

Abstract

The conversion of lignin to biofuels and biobased chemicals is currently attracting a lot of attention. We here report on the valorization of Kraft lignin by a catalytic hydrotreatment using Ni, Mo, and W phosphide catalysts supported on activated carbon in the absence of an external solvent. Experiments were carried out in a batch setup in the temperature range of 400–500 °C and 100 bar initial H2 pressure. The synthesized catalysts were characterized by X-ray diffraction, nitrogen physisorption, and transmission electron microscopy. The lignin oils were analyzed extensively by different techniques such as GPC, GC-MS-FID, 13C NMR, and elemental analysis. Two-dimensional gas chromatography (GC×GC-FID) was applied to identify and quantify distinct groups of compounds (aromatics, alkylphenolics, alkanes, etc.). Mo-based catalysts displayed higher activity compared to the W-containing catalysts. The reaction parameters such as the effect of reaction temperature, reaction time, and catalyst loading were studied for two catalysts (15MoP/AC and 20NiMoP/AC), and optimized reaction conditions regarding yields of monomeric components were identified (400 °C, 100 bar H2 at RT, 10 wt % catalyst loading on lignin intake). The highest monomer yield (45.7 wt % on lignin) was obtained for the 20NiMoP/AC (Ni 5.6 wt %, Mo 9.1 wt %, P 5.9 wt %) catalyst, which includes 25% alkylphenolics, 8.7% aromatics, and 9.9% alkanes. Our results clearly reveal that the phosphide catalysts are highly efficient catalyst to depolymerize the Kraft lignin to valuable biobased chemicals and outperform sulfided NiMo catalysts (monomer yield on lignin < 30 wt %).
Keywords: Kraft lignin, Hydrotreatment, Depolymerization, Phosphided catalysts, Biobased chemicals
Short abstract
Depolymerization of Kraft lignin to high amounts (45.7 wt % on lignin) of low molecular biobased chemicals like alkylphenolics and aromatics has been achieved using a phosphided NiMo catalyst on activated carbon.
Introduction
Lignin is one of the major components in lignocellulosic biomass and has great potential to be used as a feedstock for biofuels and biobased chemicals.1−4 It consist of a complex 3-D structure with substituted aromatic rings linked by C–C and C–O bonds.5−7 Cleavage of the linkages is in theory an attractive way to obtain low molecular weight aromatics and phenolics.8 However, its high structural heterogeneity and low reactivity of particularly the C–C linkages combined with the typically harsh reaction conditions required to breakdown the polymer hamper effective depolymerization strategies.
Lignin can be obtained from lignocellulosic biomass by a range of processes.9−12 Kraft and lignosulfonates lignins are commercially produced by the pulp and paper industry. Due to the use of sulfur reagents in the process, sulfur (1–2%) is incorporated in these lignin.13 About 55 million tons of such sulfur-containing lignins are produced annually, yet these are currently only used as an energy source in the paper mill.14 It is estimated that about 8–11 Mt·y–1 of these lignins can be used to produce aromatic platform chemicals like phenols or aromatics (benzene, toluene, xylenes) without affecting the operation of the paper mills.15−17
Several methodologies have been explored for Kraft lignin depolymerization such as oxidation,18−20 reduction,21−24 and pyrolysis.25−27 Lignin depolymerization by reductive methods is generally carried out using hydrogen and a heterogeneous catalyst in the presence of an acid or base23,28−30 and typically in a protic solvent such as methanol,31 ethanol,32−34 isopropanol,35 and water (hydrothermal method).1,29,36−41 The catalytic reductive depolymerization with hydrogen (hydrotreatment) without an external solvent has been studied as well (Table 1).42−48 This does not imply the occurrence of solid–gas reactions only, as Kraft lignin is known to start to liquefy at relatively low temperatures (175–200 °C) and as such acts as the (reactive) solvent at hydrotreatment reaction conditions. In addition, monomers (phenolics, aromatics, etc.) formed by thermal and catalytic reactions already at an early stage of the batch process will also act as the solvent.44 Typically, relatively harsh conditions are used with temperatures between 350 and 450 °C and initial hydrogen pressures at room temperature up to 100 bar. However, this strategy has advantages compared to solvent-based routes. For instance, solvents like methanol or ethanol are typically not inert and are incorporated in the products (alkylation).40 Furthermore, the use of a solvent complicates product workup and needs the introduction of an efficient solvent recycling strategy to improve the techno-economic viability of the process.
Table 1. Literature Data for the Catalytic Hydrotreatment of Various Lignins in the Absence of an External Solvent.
| lignin | catalyst | temp. (°C) | H2 pressure (bar) | time (h) | total yield of monomers (%)a | oil yield (%)a | ref |
|---|---|---|---|---|---|---|---|
| Organocell | 21%NiMo/Al2SiO5 | 420 | 100 | 1 | 21.8 | 61.6 | (42) |
| Kraft | S-21%NiMo/Al2SiO5 + S-20%Cr2O3/Al2O3 (1:1) | 430 | 90 | 1 | 38.4 | 57 | (43) |
| pyrolytic lignin | Ru/C | 400 | 100d | 4 | 39.8b | 75.8 | (44) |
| 51.3c | 75.4 | ||||||
| Alcell | Ru/C | 400 | 100d | 4 | 22.1 | 63.9 | (45) |
| 8 | 29.7 | 72.8 | |||||
| Kraft | S-NiMo/MgO-La2O3 | 350 | 100d | 4 | 26.4 | 48.2 | (46) |
| Kraft | limonite | 450 | 100d | 4 | 31 | 33.7 | (47) |
Yield is in wt % on lignin intake.
Lignin from pine wood.
Forestry residue.
Initial pressure at room temperature, up to 200 bar at actual reaction temperature.
Early studies on the catalytic hydrotreatment of a number of lignins (including Kraft lignin) using NiMo catalysts on aluminosilica as the support in the absence of an external solvent were reported by Meier et al.42 Oil yields of up to 61.6 wt % were reported. In the case of organocell lignin, the monomer yield was 21.8 wt %. Related hydrotreatment studies were reported by Oasmaa et al. using a variety of technical lignins.43 The highest oil yield was 71 wt %, obtained for an organosolv lignin using a physical mixture of NiMo on aluminosilica and Cr2O3. The amount of low molecular weight compounds was also detemined and was between 14 and 38 wt % on lignin intake. Best results were obtained using Kraft lignin. Recently, Kloekhorst et al. reported catalytic hydrotreatment studies using a pyrolytic lignin from a forestry residue and Alcell lignin with Ru/C as the catalyst.44 For forestry residue pyrolytic lignin 75 wt % of lignin oil was obtained. Detailed analysis by advanced GC methods showed that the oil contained high amounts of monomeric alkyl phenolics (20.5 wt %) and aromatics (14.1 wt %). Supported noble metal catalysts (Ru, Pd ,and Cu catalysts) have also been applied for the catalytic hydrotreatment of Alcell lignin (batch, 400 °C, 100 bar H2 at RT for 4–8 h).45 Ru/C gave the best results in terms of lignin oil yield (72.8 wt % yield based on lignin intake) and total monomers yield (29.7%, of which 12.2% alkylphenolics, 5.2% aromatics, 10.1% overhydrogenated product (alkanes)). Recently, we reported the use of bimetallic sulfided NiMo and CoMo catalysts on various supports (Al2O3, ZSM-5, AC, MgO-La2O3) for Kraft lignin hydrotreatment in the absence of an external solvent (batch, 350 °C, 100 bar H2 at RT for 4 h).46 Best results in terms of oil and monomer yield were obtained with the sulfided NiMo/MgO–La2O3 catalyst, giving 87% lignin conversion and 26.4 wt % of monomers on lignin intake, of which 15.7% were phenolics and 5.9% aromatics. Very recently, we reported the use of iron-based catalysts for the hydrotreatment of Kraft lignin in the absence of the external solvent.47 Various types of iron catalysts were explored; examples include limonite ore, Goethite, iron–nickel oxide (Fe2O3–NiO), iron oxide (Fe2O3), and iron disulfide (FeS2). The best results were obtained with limonite at 450 °C, giving 31 wt % of monomers on lignin intake, of which 17% were alkylphenolics and 8% aromatics.
The major disadvantage of the use of the sulfided NiMo/CoMo catalysts is the requirement to introduce sulfided reagents to maintain activity and stability of the catalyst. Recently, transition metal phosphide catalysts have been introduced for hydrotreatment reactions49−52 and shown to be attractive alternatives for expensive noble metal catalysts.53,54 One of the advantages of this class of catalysts is that the use of a sulfur-introducing reagent is not required to maintain activity.
