

Abstract
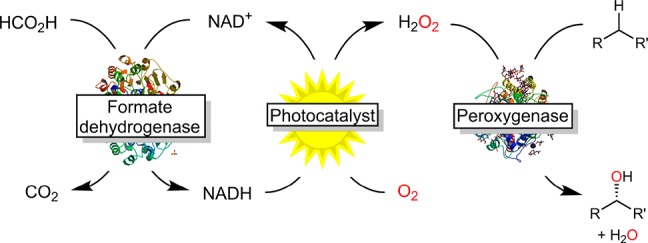
Peroxygenases require a controlled supply of H2O2 to operate efficiently. Here, we propose a photocatalytic system for the reductive activation of ambient O2 to produce H2O2 which uses the energy provided by visible light more efficiently based on the combination of wavelength-complementary photosensitizers. This approach was coupled to an enzymatic system to make formate available as a sacrificial electron donor. The scope and current limitations of this approach are reported and discussed.
Keywords: biocatalysis, formate dehydrogenase, hydrogen peroxide formation, oxyfunctionalization, peroxygenases, photocatalysis
Peroxygenases are receiving tremendous interest as catalysts for selective oxyfunctionalization reactions.1−3 Compared to the well-known P450 monooxygenases, peroxygenases exhibit a comparable reactivity pattern as both rely on an oxyferryl species (Compound I) as catalytically active compound. To generate Compound I, P450 monooxygenases rely on complicated electron transport chains while peroxygenases require only hydrogen peroxide.
Nevertheless, peroxygenases are rapidly inactivated in the presence of even small concentrations of H2O2. The peroxygenase from Caldariomyces fumago, for example, exhibits a half-life time of 38 min in the presence of 50 μM H2O2.4 Therefore, careful control of the in situ H2O2 concentration is required. Slow addition of H2O2 is possible but results in significant dilution of the reaction mixtures. More elegantly, H2O2 is generated within the reaction mixture through reduction of ambient molecular oxygen. For example, enzymatic H2O2 generation systems have been developed.5−8 More recently, also electrochemical4,9−12 and photocatalytic alternatives have moved into the focus.13−18 The latter bear the promise of utilizing sunlight as a thermodynamic driving force to promote selective oxyfunctionalization reactions. Today, however, these systems suffer from some major drawbacks such as formation of large (and toxicologically and environmentally questionable) amounts of waste (Table S4)16−18 or sluggish reaction kinetics.13,14 Also, so far, only flavin- or TiO2-based photocatalysts have been used, limiting the wavelength range to the near UV and blue light (450 nm). Hence, only a fraction of wavelengths is used, leaving a significant amount of sunlight energy unexploited.
In this study, we aimed at addressing both issues and provide the proof-of-concept for more efficient photocatalytic oxyfunctionalization reactions. We envisioned using simple formate as a sacrificial electron donor, producing CO2 as stoichiometric byproduct. Furthermore, by employing several, wavelength-complementary photocatalysts we aimed at providing a more efficient use of the energy of polychromatic light.
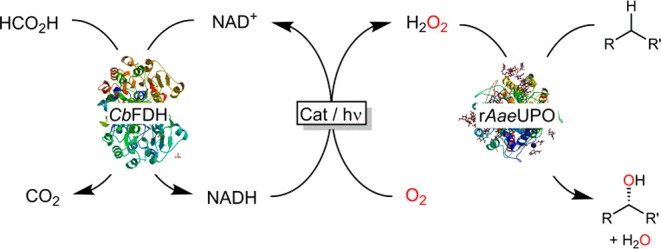
To couple formate oxidation to photocatalytic H2O2 generation, we envisioned an enzymatic relay system comprising formate dehydrogenase from Candida boidinii (CbFDH) to mediate the hydride transfer from formate to NAD+.19 The resulting NADH has previously been shown to be prone to photocatalytic, aerobic oxidation yielding H2O2 (Scheme 1).20−22 The peroxygenase used in this study was the recombinant, evolved peroxygenase from Agrocybe aegerita (rAaeUPO).23,24
Scheme 1. Proposed Photoenzymatic System for in Situ Generation of H2O2 To Promote Peroxygenase-Catalyzed Hydroxylation Reactions.
