

ABSTRACT
FSTL1 is a protein coding gene associated with cell signaling pathway regulation and the progression of a variety of disorders. In this study, we hypothesized that FSTL1 increases oncogenesis in breast cancer by enhancing stemness and chemoresistance. RT-PCR and IHC revealed significantly higher FSTL1 mRNA and protein levels in TNBC than in non-TNBC specimens and in breast cancer cell lines. We then found that FSTL1 levels were significantly increased in chemoresistant cells. LIVE/DEAD, MTT cell viability and colony formation assays did in fact demonstrate that FSTL1 is required for CDDP and DOX chemoresistance in breast cancer cell lines. FSTL1 overexpression caused significant elevation of stem cell biomarkers, as well as breast cancer cell proliferation. To determine whether the Wnt/β-catenin signaling pathway is involved in the observed effects of FSTL1, we assessed levels of pathway target. TOP/FOP flash, colony formation, and tumor sphere formation assays indicated that FSTL1 activates Wnt/β-catenin signaling through integrin β3. We then sought to identify a microRNA (miRNA) that regulates FSTL1 activity. Luciferase assays demonstrated that miR-137 reduces FSTL1 mRNA and protein levels. Ultimately, our findings indicate that there is an miR-137/FSTL1/integrin β3/Wnt/β-catenin signaling axis in breast cancer cells that regulates stemness and chemoresistance.
Keywords: FSTL1, chemoresistance, stemness, breast cancer, integrin β3, Wnt signaling, miR-137
Background
Breast cancer is the most prevalent cancer in women and the leading cause of cancer death in women worldwide.1 Since the 1990s, however, breast cancer death rates have been stable or declining in more developed nations due to earlier detection with mammography screening and improved treatment.2 There are a variety of breast cancer types that express tumor-associated antigens and hormone receptors. The development of targeted therapies and hormone therapy in particular account for major recent treatment advances.3 However, triple negative breast cancer (TNBC), which constitutes around 15% to 25% of patients, is a cluster of heterogeneous diseases that do not express the estrogen receptor, progesterone receptor, or human epidermal growth factor receptor 2 (HER2/neu).4 While there have been remarkable improvements in the treatment of a variety of breast cancers, successfully treating TNBC remains a challenge due to its aggressive nature, limited treatments options, and therapeutic resistance.5 There are currently no effective targeted antibody or hormone therapies for TNBC.6 The current standard of treatment for TNBC is surgery, radiotherapy, and chemotherapy. Because of the non-personalized nature of these therapeutic approaches, the greatest barrier to successful treatment is treatment resistance.7 In the present study, we have explored the role of Follistatin like 1 (FSTL1), a protein that has been shown to regulate breast cancer cell proliferation,8,9 in promoting the stemness and chemoresistance underlying the aggressive characteristics of TNBC.
Personalized breast cancer treatments are currently based on factors such as gene expression signatures, spread of disease, and recurrence.10-12 Patients with early stage breast cancer usually receive primary surgery at the initiating tumor site and at regional lymph nodes, with or without breast irradiation.13 They then may receive neoadjuvant systemic therapy (AST) based on individual tumor characteristics.14 Even though AST and targeted immune and hormone therapies have substantially improved treatment outcomes, there is a high risk of recurrence for patients diagnosed even in early stages.15 For TNBC, which is poorly differentiated and more prone to metastasis than other cancer types, platinum salts such as cisplatin (CDDP) are used in combination with systemic chemotherapeutics.16 Doxorubicin (DOX), an anthracycline antibiotic, is also used to treat metastatic and aggressive subtypes of breast cancer, either alone or combined with taxanes such as docetaxel.17-19 Because developing resistance to CDDP or DOX represents a serious threat to aggressive forms of breast cancer, especially TNBC, in this study we sought to identify a major gene and signaling pathway responsible for chemoresistance.