We here report the use of mono- and bimetallic phosphide catalysts with Ni, Mo, and W as the active metals for the catalytic hydrotreatment of Kraft lignin in the absence of an external solvent to obtain value-added chemicals like phenols and aromatics. Activated carbon (AC) was used as the support as previous research from our group on the hydrotreatment of Kraft lignin using sulfided NiMo and CoMo catalyst on different supports showed that char formation is lowest when using AC.46 A series of mono- and bimetallic phosphide catalysts was prepared and characterized by XRD, nitrogen physisorption, and TEM analysis. The catalysts were evaluated for the catalytic hydrotreatment of Kraft lignin, and process conditions such as temperature, reaction time, and catalyst loading were optimized to maximize monomer yields. The lignin oils after hydrotreatment were analyzed in detail by various techniques such as GPC, GC-MS/FID, GC×GC, 13C NMR, and CHNS analysis. Finally, the performance of the phosphide-based catalyst is compared with data available in the literature for lignin hydrotreatments without the use of an external solvent.
Experimental Section
Materials
Chemicals used in the study were of analytical grade and used as received. The activated carbon (AC) support was obtained from Merck, Germany. Ni(NO3)2·6H2O (99%), (NH4)6Mo7O24·4H2O (>99%), (NH4)6H2W12O40·xH2O, and (NH4)2HPO4 were purchased from Sigma-Aldrich. Dichloromethane, di-n-butylether (DBE), nitric acid, acetone, and THF were obtained from Boom B.V. Hydrogen (>99.99%), nitrogen (>99.8%), and 2% O2/Ar were purchased from Hoekloos. Indulin-AT (Kraft lignin) was from MWV specialty chemicals and provided by the Wageningen University and Research Center, The Netherlands (Dr. R. Gosselink). Indulin-AT is a purified form of Kraft pine lignin. The lignin content is 97 wt % on dry basis; the remainder is mainly ash. The elemental compostion is as follows: C = 61.87 wt %, H = 5.98 wt %, N = 0.69 wt %, S = 1.34 wt %, O = 30.12 wt %. The molecular weight is reported to be about 4000 g/mol.55
Synthesis of the Metal Phosphide Catalysts on Activated Carbon
The NiP, MoP, WP, NiMoP, and NiWP supported on AC catalysts were prepared according to a literature procedure.53,56,57 The monometallic Ni–P catalysts with 2.5 wt % of Ni and 2.6 wt % of P was prepared as follows: Ni(NO3)2·6H2O (0.61 g) was dissolved in deionized water (10 mL). (NH4)2HPO4 (0.55 g) dissolved in deionized water (10 mL) was added to the nickel nitrate solution. The resulting precipitate was dissolved by the addition of a few drops of nitric acid. AC (4.75 g) was added to the solution, and subsequently, the excess of water was removed by evaporation. The resulting solid was dried in an oven overnight at 110 °C. The catalyst was reduced in pure hydrogen flow (100 mL min–1 g–1) at 650 °C for 2 h with a heating rate of 5 °C min–1 and cooled to RT in a hydrogen flow. The catalyst was subsequently passivated under a flow of 2% O2/Ar for 2 h. The resulting catalyst is denoted as 5NiP/AC, where 5 indicates the sum of the weight percentages of Ni and P (Ni = 2.5 wt %, P = 2.6 wt %, corresponding with a molar ratio of Ni:P = 1:2). Similarly, 15MoP/AC (Mo 9.1 wt %, P 5.9 wt % with a mole ratio of Mo:P = 1:2), 15WP/AC (mole ratio W:P = 1:2; W 11.2 wt %, P 3.8 wt %), 20NiMoP/AC (Ni 5.6 wt %, Mo 9.1 wt %, P 5.9 wt % with a mole ratio of Ni:Mo:P = 1:1:2), and 20NiWP/AC (Ni 3.9 wt %, W 12.3 wt %, P 4.1 wt % with a mole ratio of Ni:W:P = 1:1:2) catalysts were prepared with the appropriate precursors. In the case of bimetallic phosphide catalysts, the metal precursor solutions were mixed together, followed by addition of an aqueous solution of (NH4)2HPO4.
Experimental Procedure for the Catalytic Hydrotreatment of Kraft Lignin
All catalytic hydrotreatment reactions were performed in a batch autoclave (100 mL, Parr Instruments Co., stainless steel type 316). The reactor was surrounded by an aluminum block containing electrical heating elements and channels allowing the flow of cooling water. The reactor content was stirred mechanically using a gas-induced Rushton turbine. The temperature and pressure in the reactor were monitored online and logged on a PC.
The hydrotreatment and workup procedure is schematically shown in Figure 1. Initially, the reactor was loaded with Kraft lignin (15 g) and catalyst (0.75 g, 5 wt.% on lignin). Subsequently, the reactor was flushed with hydrogen to expel air and pressurized to 120 bar at room temperature for leak testing. After the leak test, the pressure was reduced to 100 bar. Stirring was started (1200 rpm), and the reactor content was heated to the desired temperature (400–500 °C) with a heating rate of about 10 °C min–1. The reaction time was set to zero when the desired temperature was reached. After completion of the reaction, the reactor was cooled to room temperature with a rate of about 10 °C min–1. The pressure at room temperature was recorded to determine the amount of gas-phase components produced during the reaction. The produced gas was collected in a 3 L Tedlar gas bag to determine the composition. For all reactions, the clear formation of two separate liquid phases was observed viz. a lignin oil (light phase) and a water phase (see the Supporting Information, Figure S1). The product workup procedure is given in Figure 1. After reaction, the organic and water phases were filtered through a microfilter and separated by decanting. The solid was thoroughly washed with acetone and dried to determine the solid yield.
Figure 1.

Overview of the experimental procedure for the hydrotreatment of Kraft lignin including product workup.
The product yield, solid (i.e., unconverted lignin and/or repolymerized product) and mass balances were calculated based on initial lignin intake using eqs 1–4. All product yields (lignin oil, char, and gas) are given as wt % on lignin intake.
| 1 |
| 2 |
| 3 |
| 4 |
Analytical Procedures
The gas phase after reaction was collected and stored in a gas bag (SKC Tedlar 3 L sample bag (9.5″ × 10″)) with a polypropylene septum fitting. The gas-phase composition was analyzed using a GC-TCD (Hewlett-Packard 5890 Series II GC equipped with a Poraplot Q Al2O3/Na2SO4 column and a molecular sieve (5 Å) column). The injector temperature was set at 150 °C and the detector temperature at 90 °C. The oven temperature was kept at 40 °C for 2 min, then heated up to 90 °C at 20 °C min–1, and kept at this temperature for 2 min. A reference gas was used to quantify the results (55.19% H2, 19.70% CH4, 3.00% CO, 18.10% CO2, 0.51% ethylene, 1.49% ethane, 0.51% propylene, and 1.5% propane).
Lignin oils were analyzed by GC-MS-FID using a Quadruple Hewlett-Packard 6890 MSD attached to a Hewlett-Packard 5890 GC equipped with a sol–gel capillary column (60 m × 0.25 mm i.d. and a 0.25 μm). The injector temperature was set at 250 °C. The oven temperature was kept at 40 °C for 5 min, heated to 250 °C at a rate of 3 °C min–1, and then held at 250 °C for 10 min.
GC×GC-FID analysis was performed using a trace GC×GC from Interscience equipped with a cryogenic trap system and two columns (a 30 m × 0.25 mm i.d. and a 0.25 μm film of RTX-1701 capillary column connected by a meltfit to a 120 cm × 0.15 mm i.d. and a 0.15 μm film Rxi-5Sil MS column). An FID detector was used. A dual-jet modulator was applied using carbon dioxide to trap the samples. Helium was used as the carrier gas (continuous flow 0.8 mL/min). The injector temperature and FID temperature were set at 280 °C. The oven temperature was kept at 40 °C for 5 min and then heated up to 280 °C at a rate of 3 °C min–1. The pressure was set at 70 kPa at 40 °C. The modulation time was 6 s. For both GC×GC-FID and GC-MS-FID analyses, the samples were diluted with tetrahydrofuran (THF) and 500 ppm of di-n-butyl ether (DBE) was added as an internal standard. Detailed information on quantification of the amounts of monomers is given in ref (46) and the Supporting Information.
GPC analysis of the samples was performed using a HP1100 equipped with three MIXED-E columns (300 × 7.5 mm PL gel 3 μm) in series using a GBC LC 1240 RI detector. Average molecular weight calculations were performed using the PSS WinGPC Unity software from Polymer Standards Service. The following conditions were used: THF as the eluent at a flow rate of 1 mL min–1, 140 bar, a column temperature of 40 °C, 20 μL injection volume, and a 10 mg mL–1 sample concentration. Toluene was used as a flow marker. Polystyrene samples with different molecular weights were used as the calibration standards.
TGA analyses were performed using a TGA 7 from PerkinElmer. The samples were heated under a nitrogen atmosphere (flow of 50 mL/min) with a heating rate of 10 °C/min and temperature ramp of 30–900 °C.