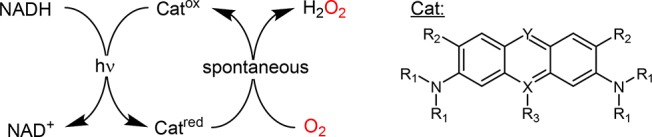
First, we screened 23 commercially available dyes for their capability of oxidizing NADH and delivering the reducing equivalents to O2 to yield H2O2 (Table S1). Out of these, 17 were discarded either because they did not oxidize NADH or because their reduced form was stable in the presence of O2 and therefore unsuitable for the aim of this study.
The remaining candidates (all acridine derivatives) were investigated further with respect to their activity in photochemical NADH oxidation and H2O2 generation. The catalytic performance (expressed as turnover frequency, TF) of the photocatalysts is shown in Table 1.
Table 1. Comparison of the H2O2 Formation Rates of Selected Acridine Derivatives in the Photochemical Oxidation of NADHa.
| catalyst | λmax/λex [nm]b | TF [h–1]c |
|---|---|---|
| acridine orange (R1 = CH3, R2 = H, X = N, Y = CH) | 480/450 | 66 ± 3 |
| proflavin (R1 = H, R2 = H, X = N, Y = CH) | 445/450 | 207 ± 17 |
| methylene blue (R1 = CH3, R2 = H, X = S+, Y = N) | 664 and 613shoulder/662 | 95 ± 3 |
| phenosafranine (R1 = H, R2 = H, R3 = phenyl, X = N+, Y = N) | 522/512 | 99 ± 2 |
| Safranine O (R1 = H, R2 = CH3, R3 = phenyl, X = N+, Y = N) | 507/519 | 97 ± 3 |
| FMN | 450/450 | 154 ± 18 |
General conditions: 50 μM catalysts, 50 mM KPi, pH 7.0, 1 mM NADH, 30 °C, 300 rpm.
λmax = wavelength exhibiting the maximal photoabsortion; λex = wavelength of the LED light source used for photoexcitation.
TF = turnover frequency of the catalyst = (H2O2 formation rate) [mM h–1]/(concentration of the photocatalyst) [mM].
Drawing correlations between the physicochemical and structural properties of the photocatalysts and their activity (Table 1) is difficult as factors such as redox potential, photoexcitability, and reactivity of the reduced form with O2 contribute to the macroscopically observed H2O2 generation rate. Future work will aim at an in-depth understanding of the influence of the substitution pattern on aspects such as reactivity and stability of the photocatalysts.
Even though all photocatalysts mentioned in Table 1 were suitable to promote the rAaeUPO-catalyzed hydroxylation reaction, we focused on phenosafranine, methylene blue, and FMN, because this combination offers a broad coverage of the visible light spectrum (Figure 1). Additionally, as permanent ions, these photocatalysts ensure high and pH-independent solubility in the aqueous reaction mixture.
Figure 1.
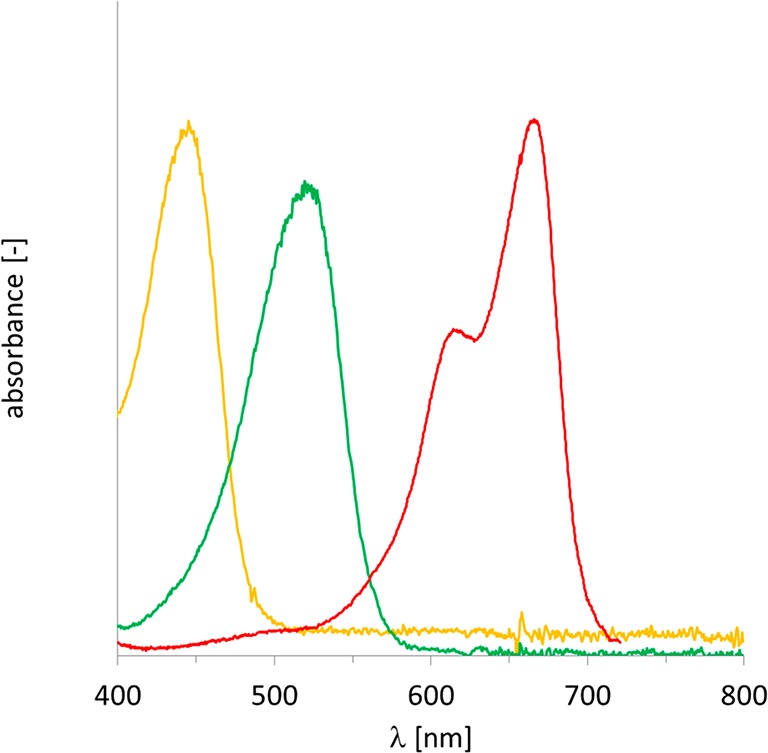
Coverage of the visible light spectrum by combining flavin mononucleotide (orange), phenosafranine (green), and methylene blue (red).
