

A better understanding of the underlying disease biology that leads to improvement in treatment outcomes is needed. Investigation of the genomic landscape of Hodgkin lymphoma has been difficult because of the low tumor content in these inflammatory cell‐ and stroma‐rich tissue samples. A comprehensive genomic profiling with targeted next‐generation sequencing panel was performed to test for genomic aberrations in archival tumor samples from patients with Hodgkin lymphoma to identify potentially actionable molecular targets.
Keywords: Genomic profiling, Hodgkin lymphoma, B2M mutation, TP53 mutation, XPO1 mutation
Abstract
Background.
The genomic landscape of Hodgkin lymphoma (HL) has been difficult to characterize due to the paucity of neoplastic cells and an abundant microenvironment. Such characterization is needed in order to improve treatment strategies.
Materials and Methods.
We performed comprehensive genomic profiling (CGP) using targeted next‐generation sequencing on archival formalin‐fixed paraffin embedded tumor samples from 63 patients to analyze the landscape of HL.
Results.
CGP was successful for 49/63 archival specimens (78%), and revealed aberrations impacting genes including B2M, TP53, and XPO1 (E571). Of the 34 patients for whom total mutation burden (TMB; mutations/megabase [Mb]) was assessed, 5 (15%) had high TMB (≥20 mutations/Mb), 18 (53%) had intermediate TMB (6–19 mutations/Mb), and 11 (32%) had low TMB (≤5 mutations/Mb). We next tested 13 patients' plasma cell‐free DNA with droplet digital polymerase chain reaction for the presence of XPO1 E571 mutation, which was confirmed in the plasma of 31% of patients. In three patients with serially collected plasma samples, XPO1 E571K allelic frequency changes corresponded with changes in tumor size on conventional radiographic imaging.
Conclusion.
The study demonstrates that comprehensive genomic profiling of archival Hodgkin lymphoma tumor samples is feasible and leads to the identification of genes that are recurrently mutated and that Hodgkin lymphoma has increased mutation burden in the majority of samples analyzed. Furthermore, tracking of XPO1 E571 mutant allele frequency in a subset of patients may also represent a potential disease‐monitoring strategy and warrants further investigation.
Implications for Practice.
This study provides the first evidence that comprehensive genomic profiling can be performed to map the genomic landscape of Hodgkin lymphoma and that a subpopulation of patients has mutations in TP53, B2M, XPO1, and other genes. It was found that 15% of patients have high mutation burden, which, in cancers such as melanoma, may indicate sensitivity to immune checkpoint inhibitors, and may thus be explored for Hodgkin lymphoma. Lastly, this work demonstrates that changes in the mutant allele frequency of XPO1 in serially collected plasma cell‐free DNA samples correspond with treatment outcomes measured with conventional radiographic imaging.
Introduction
Advances in genomic technologies have led to identification of genes commonly perturbed in cancer. Genomic testing has been quickly adopted and widely implemented in cancer care to leverage treatment decision‐making and improve treatment outcomes [1], [2]. Although technologies such as next‐generation sequencing (NGS) are increasingly accepted in nonhematologic malignancies, the genomic landscape of Hodgkin lymphoma (HL) remains to be described.
In 2016, an estimated 8,500 new cases of HL were diagnosed in the U.S. [3]. Although rates of survival have improved over the years, approximately 10% of early‐stage HL patients and 20%–30% of advanced‐stage patients become refractory after initial therapy [4]. Furthermore, late treatment‐related sequelae may lead to significant morbidity and mortality. Therapeutic options for patients with relapsed or refractory disease include salvage chemotherapy, followed by high‐dose chemotherapy and autologous stem cell transplantation (ASCT). For patients refractory to primary therapy, even with aggressive approaches such as ASCT, overall survival rates do not exceed 30% [5]. Patients who relapse after ASCT may be candidates for allogeneic stem cell transplantation (alloSCT), which yields a median overall survival of 29 months with an approximate 20% treatment‐related mortality [6]. Patients relapsing after ASCT and/or alloSCT have only limited treatment options, which include brentuximab vedotin, lenalidomide, bendamustine, or programmed cell death protein 1 (PD‐1) antibodies, with median survival usually not exceeding 24 months [4], [7]. Therefore, a pressing need exists to better understand the underlying disease biology, which may lead to improvement in treatment outcomes. Investigation of the genomic landscape of HL has been difficult because of the low tumor content in these inflammatory cell‐ and stroma‐rich tissue samples. Therefore, we performed a study using comprehensive genomic profiling (CGP) with a targeted NGS panel to test for genomic aberrations in archival tumor samples from patients with HL in order to identify potentially actionable molecular targets.