FSTL1 is a secreted follistatin-module-containing glycoprotein that was first identified as a TGF-β1-inducible gene and member of the Follistatin-SPARC family.20 Its interactions with cell signaling pathways are not limited to TGFβ, however, as a number of studies have since shown that FSTL1 engages in auto-regulatory feedback loops with several other factors, including bone morphogenic proteins (BMPs), matrix metalloproteinase-2 (MMP-2), and IL1β.21 FSTL1 also affects cell proliferation,22 apoptosis,23 and migration.24 However, it is not always clear whether FSTL1 positively or negatively regulates these cell processes, and the physiological or disease context matters greatly in determining the effects of FSTL1 activity.21 In 2017, Yang Yang et al. found that FSTL1 moderately impacts breast cancer cell and vascular endothelial cell proliferation.9 Also in 2017, Jiaqiang An et al. determined that FSTL1 inhibits cell proliferation of breast cancer cell line MDA-MB-231.8 These two studies offer potentially conflicting results that do not resolve the question of FSTL1’s effects on breast cancer aggressiveness. In the present study, we explored the role of FSTL1 in human breast cancer specimens and cell lines, ultimately finding an miR-137/FSTL1/integrin β3/Wnt/β-catenin signaling axis in breast cancer cells that regulates stemness and chemoresistance.
Materials and methods
Breast cancer samples
A total of 87 breast cancer patients, 51 with TNBC (triple negative breast cancer) and 36 with non-TNBC, were enrolled in this study. All patients attended Harbin Medical University Cancer Hospital, and diagnoses were histologically confirmed. Breast cancer tissue specimens were obtained from patients undergoing primary mastectomies at the institution. The tissues were examined diagnostically by pathologists. The samples were collected immediately, snap-frozen in liquid nitrogen, and stored at −80°C for analysis. All protocols were reviewed and approved by the Ethical Committee of Harbin Medical University. Informed and written consent was obtained from all participating patients.
Immunohistochemical staining
One representative section of the tissue was cut at 4 mm and placed on poly-L-lysine coated slides. The slides were deparaffinized, dehydrated, immersed in sodium citrate buffer (pH 6.0) or Tris-EDTA buffer (pH 9.0), pretreated in a microwave oven for 10 min, and rinsed with phosphate-buffered saline (PBS) for 10 minutes. After blocking with 3% hydrogen peroxide for 10 min at room temperature, the slides were incubated at 4°C overnight with primary anti-FSTL1 antibody (Santa Cruz Biotechnology, Santa Cruz, USA). The slides were then stained with the 2-step plus Poly-HRP anti-Rabbit IgG Detection System (ZSGB-Bio, Beijing, China). After visualization of the reaction with the DAB chromogen, the slides were counterstained with haematoxylin and covered with a glycerin gel. For negative controls, the primary antibody was substituted with PBS.
Cell culture, transfection, and treatment
Normal mammary epithelial cell line MCF-10A was maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) containing 5% horse serum (Life Technologies, Carlsbad, CA, USA), 20 ng/mL human epidermal growth factor (hEGF) (R&D Systems, Minneapolis, MN, USA), 0.5 mg/mL hydrocortisone (Sigma, St. Louis, MO, USA), 10 µg/mL insulin, 100 U/mL penicillin, and 100 U/mL streptomycin. Breast cancer cell lines (HCC38, MDA-MB-231, and MDA-MB-468) were cultured in DMEM with 10% heat-inactivated fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, 100 U/mL streptomycin, and 2 mmol/L L-glutamine. All cells were maintained at 37°C in a humidified 5% CO2 atmosphere cell incubator. For stemness analysis, breast cancer cells (4 × 104 cells/well) were seeded onto 6-well ultralow attachment plates (Corning, Corning, NY, USA) in serum-free DMEM supplying B27 supplement (Invitrogen, Carlsbad, CA, USA), 20 ng/mL human fibroblast growth factor-basic (hFGF) (R&D Systems), 20 ng/mL hEGF (R&D Systems), and heparin (Sigma). The tumor spheres were counted after 7 days.