Using these photocatalysts, either individually or in combination, together with the CbFDH/NAD/HCO2H system generated H2O2 to promote the rAaeUPO-catalyzed stereospecific hydroxylation of ethylbenzene to (R)-1-phenylethanol (Figure 2).
Figure 2.
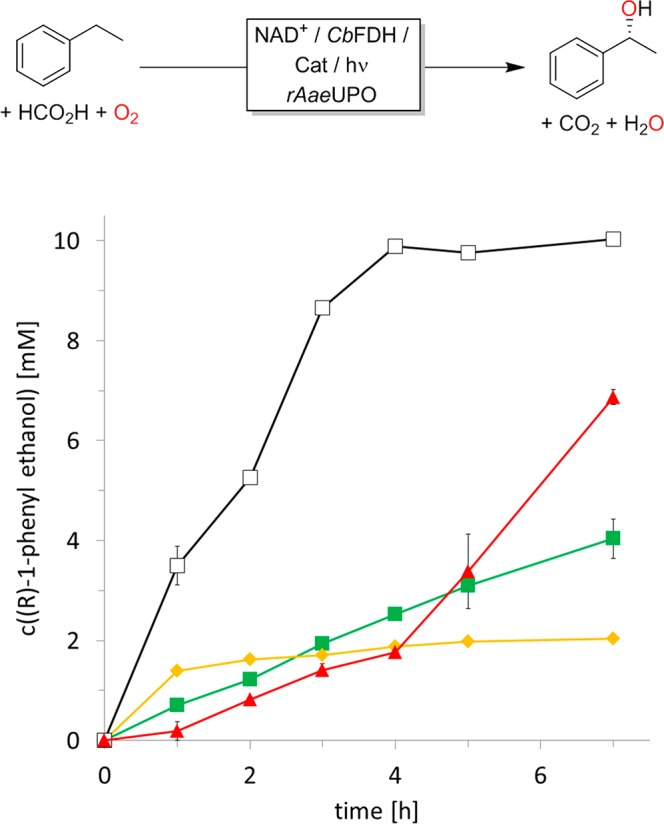
Photoenzymatic hydroxylation of ethylbenzene to (R)-1-phenylethanol using 5 μM FMN (orange ⧫), 10 μM methylene blue (red ▲), 5 μM phenosafranine (green ■), or 10 μM methylene blue + 5 μM phenosafranine + 5 μM FMN (□) as photocatalysts. Conditions: c(rAaeUPO) = 100 nM, c(CbFDH) = 4.8 μM, c(NAD+) = 0.4 mM, c(NaHCO2) = 75 mM, c(ethylbenzene) = 10 mM, 50 mM KPi buffer (pH 7, 0.8% MeOH (v/v)), T = 30 °C, λ = 450, 520, and 630 nm (blue, green, and red LED light). Please note: using a broadspectrum (sunlight-imitating) light source (Lightincure LC8 L9566, Hamamatsu) gave comparable results (Figure S9).
In the absence of either photocatalyst or rAaeUPO, no product formation was observed. The same is true for experiments performed in the darkness with the exception of methylene blue where upon prolonged reaction times some product traces were found (0.4 mM after 48 h). This is in line with previous observations that methylene blue is capable of a “dark-reaction” with NADH.21,25 Some product formation (approximately 5–20% of the “normal” product formation rate) was observed in the absence of either component of the NADH regeneration system (i.e., in the absence of formate, CbFDH, or NAD+). The latter observation most likely can be attributed to an undesired reductive quenching of the excited photocatalysts by oxidizable components in the reaction mixture (i.e., proteins, amino acids, etc.; see also Tables S2 and S3).26 It is worth mentioning that the optical purity of the product always exceeded 95% enantiomeric excess (ee).