13C NMR spectra were acquired at 25 °C using an Agilent 400 MHz spectrometer. Approximately 0.1 g of lignin oil was dissolved in 0.6 mL of dimethylsulfoxide-d6 (DMSO). The number of scans was 2048 with a relaxation time of 5 s. The data were processed using the MestReNova software.
Elemental analysis (C, H, N, and S) were performed using a Euro Vector 3400 CHN-S analyzer. The oxygen content was determined by difference. All experiments were carried out in duplicate, and the average value is provided.
TOC (total organic carbon) in the aqueous phase was determined with a Shimadzu TOC-VCSH TOC analyzer equipped with an OCT-1 sampler port.
Transmission electronic microscopy (TEM) images were obtained using a Philips CM12 operated at an acceleration voltage of 120 kV. Samples for TEM measurements were ultrasonically dispersed in ethanol and subsequently deposited on mica grid coated with carbon.
X-ray diffraction data of the catalysts were recorded on a Bruker D8 advance diffractometer using Cu Kα radiation (λ = 0.1544 nm) at 40 kV. XRD patterns were measured in reflection geometry in the 2θ range between 5° and 80° with a step size of 0.04°.
Statistical Modeling
Multivariable regression was used to model the experimental data, and for this purpose the Design Expert Version 8.0.0 software package was used. The experimental data were modeled using eq 5.
| 5 |
Here y is a dependent variable (lignin oil yield), xi and xj are the independent variables (temperature (°C) and reaction time (h)), bo, bi, bii, and bij are the regression coefficients of the model, and e is the error of the model. The regression equations were obtained by backward elimination of statistically nonsignificant parameters. A parameter was considered statistically relevant when the p value was less than 0.05.
Results and Discussion
Catalyst Synthesis and Characterization
The NiP, MoP, WP, NiMoP, and NiWP supported on AC catalysts were prepared according to a procedure reported in the literature53,56,57 and involves a wet impregnation procedure with (NH4)2HPO4 as the phosphide source. The molar ratio of metal to P was in all cases set to 1:2. For the bimetallic compounds, the soluble metal precursors were mixed before addition to the AC. All catalysts were reduced with hydrogen at 650 °C for 2 h, followed by a passivation step at room temperature with 2% O2 in air. Catalyst coding, the exact elemental composition of the catalysts, and relevant properties are given in Table 2. XRD patterns of Ni-, Mo-, and W-containing mono- and bimetallic catalysts are shown in the Supporting Information (Figure S2) as well as representative TEM images (Figure S3). Nitrogen adsorption–desorption isotherms and pore size distriution curves are shown in Figure S4 (Supporting Information).
Table 2. Composition and Textural Properties of the Metal Phosphide Catalysts.
| catalyst | molar ratio Ni:(Mo or W):P | metal and P content (wt %) | avg. pore diameter (nm) | BET surface area (m2/g) |
|---|---|---|---|---|
| AC | 3.27 | 752 | ||
| 5NiP/AC | 1:0:2 | Ni 2.5, P 2.6 | 3.24 | 750 |
| 15MoP/AC | 0:1:2 | Mo 9.1, P 5.9 | 3.26 | 502 |
| 15WP/AC | 0:1:2 | W 11.2, P 3.8 | 3.33 | 540 |
| 20NiMoP/AC | 1:1:2 | Ni 5.6, Mo 9.1, P 5.9 | 3.35 | 381 |
| 20NiWP/AC | 1:1:2 | Ni 3.9, W 12.3, P 4.1 | 3.25 | 540 |
Catalytic Hydrotreatment of Kraft Lignin Using Metal Phosphide Catalysts
Product Yields and Mass Balances for Experiments at 400 °C
In the first phase of the research, hydrotreatment experiments of Kraft lignin were performed at 400 °C using the Ni-, W-, and Mo-based mono- and bimetallic phosphides supported on activated carbon. The major product is a lignin oil with yields in the range of 39.2–64.3% on lignin intake (Table 3). Other products include a water phase (about 20%), solid residue (char 5.1–22.9%), and gas phase (8–10%). The mass balances closure excluding hydrogen consumption is very good with values between 90% and 99% (Table 3, see also Supporting Information (Table S1) for a mass balance including hydrogen consumption). Carbon balances were also calculated based on the measured carbon content in the gas phase (GC), lignin oil (elemental analyses), and water phase (total organic carbon, TOC), though excluding the carbon content of the solid phase. The TOC in the water phase was very low (less than 0.07 wt % on lignin) and as such is not a major loss of carbon for the process. The carbon balance closure is >90% for most of the catalysts (Table 3), the only exception being 5NiP/AC (56%), which is due to the high amount of solids formed when using this catalyst.
Table 3. Lignin Oil Yields and Mass Balances for Catalytic Hydrotreatment of Kraft Lignin Using Mono- and Bimetallic Phosphide Catalystsa.
| catalyst | oil yield (%)b | gas phase (%)b | water (%)b | solids (%)b | mass balance (%)b | carbon balance (%)c |
|---|---|---|---|---|---|---|
| 5NiP/AC | 39.2 | 9.4 | 18.4 | 22.9 | 90 | 56 |
| 15MoP/AC | 61.2 | 8.4 | 21.0 | 5.1 | 96 | 90 |
| 15WP/AC | 60.8 | 9.5 | 20.4 | 6.2 | 97 | 91 |
| 20NiMoP/AC | 64.3 | 10.1 | 19.8 | 5.1 | 99 | 96 |
| 20NiWP/AC | 59.3 | 10.3 | 20.2 | 8.2 | 98 | 90 |
Reaction conditions: Kraft lignin, 15 g; catalyst, 0.75 g; 400 °C; hydrogen pressure of 100 bar at RT; 4 h; 1200 rpm.
Percent is on weight basis of lignin intake.
Including carbon content of gas phase, lignin oil, and water phase, excluding carbon content of solid phase.
In the absence of a catalyst, the main product is a solid residue (>50 wt %), with by far lower amounts of a lignin oil (9.4 wt % on ligin) than obtained for the catalytic recations. As such, a catalyst is required for good depolymerization activity, though some thermal depolymerization also occurs.
The worst performance was found for the monometallic 5NiP/AC catalyst, giving a low lignin oil yield (39.2%) and high amounts of solids (22.9%). In the case of Mo- and W-containing monometallic catalysts, the lignin oil yields are considerably higher (about 60%) and also less residue was observed after reaction. However, precise comparsion of the data is not possible at this stage due to the difference in metal content.
When comparing the bimetallic catalysts, the highest lignin oil yield (64.3%) was obtained for bimetallic 20NiMoP/AC catalyst, indicating that Mo-containing phosphide catalysts exhibit better performance than the W-based catalyst.
Characterization of the Lignin Oils
The elemental composition of the lignin oils obtained at 400 °C were determined by elemental analysis. The oxygen and hydrogen contents are provided in the form of a van Krevelen diagram in Figure 2. The O/C and H/C values for the lignin oils are all present in a relatively narrow range with an O/C of about 0.05 and an H/C between 1.02 and 1.07. The O/C value is considerably lower than for the Kraft lignin feed (O/C = 0.36), indicating substantial removal of bound oxygen by, for instance, hydrodeoxygenation reactions to form water. The range of H/C and O/C values for lignin oils are in between that of alkylphenolics and aromatics, which indicates the presence of significant amounts of such component classes. This is confirmed by detailed analysis of the lignin oils (vide infra). The sulfur content in all of the lignin oils was determined and was shown to be about 0.01 wt % for all samples. The Kraft lignin used for this study contains 1.34 wt % sulfur, indicating that hydrodesulfurization takes place to a considerable extent. Furthermore, incorporation of S in the solid residue is also an option, though this was not investigated.
Figure 2.

van Krevelen plot for lignin oils obtained at 400 °C. Reaction conditions: Kraft lignin, 15 g; catalyst, 0.75 g; 400 °C; hydrogen pressure of 100 bar at RT; 4 h; 1200 rpm.
GPC chromatograms for the lignin oils obtained for the various metal phosphide catalysts at 400 °C are presented in Figure 3. For all of the lignin oils sharp intense peaks are observed in the low molecular weight region (80–150 g/mol), indicating the presence of a significant amount of low molecular weight monomers. The average molecular weight for the original Kraft lignin determined by our GPC method is 985 g/mol, indicating that the lignin oils are considerably depolymerized. However, the extent of depolymerization is underestimated as our molecular weight data for the Kraft lignin are by far lower than reported in the literature (4000 g/mol). The low value for the Kraft lignin feed found by us is due to a limited solubilty in the eluent used for the GPC analysis (THF).
Figure 3.
