

This article reports the results of the CONSIGN study, which was designed to characterize the safety of regorafenib in a large patient population, estimate progression‐free survival, and provide patients with treatment‐refractory metastatic colorectal cancer access to regorafenib before market authorization.
Keywords: Regorafenib, Metastatic colorectal cancer, Prospective studies, Toxicities
Abstract
Background.
In the phase III CORRECT trial, regorafenib significantly improved survival in treatment‐refractory metastatic colorectal cancer (mCRC). The CONSIGN study was designed to further characterize regorafenib safety and allow patients access to regorafenib before market authorization.
Methods.
This prospective, single‐arm study enrolled patients in 25 countries at 186 sites. Patients with treatment‐refractory mCRC and an Eastern Cooperative Oncology Group performance status (ECOG PS) ≤1 received regorafenib 160 mg once daily for the first 3 weeks of each 4‐week cycle. The primary endpoint was safety. Progression‐free survival (PFS) per investigator assessment was the only efficacy evaluation.
Results.
In total, 2,872 patients were assigned to treatment and 2,864 were treated. Median age was 62 years, ECOG PS 0/1 was 47%/53%, and 74% had received at least three prior regimens for metastatic disease. Median treatment duration was three cycles. Treatment‐emergent adverse events (TEAEs) led to dose reduction in 46% of patients. Regorafenib‐related TEAEs led to treatment discontinuation in 9%. Grade 5 regorafenib‐related TEAEs occurred in <1%. The most common grade ≥3 regorafenib‐related TEAEs were hypertension (15%), hand–foot skin reaction (14%), fatigue (13%), diarrhea (5%), and hypophosphatemia (5%). Treatment‐emergent grade 3–4 laboratory toxicities included alanine aminotransferase (6%), aspartate aminotransferase (7%), and bilirubin (13%). Ongoing monitoring identified one nonfatal case of regorafenib‐related severe drug‐induced liver injury per DILI Working Group criteria. Median PFS (95% confidence interval [CI]) was 2.7 months (2.6–2.7).
Conclusion.
In CONSIGN, the frequency and severity of TEAEs were consistent with the known safety profile of regorafenib. PFS was similar to reports of phase III trials. ClinicalTrials.gov: NCT01538680.
Implications for Practice.
Patients with metastatic colorectal cancer (mCRC) who fail treatment with standard therapies, including chemotherapy and monoclonal antibodies targeting vascular endothelial growth factor or epidermal growth factor receptor, have few treatment options. The multikinase inhibitor regorafenib was shown to improve survival in patients with treatment‐refractory mCRC in the phase III CORRECT (N = 760) and CONCUR (N = 204) trials. However, safety data on regorafenib for mCRC in a larger number of patients were not available. The CONSIGN trial, carried out prospectively in more than 2,800 patients across 25 countries, confirmed the safety profile of regorafenib from the phase III trials and reinforced the importance of using treatment modifications to manage adverse events.
摘要
背景。在 III 期 CORRECT 试验中,瑞戈非尼显著提高了难治性转移性结肠直肠癌 (mCRC) 的生存率。CONSIGN 研究旨在进一步彰显瑞戈非尼的安全性,以及让患者在获得上市许可前使用此药品。
方法。此项前瞻性单臂研究在 25 个国家的 186 个地点招募患者。患有难治性 mCRC 且东部肿瘤协作组体能状态 (ECOG PS) <1 的患者以 160 mg 的剂量服用瑞戈非尼,每天一次,每 4 星期周期中的前 3 周服用。主要疗效指标为安全性。每位研究员的无进展生存期 (PFS) 评估是唯一的疗效评估。
结果。总计有 2,872 名患者被分配治疗,2,864 名患者接受治疗。中值年龄为 62 岁,ECOG PS 0/1 为 47%/53%,有 74% 已收到至少三个转移性疾病的前治疗方案。平均治疗时间为三个周期。治疗突发不良反应事件 (TEAE) 导致 46% 的患者减少剂量。9% 的患者因瑞戈非尼相关 TEAE 而停止治疗。少于 1% 的患者出现 5 级瑞戈非尼相关 TEAE。最常见的 3 级及以上瑞戈非尼相关 TEAE 为高血压 (15%)、手足皮肤反应 (14%)、疲劳 (13%)、腹泻 (5%) 和低磷血症 (5%)。治疗突发 3‐4 级实验室毒性包括丙氨酸转氨酶 (6%)、天冬氨酸转氨酶 (6%) 和胆红素 (6%)。正在进行的监测确定了每个 DILI 工作组标准的瑞戈非尼相关药物所致重度肝损伤的非致命病例。中位 PFS(95% 置信区间 [CI])为 2.7 个月 (2.6–2.7)。
结论。在 CONSIGN 中,TEAE 的频率和严重程度与瑞戈非尼已知的安全曲线一致。PFS 与 III 期试验报告相似。ClinicalTrials.gov:NCT01538680。
对实践的启示:采用标准治疗(包括化疗和针对血管内皮生长因子或表皮生长因子受体的单克隆抗体)而未达到治疗效果的转移性结肠直肠癌 (mCRC) 患者所能选择的治疗方案极少。在 III 期 CORRECT (N = 760) 和 CONCUR (N = 204) 试验中,多重激酶抑制剂瑞戈非尼被证明可以提高难治性 mCRC 患者的生存率。但是,无法提供大量患者针对 mCRC 的瑞戈非尼安全数据。在 25 个国家的超过 2,800 名患者中进行的前瞻性 CONSIGN 试验,从 III 期试验中证实了瑞戈非尼的安全性,并加强了使用治疗修改来管理不良反应事件的重要性。