Materials and Methods
Patients and Tumor Samples
Formalin‐fixed paraffin‐embedded (FFPE) archival tumor samples, procured during routine clinical procedures for 63 HL patients, were reviewed and evaluated.
Comprehensive Genomic Profiling
A single hematoxylin and eosin‐stained slide was examined in order to confirm the presence of tumor. CGP was performed from FFPE tissue using the Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists‐accredited, New York State‐approved FoundationOne (DNA‐seq) [8] or FoundationOne Heme (DNA/RNA‐seq) [9] assay. A minimum of 50 ng of DNA and 300 ng of RNA were extracted, RNA was converted to complementary DNA, and DNA was sonicated with subsequent adaptor ligation and hybridization capture, as described previously [8], [9]. DNA‐based captured libraries (up to 405 cancer‐related genes plus select introns from 31 genes) were sequenced to a median unique exon coverage depth of 500× using Illumina sequencing, and, following removal of duplicate reads, were analyzed for base substitutions, insertions, deletions, copy number alterations (focal gene amplifications and homozygous deletions), and select gene fusions. RNA‐based captured libraries (265 genes) achieved ≥3M on‐target unique pairs and were analyzed only for the presence of rearrangements. Total mutation burden (TMB) was defined as the number of somatic coding base substitutions and indel alterations minus known driver alterations per megabase (Mb) of genome examined, adjusted for the tumor content to allow for cross‐study comparisons. A TMB of ≥20 mutations/Mb was considered high; 6–19 mutations/Mb intermediate; and ≤5 mutations/Mb low.
Plasma Collection and Cell‐Free DNA Testing for the XPO1E571 Mutation
Whole blood was collected in ethylenediaminetetraacetic acid‐containing tubes and centrifuged and spun twice within 2 hours to yield plasma. We extracted cell‐free DNA (cfDNA) from plasma samples collected from 13 relapsed/refractory Hodgkin lymphoma patients before initiation of systemic therapy using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. In order to identify common XPO1 mutations in plasma cfDNA, we designed droplet digital polymerase chain reaction (ddPCR) probes against the E571K and E571V mutations. Sixteen nanograms of unamplified cfDNA was tested using ddPCR for the presence of the XPO1 E571K or XPO1 E571V mutation to distinguish the wild‐type allele from the mutant using the QX200 Droplet Digital PCR platform (Bio‐Rad, Pleasanton, CA) according to the manufacturer's standard protocol. The lower limit of detection was approximately <0.1% mutant allele frequency (MAF) per single well.
Results
Comprehensive Genomic Profiling of Archival Tumor Tissue
Molecular profiling was successfully performed in 49 (78%) of the 63 classical Hodgkin lymphoma FFPE specimens (Fig. 1; Table 1). Of these 49 specimens, 9 (18%) had no alterations, and in 40 specimens, a total of 44 genes with 95 alterations, including nonsynonymous mutations, splice site changes, rearrangements, or copy number alterations, were identified. Detailed patient characteristics were available for 18 patients treated at MD Anderson Cancer Center (Table 2).
Figure 1.

Enrollment of Hodgkin lymphoma patients whose archival tumor tissue was submitted for comprehensive genomic profiling.
Table 1. Comprehensive genomic profiling of archival tumor tissue in Hodgkin lymphoma patients.