FSTL1 cDNA was amplified from MDA-MB-231 mRNA by polymerase chain reaction (PCR) and cloned into the pcDNA3.1 vector to generate FSTL1 overexpression plasmids. Integrin β3 siRNA and control siRNA were purchased from OriGene (Beijing, China). Wnt/β-catenin signaling reporter assays were carried out in 24-well plates with 100 ng of TOP Flash, FOP Flash, and Renilla plasmid transfection. The dual-luciferase reporter assay system (Promega, Madison, WI, USA) was used to detect luciferase activity. Lipofectamine 2000 (Invitrogen) was used for plasmid and siRNA transfection according to the manufacturer’s instructions. Breast cancer cells were treated with different doses of cisplatin (CDDP) or doxorubicin (DOX) (Sigma) after transfection.
Real-time PCR (RT-PCR)
Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. RT-PCR assays were carried out in an ABI Prism 7900HT thermal cycler (Applied Biosystems, Foster City, CA, USA). RT-PCR amplification was performed in 20 μL reaction mixture containing 2 μL cDNA sample, 10 μL Quanti-Tect SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA), and specific primer sets. PCR began with a 15-min hot start at 95°C followed by 40 cycles of denaturation at 94°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 1 min. Dissociation curve analysis (95°C for 15 s, 60°C for 15 s, and 95°C for 15 s) was performed at the end of the 40 cycles to verify the PCR product identity. Data were analyzed using Sequence Detector Systems version 2.0 software (Applied Biosystems). Finally, relative gene expression levels were normalized to an internal reference gene (β-actin). The primers used are as follows: FSTL1 sense: 5ʹ-CCTGTGTGTGGCAGTAATGG-3ʹ, antisense: 5ʹ-TCAGGAGGGTTGAAAGATGG-3ʹ; CD133 sense: 5ʹ- CAGAGTACAACGCCAAACCA-3ʹ, antisense: 5ʹ-AAATCACGATGAGGGTCAGC-3ʹ; Nanog sense: 5ʹ-ACAACTGGCCGAAGAATAGC-3ʹ, antisense: 5ʹ-AGTGTTCCAGGAGTGGTTGC-3ʹ; SOX2 sense: 5ʹ-CGGTACCCGGGGATCCCCGCATGTACAACATGATGG-3ʹ, antisense: 5ʹ-CATAATGGCCGTCGACCACATGTGTGAGAGGGGCA-3ʹ; Integrin β3 sense: 5ʹ-GACTTTGGCAAGATCACGGG-3ʹ, antisense: 5ʹ-GCACATCTCCCCCTTGTAGC-3ʹ; β-catenin sense: 5ʹ-GCTGATTTGATGGAGTTGGA-3ʹ, antisense: 5ʹ-TCAGCTACTTGTTCTTGAGTGAA-3ʹ; myc sense: 5ʹ-TTGCAGCTGCTTAGACGCTG-3ʹ, antisense: 5ʹ-CCACATACAGTCCTGGATGA-3ʹ; cyclin D1 sense: 5ʹ-GGATGCTGGAGGTCTGCGAG-3ʹ, antisense: 5ʹ-GAGAGGAAGCGTGTGAGGCG-3ʹ; β-actin sense: 5ʹ-TTGCCGACAGGATGCAGAA-3ʹ, antisense: 5ʹ-GCCGATCCACACGGAGTACT-3ʹ.
Western blot
Total proteins of cultured cells were extracted using RIPA buffer at 4°C for 30 min. The cell lysates were centrifuged at 4°C for 10 min at 12,000 g to separate soluble proteins. Proteins were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% (w/v) non-fat powdered milk in Tris Buffered Saline (TBS) containing 0.1% Tween for 1 h, washed with TBS/Tween, incubated overnight at 4°C with antibodies against FSTL1, CD133, Nanog, SOX2, Integrin β3, β-catenin, myc, cyclin D1, or β-actin, and then incubated for 2 h with appropriate secondary antibodies. The primary antibodies were all from Santa Cruz Biotechnology. Signals were visualized by chemiluminescence (Beyotime, Beijing, China).
MTT assay
Breast cancer cells were grown on 96-well plates at an initial concentration of 5 × 103 cells/mL per well. 20 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/ml) were added to the growing medium of each well at days 0, 1, 2, 3, 4, 5, and 6. Cells were then incubated at 37 °C in the dark. After 4 h, the MTT solution was removed and replaced with 100 µL of dimethyl sulfoxide (DMSO). Absorbance values were determined at a wavelength of 590 nm in a microplate reader.
