (2017) Parasites lacking the micronemal protein MIC2 are deficient in surface attachment and host cell egress, but remain virulent in vivo. Wellcome Open Res. 2, 32 10.12688/wellcomeopenres.11594.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huynh M. H., Rabenau K. E., Harper J. M., Beatty W. L., Sibley L. D., and Carruthers V. B. (2003) Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 22, 2082–2090 10.1093/emboj/cdg217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harper J. M., Huynh M.-H., Coppens I., Parussini F., Moreno S., and Carruthers V. B. (2006) A cleavable propeptide influences Toxoplasma infection by facilitating the trafficking and secretion of the TgMIC2-M2AP invasion complex. Mol. Biol. Cell. 17, 4551–4563 10.1091/mbc.e06-01-0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huynh M.-H., Liu B., Henry M., Liew L., Matthews S. J., and Carruthers V. B. (2015) Structural basis of Toxoplasma gondii MIC2-associated protein interaction with MIC2. J. Biol. Chem. 290, 1432–1441 10.1074/jbc.M114.613646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams J. C., and Lawler J. (2011) The thrombospondins. Cold Spring Harb. Perspect. Biol. 3, a009712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hofsteenge J., Huwiler K. G., Macek B., Hess D., Lawler J., Mosher D. F., and Peter-Katalinic J. (2001) C-Mannosylation and O-fucosylation of the thrombospondin type 1 module. J. Biol. Chem. 276, 6485–6498 10.1074/jbc.M008073200 [DOI] [PubMed] [Google Scholar]
- 19. Buettner F. F. R., Ashikov A., Tiemann B., Lehle L., and Bakker H. (2013) C. elegans DPY-19 is a C-mannosyltransferase glycosylating thrombospondin repeats. Mol. Cell. 50, 295–302 10.1016/j.molcel.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 20. Hofsteenge J., Müller D. R., de Beer T., Löffler A., Richter W. J., and Vliegenthart J. F. G. (1994) New type of linkage between a carbohydrate and a protein: C-glycosylation of a specific tryptophan residue in human RNase Us. Biochemistry 33, 13524–13530 10.1021/bi00250a003 [DOI] [PubMed] [Google Scholar]
- 21. Swearingen K. E., Lindner S. E., Shi L., Shears M. J., Harupa A., Hopp C. S., Vaughan A. M., Springer T. A., Moritz R. L., Kappe S. H. I., and Sinnis P. (2016) Interrogating the plasmodium sporozoite surface: identification of surface-exposed proteins and demonstration of glycosylation on CSP and TRAP by mass spectrometry-based proteomics. PLoS Pathog. 12, e1005606 10.1371/journal.ppat.1005606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo Y., Koles K., Vorndam W., Haltiwanger R. S., and Panin V. M. (2006) Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J. Biol. Chem. 281, 9393–9399 10.1074/jbc.M511975200 [DOI] [PubMed] [Google Scholar]
- 23. Vasudevan D., Takeuchi H., Johar S. S., Majerus E., and Haltiwanger R. S. (2015) Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol. 25, 286–295 10.1016/j.cub.2014.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kozma K., Keusch J. J., Hegemann B., Luther K. B., Klein D., Hess D., Haltiwanger R. S., and Hofsteenge J. (2006) Identification and characterization of a β1,3-glucosyltransferase that synthesizes the Glc-β1,3-Fuc disaccharide on thrombospondin type 1 repeats. J. Biol. Chem. 281, 36742–36751 10.1074/jbc.M605912200 [DOI] [PubMed] [Google Scholar]
- 25. Hoppe C. M., Albuquerque-Wendt A., Bandini G., Leon D. R., Shcherbakova A., Buettner F. F. R., Izquierdo L., Costello C. E., Bakker H., and Routier F. H. (2018) Apicomplexan C-mannosyltransferases modify thrombospondin type I-containing adhesins of the TRAP family. Glycobiology 28, 333–343 10.1093/glycob/cwy013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doucey M.-A., Hess D., Cacan R., and Hofsteenge J. (1998) Protein C-mannosylation is enzyme-catalysed and uses dolichyl-phosphate-mannose as a precursor. Mol. Biol. Cell. 9, 291–300 10.1091/mbc.9.2.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shcherbakova A., Tiemann B., Buettner F. F. R., and Bakker H. (2017) Distinct C-mannosylation of netrin receptor thrombospondin type 1 repeats by mammalian DPY19L1 and DPY19L3. Proc. Natl. Acad. Sci. U.S.A. 114, 2574–2579 10.1073/pnas.1613165114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang L. W., Dlugosz M., Somerville R. P. T., Raed M., Haltiwanger R. S., and Apte S. S. (2007) O-Fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J. Biol. Chem. 282, 17024–17031 10.1074/jbc.M701065200 [DOI] [PubMed] [Google Scholar]