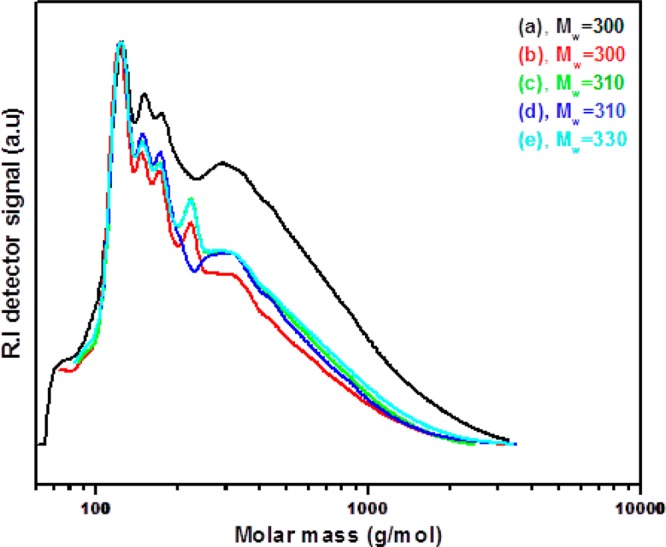
Gel permeation chromatograms of lignin oils obtained for over various phosphide catalysts at 400 °C: (a) 5NiP/AC, (b) 15MoP/AC, (c) 15WP/AC, (d) 20NiMoP/AC, and (e) 20NiWP/AC.
GC analysis was performed on the product oils to gain insights into the molecular composition. A representative GC-MS spectrum is given in the Supporting Information (Figure S5) and shows the presence of a large number of compounds belonging to various organic component classes. Quantification of the monomers present in the lignin oils was done by GC×GC analysis using n-dibutylether as an internal standard. The main advantage of this technique is better separation of products allowing clustering of component classes (alkylphenolics, aromatics, alkanes, etc.). A typical GC×GC chromatogram is shown in the Supporting Information (Figure S6), where the different regions for the various classes of monomers can be clearly seen. The total monomer yield (GC×GC analysis) for the catalytic hydrotreatment reactions performed at 400 °C is shown in Figure 4. Highest total monomer yields were obtained for the Mo-containing phosphide catalysts, with values up to 40 wt % on lignin intake. The main product class is alkylphenolics, with yields of about 22 wt % on lignin intake for the Mo-containing catalysts, followed by aromatics, with yields of about 8 wt %. In addition, some overhydrogenated products like cyclic and linear alkanes are present.
Figure 4.

Monomer yield (wt % on lignin intake) for hydrotreatment reactions carried out at 400 °C.
The lignin oil samples were also characterized using 13C NMR. This method has the advantage over GC methods that all of the components in the sample are identifiable and not only the low molecular weight fraction detectable by GC. Figure 5 shows the spectra of the parent Kraft lignin and the lignin oil obtained at 400 °C using the bimetallic 20NiMoP/AC catalyst. Information about chemical reactions occurring during the hydrotreatment process can be obtained by integration of peak intensities in chemical shift ranges belonging to carbon atoms with different chemical environments (aliphatics, δ 0–36 ppm; methoxy, δ 52–58 ppm; ether bonds, δ 58–100 ppm; aromatics, δ 100–160 ppm44). Kraft lignin exhibits peaks at δ 50.1 and δ 60.5 ppm, corresponding to methoxy and ether carbons, respectively. A number of peaks are observed in the range from δ 107 to 152 ppm, related to aromatic carbons in the lignin polymer. After the hydrotreatment reaction using the 20NiMoP/AC catalyst, the peaks related to methoxy groups were not observed, indicating that most of the methoxy groups are removed during the process. In addition, typical resonances from the C–O–C linkages have also disappeared, suggesting that ether linkages are broken, leading to the formation of lower molecular weight components. Also, the intensity of the peaks in the aliphatic and aromatic region is relatively high in the hydrotreated Kraft lignin. The intense peaks in the δ 0–36 ppm region are due to presence of alkyl chains (methyl, ethyl, and propyl, etc.) on the depolymerized products such as alkylphenolics, alkyl-substituted aromatics, and over-hydrogenated products.
Figure 5.
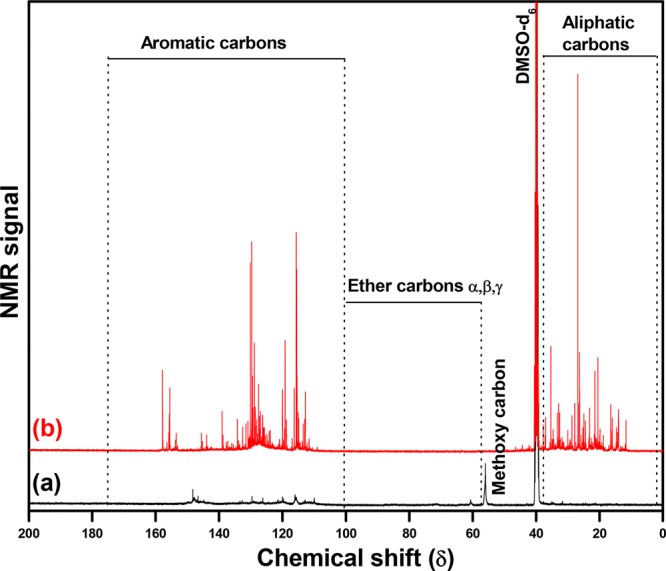
13C NMR spectra in DMSO-d6 of (a) Kraft lignin and (b) lignin oil obtained using 20NiMoP/AC at 400 °C.
Characterization of the Gas Phase
The gas-phase composition after reaction was determined by GC for all experiments performed at 400 °C (Table 3), and the data are given in the Supporting Information (Table S2). It was shown to consist of mainly unconverted hydrogen, CO2 (2.7–4.1 wt % on lignin), CO (<0.2 wt % on lignin), and hydrocarbons, mainly in the form of CH4 (3.6–5.3 wt % on lignin) and some of ethane (0.7–0.9 wt % on lignin) and propane (0.5–0.7 wt % on lignin). The gas-phase components may be formed by reactions involving the lignin (e.g., methoxy removal and formation of methane, decarbonylation), as well as subsequent gas-phase reactions (water–gas shift and CO/CO2 hydrogenation). The formation of H2S is anticipated based on the relatively harsh conditions and the presence of organic sulfur in the Kraft lignin feed, though it could not quantified by the GC method used in this study.
Optimization of Reaction Parameters
The mono- and bimetallic Mo-containing catalysts (15MoP/AC and 20NiMoP/AC) gave the highest amount of monomers based on GC×GC analysis. Hence, these catalysts were selected for further optimization studies with an emphasis on reaction temperature, reaction time, and catalyst loading. The yields of the various products (lignin oil, gas-phase components, and solid residue) were determined, and the lignin oils were characterized in detail.
Effect of Temperature and Reaction Time
The effect of temperature and reaction time was studied for temperatures in the range of 400–500 °C and reaction times between 0 and 8 h. A reaction time of 0 h means that the batch reactor was heated to the predetermined reaction temperature and then immediately cooled to room temperature. The product yields and mass balances are provided in Table 4. Very good mass balance closures (>93%) were obtained for all reactions. It is evident that when the severity is increased (higher temperature, longer batch times), the lignin oil yield is decreased. This is particularly evident when comparing the lignin oil yield for the monometallic Mo catalyst, viz from 80.5 wt % at the lowest severity (400 °C, 0 h) to 37.2 wt % at a high severity (450 °C, 4 h). At high severity, the amount of solid increases, indicating that repolymerization of reactive fragments to solids becomes more prominent at higher severity. In addition, the formation of larger amounts of water at higher severities suggests a higher rate of hydrodeoxygenation reactions at these conditions. When comparing both catalysts, char formation was the lowest for the bimetallic 20NiMoP/AC catalyst at all reaction temperatures and batch times. The only difference between both catalysts when regarding composition (Table 2) is the presence of 5 wt % of Ni in the bimetallic catalyst. As such, it appears that the addition of Ni to the MoP/AC catalyst has a positive effect on performance.