Cell cycle
Breast cancer cells were seeded at a density of 0.5 × 106 cells per well in 6-well plates and incubated overnight. The cells were harvested, washed with PBS and fixed with 70% ethanol for 1 h on ice. The cells were then resuspended in 0.2 mL of PI staining solution containing 100 μg/mL RNase A and 50 μg/mL PI at 37 °C in an incubator for 30 min after washing in PBS. A flow cytometer (BD Biosciences, Bedford, MA) was used for cell cycle analysis.LIVE/DEAD Cell Viability Assays
We performed cell viability assays using LIVE/DEAD Cell Viability kit (ThermoFisher, San José, CA) according to manufacturers’ instruction.
Colony formation assays
Breast cancer cells (500 per well) were plated onto each well of a 6-well plate and incubated at 37 °C for 2 weeks. The cells were then fixed with 4% paraformaldehyde and stained with 1% crystal violet. Colony numbers were then counted.
Statistics
All analyses were performed using the statistical software GraphPad Prism 5.0 (GraphPad Software, Inc.; La Jolla, CA). For experimental data, one-way analysis of variance (ANOVA) was performed for serial analysis, while two treatment groups were compared using the unpaired Student’s t-test. All experiments were performed at least three times. Data were expressed as mean ± standard deviation (SD). P values of < 0.05 were considered statistically significant in all analyses.
Results
FSTL1 expression is increased in TNBC specimens, cells, and drug-resistant TNBC cells
FSTL1 has been reported to play a pivotal role in breast cancer cell proliferation, angiogenesis, and migration.8,9 To explore the function of FSTL1 in breast cancer chemoresistance, we analyzed FSTL1 expression in 56 human TNBC and 21 human non-TNBC tissues. IHC showed stronger FSTL1 staining in TNBC than in non-TNBC specimens (Figure 1a). RT-PCR revealed higher FSTL1 mRNA expression in TNBC tissues than in non-TNBC tissues (Figure 1b). We further examined FSTL1 expression in three breast cancer cell lines (MDA-MB-231, MDA-MB-468, and HCC38). As compared to one normal mammary epithelial cell line (MCF-10A), both mRNA and protein levels in breast cancer cells were elevated (Figure 1c and d). We then generated CDDP or DOX resistant MDA-MB-231 and MDA-MB-468 cells. FSTL1 mRNA and protein levels were significantly increased in these chemoresistant cells (Figure 1e and f). Overall, these results suggest that FSTL1 contributes to TNBC by promoting chemoresistance.
Figure 1.
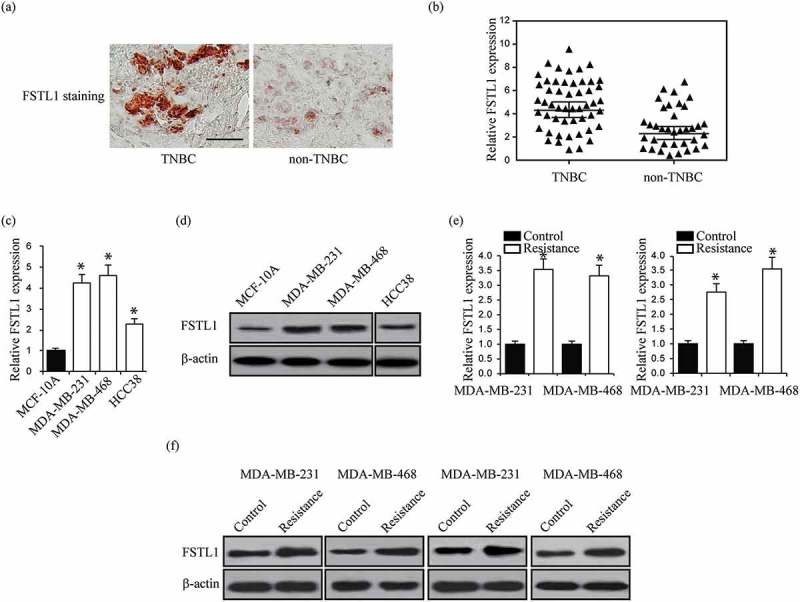
FSTL1 expression is increased in TNBC tissues, TNBC cells, and TNBC cells of multiple drug resistance. (a) Representative immunostaining for FSTL1 expression in TNBC and non-TNBC tissues. (b) Relative FSTL1 mRNA expression in TNBC and non-TNBC tissues. p < 0.001. (c, d) Relative FSTL1 mRNA (c) and protein (d) expression in one normal mammary epithelial cell line MCF-10A and four breast cancer cell lines MDA-MB-231, MDA-MB-468, and HCC38. * p < 0.05. (e, f) Relative FSTL1 mRNA (e) and protein (f) expression in CDDP or DOX resistant MDA-MB-231 and MDA-MB-468 cells analyzed by RT-PCR and western blot, respectively. * p < 0.05.