Abbreviations: F1, FoundationOne (DNA‐seq); F1H, FoundationOne Heme (DNA/RNA); NA, not available; ND, not done; TMB, total mutation burden; X, a test was performed for this patient (DNA test, or RNA test).
Table 2. Patients’ characteristics for 18 patients treated at MD Anderson Cancer Center.

Abbreviations: EBER, EBV‐encoded RNA; EBV, Epstein‐Barr virus; HL, Hodgkin lymphoma; NOS, not otherwise specified.
The most commonly mutated gene was TP53 (tumor protein p53) with 13 mutations in 11 patients (22%; Table 1; Fig. 2). The next most commonly mutated gene was B2M (beta‐2 microglobulin) with 12 mutations in 11 patients (22%; Table 1; Fig. 2). For 82% (9/11) of patients with B2M alterations (with patient 2 demonstrating two B2M mutations), mutations occurred at the M1 position (M1I, n = 2; M1K, n = 5; M1R, n = 1; M1T, n = 1; M1V, n = 1; Table 1). The next most frequently mutated gene was XPO1 (exportin 1; CRM1), for which nine (18%) cases harbored an E571 mutation (E571K: n = 8, E571V: n = 1; Table 1; Fig. 2). These mutations were not mutually exclusive of TP53 or B2M aberrations. For two of these nine patients, an XPO1 mutation (E571K: n = 1, E571V: n = 1) was the only panel gene demonstrating an aberration, whereas the remaining seven patients demonstrated 1 through 20 additional mutations. The next most frequently mutated genes include TNFAIP3 (tumor necrosis factor, alpha‐induced protein 3) and SOCS1 (suppressor of cytokine signaling 1). Eight aberrations in TNFAIP3 were identified in seven patients (14%; Table 1; Fig. 2). Notably, two events were nonsense mutations and the remaining six were frameshift events, with two of these occurring in patient 23. Five events were additionally identified in SOCS1 across five patients (10%; Table 1; Fig. 2). Three missense mutations, each occurring in three separate patients, were identified, along with rearrangements in patients 7 and 39. In both patients 7 and 39, an IGH‐SOCS1 rearrangement was identified, with a breakpoint occurring in exon two, the only coding exon, of SOCS1. In three of the five patients demonstrating SOCS1 aberrations, the identified SOCS1 mutation was the only event identified from CGP (Fig. 2). Programmed death‐ligand 1 (PD‐L1) amplification was also uncommon with only one case (patient 2) whose tumor harbored amplification of CD274.
Figure 2.

Molecular alterations in archival tumor samples from 40 Hodgkin lymphoma patients. The nine patients for whom comprehensive genomic profiling did not identify alterations are not shown. Split squares are shown if two events were identified in the same gene (e.g., B2M and TP53 for patient 2).
Of the 34 patients for whom TMB was assessed, 5 (15%) had high TMB, 18 (53%) had intermediate TMB, and 11 (32%) had low TMB (Fig. 3). Only two patients, both with intermediate TMB, received anti‐PD‐1 antibodies, and both patients responded to treatment.
Figure 3.

Total mutation burden for 34 Hodgkin lymphoma patients. High mutation burden is depicted in red, intermediate in orange, and low in blue. Two patients (yellow stars) received anti‐programmed cell death protein 1 antibodies and responded to treatment. Abbreviation: Mb, megabase.
ddPCR of Plasma cfDNA for XPO1 E571 Mutations
To confirm if recurrent XPO1 E571 mutations are measureable in blood plasma and have the potential to act as a biomarker, ddPCR of cfDNA was performed. Of the 13 patients whose tumor samples were subjected to ddPCR analysis, 3 had XPO1 E571K mutations (MAFs of 6.10%, 2.07%, and 6.10%, respectively) and 1 had the XPO1 E571V mutation (MAF of 2.70%). Among these four patients’ cases, CGP of the tumor tissue had not been performed in one case, XPO1 mutation was not detected by profiling in one case, no results were obtained by profiling for one case, and the XPO1 E571V mutation was confirmed in the tumor in one case (Table 3). Among the remaining nine cases in which ddPCR did not detect XPO1 E571K or E571V mutations in cfDNA, CGP did not produce results in one case, demonstrated no XPO1 mutation in one case, and was not performed for seven cases (Table 3). All three patients whose cfDNA had XPO1 E571K mutations had longitudinal collection of plasma during systemic therapy, and changes in XPO1 E571K MAFs corresponded to changes in tumor size on conventional radiographic imaging with positron emission tomography‐computed tomography (Fig. 4).