Table 4. Lignin Oil Yields and Mass Balances for the Hydrotreatment of Kraft Lignin Using Mono- and Bimetallic Mo-Based Catalystsa.
| catalyst | temp. (°C) | time (h) | oil yield (%)b | gas phase (%)b | water (%)b | solid (%)b | mass balance (%)b |
|---|---|---|---|---|---|---|---|
| 20NiMoP/AC | 350 | 4 | 61.7 | 5.4 | 11.8 | 4.5 | 85c |
| 15MoP/AC | 400 | 0 | 80.5 | 7.5 | 7.8 | 2.9 | 99 |
| 2 | 67.2 | 9.0 | 20.3 | 1.7 | 97 | ||
| 4 | 61.2 | 8.4 | 21.0 | 5.1 | 96 | ||
| 8 | 59.8 | 10.7 | 23.0 | 4.9 | 98 | ||
| 20NiMoP/AC | 400 | 0 | 77.4 | 6.4 | 12.0 | 1.7 | 97 |
| 2 | 67.2 | 8.6 | 19.7 | 4.0 | 99 | ||
| 4 | 64.3 | 10.1 | 19.8 | 5.1 | 99 | ||
| 8 | 64.5 | 9.8 | 20.3 | 3.5 | 98 | ||
| 15MoP/AC | 425 | 4 | 49.5 | 9.7 | 20.7 | 13.2 | 93 |
| 20NiMoP/AC | 425 | 4 | 55.7 | 14.4 | 21.5 | 7.6 | 99 |
| 15MoP/AC | 450 | 0 | 64.7 | 9.8 | 18.8 | 4.2 | 97 |
| 1 | 51.1 | 11.5 | 20.0 | 12.5 | 95 | ||
| 4 | 37.2 | 15.4 | 22.5 | 20.4 | 95 | ||
| 20NiMoP/AC | 450 | 0 | 62.7 | 9.2 | 20.3 | 5.1 | 97 |
| 1 | 52.1 | 11.8 | 22.4 | 11.5 | 98 | ||
| 4 | 42.1 | 13.7 | 22.8 | 17.7 | 96 | ||
| 15MoP/AC | 500 | 0 | 41.4 | 12.4 | 22.5 | 19.8 | 96 |
| 20NiMoP/AC | 500 | 0 | 47.4 | 12.8 | 22.7 | 15.2 | 98 |
Reaction conditions: Kraft lignin, 15 g; catalyst, 0.75 g; hydrogen pressure of 100 bar at RT; 1200 rpm.
Percent is wt % on lignin intake.
Lower mass balance closure than experiments at higher temperatures due to the viscous nature of the product oil, which hampers isolation and separation.
The experimental data given in Table 4 for the best catalyst (20NiMoP/AC) were used as input for the development of a multivariable regression model for the lignin oil yield as a function of the temperature and batch time. The coefficients for the regression model are provided in Table S3 (Supporting Information), and relevant statistical data are given in Table S4 (Supporting Information). The model relation between process conditions and lignin oil yield is given by eq 6.
| 6 |
The p value of the model is low (<0.0024), which indicates that the model is statistically significant. The effects of the relevant process variables on the lignin oil yield are provided in the contour plot provided in Figure 6.
Figure 6.

Lignin oil yield (wt % on lignin) versus temperature (°C) and reaction time (h).
The data in Figure 6 clearly show that lowest severity is preferred for high lignin oil yields. However, the amount of lignin oil is not the only catalyst performance indicator; of higher interest is the amount of monomeric alkylphenolics and aromatics in the oil, the target product classes of this study. Therefore, all lignin oils were subjected to GPC and GC×GC analyses. The GPC chromatograms of the lignin oils obtained at various reaction temperatures and times for the bimetallic 20MoP/AC catalyst are given in Figure 7, whereas the ones for 15MoP/AC are provided in the Supporting Information (Figure S7). The weight-average molecular weight values for all oils are given in Table 5.
Figure 7.
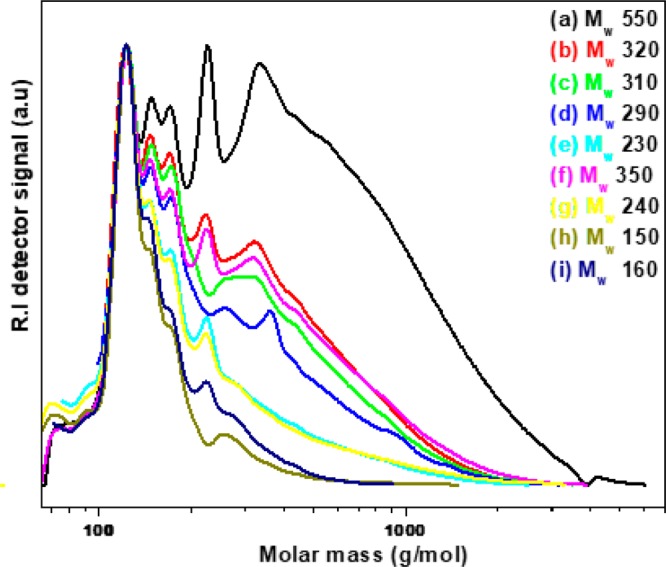
Gel permeation chromatograms of the lignin oils obtained using the 20NiMoP/AC catalysts at different temperatures and reaction times: (a) 400 °C-0 h, (b) 400 °C-2 h, (c) 400 °C-4 h, (d) 400 °C-8 h, (e) 425 °C-4 h, (f) 450 °C-0 h, (g) 450 °C-1 h, (h) 450 °C-4 h, and (i) 500 °C-0 h.
Table 5. Monomer Yield (wt % on lignin) and Component Class Distribution for the Lignin Oils Obtained Using the Phosphided Mono- and Bimetallic Mo-Based Catalystsa.
| catalyst | temp. (°C) | time (h) | total monomer yield (%)b | alkylphenolics (%)b | aromatics (%)b | cyclic + linear alkanes (%)b | GPC (Mw) |
|---|---|---|---|---|---|---|---|
| 20NiMoP/AC | 350 | 4 | 20.5 | 11.4 | 2.7 | 5.2 | 860 |
| 15MoP/AC | 400 | 0 | 20.5 | 13.5 | 2.2 | 2.1 | 550 |
| 2 | 36.3 | 21.8 | 6.0 | 6.6 | 350 | ||
| 4 | 38.7 | 22.4 | 8.0 | 7.4 | 300 | ||
| 8 | 39.3 | 20.5 | 9.1 | 8.3 | 280 | ||
| 20NiMoP/AC | 400 | 0 | 24.5 | 14.1 | 2.4 | 2.6 | 550 |
| 2 | 38.5 | 22.5 | 6.6 | 7.8 | 320 | ||
| 4 | 39.9 | 22.5 | 7.6 | 8.0 | 310 | ||
| 8 | 39.5 | 21.0 | 9.1 | 8.3 | 290 | ||
| 20MoP/AC | 425 | 4 | 39.6 | 19.0 | 10.4 | 7.2 | 220 |
| 20NiMoP/AC | 425 | 4 | 38.9 | 22.2 | 10.1 | 6.1 | 230 |
| 15MoP/AC | 450 | 0 | 31.2 | 21.3 | 4.3 | 2.9 | 300 |
| 1 | 38.2 | 24.2 | 9.2 | 2.9 | 220 | ||
| 4 | 32.9 | 16.6 | 11.6 | 3.4 | 170 | ||
| 20NiMoP/AC | 450 | 0 | 34.8 | 22.6 | 4.9 | 3.7 | 350 |
| 1 | 39.2 | 22.9 | 8.9 | 4.9 | 240 | ||
| 4 | 37.7 | 20.4 | 11.2 | 3.4 | 150 | ||
| 15MoP/AC | 500 | 0 | 33.0 | 20.4 | 8.1 | 3.2 | 150 |
| 20NiMoP/AC | 500 | 0 | 36.6 | 22.6 | 8.9 | 3.4 | 160 |
Reaction conditions: Kraft lignin, 15 g; catalyst, 0.75 g; hydrogen pressure of 100 bar at RT; 1200 rpm.
Percent is on weight basis of lignin intake.
It is clear that the weight-average molecular weight is a function of the severity and that higher severity leads to lower molecular weight values. As such, depolymerization is more pronounced at higher temperatures. However, the amount of lignin oil is also reduced at higher severity due to repolymerization and gasification reactions. As such, a delicate balance between depolymerization and repolymerization/gasification determines the amount and molecular weight of the lignin oil. At the highest severity, very sharp peaks were observed without tailing, indicating the presence of large amounts of lower molecular weight components.
This was confirmed by GC×GC analysis (Table 5). The total monomer yield ranged from 20.5 to 39.9 wt % on lignin intake, with slightly higher yields for the bimetallic 20NiMoP/AC catalysts. The highest value for 20NiMoP/AC was found at low/intermediate severity, 400 °C, and 4 h batch time. In combination with an oil yield of 64 wt % at these conditions, the lignin oil contains 62 wt % of monomers, in line with the GPC data.
This maximum oil yield and amount of monomers for the 20NiMoP/AC catalyst was found at the lowest temperature within the range of temperatures selected, and it is possible that better resuslts are attainable at lower temperatures. As such, a separate experiment was carried out with 20NiMoP/AC at 350 °C and a 4 h bath time. In this case, the oil yield was 61.7 wt % and the total monomer yield was 20.5 wt % (GC×GC). In particular, the monomer yield is a factor of 2 lower than at 400 °C, implying that the latter is indeed better than 350 °C when considering catalyst performance.