Introduction
Treatment for metastatic colorectal cancer (mCRC) consists of chemotherapy based on a fluoropyrimidine, oxaliplatin, and irinotecan, in combination or in sequence, and monoclonal antibodies targeting vascular endothelial growth factor [1], [2]. Epidermal growth factor receptor antibodies are used in patients with RAS wild‐type tumors. Many patients still have good performance status when they progress on these regimens, so additional treatment options are needed.
Regorafenib is an oral multikinase inhibitor [3] approved for patients with mCRC previously treated with standard therapies, based on results of the phase III CORRECT trial (NCT01103323), which showed an overall survival benefit versus placebo (6.4 vs. 5.0 months; hazard ratio [HR] 0.77; one‐sided p = .0052) [4]. The most common grade ≥3 regorafenib‐related treatment‐emergent adverse events (TEAEs) were hand–foot skin reaction (HFSR), fatigue, diarrhea, hypertension, and rash or desquamation. The phase III CONCUR trial (NCT01584830) confirmed the overall survival benefit of regorafenib in Asian patients [5]. The CONSIGN study (NCT01538680) was designed to characterize regorafenib safety in a larger patient population, to provide patients with treatment‐refractory mCRC access to regorafenib prior to market authorization, and to estimate progression‐free survival (PFS).
Materials and Methods
Study Design and Participants
CONSIGN was a prospective, open‐label, single‐arm, phase IIIb study carried out in 25 countries in 186 centers in Europe, North America, Israel, and Australia. Eligible patients had documented adenocarcinoma of the colon or rectum and an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤1, had received approved standard therapies including a fluoropyrimidine, oxaliplatin, irinotecan, bevacizumab, and cetuximab/panitumumab for patients with KRAS wild‐type tumors, and had disease progression within 3 months of treatment. A protocol amendment allowed inclusion of patients from Mexico and Russia who had not received bevacizumab or cetuximab/panitumumab.
The study was approved by the independent ethics committee or institutional review board at each site and was conducted in accordance with the guidelines of the Declaration of Helsinki and good clinical practice. All participants were required to provide written informed consent.
Procedures
Patients received oral regorafenib 160 mg once daily for 3 weeks on/1 week off in 4‐week cycles. Treatment continued until patients met one of the predefined withdrawal criteria, including disease progression, unacceptable toxicity, withdrawal of consent, or investigator decision to discontinue. Treatment beyond progression was at the investigator's discretion. Other systemic anticancer therapies including cytotoxic therapy, immunotherapy, hormonal therapy, or other investigational antitumor agents were not allowed during the study or within 4 weeks before study start (6 weeks for mitomycin C).
The regorafenib dose could be reduced, interrupted, or permanently discontinued to manage treatment‐related toxicities. The dose could be re‐escalated to 160 mg maximum once toxic effects resolved.
The primary endpoint was safety. TEAEs, graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‐CTCAE) version 4.0, were assessed through 30 days after treatment. Laboratory parameters, including complete blood count, and sodium, potassium, chloride, aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, alkaline phosphatase, calcium, lipase, phosphate, and creatinine levels, were assessed every 2 weeks during the first two cycles and every 2 or 4 weeks (at the investigator's discretion) thereafter. Laboratory abnormalities were considered TEAEs if they required treatment, caused withdrawal, caused clinical manifestations, or were judged clinically relevant by the investigator.