Table 3. XPO1 mutations in plasma cfDNA.
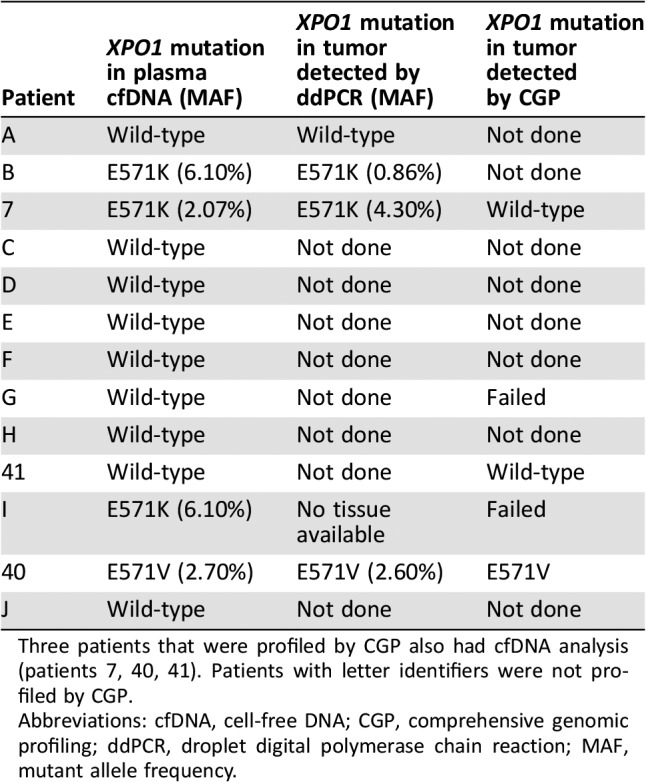
Three patients that were profiled by CGP also had cfDNA analysis (patients 7, 40, 41). Patients with letter identifiers were not profiled by CGP.
Abbreviations: cfDNA, cell‐free DNA; CGP, comprehensive genomic profiling; ddPCR, droplet digital polymerase chain reaction; MAF, mutant allele frequency.
Figure 4.

Dynamic changes in XPO1 E571K MAF. Changes in XPO1 E571K MAF in plasma cell‐free DNA are shown as blue graph lines. Changes in tumor size, represented by the sum of perpendicular diameters of target tumor lesions evaluated by Cheson criteria (orange bars), in two Hodgkin lymphoma patients treated with mTOR and HDAC inhibitors (A and B) and one patient treated with mTOR and HDAC inhibitors followed by an anti‐CTLA4 antibody plus lenalidomide (C), are also shown. Abbreviations: CTLA4, cytotoxic T‐lymphocyte associated protein 4; HDAC, histone deacetylase; MAF, mutant allele frequency; mTOR, mammalian target of rapamycin.
Discussion
Our study demonstrates that CGP using targeted NGS of FFPE archival tumor samples from Hodgkin lymphoma patients is feasible. We observed a spectrum of alterations distributed across 44 cancer‐related genes; the most commonly mutated genes were TP53 (22% of patients), B2M (22% of patients), and XPO1 (18% of patients). We also detected XPO1 E571K or E571V mutations in 31% of the assayed plasma cfDNA samples. To our knowledge, our study is the first to report analysis of the genomic landscape of Hodgkin lymphoma using archival tumor samples.