The relative rates observed with the individual photocatalysts qualitatively corresponded to the photocatalytic H2O2 generation rates shown in Table 1. Noteworthy, when using a combination of the single photocatalysts, the product formation rate was approximately the sum of the previously observed individual rates (Figure 2). The turnover numbers calculated for the catalytic components (rAaeUPO, photocatalysts, NAD, and CbFDH) were 100 000, 500, 25, and 1785, respectively.
Next, we further examined the influence of different reaction parameters on the rate of the photoenzymatic hydroxylation reaction in more detail (Figure 3). Quite expectedly, the concentration of the photocatalysts directly influenced the rate of the overall system (Figure 3A). While this correlation was linear with methylene blue over the entire concentration range investigated, a saturation-type behavior was observed with phenosafranine and FMN, which most likely can be attributed to the decreasing optical transparency of the reaction mixture at elevated concentrations of the latter photocatalysts. The overall reaction rate also directly correlated with the intensity of the light source (Figure 3B).
Figure 3.

Influence of different reaction parameters on the product formation rate of the photoenzymatic hydroxylation of ethylbenzene. (A) Concentration of the photocatalyst (red ■: methylene blue; green ⧫: phenosafranine; orange ▲: FMN); (B) intensity of broadband light source (c(rAaeUPO) = 100 nM, c(CbFDH) = 4.8 μM, c(NAD+) = 0.4 mM); concentration of rAaeUPO (C), CbFDH (D), or NAD+ (E). Conditions (unless indicated otherwise): c(rAaeUPO) = 50 nM, c(CbFDH) = 2.4 μM, c(NAD+) = 0.2 mM, c(NaHCO2) = 75 mM, c(ethylbenzene) = 10 mM, 50 mM KPi buffer (pH 7, 0.8% MeOH (v/v)), T = 30 °C.
Varying the concentration of either NAD+ (Figure 3E) or CbFDH (Figure 3D) directly influenced the reaction rate while the concentration of rAaeUPO (Figure 3C) had no clear influence.
Overall, we conclude that the photochemical oxidation of NADH (being influenced by the in situ concentration of NADH and the concentration of the photoexcited photocatalyst(s)) was overall rate-limiting under the conditions investigated.
Interestingly, with methylene blue, an acceleration of the reaction rate was observed over time (Figure 2). This acceleration could be assigned to a photochemical activation of the photocatalyst as a similar observation was made upon preincubation of methylene blue alone by red light (Figure S6); blue light did not induce this acceleration. Currently, we are lacking a plausible explanation for this activation effect, and further studies will be necessary to understand (and synthetically exploit) this observation.
One major challenge observed, especially using FMN as photocatalyst (Figure 2 ⧫), was the poor long-term stability of the overall reaction. Particularly, the NADH regeneration reaction was impaired (Figure S7). We therefore investigated the stability of CbFDH in the presence of the photocatalysts upon illumination (Figure 4).
Figure 4.
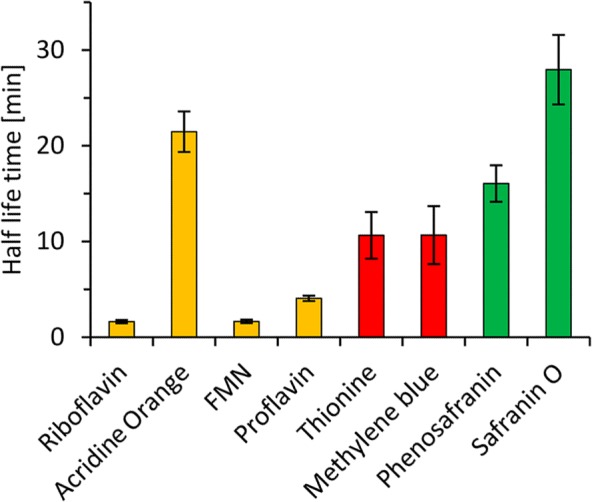
Stability of CbFDH in the presence of some photocatalysts upon illumination. General conditions: 2.4 μM CbFDH, 50 μM photocatalysts, 50 mM KPi buffer, pH 7, 300 rpm, 30 °C. For comparison, the half-life time of CbFDH in the presence of the photocatalysts but under dark conditions was 433 min.