Deaths up to 30 days after treatment were recorded. Information about deaths was not formally collected after this point. The by‐cycle frequency of a TEAE is defined as the number of patients with the TEAE starting or worsening during a cycle and is expressed as a percentage of patients treated during that cycle.
The only efficacy variable assessed was PFS, defined as the time from treatment assignment to first observed disease progression or death by any cause. Tumor measurements were made at intervals and with methods that complied with each institution's best standard of care. Only the date of disease progression was collected. If radiographic imaging was not possible, use of clinical progression was allowed.
Statistical Analysis
Statistical analyses used SAS version 9.1.3 or higher (SAS Institute Inc., Cary, NC). Data were analyzed descriptively. Efficacy analyses were based on the full analysis set, comprising all patients assigned to treatment. PFS was estimated by the Kaplan‐Meier method. PFS for patients without disease progression or death before or at the last visit was censored at the date of the last clinical or radiological assessment.
Results
Patient Disposition, Baseline Characteristics, and Dosing
Between April 2012 and December 2013, 3,309 patients were screened in 25 countries at 186 centers; 2,872 were assigned to treatment (supplemental online Fig. 1). Of those, 2,864 patients initiated treatment (safety population). Eight patients were not treated: four had adverse events not associated with clinical disease progression, three withdrew, and one had disease progression between treatment assignment and planned treatment start. At the analysis cutoff date, January 2, 2015, 13 patients remained on treatment.
The median age was 62 years (range 19–89); 268 patients (9%) were aged ≥75 years (Table 1). Approximately half had ECOG PS 1 (53%) and half had KRAS mutations (51%). Twenty‐six percent had received fewer than three prior regimens for metastatic disease, and 96% had received prior bevacizumab.
Table 1. Baseline characteristics of patients.
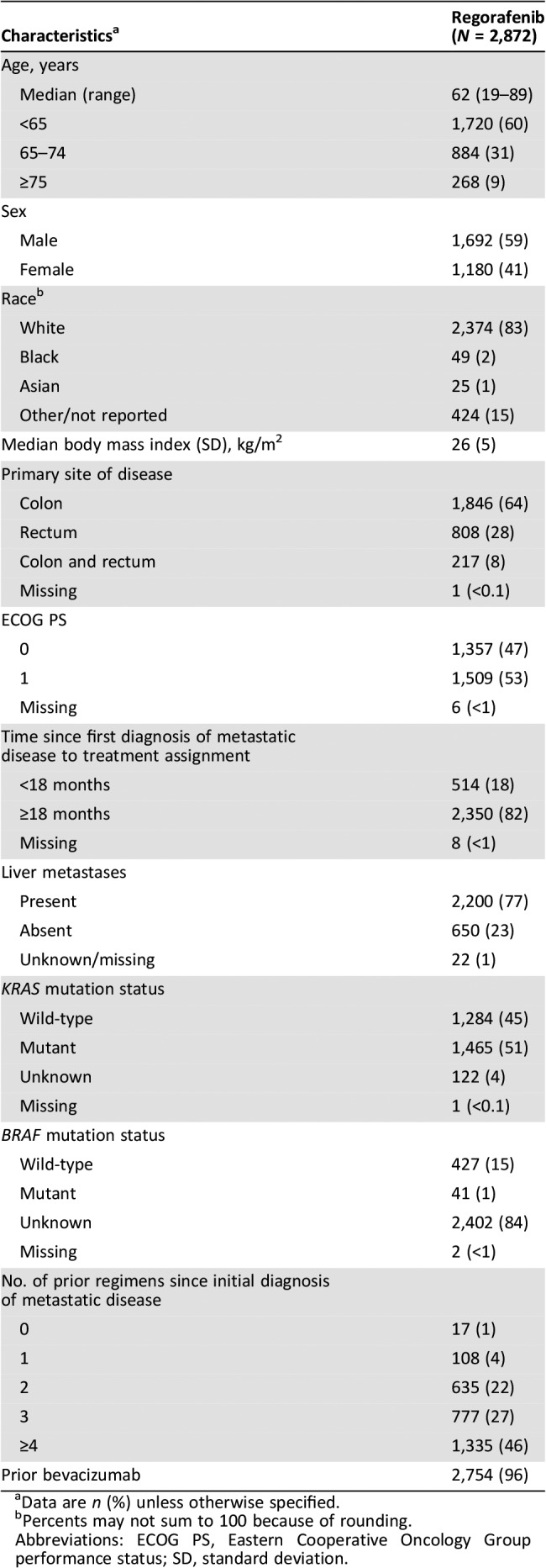
Data are n (%) unless otherwise specified.
Percents may not sum to 100 because of rounding.