Large‐scale genomic profiling has been challenging because of the nature of the disease, as Hodgkin and Reed‐Sternberg (HRS) cells are relatively sparse in the stroma‐rich environment [10]. To address this problem, researchers have performed flow sorting of these cells to prepare samples for whole exome sequencing. Reichel et al. [10] used this approach for 10 fresh tumor samples and detected B2M alterations in 80% of cases. Other alterations including XPO1 mutations have also been reported; however, the need for fresh tumor tissue limits the clinical utility of this approach. Notably, we did not perform microdissection of HRS cells in this study.
Others have also assessed the genomic profiles of tumor biopsies and plasma‐derived cfDNA. Camus et al. [11] reported XPO1 E571K mutations in 24% of tumor biopsies and in approximately 50% of plasma cfDNA samples from Hodgkin lymphoma patients; however, the investigators used ddPCR, which was not designed to detect alterations in cancer‐related genes other than XPO1. We improve upon these studies by performing genomic profiling of more readily available archival tumor specimens in order to evaluate the genomic landscape of Hodgkin lymphoma.
In our study, TP53 mutations were among the most frequent molecular abnormalities, occurring in 22% of patients. Previous studies have reported lower frequency of TP53 mutations, which occurred in approximately 9%–11% of Hodgkin lymphoma patients [12], [13]. However, previous studies suggested that TP53 mutation frequency might be associated with enrichment for HRS cells [14], [15]. Arguably, samples meeting quality standards for the CGP could have been plausibly enriched for HRS cells. Unfortunately, a small sample size precluded analysis of prognostic significance. Molecular alterations in B2M were also among the most frequently detected molecular abnormalities. B2M inactivating mutations result in a loss of expression of the major histocompatibility complex class I complex, which is involved in antigen presentation. Decreased expression of B2M has also been reported to be associated with worse prognosis in non‐Hodgkin lymphoma patients [16]. Notably, inactivating B2M mutations have been observed in Hodgkin lymphoma patients, and lack of B2M expression has been associated with a favorable prognosis [10]. With respect to therapeutic insights, one recent study suggested that truncating B2M mutations may be associated with acquired resistance to PD‐1 blockade in metastatic melanoma [10]. These studies, along with our findings, demonstrate the benefits of genomic profiling to improve our understanding of the genomic landscape of Hodgkin lymphoma tumors, as well as to improve therapeutic selection.
Other frequent recurrent alterations in the present study were XPO1 E571 mutations, which were detected in 18% of archival tumor samples and 31% of plasma cfDNA samples. Small numbers precluded assessment of concordance between plasma cfDNA and tumor tissue testing. XPO1 regulates the nuclear export and localization of proteins involved in cell proliferation and the cell cycle. Increased expression of XPO1 in cancer patients has been shown to be associated with worse survival and increased metastasis [17], and mutations in this gene have been reported in B‐cell lymphoma [18]. A recent study using ddPCR reported recurrent XPO1 E571K mutations in 24% of tumor biopsies and about 50% of plasma cfDNA samples from Hodgkin lymphoma patients [11]. Currently, XPO1 inhibitors such as selinexor are being evaluated in solid tumors and hematologic malignancies in clinical trials; however, the possible relationship between XPO1 mutations and sensitivity or resistance to XPO1 inhibition remains unknown because preclinical data on B‐cell lymphoma and osteosarcoma cell lines demonstrated efficacy independent of XPO1 mutation status [18].
Other potentially targetable molecular alterations identified in our study include loss of TSC2 (tuberous sclerosis 2), for which mammalian target of rapamycin inhibitors may be effective [19]; and a BRCA1 mutation, which suggests treatment with DNA‐damaging platinum agents or poly(ADP‐ribose) polymerase inhibitors [20], [21]. However, the one BRCA1 mutation we identified may be a germline event given the event's allelic frequency of 46% and the patient's family history of breast cancer. Previous studies have reported that germline BRCA1 mutations are infrequent in Hodgkin lymphoma [22].