Especially, the flavin derived photocatalysts rapidly inactivated CbFDH. Most probably, this occurred due to oxidative modification of surface-bound amino acids leading to enzyme inactivation/denaturation. Further experiments identifying the amino acids involved are currently ongoing. On the basis of this, CbFDH mutants exhibiting increased robustness in the presence of the exited photocatalysts can be conceived. Also, physical separation of photocatalysts and CbFDH may represent a solution to this inactivation issue. In similar cases, this strategy resulted in significant stabilizations of the overall reaction.27−30
Another issue of the photoenzymatic reaction system may be the well-known photobleaching of the organic photocatalysts.31,32 Particularly, the flavin-based photocatalysts exhibited a rather modest stability upon illumination with 450 nm (Figure 5). Safranine derivatives excelled in this respect by more than 100-fold longer half-life times as compared to, e.g., FMN.
Figure 5.
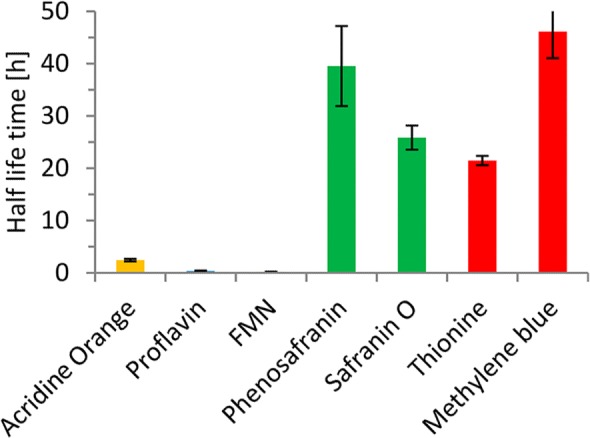
Stability of the photocatalysts upon illumination. General conditions: 100 μM photocatalysts, 50 mM KPi buffer, pH 7, 300 rpm, 30 °C.
These findings also were confirmed in photoenzymatic reactions using FMN, phenosafranine, or methylene blue, respectively (Table 2). Compared to the first, the latter two gave significantly higher turnover numbers for all catalysts employed.
Table 2. Comparison of the Turnover Numbers (TON) of the Different Catalystsa.
| photocatalyst | rAaeUPO | CbFDH | |
|---|---|---|---|
| FMN | 649 | 6490 | 135 |
| phenosafranine | 2500 | 25000 | 520 |
| methylene blue | 3992 | 39920 | 832 |
Conditions: c(rAaeUPO) = 100 nM, c(CbFDH) = 4.8 μM, c(NAD+) = 0.4 mM, c(NaHCO2) = 75 mM, c(ethylbenzene) = 10 mM, c(photocatalyst) = 1 μM, 50 mM KPi buffer (pH 7, 0.8% MeOH (v/v)), T = 30 °C, polychromatic light source.
Overall, we have demonstrated that simple electron donors such as formate can drive peroxygenase-catalyzed oxyfunctionalization reactions. Furthermore, in this study, we have demonstrated that a more efficient usage of the visible light spectrum is possible by combining complementary photocatalysts.
Admittedly, this system still is far from preparative applicability. Especially, the robustness of the formate dehydrogenase used represents a practical limitation, which may be overcome by evaluating FDHs from other sources or CbFDH mutants with improved resistance. Nevertheless, it should be noted that despite the early stage of development the proposed reaction scheme already compares well with the state-of-the-art system.
We are convinced that with this study we are paving the way toward more efficient photoenzymatic reaction schemes.
Acknowledgments
We thank The Netherlands Organisation for Scientific Research for financial support through a VICI grant (No. 724.014.003).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b03752.