FSTL1 enhances multiple drug resistance in breast cancer cells
To test our hypothesis that FSTL1 plays a crucial role in breast cancer cell chemoresistance, we transfected MDA-MB-231, MDA-MB-468 and HCC38 cells with FSTL1 overexpression plasmid (Figure 2a). These cells were subjected to LIVE/DEAD cell viability assays under CDDP (5 μ m) and DOX (100 μ m) treatment. As shown in Figure 2b and c, more cells were induced death by CDDP and DOX. We then evaluated drug resistance by treating the cells with different doses of CDDP or DOX. MTT cell viability assays demonstrated that FSTL1 overexpression significantly increased cell viability under CDDP or DOX treatment in both MDA-MB-231, MDA-MB-468 and HCC38 cells (Figure 2c and d). Colony formation assays showed that FSTL1 overexpression also significantly increased colony formation in these treated cell lines (Figure 2d and e). We further knocked down FSTL1 expression in MDA-MB-231, MDA-MB-468 and their chemoresistant MDA-MB-231 and MDA-MB-468 cells (Supplemental Figure 1a). We treated these breast cancer cells with CDDP and DOX, and performed LIVE/DEAD and colony formation assays. The results showed that knockdown of FSTL1 significantly reduced cell chemoresistance (Supplemental Figure 1b and e). Taken together, these findings indicate that FSTL1 expression is required for breast cancer cell resistance to CDDP and DOX.
Figure 2.
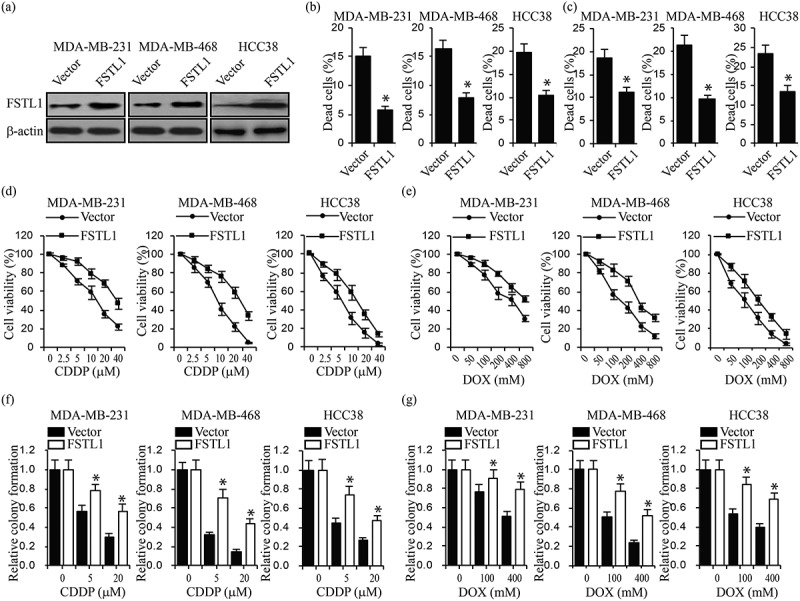
FSTL1 enhances multiple drug resistance in breast cancer cells. (a) FSTL1 protein expression in MDA-MB-231, MDA-MB-468 and HCC38 cells transfected with control vector or FSTL1 plasmids by western blot. (b, c) Cell LIVE/DEAD assays in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells treated with CDDP (b) or DOX (c), respectively. *p < 0.05. (d, e) Cell viability in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells treated with different doses of CDDP (d) or DOX (e) indicated, respectively. (f, g) Colony formation in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells treated with different doses of CDDP (f) or DOX (g) indicated, respectively. *p < 0.05.