We also identified recurrent events in TNFAIP3 (14% of patients) and SOCS1 (10% of patients). TNFAIP3 is involved in the regulation of apoptosis and nuclear factor κB signaling, and the nonsense and frameshift mutations we identified are predicted to be inactivating due to the predicted translation of a truncated protein. Deletions, mutations, and decreased protein expression of TNFAIP3 have been reported in approximately 24%–44% of Hodgkin lymphoma patients and in 10%–38% of non‐Hodgkin lymphoma patients [23], [24]. With respect to SOCS1, S116N has been reported in diffuse large B‐cell lymphoma [25], and A17T has been reported in B‐cell lymphoma [26]. A17T is outside of the gene's functional domains, but S116N is in the gene's conserved Src Homology 2 domain and thus may affect protein interactions if expressed. Mutations in SOCS1, which has roles in apoptosis, and in cell survival and growth through JAK/STAT signaling, have also been reported in Reed‐Sternberg cells and Hodgkin cell lines as inactivating mutations [27]. The SOCS1 rearrangement we identified in patients 7 and 39 is also predicted to be inactivating as it disrupts the only coding exon of SOCS1. This event is similar to a t(14;16)(q32;p13.1) event that was previously described in a Hodgkin lymphoma patient [28].
High TMB was recently found to be predictive of favorable response to PD‐1‐ and PD‐L1‐targeted therapies in lung cancer [29]. In our study, 15% patients with TMB data had high TMB. However, only two patients with known TMB received PD‐1 antibodies, and both responded despite having intermediate TMB. Thus, the relationship between TMB and response to immune checkpoint inhibitors remains to be clarified.
We further show that ddPCR is a highly sensitive approach that can detect XPO1 E571 mutations in plasma cfDNA samples from Hodgkin lymphoma patients. Using ddPCR, we detected XPO1 E571 mutations in 31% of cases, and we were able to confirm the mutation in archival tumor samples in three of the four cases for which tumor tissue was available. In addition, dynamic tracking of XPO1 MAF corresponded with changes observed during routine radiographic imaging. Previously, Camus et al. [11] reported that ddPCR detected XPO1 E571K mutations in about 50% of plasma cfDNA samples; however, in nearly half those cases, the XPO1 mutation was not confirmed in the tumor tissue. Also, patients with detectable XPO1 mutation in plasma cfDNA at the end of therapy had a trend toward shorter progression‐free survival.
Our study has several potential limitations. First, the sample size was relatively small, and second, CGP encompassed up to 405 selected cancer‐related genes such that novel events may be missed. In addition, our method did not include testing of normal DNA to reliably exclude possible germline alterations. Third, our method did not include sorting the HRS cells. Therefore, it is unclear if all reported variants are truly from HRS cells. Fourth, although we were able to obtain TMB data for a considerable number of patients, only a few patients received checkpoint inhibitors; thus, the value of TMB in predicting immunotherapy response remains unknown. In addition, whether any of the recurrent molecular alterations we identified may be used for therapeutic decision‐making remains unclear. Furthermore, profiling of degraded FFPE DNA lends itself to the possibility of false negatives. For example, previous studies have reported a recurrence of PD‐L1/CD274 and PD‐L2/PDCD1LG2 amplification in newly diagnosed HL cases [30] and nodular sclerosis HL [31]. However, only one patient profiled here (33 of 49 were tested for PD‐L1/CD274 and PD‐L2/PDCD1LG2 amplification as these genes were not initially included in the panel) demonstrated PD‐L1 amplification such that this caveat must be considered during data interpretation. Finally, the roles of B2M and XPO1 mutations as possible biomarkers of response to XPO1 inhibitors or immune checkpoint inhibitors, respectively, remain to be evaluated in preclinical and prospective clinical studies.
Conclusion
Overall, we demonstrate the feasibility of performing CGP on archival tumor specimens from Hodgkin lymphoma patients. In addition to improving our understanding of the genomic landscape of Hodgkin lymphoma, we identified recurrent alterations in the TP53, B2M, XPO1, and TNFAIP3 genes. Fifteen percent of patients demonstrated high TMB, which in other cancers was found to be associated with activity of immune checkpoint inhibitors. We further show that XPO1 mutations may be detected in patient cfDNA, and that changes in the MAF of cfDNA XPO1 mutations correlate with changes in tumor size. As a result, these findings provide potential strategies for therapeutic selection and molecular monitoring of patients. Continued genomic analysis of Hodgkin lymphoma patients thus creates a foundation for improving patient care.