Complete description of the experimental and analytical procedures as well as some additional results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wang Y.; Lan D.; Durrani R.; Hollmann F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges?. Curr. Opin. Chem. Biol. 2017, 37, 1–9. 10.1016/j.cbpa.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Hofrichter M.; Ullrich R. Oxidations catalyzed by fungal peroxygenases. Curr. Opin. Chem. Biol. 2014, 19, 116–125. 10.1016/j.cbpa.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Dong J.; Fernández-Fueyo E.; Hollmann F.; Paul C.; Pesic M.; Schmidt S.; Wang Y.; Younes S.; Zhang W. Biocatalytic oxidation reactions - a Chemist’s perspective. Angew. Chem., Int. Ed. 2018, 57, 9238–9261. 10.1002/anie.201800343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S.; Steckhan E.; Liese A. First asymmetric electroenzymatic oxidation catalyzed by a peroxidase. Electrochem. Commun. 2004, 6, 583–587. 10.1016/j.elecom.2004.04.009. [DOI] [Google Scholar]
- Bankar S. B.; Bule M. V.; Singhal R. S.; Ananthanarayan L. Glucose oxidase - An overview. Biotechnol. Adv. 2009, 27, 489–501. 10.1016/j.biotechadv.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Ni Y.; Fernández-Fueyo E.; Baraibar A. G.; Ullrich R.; Hofrichter M.; Yanase H.; Alcalde M.; van Berkel W. J. H.; Hollmann F. Peroxygenase-catalyzed oxyfunctionalization reactions promoted by the complete oxidation of methanol. Angew. Chem., Int. Ed. 2016, 55, 798–801. 10.1002/anie.201507881. [DOI] [PubMed] [Google Scholar]
- Pesic M.; Willot S. J.-P.; Fernández-Fueyo E.; Tieves F.; Alcalde M.; Hollmann F. Multienzymatic in situ hydrogen peroxide generation cascade for peroxygenase-catalysed oxyfunctionalisation reactions. Z. Naturforsch., C: J. Biosci. 2018, 10.1515/znc-2018-0137. [DOI] [PubMed] [Google Scholar]
- Pereira P. C.; Arends I.; Sheldon R. A. Optimizing the chloroperoxidase-glucose oxidase system: The effect of glucose oxidase on activity and enantioselectivity. Process Biochem. 2015, 50, 746–751. 10.1016/j.procbio.2015.02.006. [DOI] [Google Scholar]
- Holtmann D.; Krieg T.; Getrey L.; Schrader J. Electroenzymatic process to overcome enzyme instabilities. Catal. Commun. 2014, 51, 82–85. 10.1016/j.catcom.2014.03.033. [DOI] [Google Scholar]
- Getrey L.; Krieg T.; Hollmann F.; Schrader J.; Holtmann D. Enzymatic halogenation of the phenolic monoterpenes thymol and carvacrol with chloroperoxidase. Green Chem. 2014, 16, 1104–1108. 10.1039/C3GC42269K. [DOI] [Google Scholar]
- Krieg T.; Huttmann S.; Mangold K.-M.; Schrader J.; Holtmann D. Gas diffusion electrode as novel reaction system for an electro-enzymatic process with chloroperoxidase. Green Chem. 2011, 13, 2686–2689. 10.1039/c1gc15391a. [DOI] [Google Scholar]
- Horst A. E. W.; Bormann S.; Meyer J.; Steinhagen M.; Ludwig R.; Drews A.; Ansorge-Schumacher M.; Holtmann D. Electro-enzymatic hydroxylation of ethylbenzene by the evolved unspecific peroxygenase of Agrocybe aegerita. J. Mol. Catal. B: Enzym. 2016, 133, S137–S142. 10.1016/j.molcatb.2016.12.008. [DOI] [Google Scholar]
- Zhang W.; Fernández-Fueyo E.; Ni Y.; van Schie M.; Gacs J.; Renirie R.; Wever R.; Mutti F. G.; Rother D.; Alcalde M.; Hollmann F. Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalizations. Nat. Catal. 2018, 1, 55–62. 10.1038/s41929-017-0001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Burek B. O.; Fernández-Fueyo E.; Alcalde M.; Bloh J. Z.; Hollmann F. Selective activation of C-H bonds by cascading photochemistry with biocatalysis. Angew. Chem., Int. Ed. 2017, 56, 15451–15455. 10.1002/anie.201708668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. S.; Ni Y.; Fernández-Fueyo E.; Lee M.; Hollmann F.; Park C. B. Photoelectroenzymatic Oxyfunctionalization on Flavin-Hybridized Carbon Nanotube Electrode Platform. ACS Catal. 2017, 7, 1563–1567. 10.1021/acscatal.6b03453. [DOI] [Google Scholar]