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; SD, standard deviation.
Median (range) and mean (standard deviation [SD]) treatment durations were 2.5 months (0.03–30.4) and 3.5 months (3.8), respectively (Table 2). The mean (SD) daily dose (assessed only on days when a dose was given) was 146 (19) mg. The mean (SD) percent of planned dose (including dose interruptions) was 75% (20%). Eighty‐four percent of patients had at least one treatment interruption/delay, and 49% had at least one dose reduction. Most dose‐reduction events (91%; 1,739/1,918) were for TEAEs. The most common reasons for dose‐interruption events were TEAEs (3,942/6,451 events; 61%) and error by the patient (1,737/6,451 events; 27%).
Table 2. Dosing, treatment duration, and modifications.

Includes time off drug/interruptions.
Based on patients starting each cycle.
Three patients received doses >160 mg.
Modifications include reductions, interruptions/delays, and re‐escalations; a dose re‐escalation was defined as a dose higher than the previous dose reported or as a dose after a dose interruption (unless a dose reduction was prescribed following an interruption) or as a dose increase after a dose reduction until the protocol dose of 160 mg was reached.
Based on the total number of events.
Abbreviations: IQR, interquartile range; SD, standard deviation.
Dose re‐escalations, defined as either receiving a higher dose than a previous dose or receiving a dose after a treatment interruption (unless a dose reduction was prescribed after an interruption), were reported in 53% of patients (Table 2; the numbers of patients in each separate category were not captured). There was a total of 3,462 dose re‐escalation events. Reasons were not recorded for most dose re‐escalations because providing the reason was not required.
At data cutoff, more than 1 year after all patients had completed the first visit, 2,851/2,864 patients had discontinued treatment, most frequently because of progressive disease/TEAEs secondary to clinical progression (n = 2,240; 78%). Three hundred twenty‐seven patients (11%) discontinued for TEAEs not associated with clinical progression (supplemental online Fig. 1).
Safety
Overall, 2,847 patients (99%) had at least one TEAE; TEAEs were regorafenib related in 2,613 patients (91%) (Table 3). The most frequent grade ≥3 regorafenib‐related TEAEs were hypertension (15%), HFSR (14%), fatigue (13%), diarrhea (5%), and hypophosphatemia (5%). Serious TEAEs occurred in 1,251 patients (44%) and were regorafenib related in 251 (9%). For the most common regorafenib‐related TEAEs (of any grade), the by‐cycle frequency, defined as the number of patients with that TEAE starting or worsening during a cycle among all patients treated during that cycle, was generally highest in cycles 1 and 2 (supplemental online Fig. 2).
Table 3. Treatment‐emergent and drug‐related treatment‐emergent adverse events (safety population)a .

Events listed are TEAEs occurring in at least 10% of patients in either treatment group.
Laboratory abnormalities were reported as TEAEs if they required treatment, caused the patient to withdraw, caused apparent clinical manifestations, or were judged clinically relevant by the investigator—not all laboratory abnormalities were reported as TEAEs (see also supplemental online Table 3).
The most common TEAEs and drug‐related TEAEs of any grade in this category included general physical health deterioration, asthenia, night sweats, and metastatic colorectal cancer.
Of the 4% of patients who experienced grade 3 events (n = 105; drug related, n = 19), the most common events were general physical health deterioration (n = 87; drug related, n = 15), asthenia (n = 8; drug related, n = 4), and performance status decreased (n = 5; drug related, n = 0). Patients with grade 4 events (n = 18; drug related, n = 0) had general physical health deterioration (n = 15), asthenia (n = 2), and metastatic colorectal cancer (n = 1).
Adverse events were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
Abbreviations: AST, aspartate aminotransferase; HFSR, hand–foot skin reaction; NA, not applicable; TEAE, treatment‐emergent adverse event.
TEAEs (regardless of relation to regorafenib) led to treatment discontinuation in 25% of patients, dose reduction in 46%, and treatment interruption/delay in 68%. TEAEs judged regorafenib related led to treatment discontinuation in 266 patients (9%) and treatment modifications in 1,732 (60%).
The most common TEAEs regardless of relation to regorafenib leading to treatment discontinuation were general disorders and administrative site conditions—other (6%; most frequently specified as general physical health deterioration), fatigue (3%), and increased bilirubin (2%; supplemental online Table 1). HFSR led to treatment discontinuation in 1% of patients (supplemental online Table 1). The most common TEAEs (of any grade) regardless of relation to regorafenib leading to dose reduction were HFSR (17%), fatigue (9%), diarrhea (5%), and hypertension (4%; supplemental online Table 2). Of the 17% of patients who had a dose reduction due to HFSR, 13% had a dose reduction due to grade 1 or 2 HFSR, and 4% had a dose reduction due to grade 3 HFSR (supplemental online Table 2).