FSTL1 augments breast cancer cell stemness
Because multiple drug resistance is a property of breast cancer stem cells, we hypothesized that FSTL1 expression increases breast cancer cell stemness. Sphere formation assays showed that upregulation of FSTL1 in MDA-MB-231, MDA-MB-468 and HCC38 cells led to a significant increase in sphere number (Figure 3a). To confirm that these increases were accompanied by increases in stem cell biomarkers, we analyzed CD133, Nanog, and SOX2 levels by RT-PCR and western blot. Indeed, FSTL1 overexpression significantly elevated expression levels of all stem cell biomarkers in both MDA-MB-231, MDA-MB-468 and HCC38 cells (Figure 3b). We performed cell cycle assays to analyze cell proliferation and self-renewal. As shown in Figure 3c, overexpression of FSTL1 significantly decreased cells in G0/G1 and increased cells in M phase (Figure 3c). Furthermore, MTT assays showed that upregulation of FSTL1 promotes breast cancer cell proliferation (Figure 3d). These findings support the hypothesis that FSTL1 enhances breast cancer cell stemness.
Figure 3.
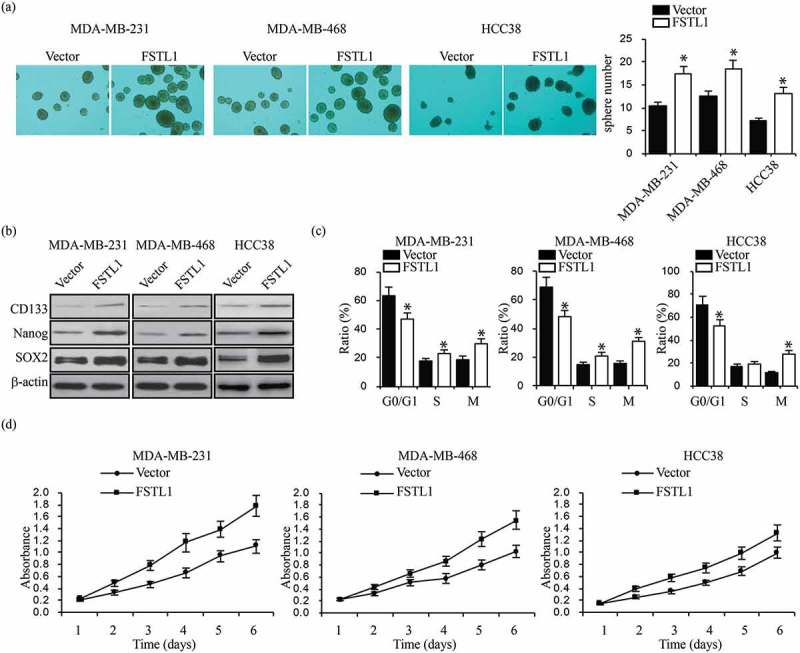
FSTL1 augments breast cancer cell stemness traits. (a) Tumor sphere formation assays in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells. Sphere numbers were quantified. *p < 0.05. (b) Protein expression of stem cell markers (CD133, Nanog, and SOX2) in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells. *p < 0.05. (c) Cell cycle analysis in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells. *p < 0.05. (d) MTT assays were performed to analyze cell proliferation in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells.