- Churakova E.; Arends I. W. C. E.; Hollmann F. Increasing the Productivity of Peroxidase-Catalyzed Oxyfunctionalization: A Case Study on the Potential of Two-Liquid-Phase Systems. ChemCatChem 2013, 5, 565–568. 10.1002/cctc.201200490. [DOI] [Google Scholar]
- Churakova E.; Kluge M.; Ullrich R.; Arends I.; Hofrichter M.; Hollmann F. Specific photobiocatalytic oxyfunctionalization reactions. Angew. Chem., Int. Ed. 2011, 50, 10716–10719. 10.1002/anie.201105308. [DOI] [PubMed] [Google Scholar]
- Perez D. I.; Mifsud Grau M.; Arends I. W. C. E.; Hollmann F. Visible light-driven and chloroperoxidase-catalyzed oxygenation reactions. Chem. Commun. 2009, 6848–6850. 10.1039/b915078a. [DOI] [PubMed] [Google Scholar]
- Popov V. O.; Lamzin V. S. NAD+-dependent formate dehydrogenase. Biochem. J. 1994, 301, 625–643. 10.1042/bj3010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch M.; Schmidt S.; Arends I. W. C. E.; Oppelt K.; Kara S.; Hollmann F. Photobiocatalytic alcohol oxidation using LED light sources. Green Chem. 2017, 19, 376–379. 10.1039/C6GC02008A. [DOI] [Google Scholar]
- Kochius S.; Ni Y.; Kara S.; Gargiulo S.; Schrader J.; Holtmann D.; Hollmann F. Light-Accelerated Biocatalytic Oxidation Reactions. ChemPlusChem 2014, 79, 1554–1557. 10.1002/cplu.201402152. [DOI] [Google Scholar]
- Gargiulo S.; Arends I. W. C. E.; Hollmann F. A Photoenzymatic System for Alcohol Oxidation. ChemCatChem 2011, 3, 338–342. 10.1002/cctc.201000317. [DOI] [Google Scholar]
- Molina-Espeja P.; Garcia-Ruiz E.; Gonzalez-Perez D.; Ullrich R.; Hofrichter M.; Alcalde M. Directed Evolution of Unspecific Peroxygenase from Agrocybe aegerita. Appl. Environ. Microbiol. 2014, 80, 3496–3507. 10.1128/AEM.00490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich R.; Nüske J.; Scheibner K.; Spantzel J.; Hofrichter M. Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Appl. Environ. Microbiol. 2004, 70, 4575–4581. 10.1128/AEM.70.8.4575-4581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könst P.; Kara S.; Kochius S.; Holtmann D.; Arends I. W. C. E.; Ludwig R.; Hollmann F. Expanding the Scope of Laccase-Mediator Systems. ChemCatChem 2013, 5, 3027–3032. 10.1002/cctc.201300205. [DOI] [Google Scholar]
- Frisell W. R.; Chung C. W.; Mackenzie C. G. Catalysis of Oxidation of Nitrogen Compounds by Flavin Coenzymes in the Presence of Light. J. Biol. Chem. 1959, 234, 1297–1302. [PubMed] [Google Scholar]
- Hildebrand F.; Lütz S. Stable Electroenzymatic Processes by Catalyst Separation. Chem. - Eur. J. 2009, 15, 4998–5001. 10.1002/chem.200900219. [DOI] [PubMed] [Google Scholar]
- Rudroff F.; Mihovilovic M. D.; Gröger H.; Snajdrova R.; Iding H.; Bornscheuer U. T. Opportunities and challenges for combining chemo- and biocatalysis. Nature Catal. 2018, 1, 12–22. 10.1038/s41929-017-0010-4. [DOI] [Google Scholar]
- Sato H.; Hummel W.; Gröger H. Cooperative Catalysis of Noncompatible Catalysts through Compartmentalization: Wacker Oxidation and Enzymatic Reduction in a One-Pot Process in Aqueous Media. Angew. Chem., Int. Ed. 2015, 54, 4488–4492. 10.1002/anie.201409590. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Fernandez Fueyo E.; Hollmann F.; Leemans Martin L.; Pesic M.; Wardenga R.; Höhne M.; Schmidt S. Combining Photo-Organo Redox- and Enzyme Catalysis Facilitates Asymmetric C-H Bond Functionalization. Eur. J. Org. Chem. 2018, 10.1002/ejoc.201801692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinska-Rak M.; Golczak A.; Sikorski M. Photochemistry of Riboflavin Derivatives in Methanolic Solutions. J. Phys. Chem. A 2012, 116, 1199–1207. 10.1021/jp2094593. [DOI] [PubMed] [Google Scholar]
- Holzer W.; Shirdel J.; Zirak P.; Penzkofer A.; Hegemann P.; Deutzmann R.; Hochmuth E. Photo-induced degradation of some flavins in aqueous solution. Chem. Phys. 2005, 308, 69–78. 10.1016/j.chemphys.2004.08.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.