- 29. Wang L. W., Leonhard-Melief C., Haltiwanger R. S., and Apte S. S. (2009) Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. J. Biol. Chem. 284, 30004–30015 10.1074/jbc.M109.038059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siupka P., Hamming O. T., Kang L., Gad H. H., and Hartmann R. (2015) A conserved sugar bridge connected to the WSXWS motif has an important role for transport of IL-21R to the plasma membrane. Genes Immun. 16, 405–413 10.1038/gene.2015.22 [DOI] [PubMed] [Google Scholar]
- 31. Benz B. A., Nandadasa S., Takeuchi M., Grady R. C., Takeuchi H., LoPilato R. K., Kakuda S., Somerville R. P. T., Apte S. S., Haltiwanger R. S., and Holdener B. C. (2016) Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol. 416, 111–122 10.1016/j.ydbio.2016.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lopaticki S., Yang A. S. P., John A., Scott N. E., Lingford J. P., O'Neill M. T., Erickson S. M., McKenzie N. C., Jennison C., Whitehead L. W., Douglas D. N., Kneteman N. M., Goddard-Borger E. D., and Boddey J. A. (2017) Protein O-fucosylation in Plasmodium falciparum ensures efficient infection of mosquito and vertebrate hosts. Nat Commun. 8, 561 10.1038/s41467-017-00571-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu H., Khatri K., Klein J., Leymarie N., and Zaia J. (2016) A review of methods for interpretation of glycopeptide tandem mass spectral data. Glycoconj. J. 33, 285–296 10.1007/s10719-015-9633-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wuhrer M., Deelder A. M., and van der Burgt Y. E. M. (2011) Mass spectrometric glycan rearrangements. Mass Spectrom. Rev. 30, 664–680 10.1002/mas.20337 [DOI] [PubMed] [Google Scholar]
- 35. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinez-Duncker I., Mollicone R., Candelier J. J., Breton C., and Oriol R. (2003) A new superfamily of protein-O-fucosyltransferases, 2-fucosyltransferases, and 6-fucosyltransferases: phylogeny and identification of conserved peptide motifs. Glycobiology 13, 1C–5C 10.1093/glycob/cwg113 [DOI] [PubMed] [Google Scholar]
- 37. Valero-González J., Leonhard-Melief C., Lira-Navarrete E., Jiménez-Osés G., Hernández-Ruiz C., Pallarés M. C., Yruela I., Vasudevan D., Lostao A., Corzana F., Takeuchi H., Haltiwanger R. S., and Hurtado-Guerrero R. (2016) A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 12, 240–246 10.1038/nchembio.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sidik S. M., Hackett C. G., Tran F., Westwood N. J., and Lourido S. (2014) Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9, e100450 10.1371/journal.pone.0100450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huynh M.-H., and Carruthers V. B. (2009) Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 10.1128/EC.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ouologuem D. T., and Roos D. S. (2014) Dynamics of the Toxoplasma gondii inner membrane complex. J. Cell Sci. 127, 3320–3330 10.1242/jcs.147736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parker J. L., and Newstead S. (2017) Structural basis of nucleotide sugar transport across the Golgi membrane. Nature 551, 521–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saeij J. P. J., Boyle J. P., Grigg M. E., Arrizabalaga G., and Boothroyd J. C. (2005) Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73, 695–702 10.1128/IAI.73.2.695-702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bandini G., Haserick J. R., Motari E., Ouologuem D. T., Lourido S., Roos D. S., Costello C. E., Robbins P. W., and Samuelson J. (2016) O-Fucosylated glycoproteins form assemblies in close proximity to the nuclear pore complexes of Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 113, 11567–11572 10.1073/pnas.1613653113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sidik S. M., Huet D., Ganesan S. M., Huynh M.-H., Wang T., Nasamu A. S., Thiru P., Saeij J. P. J., Carruthers V. B., Niles J. C., and Lourido S. (2016) A Genome-wide CRISPR screen in Toxoplasma identifies essential apicomplexan genes. Cell 166, 1423–1435.e12 10.1016/j.cell.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033 10.1093/nar/gkw880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gajria B., Bahl A., Brestelli J., Dommer J., Fischer S., Gao X., Heiges M., Iodice J., Kissinger J. C., Mackey A. J., Pinney D. F., Roos D. S., Stoeckert C. J. Jr., Wang H., and Brunk B. P. (2008) ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 36, D553–D556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gas-Pascual E., Ichikawa H. T., Sheikh M. O., Serji M. I., Deng B., Mandalasi M., Bandini G., Samuelson J., Wells L., and West C. M. (2019) CRISPR/Cas9 and glycomics tools for Toxoplasma glycobiology. J. Biol. Chem. 294, 1104–1125 10.1074/jbc.RA118.006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y., Shao L., Shi S., Harris R. J., Spellman M. W., Stanley P., and Haltiwanger R. S. (2001) Modification of epidermal growth factor-like repeats with O-fucose: molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J. Biol. Chem. 276, 40338–40345 10.1074/jbc.M107849200 [DOI] [PubMed] [Google Scholar]