Acknowledgments
The study was conducted in accordance with the guidelines of MD Anderson's Institutional Review Board. Additionally, approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817) per protocol at Foundation Medicine. We thank the patients who participated in this study, as well as Vivianne Velez‐Bravo, Anne T. James, Anjali Raina, and Goran Cabrilo for their contributions to this study. We thank The Sheikh Khalifa Al Nahyan Ben Zayed Institute for Personalized Cancer Therapy, the National Center for Advancing Translational Sciences (#UL1 TR000371), the National Institutes of Health through MD Anderson's Cancer Center Support Grant (P30‐CA016672), MD Anderson Non‐Standard of Care Clinical Charges, and TGen for support.
Author Contributions
Conception/design: Winnie S. Liang; Filip Janku
Provision of study material or patients: Winnie S. Liang, Jo‐Anne Vergilio, Helen J. Huang, Yasuhiro Oki, Ignacio Garrido‐Laguna, Haeseong Park, Jason R. Westin, Funda Meric‐Bernstam, David Fabrizio, Michelle A. Fanale, Filip Janku
Collection and/or assembly of data: Winnie S. Liang, Jo‐Anne Vergilio, Helen J. Huang, Yasuhiro Oki, Ignacio Garrido‐Laguna, Haeseong Park, Jason R. Westin, Funda Meric‐Bernstam, David Fabrizio, Michelle A. Fanale, Filip Janku
Data analysis and interpretation: Winnie S. Liang, Jo‐Anne Vergilio, Helen J. Huang, Yasuhiro Oki, Ignacio Garrido‐Laguna, Haeseong Park, Jason R. Westin, Funda Meric‐Bernstam, David Fabrizio, Michelle A. Fanale, Filip Janku
Manuscript writing: Winnie S. Liang, Jo‐Anne Vergilio, Bodour Salhia, Helen J. Huang, Yasuhiro Oki, Ignacio Garrido‐Laguna, Haeseong Park, Jason R. Westin, Funda Meric‐Bernstam, David Fabrizio, Vincent A. Miller, Philip J. Stephens, Michelle A. Fanale, Jeffrey S. Ross, Filip Janku
Final approval of manuscript: Winnie S. Liang, Jo‐Anne Vergilio, Bodour Salhia, Helen J. Huang, Yasuhiro Oki, Ignacio Garrido‐Laguna, Haeseong Park, Jason R. Westin, Funda Meric‐Bernstam, David Fabrizio, Vincent A. Miller, Philip J. Stephens, Michelle A. Fanale, Jeffrey S. Ross, Filip Janku
Disclosures
Jo‐Anne Vergilio: Foundation Medicine, Inc. (E, OI); David Fabrizio: Foundation Medicine, Inc. (E, OI); Vincent A. Miller: Foundation Medicine, Inc. (E, OI); Philip J. Stephens: Foundation Medicine, Inc. (E, OI); Jeffrey S. Ross: Foundation Medicine, Inc. (E, OI); Filip Janku: Biocartis, Trovagene, Foundation Medicine, Inc. (RF), Trovagene, Guardant Health (SAB). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Janku F, Wheler JJ, Westin SN et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol 2012;30:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 4.Townsend W, Linch D. Hodgkin's lymphoma in adults. Lancet 2012;380:836–847. [DOI] [PubMed] [Google Scholar]
- 5.Josting A, Rueffer U, Franklin J et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: A report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–1286. [PubMed] [Google Scholar]
- 6.Sureda A, Robinson S, Canals C et al. Reduced‐intensity conditioning compared with conventional allogeneic stem‐cell transplantation in relapsed or refractory Hodgkin's lymphoma: An analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–462. [DOI] [PubMed] [Google Scholar]
- 7.Ansell SM, Lesokhin AM, Borrello I et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J, Abdel‐Wahab O, Nahas MK et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichel J, Chadburn A, Rubinstein PG et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed‐Sternberg cells. Blood 2015;125:1061–1072. [DOI] [PubMed] [Google Scholar]