Effect of Catalyst Loading
The effect of catalyst loading (5 and 10 wt %) on lignin oil yield and composition was studied at 400 and 450 °C using the 20NiMoP/AC catalyst, and the results are given in Table 6. The mass balances closures are good and in the range of 94–100%. For the reactions performed at 400 °C, the lignin oil yield increased from 67.2% to 70.6% upon increasing the catalyst loading from 5 to 10 wt %. Char formation is reduced considerably, and actually no char is observed at the highest catalyst loading. As such, this implies that the repolymerization reactions leading ultimately to char are likely noncatalytic and thus thermal in nature, while the depolymerization reactions are metal catalyzed. Performance at 450 °C is worse, and more char and less oil are observed.
Table 6. Effect of Catalyst Loading on Lignin Oil Yielda.
| catalyst loading (wt % on lignin) | temp. (°C) | time (h) | oil yield (%)b | gas phase (%)b | water (%)b | solids (%)b | mass balance (%)b |
|---|---|---|---|---|---|---|---|
| 5 | 400a | 2 | 67.2 | 8.6 | 19.7 | 4.0 | 99 |
| 10 | 400a | 2 | 70.6 | 9.4 | 20.7 | 100 | |
| 5 | 450b | 1 | 52.1 | 11.8 | 22.4 | 11.5 | 98 |
| 10 | 450b | 1 | 49.5 | 11.7 | 22.0 | 10.8 | 94 |
Reaction conditions: Kraft lignin, 15 g; hydrogen pressure of 100 bar at RT; 1200 rpm.
Percent is on weight basis of lignin intake.
The lignin oils were further characterized by GPC, and the results are shown in Figure 8. Higher catalyst loadings at both temperatures lead to a reduction in the molecular weight of the lignin oils, indicative of a catalytic effect on the depolymerization reactions.
Figure 8.
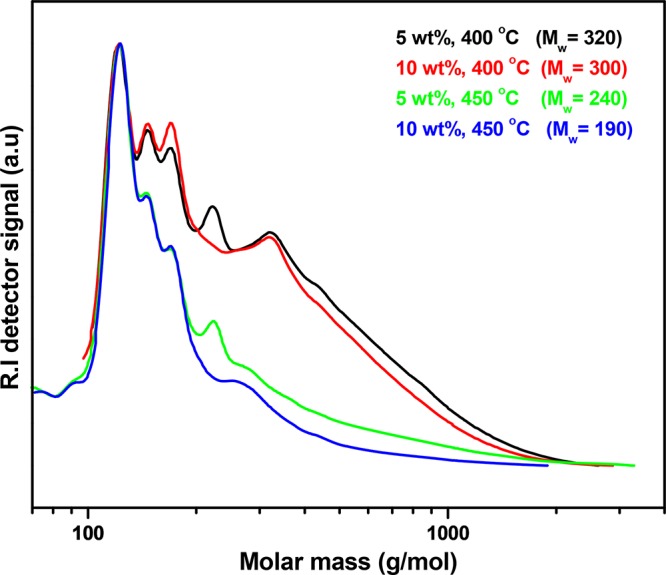
Effect of catalyst (20NiMoP/AC) loading on lignin oil average molecular weights.
The volatility of the product oil obtained at 400 °C, 100 bar, and 10 wt % catalyst loading was determined using TGA, and the results are given in Figure S8 (Supporting Information). It shows that more than 80% of the sample weight is lost when increasing the temperature from room temperature to 350 °C, illustrating indeed that the amount of low molecular weight compounds is high in the sample, in line with GPC and GC×GC data.
The monomer yield and the amounts of alkylphenolics, aromatics, and alkanes in the lignin oil, as determined by GC×GC analysis, are given in Figure 9. For reactions performed at 400 °C, an increase in catalyst loading from 5 to 10 wt % leads to a higher monomers yield from 38.5% to 45.7%, the highest value obtained in this study. In this case, the alkylphenolics yield is 25 wt % on lignin.
Figure 9.

Effect of catalyst loading (20NiMoP/AC) and temperature on the monomer yield (wt % on lignin) and amounts of important product classes (wt % on lignin).
Reaction Network
On the basis of the product yields and composition of the lignin oil (elemental analysis, GPC, GC-MS, GC×GC, and 13C NMR) discussed in the previous sections and literature data, a reaction network is proposed for the hydrotreatment of Kraft lignin using metal phosphide catalysts and hydrogen (Scheme 1). It involves a number of serial and parallel reactions occurring in the liquid and gas phase. The desired reactions to low molecular weight alkylphenolics and aromatics involves thermal and catalytic depolymerization (hydrocracking) of the Kraft lignin by cleavage of linkages. The most reactive linkages are the ether linkages, though these are not highly abundant in Kraft lignin. Catalyst promotes depolymerization reactions, though reactions in the absence of a catalyst also give (limited) amounts of lignin oils, indicating that thermal depolymerization reactions also play a role (vide supra). The oligomeric fragments can either be further depolymerized to low molecular weight compounds in the form of alkylphenolics or repolymerize (also together with already formed low molecular weight compounds) to higher condensed structures, ultimately leading to char. The latter pathway is likely a thermal reaction and thus can be suppressed by the use of very active catalysts that reduce the amounts of reactive intermediates by catalyzing subsequent conversions. The intermediate low molecular weight alkylphenolics are not inert under reaction conditions and may be further hydrodeoxygenated to aromatics and alkanes, as is evident from the GC×GC results. Two possible pathways may be distinguished: (i) hydrodeoxygenation of alkylphenolics to aromatics and (ii) hydrogenation of the aromatic rings of the alkylphenolics followed by hydrodeoxygenation to form alkanes. The latter is undesirable as alkanes are less valuable than aromatics and typically only have fuel value. In addition, methoxy removal by hydrogenolysis reactions may also occur, as shown by model component studies,58,59 leading to the formation of methanol. The latter is likely not inert under the prevailing reaction conditions and may be converted to gas-phase components.60,61
Scheme 1. Proposed Reaction Network for the Hydrotreatment of Kraft Lignin Using the Metal Phosphide Catalysts.

When using the metal phosphide catalysts and particularly 20NiMoP/AC at optimized conditions, high yields of alkylphenolics are obtained, with smaller amounts of aromatics, low amounts of overhydrogenated alkanes, and essentially no char. This means that the catalysts are very reactive at reported optimized conditions and promote hydrocracking reactions as well as methoxy removal reactions while being less reactive for hydrodeoxygention of alkylphenolics, leading to aromatics and hydrogenation reactions of the C–C bonds in the aromatic rings to give alkanes.
Comparison of Catalytic Performance of the 20NiMoP/AC with Literature Data
The monomer yield for the best catalyst in this study (20NiMoP/AC) was compared with the data provided in the literature regarding the catalytic hydrotreatment of various lignins in the absence of an external solvent, and the results are given in Figure 10 and Table 1. When considering sulfur-containing lignins like Kraft lignin, the phosphided NiMo catalyst reported here performs best among the catalyst reported in the literature, and 45.7 wt % of monomers on lignin intake was obtained (400 °C, 2 h batch time, and 10 wt % of catalyst loading). Interestingly, performance is better than reported for sulfided NiMo catalysts on various supports, indicating the potential of phosphide catalyst for the catalytic hydrotreatment of (sulfur-containing) lignins. Monomer yield is lower than found for sulfur-free pyrolytic lignins, which is not surprising as this class of lignins has a considerably lower molecular weight than typical Kraft lignins.
Figure 10.

Overview of monomer yields for the solvent-free catalytic hydrotreatment of lignins (literature references to individual entries are given in Table 1; last column is the best result from this study).
Conclusions
A series of mono- and bimetallic Ni, Mo, and W phosphides supported on activated carbon was tested for the solvent-free catalytic hydrotreatment of Kraft lignin. Catalytic experiments showed that the Mo-containing phosphide catalysts exhibit better performance in terms of oil, char, and monomer yield compared to W-containing metal phosphides. The effect of process conditions on catalytic performance of the Mo-containing mono- and bimetallic catalysts was investigated (400–500 °C, batch times between 0 and 8 h, catalyst loadings of 5 and 10 wt %). The optimized reaction conditions for the 20NiMoP/AC catalyst to obtain high monomer yields were determined to be 400 °C, 2 h batch time, and 10 wt % of catalyst loading. At these conditions, the monomer yield was 45.7% on lignin intake, which is significantly higher than values reported in the literature for the catalytic hydrotreatment of Kraft lignin without the use of an external solvent, showing the potential of this class of metal phosphides for the hydrotreatment of sulfur-rich lignins. The composition of the lignin oils was determined by GPC, GC-MS, GC×GC, and 13C NMR and shown to consist of low molecular weight components as well as lignin oligomers (GPC). GC×GC analysis shows that the most abundant monomers are alkylphenolics, with yields up to 25 wt % on lignin. To the best of our knowledge, we are the first to demonstrate that bimetallic NiMo phosphide-based catalysts are suitable for the hydrotreatment of Kraft lignin without the need for an external solvent. The main advantage compared to conventional sulfided NiMo catalysts on alumina supports is that the need of a sulfiding agent for good catalyst performance is not required.