A total of 405 of 2,864 patients (14%) had grade 5 TEAEs. Most fatal outcomes were related to disease progression; only 13 (<1%) were judged regorafenib related: worsening of general condition (2), sepsis (2), asthenia (1), cardiac arrest (1), heart failure (1), intracranial hemorrhage (1), gastric hemorrhage (1), thromboembolic event (1), colonic perforation (1), hepatic failure (1), and death not otherwise specified (1).
Treatment‐emergent grade 3 or 4 laboratory abnormalities included increased bilirubin (13%), increased AST (7%), and increased ALT (6%; supplemental online Table 3). One nonfatal case consistent with regorafenib‐related severe drug‐induced liver injury by DILI Working Group criteria [6] was identified from ongoing hepatotoxicity monitoring: a 65‐year‐old male with liver metastases and no history of viral hepatitis or acute viral infection had transient elevations in ALT (grade 3) and AST (grade 2) at cycle 2, day 15, which decreased to grade 0 during cycle 3. At cycle 4, day 15, the patient had grade 4 transaminase increases, and regorafenib was interrupted. At cycle 5, day 1, there were further increases in transaminases, an increase in gamma‐GT, and icterus, and regorafenib was permanently discontinued. The events resolved approximately 3 months after onset without further therapy.
Overall rates of regorafenib‐related TEAEs were generally slightly higher in older patients. Regorafenib‐related grade ≥3 TEAEs occurred in 55%, 59%, and 64% of patients aged <65, 65–74, and ≥75 years, respectively (Table 4). Among patients aged ≥75 years, grade ≥3 regorafenib‐related fatigue and hypertension were higher and grade 3 regorafenib‐related HFSR lower than in younger age groups (Table 4). The treatment duration, median number of cycles, and daily dose were not markedly different in subgroups of patients defined by age (supplemental online Table 4). In patients aged <65, 65–74, and ≥75 years, 22%, 21%, and 16%, respectively, received ≥6 cycles, 11%, 9%, and 8%, respectively, received ≥9 cycles.
Table 4. Drug‐related treatment‐emergent adverse events by age subgroupa .

TEAEs listed are regorafenib‐related TEAEs occurring at grade ≥3 in ≥5% of patients overall, graded by National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
Abbreviations: HFSR, hand–foot skin reaction; TEAE, treatment‐emergent adverse event.
Progression‐Free Survival
Median PFS (95% CI) was 2.7 months (2.6, 2.7) overall (Fig. 1A): 2.8 months (2.7, 2.9) for patients with KRAS wild‐type tumors and 2.5 months (2.4, 2.6) for patients with KRAS mutant tumors (Fig. 1B). Among patients with baseline ECOG PS 0 and 1, median PFS was 3.0 (2.8, 3.1) and 2.3 months (2.2, 2.5), respectively. Among patients aged ≥65 years and ≥75 years, median PFS was 2.6 (2.5, 2.7) and 2.5 months (2.1, 2.7), respectively.
Figure 1.

Kaplan‐Meier analyses of PFS for all patients (A) and by baseline KRAS mutation status (B). PFS was by investigator assessment, and intervals for radiological or clinical tumor assessments were not predetermined. Abbreviations: CI, confidence interval; PFS, progression‐free survival; wt, wild‐type.
Twenty‐three percent of patients (674/2,872) had PFS >4 months. Estimated PFS rates (95% CI) at 6, 9, and 12 months were 15% (14, 16), 8% (6, 9), and 4% (3, 5), respectively. Exploratory analyses suggested that the subgroup with PFS >4 months tended to have a higher proportion of patients with ECOG PS 0, no liver metastases, and a longer time since diagnosis of metastatic disease versus the subgroup with short PFS (supplemental online Table 5). There was no apparent difference in KRAS mutation status between the long and short PFS groups. Among patients aged ≥65, ≥70, and ≥75 years, similar proportions had PFS >4 months and ≤4 months. Patients with PFS >4 months received a lower median regorafenib dose (138 mg) than those with shorter PFS (160 mg) and had higher rates of treatment modifications and dose reductions (supplemental online Table 6).