- 50. Sheiner L., Santos J. M., Klages N., Parussini F., Jemmely N., Friedrich N., Ward G. E., and Soldati-Favre D. (2010) Toxoplasma gondii transmembrane microneme proteins and their modular design. Mol Microbiol. 77, 912–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ishikawa H. O., Ayukawa T., Nakayama M., Higashi S., Kamiyama S., Nishihara S., Aoki K., Ishida N., Sanai Y., and Matsuno K. (2010) Two pathways for importing GDP-fucose into the endoplasmic reticulum lumen function redundantly in the O-fucosylation of Notch in Drosophila. J. Biol. Chem. 285, 4122–4129 10.1074/jbc.M109.016964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ashikov A., Routier F., Fuhlrott J., Helmus Y., Wild M., Gerardy-Schahn R., and Bakker H. (2005) The human solute carrier gene SLC35B4 encodes a bifunctional nucleotide sugar transporter with specificity for UDP-xylose and UDP-N-acetylglucosamine. J. Biol. Chem. 280, 27230–27235 10.1074/jbc.M504783200 [DOI] [PubMed] [Google Scholar]
- 53. Lu L., Hou X., Shi S., Körner C., and Stanley P. (2010) Slc35c2 promotes Notch1 fucosylation and is required for optimal Notch signaling in mammalian cells. J. Biol. Chem. 285, 36245–36254 10.1074/jbc.M110.126003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vasudevan D., and Haltiwanger R. S. (2014) Novel roles for O-linked glycans in protein folding. Glycoconj. J. 31, 417–426 10.1007/s10719-014-9556-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huynh M.-H., and Carruthers V. B. (2016) A Toxoplasma gondii ortholog of Plasmodium GAMA contributes to parasite attachment and cell invasion. mSphere e00012–16 10.1128/mSphere.00012-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khurana S., Coffey M. J., John A., Uboldi A. D., Huynh M.-H., Stewart R. J., Carruthers V. B., Tonkin C., Goddard-Borger E. D., and Scott N. (2019) Protein O-fucosyltransferase 2-mediated O-glycosylation of MIC2 is dispensable for Toxoplasma gondii tachyzoites. J. Biol. Chem. 294, 1541–1553 10.1074/jbc.RA118.005357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rahman K., Mandalasi M., Zhao P., Sheikh M. O., Taujale R., Kim H. W., van der Wel H., Matta K., Kannan N., Glushka J. N., Wells L., and West C. M. (2017) Characterization of a cytoplasmic glucosyltransferase that extends the core trisaccharide of the Toxoplasma Skp1 E3 ubiquitin ligase subunit. J. Biol. Chem. 292, 18644–18659 10.1074/jbc.M117.809301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rahman K., Zhao P., Mandalasi M., van der Wel H., Wells L., Blader I. J., and West C. M. (2016) The E3 ubiquitin ligase adaptor protein Skp1 is glycosylated by an evolutionarily conserved pathway that regulates protist growth and development. J. Biol. Chem. 291, 4268–4280 10.1074/jbc.M115.703751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., and Barton G. J. (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roos D. S., Donald R. G. K., Morrissette N. S., and Moulton A. L. C. (1994) Molecular Tools for Genetic Dissection of the Protozoan Parasite Toxoplasma gondii. Methods Cell Biol. 45, 27–63 10.1016/S0091-679X(08)61845-2 [DOI] [PubMed] [Google Scholar]
- 62. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaltgrad E., Sen Gupta S., Punna S., Huang C.-Y., Chang A., Wong C.-H., Finn M. G., and Blixt O. (2007) Anti-carbohydrate antibodies elicited by polyvalent display on a viral scaffold. Chembiochem 8, 1455–1462 10.1002/cbic.200700225 [DOI] [PubMed] [Google Scholar]
- 64. Güther M. L. S., Lee S., Tetley L., Acosta-Serrano A., and Ferguson M. A. J. (2006) GPI-anchored proteins and free GPI glycolipids of procyclic form Trypanosoma brucei are nonessential for growth, are required for colonization of the tsetse fly, and are not the only components of the surface coat. Mol. Biol. Cell. 17, 5265–5274 10.1091/mbc.e06-08-0702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Duk M., Ugorski M., and Lisowska E. (1997) β-Elimination of O-glycans from glycoproteins transferred to immobilon P membranes: method and some applications. Anal. Biochem. 253, 98–102 10.1006/abio.1997.9994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.