Acknowledgments
This research has been performed within the framework of the CatchBio program, project 053.70.732. The authors gratefully acknowledge the financial support of the Smart Mix program of the Ministry of Economic Affairs and The Netherlands Ministry of Education, Culture and Science. We also thank Anne Appeldoorn, Erwin Wilbers, Marcel de Vries, Hans van der Velde, Leon Rohrbach, and Jan Henk Marsman for technical and analytical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssuschemeng.8b04411.
Visual appearance of a liquid product, catalyst characterization details (XRD, N2 physisorption, TEM), GC (GC-MS, GC×GC), GPC and TGA data of product oils, tables with mass balances including hydrogen consumption, gas-phase composition, model analysis data (coefficients and ANOVA), details about quantification of GC×GC chromatograms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Huber G. W.; Iborra S.; Corma A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106 (9), 4044–4098. 10.1021/cr068360d. [DOI] [PubMed] [Google Scholar]
- Melero J. A.; Iglesias J.; Garcia A. Biomass as renewable feedstock in standard refinery units. Feasibility, opportunities and challenges. Energy Environ. Sci. 2012, 5, 7393–7420. 10.1039/c2ee21231e. [DOI] [Google Scholar]
- Gallezot P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41 (4), 1538–1558. 10.1039/C1CS15147A. [DOI] [PubMed] [Google Scholar]
- Tuck C. O.; Perez E.; Horvath I. T.; Sheldon R. A.; Poliakoff M. Valorization of Biomass: Deriving More Value from Waste. Science 2012, 337, 695–699. 10.1126/science.1218930. [DOI] [PubMed] [Google Scholar]
- Hon D. N. S.; Shiraishi N.. Wood and Cellulosic Chemistry; Marcel Dekker: New York and Basel, 1991. [Google Scholar]
- Sjostrom E.Wood chemistry, fundamentals and applications, 2nd ed.; Academic Press: San Diego, 1992. [Google Scholar]
- Fengel D.; Wegener G.. Wood: Chemistry, Ultrastructure, Reactions; Walter de Gruyter: New York, 1984. [Google Scholar]
- Li C.; Zhao X.; Wang A.; Huber G. W.; Zhang T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115 (21), 11559–11624. 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- Clark J. H.; Luque R.; Matharu A. S. Green chemistry, biofuels, and biorefinery. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 183–207. 10.1146/annurev-chembioeng-062011-081014. [DOI] [PubMed] [Google Scholar]
- Watkins D.; Nuruddin Md.; Hosur M.; Tcherbi-Narteh A. T.; Jeelani S. Extraction and characterization of lignin from different biomass resources. J. Mater. Res. Technol. 2015, 4 (1), 26–32. 10.1016/j.jmrt.2014.10.009. [DOI] [Google Scholar]
- Sun X. F.; Cang R.; Fowler P.; Baird M. S. J. Agric. Extraction and Characterization of Original Lignin and Hemicelluloses from Wheat Straw. J. Agric. Food Chem. 2005, 53 (4), 860–870. 10.1021/jf040456q. [DOI] [PubMed] [Google Scholar]
- Brandt A.; Gräsvik J.; Hallett J. P.; Welton T. Deconstruction of lignocellulosic biomass with ionic liquids. Green Chem. 2013, 15, 550–583. 10.1039/c2gc36364j. [DOI] [Google Scholar]
- Laurichesse S.; Averous L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. 10.1016/j.progpolymsci.2013.11.004. [DOI] [Google Scholar]
- Gosselink R. J. A.; de Jong E.; Guran B.; Abacherli A. Co-ordination network for lignin standardisation, production and applications adapted to market requirements (EUROLIGNIN). Ind. Crops Prod. 2004, 20, 121–129. 10.1016/j.indcrop.2004.04.015. [DOI] [Google Scholar]
- Kleinert M.; Barth T. Phenols from Lignin. Chem. Eng. Technol. 2008, 31 (5), 736–745. 10.1002/ceat.200800073. [DOI] [Google Scholar]
- Thring R. W.; Katikaneni S. P. R.; Bakhshi N. N. The production of gasoline range hydrocarbons from Alcell® lignin using HZSM-5 catalyst. Fuel Process. Technol. 2000, 62, 17–30. 10.1016/S0378-3820(99)00061-2. [DOI] [Google Scholar]
- Gellerstedt G.; Li J.; Eide I.; Kleinert M.; Barth T. Chemical Structures Present in Biofuel Obtained from Lignin. Energy Fuels 2008, 22 (6), 4240–4244. 10.1021/ef800402f. [DOI] [Google Scholar]
- Rahimi A.; Ulbrich A.; Coon J. J.; Stahl S. S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515 (7526), 249–252. 10.1038/nature13867. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Chen Y.; Qin T.; Wang L.; Tang Y.; Sun Y.; Wan P. Lignin depolymerization via an integrated approach of anode oxidation and electro-generated H2O2 oxidation. RSC Adv. 2014, 4, 6232–6238. 10.1039/c3ra47516f. [DOI] [Google Scholar]
- Chatel G.; Rogers R. D. Review: Oxidation of Lignin Using Ionic Liquids—An Innovative Strategy To Produce Renewable Chemicals. ACS Sustainable Chem. Eng. 2014, 2, 322–339. 10.1021/sc4004086. [DOI] [Google Scholar]
- Zakzeski J.; Bruijnincx P. C. A.; Jongerius A. L.; Weckhuysen B. M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110 (6), 3552–3599. 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Ekhe J. D. Cu–Mo doped zeolite ZSM-5 catalyzed conversion of lignin to alkyl phenols with high selectivity. Catal. Sci. Technol. 2015, 5, 2117–2124. 10.1039/C4CY01700E. [DOI] [Google Scholar]
- Deepa A. K.; Dhepe P. L. Lignin Depolymerization into Aromatic Monomers over Solid Acid Catalysts. ACS Catal. 2015, 5 (1), 365–379. 10.1021/cs501371q. [DOI] [Google Scholar]
- Parsell T.; Yohe S.; Degenstein J.; Jarrell T.; Klein I.; Gencer E.; Hewetson B.; Hurt M.; Kim J. I.; Choudhari H.; Saha B.; Meilan R.; Mosier N.; Ribeiro F.; Delgass W. N.; Chapple C.; Kenttamaa H. I.; Agrawal R.; Omar M. M. A. A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass. Green Chem. 2015, 17, 1492–1499. 10.1039/C4GC01911C. [DOI] [Google Scholar]
- Olcese R. N.; Lardier G.; Bettahar M.; Ghanbaja J.; Fontana S.; Carre V.; Aubriet F.; Petitjean D.; Dufour A. Aromatic Chemicals by Iron-Catalyzed Hydrotreatment of Lignin Pyrolysis Vapor. ChemSusChem 2013, 6, 1490–1499. 10.1002/cssc.201300191. [DOI] [PubMed] [Google Scholar]
- Li X.; Su L.; Wang Y.; Yu Y.; Wang C.; Li X.; Wang Z. Catalytic fast pyrolysis of Kraft lignin with HZSM-5 zeolite for producing aromatic hydrocarbons. Front. Environ. Sci. Eng. 2012, 6 (3), 295–303. 10.1007/s11783-012-0410-2. [DOI] [Google Scholar]
- Lazaridis P. A.; Fotopoulos A. P.; Karakoulia S. A.; Triantafyllidis K. S. Catalytic Fast Pyrolysis of Kraft Lignin With Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. 10.3389/fchem.2018.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Arancon R. A. D.; Labidi J.; Luque R. Lignin depolymerisation strategies: towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43 (22), 7485–7500. 10.1039/C4CS00235K. [DOI] [PubMed] [Google Scholar]
- Jongerius A. L.; Bruijnincx P. C. A.; Weckhuysen B. M. Liquid-phase reforming and hydrodeoxygenation as a two-step route to aromatics from lignin. Green Chem. 2013, 15, 3049–3056. 10.1039/c3gc41150h. [DOI] [Google Scholar]
- Azadi P.; Inderwildi O. R.; Farnood R.; King D. A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renewable Sustainable Energy Rev. 2013, 21, 506–523. 10.1016/j.rser.2012.12.022. [DOI] [Google Scholar]
- Barta K.; Ford P. C. Catalytic Conversion of Nonfood Woody Biomass Solids to Organic Liquids. Acc. Chem. Res. 2014, 47, 1503–1512. 10.1021/ar4002894. [DOI] [PubMed] [Google Scholar]