Discussion
This large, prospective study in patients with treatment‐refractory mCRC shows that the safety profile of regorafenib is consistent with results of the phase III trials [4], [5]. Regorafenib‐related TEAEs occurred in 91% of patients, with the most common including fatigue, HFSR, hypertension, and diarrhea at rates consistent with those in the CORRECT trial [4]. Regorafenib‐related TEAEs led to dose modifications in 60% of patients but to treatment discontinuation in only 9%, suggesting that regorafenib‐related TEAEs were managed with dose modifications, allowing patients to stay on therapy. The high rate of dose reductions observed in this study and in the phase III trials has motivated the exploration of new dosing strategies, including starting regorafenib at doses <160 mg and increasing the dose as tolerated; this approach reflects current clinical practice by some physicians [1], [7], [8], [9], [10]. A recently reported small, randomized phase II trial (n = 123) showed that a higher proportion of patients with mCRC starting regorafenib treatment using a rapid dose‐escalation strategy during the first cycle (starting at 80 mg daily for 1 week and escalating the daily dose by 40 mg during weeks 2 and 3 to 160 mg daily as tolerated) continued to cycle 3 compared with those starting at the 160 mg daily dose; data suggest that patients treated using the dose‐escalation strategy may have had better outcomes [11].
The patient population in CONSIGN was comparable to regorafenib‐treated patients in CORRECT [4]. The median age was similar (CONSIGN 61 years; CORRECT 61 years), and most patients were white (CONSIGN 83%; CORRECT 78%). A similar proportion of patients in CONSIGN versus CORRECT had an ECOG PS of 1 (53% vs. 48%), and in both studies, 82% of patients had been diagnosed with metastatic disease for ≥18 months. In CORRECT, all patients had received prior bevacizumab. Although not all patients in CONSIGN had received prior bevacizumab, the proportion who did not was small (n = 118; 4%).
Safety was closely monitored in CONSIGN. Both CONSIGN and CORRECT included laboratory evaluations of ALT, AST, and bilirubin every 2 weeks during the first two cycles. Rates of grade ≥3 TEAEs were similar (80%, CONSIGN; 78%, CORRECT regorafenib arm), as were rates of grade ≥3 TEAEs judged regorafenib related (CONSIGN 57%; CORRECT 55%) [4]. In both studies, regorafenib‐related grade 5 TEAEs were rare (CONSIGN <1%; CORRECT 1% [12]); the most common regorafenib‐related grade ≥3 TEAEs included hypertension, fatigue, HFSR, and diarrhea [4]. The higher rate of drug‐related grade ≥3 hypertension in CONSIGN (15% vs. 7% in CORRECT) may be related in part to the more stringent NCI‐CTCAE version 4.0 grading of hypertension used in CONSIGN (CORRECT used version 3.0).
In CONSIGN, the safety profile of regorafenib in patients aged ≥75 years was generally comparable to that in younger subgroups. We observed higher rates of grade 3 fatigue and grade ≥3 hypertension in patients aged ≥75 years compared with younger patients. These differences may be confounded by age, because older age is associated with increased risk of fatigue and hypertension. There were no marked differences in treatment duration or daily dose across age subgroups. This suggests that patients should not be considered unsuitable for treatment solely because of age.
Rates of grade 3 or 4 hepatic laboratory parameters were similar in CONSIGN and CORRECT (ALT: 6% [CONSIGN] and 5% [CORRECT]; AST: 7% and 6%; bilirubin: 13% and 12%). One case of regorafenib‐related severe drug‐induced liver injury in CONSIGN, which was identified based on DILI Working Group criteria [6], resolved after regorafenib discontinuation. One case of regorafenib‐related severe drug‐induced liver injury also occurred in CORRECT; in both cases, the event occurred in a patient with liver metastases [4]. In CONSIGN, the 13 regorafenib‐related grade 5 TEAEs included one colonic perforation and no gastrointestinal fistulas. Exploratory analyses showed that the frequency of regorafenib‐related TEAEs was highest during the first two cycles, consistent with CORRECT [13]. The early occurrence of TEAEs supports frequent monitoring and proactive management of TEAEs during initial treatment cycles [14].