- 11.Camus V, Stamatoullas A, Mareschal S et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell‐free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016;101:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forbes SA, Beare D, Gunasekaran P et al: COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maggio EM, Stekelenburg E, Van den Berg A et al. TP53 gene mutations in Hodgkin lymphoma are infrequent and not associated with absence of Epstein‐Barr virus. Int J Cancer 2001;94:60–66. [DOI] [PubMed] [Google Scholar]
- 14.Gupta RK, Patel K, Bodmer WF et al. Mutation of p53 in primary biopsy material and cell lines from Hodgkin disease. Proc Natl Acad Sci U S A 1993;90:2817–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trumper LH, Brady G, Bagg A et al. Single‐cell analysis of Hodgkin and Reed‐Sternberg cells: Molecular heterogeneity of gene expression and p53 mutations. Blood 1993;81:3097–3115. [PubMed] [Google Scholar]
- 16.Amiot L, Onno M, Lamy T et al. Loss of HLA molecules in B lymphomas is associated with an aggressive clinical course. Br J Haematol 1998;100:655–663. [DOI] [PubMed] [Google Scholar]
- 17.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 2012;83:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jardin F, Pujals A, Pelletier L et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B‐cell lymphoma. Am J Hematol 2016;91:923–930. [DOI] [PubMed] [Google Scholar]
- 19.Inoki K, Li Y, Xu T et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003;17:1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul I, Savage KI, Blayney JK et al. PARP inhibition induces BAX/BAK‐independent synthetic lethality of BRCA1‐deficient non‐small cell lung cancer. J Pathol 2011;224:564–574. [DOI] [PubMed] [Google Scholar]
- 21.Rottenberg S, Jaspers JE, Kersbergen A et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008;105:17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols KE, Heath JA, Friedman D et al. TP53, BRCA1, and BRCA2 tumor suppressor genes are not commonly mutated in survivors of Hodgkin's disease with second primary neoplasms. J Clin Oncol 2003;21:4505–4509. [DOI] [PubMed] [Google Scholar]
- 23.Nomoto J, Hiramoto N, Kato M et al. Deletion of the TNFAIP3/A20 gene detected by FICTION analysis in classical Hodgkin lymphoma. BMC Cancer 2012;12:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitz R, Hansmann ML, Bohle V et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 2009;206:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohr JG, Stojanov P, Lawrence MS et al. Discovery and prioritization of somatic mutations in diffuse large B‐cell lymphoma (DLBCL) by whole‐exome sequencing. Proc Natl Acad Sci U S A 2012;109:3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mottok A, Renne C, Seifert M et al. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B‐cell lymphoma entities. Blood 2009;114:4503–4506. [DOI] [PubMed] [Google Scholar]
- 27.Weniger MA, Melzner I, Menz CK et al. Mutations of the tumor suppressor gene SOCS‐1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho‐STAT5 accumulation. Oncogene 2006;25:2679–2684. [DOI] [PubMed] [Google Scholar]
- 28.Martin‐Subero JI, Klapper W, Sotnikova A et al. Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed‐Sternberg cells of classical Hodgkin lymphoma. Cancer Res 2006;66:10332–10338. [DOI] [PubMed] [Google Scholar]
- 29.Spigel DR, Betzig Schrock A, Fabrizio D et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD‐1/PD‐L1 targeted therapies. Presented at: 2016. ASCO Annual Meeting; 2016; Chicago.
- 30.Roemer MG, Advani RH, Ligon AH et al. PD‐L1 and PD‐L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green MR, Monti S, Rodig SJ et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD‐1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B‐cell lymphoma. Blood 2010;116:3268–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]