- Ma X.; Ma R.; Hao W.; Chen M.; Yan F.; Cui K.; Tian Y.; Li Y. Common Pathways in Ethanolysis of Kraft Lignin to Platform Chemicals over Molybdenum-Based Catalysts. ACS Catal. 2015, 5, 4803–4813. 10.1021/acscatal.5b01159. [DOI] [Google Scholar]
- Ma R.; Hao W.; Ma X.; Tian Y.; Li Y. Catalytic Ethanolysis of Kraft Lignin into High-Value Small-Molecular Chemicals over a Nanostructured α–Molybdenum Carbide Catalyst. Angew. Chem., Int. Ed. 2014, 53 (28), 7310–7315. 10.1002/anie.201402752. [DOI] [PubMed] [Google Scholar]
- Huang X.; Korányi T. I.; Boot M. D.; Hensen E. J. M. Catalytic Depolymerization of Lignin in Supercritical Ethanol. ChemSusChem 2014, 7, 2276–2288. 10.1002/cssc.201402094. [DOI] [PubMed] [Google Scholar]
- Löfstedt J.; Dahlstrand C.; Orebom A.; Meuzelaar G.; Sawadjoon S.; Galkin M. V.; Agback P.; Wimby M.; Corresa E.; Mathieu Y.; Sauvanaud L.; Eriksson S.; Corma A.; Samec J. S. M. Green Diesel from Kraft Lignin in Three Steps. ChemSusChem 2016, 9, 1392–1396. 10.1002/cssc.201600172. [DOI] [PubMed] [Google Scholar]
- Pandey M. P.; Kim C. S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. 10.1002/ceat.201000270. [DOI] [Google Scholar]
- Mohan D.; Pittman C. U.; Steele P. H. Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review. Energy Fuels 2006, 20, 848–889. 10.1021/ef0502397. [DOI] [Google Scholar]
- Roberts V.; Stein V.; Reiner T.; Lemonidou A.; Li X.; Lercher J. A Towards quantitative catalytic lignin depolymerisation. Chem. - Eur. J. 2011, 17 (21), 5939–5948. 10.1002/chem.201002438. [DOI] [PubMed] [Google Scholar]
- Onwudili J. A.; Williams P. T. Catalytic depolymerization of alkali lignin in subcritical water: influence of formic acid and Pd/C catalyst on the yields of liquid monomeric aromatic products. Green Chem. 2014, 16, 4740–4748. 10.1039/C4GC00854E. [DOI] [Google Scholar]
- Narani A.; Chowdari R. K.; Cannilla C.; Bonura G.; Frusteri F.; Heeres H. J.; Barta K. Efficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol. Green Chem. 2015, 17, 5046–5057. 10.1039/C5GC01643F. [DOI] [Google Scholar]
- Kloekhorst A.; Shen Y.; Yie Y.; Fang M.; Heeres H. J. Catalytic hydrodeoxygenation and hydrocracking of Alcell® lignin in alcohol/formic acid mixtures using a Ru/C catalyst. Biomass Bioenergy 2015, 80, 147–161. 10.1016/j.biombioe.2015.04.039. [DOI] [Google Scholar]
- Meier D.; Ante R.; Faix O. Catalytic hydropyrolysis of lignin: Influence of reaction conditions on the formation and composition of liquid products. Bioresour. Technol. 1992, 40, 171–177. 10.1016/0960-8524(92)90205-C. [DOI] [Google Scholar]
- Oasmaa A.; Alen R.; Meier D. Catalytic hydrotreatment of some technical lignins. Bioresour. Technol. 1993, 45 (3), 189–194. 10.1016/0960-8524(93)90111-N. [DOI] [Google Scholar]
- Kloekhorst A.; Wildschut J.; Heeres H. J. Catalytic hydrotreatment of pyrolytic lignins to give alkylphenolics and aromatics using a supported Ru catalyst. Catal. Sci. Technol. 2014, 4, 2367–2377. 10.1039/C4CY00242C. [DOI] [Google Scholar]
- Kloekhorst A.; Heeres H. J. Catalytic Hydrotreatment of Alcell Lignin Using Supported Ru, Pd, and Cu Catalysts. ACS Sustainable Chem. Eng. 2015, 3, 1905–1914. 10.1021/acssuschemeng.5b00041. [DOI] [Google Scholar]
- Kumar Ch. R.; Anand N.; Kloekhorst A.; Cannilla C.; Bonura G.; Frusteri F.; Barta K.; Heeres H. J. Solvent free depolymerization of Kraft lignin to alkyl-phenolics using supported NiMo and CoMo catalysts. Green Chem. 2015, 17, 4921–4930. 10.1039/C5GC01641J. [DOI] [Google Scholar]
- Agarwal S.; Chowdari R. K.; Hita I.; Heeres H. J. Experimental Studies on the Hydrotreatment of Kraft Lignin to Aromatics and Alkylphenolics Using Economically Viable Fe-Based Catalysts. ACS Sustainable Chem. Eng. 2017, 5 (3), 2668–2678. 10.1021/acssuschemeng.6b03012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wild P. J.; Huijgen W. J. J.; Kloekhorst A.; Chowdari R. K.; Heeres H. J. Biobased alkylphenols from lignins via a two-step pyrolysis – Hydrodeoxygenation approach. Bioresour. Technol. 2017, 229, 160–168. 10.1016/j.biortech.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Wu S. K.; Lai P. C.; Lin Y. C.; Wan H. P.; Lee H. T.; Chang Y. H. Atmospheric Hydrodeoxygenation of Guaiacol over Alumina-, Zirconia-, and Silica-Supported Nickel Phosphide Catalysts. ACS Sustainable Chem. Eng. 2013, 1 (3), 349–358. 10.1021/sc300157d. [DOI] [Google Scholar]
- Zhao H. Y.; Li D.; Bui P.; Oyama S. T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal., A 2011, 391 (1–2), 305–310. 10.1016/j.apcata.2010.07.039. [DOI] [Google Scholar]
- Ma X. L.; Tian Y.; Hao W. Y.; Ma R.; Li Y. D. Production of phenols from catalytic conversionof lignin over a tungsten phosphide catalyst. Appl. Catal., A 2014, 481, 64–70. 10.1016/j.apcata.2014.05.002. [DOI] [Google Scholar]
- Li C.; Zhao X.; Wang A.; Huber G. W.; Zhang T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115 (21), 11559–11624. 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- Bowker R. H.; Ilic B.; Carrillo B. A.; Reynolds M. A.; Murray B. D.; Bussell M. E. Carbazole hydrodenitrogenation over nickel phosphide and Ni-rich bimetallic phosphide catalysts. Appl. Catal., A 2014, 482, 221–230. 10.1016/j.apcata.2014.05.026. [DOI] [Google Scholar]
- Burns A. W.; Gaudette A. F.; Bussell M. E. Hydrodesulfurization properties of cobalt–nickel phosphide catalysts: Ni-rich materials are highly active. J. Catal. 2008, 260 (2), 262–269. 10.1016/j.jcat.2008.10.001. [DOI] [Google Scholar]
- Constant S.; Wienk H. L. J.; Frissen A. E.; Peinder P.d.; Boelens R.; van Es D. S.; Grisel R. J. H.; Weckhuysen B. M.; Huijgen W. J. J.; Gosselink R. J. A.; Bruijnincx P. C. A. New insights into the sructure and composition of technical lignins: a comparative characterisation study. Green Chem. 2016, 18 (9), 2651–2665. 10.1039/C5GC03043A. [DOI] [Google Scholar]
- Yang P.; Kobayashi H.; Hara K.; Fukuoka A. Phase Change of Nickel Phosphide Catalysts in the Conversion of Cellulose into Sorbitol. ChemSusChem 2012, 5, 920–926. 10.1002/cssc.201100498. [DOI] [PubMed] [Google Scholar]
- Yao Z. W. Exploration on synthesis of activated carbon supported molybdenum carbide, nitride and phosphide via carbothermal reduction route. J. Alloys Compd. 2009, 475, L38–L41. 10.1016/j.jallcom.2008.07.130. [DOI] [Google Scholar]
- Maki-Arvela P.; Murzin D. Y. Hydrodeoxygenation of Lignin-Derived Phenols: From Fundamental Studies towards Industrial Applications. Catalysts 2017, 7 (9), 265. 10.3390/catal7090265. [DOI] [Google Scholar]
- Li X.; Chen G.; Liu C.; Ma W.; Yan B.; Zhang J. Hydrodeoxygenation of lignin-derived bio- oil using molecular sieves supported metal catalysts: A critical review. Renewable Sustainable Energy Rev. 2017, 71, 296–308. 10.1016/j.rser.2016.12.057. [DOI] [Google Scholar]
- Gabriel S.; Scurrell M. S. The Conversion of Methanol into Higher Hydrocarbons Catalyzed by Gold. ChemCatChem 2016, 8 (19), 3118–3120. 10.1002/cctc.201600727. [DOI] [Google Scholar]
- Ilias S.; Bhan A. Mechanism of the Catalytic Conversion of Methanol to Hydrocarbons. ACS Catal. 2013, 3, 18–31. 10.1021/cs3006583. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.