Median PFS in CONSIGN, assessed by investigators according to best local practice standards, was in the range of that reported for regorafenib‐treated patients in phase III trials [4], [5] and was similar across patients with KRAS wild‐type and KRAS‐mutant tumors, consistent with prior reports [15], [16]. Estimated PFS rates at 6 and 9 months were 15% and 8%, respectively, because most patients had received at least three prior regimens for metastatic disease. Approximately one‐fifth of patients (21%) were treated for ≥6 cycles and smaller percentages were treated for ≥9 and ≥12 cycles, with a few patients receiving ≥24 cycles. Future work on biomarkers may help identify characteristics of patients able to receive extended treatment. CONSIGN was not designed to collect survival data. Exploratory analyses showed that the subgroup of patients with longer PFS tended to have a slightly higher proportion of patients with better performance status, no liver metastases, and a longer time since diagnosis of metastatic disease, compared with the subgroup with shorter PFS. These findings are consistent with the results of a cohort study of regorafenib, which suggested that factors including performance status, time from diagnosis of metastases, and liver involvement were associated with outcomes [17]. Limitations of CONSIGN include the lack of standardized PFS assessments across study centers, the lack of follow‐up for overall survival, and the absence of a comparator arm.
Conclusion
The results of this multicenter study of more than 2,800 patients show that the safety and efficacy profiles of regorafenib in patients with mCRC are consistent with results of the randomized, controlled, double‐blind phase III trials. The rates of dose reductions and interruptions highlight the importance of optimal patient selection, dose modifications, and adverse event management during regorafenib treatment.
See http://www.TheOncologist.com for supplemental material available online.
Acknowledgments
This study was sponsored by Bayer. Bayer provided the study medication and collaborated with the authors to design the study. Bayer worked with the investigators on the collection, analysis, and interpretation of the data, and on the preparation of this report. The authors made the final decision to submit the article for publication. Editorial assistance in the preparation of this manuscript was provided by Ann Contijoch (Bayer) and Jennifer Tobin (SuccinctChoice Medical Communications, London, UK, with financial support from Bayer). The authors thank the patients who participated in the CONSIGN trial, their families, and the CONSIGN investigators. A list of the CONSIGN investigators can be found in supplemental online Table 7. Stefano Cascinu is currently affiliated with the Department of Oncology/Hematology, Università di Modena e Reggio Emilia, Modena, Italy. Rocio Garcia‐Carbonero is currently affiliated with the Department of Medical Oncology, Hospital Universitario Doce de Octubre, IIS imas 12, CNIO, CIBERONC, Universidad Complutense de Madrid, Madrid, Spain.
Author Contributions
Conception/design: Eric Van Cutsem, Axel Grothey, Joachim Kalmus
Collection and/or assembly of data: Eric Van Cutsem, Erika Martinelli, Stefano Cascinu, Alberto Sobrero, Maria Banzi, Jean‐François Seitz, Carlo Barone, Marc Ychou, Marc Peeters, Baruch Brenner, Ralf Dieter Hofheinz, Evaristo Maiello, Thierry André, Andrea Spallanzani, Rocio Garcia‐Carbonero, Yull E. Arriaga, Udit Verma, Axel Grothey, Alfredo Falcone, Alberto Zaniboni
Data analysis and interpretation: Eric Van Cutsem, Marc Ychou, Rocio Garcia‐Carbonero, Yull E. Arriaga, Udit Verma, Axel Grothey, Christian Kappeler, Ashok Miriyala, Joachim Kalmus, Alfredo Falcone, Alberto Zaniboni
Manuscript writing: Eric Van Cutsem, Erika Martinelli, Stefano Cascinu, Alberto Sobrero, Maria Banzi, Jean‐François Seitz, Carlo Barone, Marc Ychou, Marc Peeters, Baruch Brenner, Ralf Dieter Hofheinz, Evaristo Maiello, Thierry André, Andrea Spallanzani, Rocio Garcia‐Carbonero, Yull E. Arriaga, Udit Verma, Axel Grothey, Christian Kappeler, Ashok Miriyala, Joachim Kalmus, Alfredo Falcone, Alberto Zaniboni
Final approval of manuscript: Eric Van Cutsem, Erika Martinelli, Stefano Cascinu, Alberto Sobrero, Maria Banzi, Jean‐François Seitz, Carlo Barone, Marc Ychou, Marc Peeters, Baruch Brenner, Ralf Dieter Hofheinz, Evaristo Maiello, Thierry André, Andrea Spallanzani, Rocio Garcia‐Carbonero, Yull E. Arriaga, Udit Verma, Axel Grothey, Christian Kappeler, Ashok Miriyala, Joachim Kalmus, Alfredo Falcone, Alberto Zaniboni
Disclosures
Eric Van Cutsem: Amgen, Bayer, Boehringer Ingelheim, Celgene, Ipsen, Eli Lilly & Co., Merck, Merck KGaA, Novartis, Roche, Sanofi, Servier (RF); Stefano Cascinu: Bayer (H); Alberto Sobrero: Bayer, Roche, Sanofi, Amgen, Bristol‐Myers Squibb, Merck (H); Jean‐François Seitz: Bayer, Roche, Sanofi, Amgen, Merck (H); Marc Ychou: Bayer (C/A, RF); Marc Peeters: Bayer (RF, H); Ralf Dieter Hofheinz: Bayer (H); Thierry André: Baxter, Roche, Servier, Bristol‐Myers Squibb, Amgen, Sanofi, XBiotech, GlaxoSmithKline, Eli Lilly & Co. (H); Rocio Garcia‐Carbonero: Bayer, Servier (RF, H) and her institution received grants from Bayer during the conduct of the study and other Bayer‐related studies; Yull E. Arriaga: Boston Biomedical, Genentech, AstraZeneca (H); Udit Verma: Bayer (RF); Axel Grothey: Bayer (C/A); Christian Kappeler: Bayer (E); Ashok Miriyala: Bayer (E, OI); Joachim Kalmus: Bayer (E, OI); Alfredo Falcone: Amgen, Bayer, Sanofi, Merck, Eli Lilly & Co., Roche, Servier (RF, H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Van Cutsem E, Cervantes A, Adam R et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386–1422. [DOI] [PubMed] [Google Scholar]
- 2.Network National Comprehensive Cancer. NCCN Clinical Practice Guidelines in Oncology: Colon Cancer. Version 2.2017. Fort Washington, PA: National Comprehensive Cancer Network; 2017. [DOI] [PubMed] [Google Scholar]
- 3.Wilhelm SM, Dumas J, Adnane L et al. Regorafenib (BAY 73‐4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011;129:245–255. [DOI] [PubMed] [Google Scholar]
- 4.Grothey A, Van Cutsem E, Sobrero A et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:303–312. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Qin S, Xu R et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2015;16:619–629. [DOI] [PubMed] [Google Scholar]
- 6.Aithal GP, Watkins PB, Andrade RJ et al. Case definition and phenotype standardization in drug‐induced liver injury. Clin Pharmacol Ther 2011;89:806–815. [DOI] [PubMed] [Google Scholar]
- 7.ClinicalTrials.gov . Regorafenib in metastatic colorectal cancer. https://clinicaltrials.gov/ct2/show/NCT02466009. ID: NCT02466009. Accessed August 8, 2016.
- 8.ClinicalTrials.gov. Lower or standard dose regorafenib in treating patients with refractory metastatic colorectal cancer. https://clinicaltrials.gov/ct2/show/NCT02368886. ID: NCT02368886. Accessed August 8, 2016.
- 9.Grothey A. Regorafenib in metastatic colorectal cancer: Optimal dosing and patient selection recommendations. Clin Adv Hematol Oncol 2015;13:514–517. [PubMed] [Google Scholar]
- 10.Tabchi S, Ghosn M. Regorafenib: Start low and go slow. Target Oncol 2015;10:445–447. [DOI] [PubMed] [Google Scholar]
- 11.Bekaii‐Saab TS, Ou FS, Anderson DM et al. Regorafenib dose optimization study (ReDOS): Randomized phase II trial to evaluate dosing strategies for regorafenib in refractory metastatic colorectal cancer (mCRC)—An ACCRU Network study. J Clin Oncol 2018;36 (suppl 4):611A. [Google Scholar]
- 12.Van Cutsem E, Ciardiello F, Seitz JF et al. Results from the large, open‐label phase 3b CONSIGN study of regorafenib in patients with previously treated metastatic colorectal cancer. Ann Oncol 2015;26 (suppl 4):LBA‐05. [Google Scholar]
- 13.Grothey A, Sobrero AF, Siena S et al. Time profile of adverse events (AEs) from regorafenib (REG) treatment for metastatic colorectal cancer (mCRC) in the phase III CORRECT study. J Clin Oncol 2013;31 (suppl 15):3637A. [Google Scholar]
- 14.De Wit M, Boers‐Doets CB, Saettini A et al. Prevention and management of adverse events related to regorafenib. Support Care Cancer 2014;22:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabernero J, Lenz HJ, Siena S et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: A retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol 2015;16:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teufel M, Kalmus J, Rutstein M et al. Analysis of biomarkers in circulating tumor DNA from the phase 3 CONCUR study of regorafenib in Asian patients with metastatic colorectal cancer (mCRC): Correlation with clinical outcome. Eur J Cancer 2015;51 (suppl 3):S332. [Google Scholar]
- 17.Adenis A, de la Fouchardiere C, Paule B et al. Survival, safety, and prognostic factors for outcome with regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: Results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer 2016;16:412. [DOI] [PMC free article] [PubMed] [Google Scholar]