FSTL1 activates Wnt/β-catenin signaling through integrin β3
In previous studies, we observed that FSTL1 and Wnt/β-catenin signaling interact in lung development25,26 and that Wnt/β-catenin signaling regulates breast cancer stem cell development and chemoresistance.27 To determine whether a connection between FSTL1 and Wnt/β-catenin signaling exists in breast cancer cells, we first assessed Wnt/β-catenin signaling target gene expression levels. RT-PCR and western blot of β-catenin, myc, and cyclin D1 in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells demonstrated increased target gene expression as compared to control cells (Figure 4a and b). TOP-flash Wnt signaling luciferase activity assays confirmed that FSTL1 promotes Wnt/β-catenin signaling (Figure 4c). To further probe the effects of FSTL1 on the Wnt/β-catenin signaling pathway, we evaluated the functional relationship between FSTL1 and integrin β3. We knocked down integrin β3 in both MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. Western blot revealed that downregulation of integrin β3 did not affect FSTL1 expression (Figure 4d). TOP-flash, colony formation, and tumor sphere formation assays showed that overexpression of FSTL1 partially rescued Wnt signaling activity and proliferation, which are normally inhibited by downregulation of integrin β3 (Figure 4e–g). These results suggest that FSTL1 at least partially activates Wnt/β-catenin signaling through integrin β3.
Figure 4.
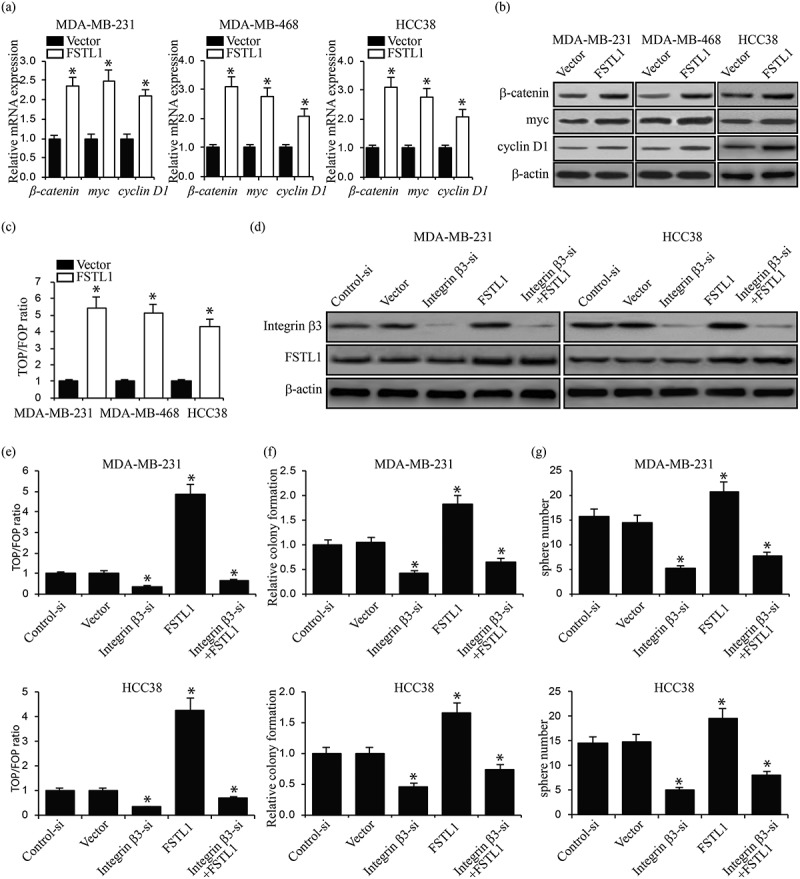
FSTL1 activates Wnt/β-catenin signaling through integrin β3. (a, b) mRNA (a) and protein (b) expression of Wnt/β-catenin signaling target genes (β-catenin, myc, and cyclin D1) in FSTL1 overexpression MDA-MB-231, MDA-MB-468 and HCC38 cells. *p < 0.05. (c) TOP/FOP ratios in FSTL1 overexpression MDA-MB-231, MDA-MB-468 cells and HCC38. *p < 0.05. (d) Western blots of integrin β3 and FSTL1 expression in integrin β3 knockdown MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. (e) TOP/FOP ratios in integrin β3 knockdown MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. *p < 0.05. (f) Relative colony formation in integrin β3 knockdown MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. *p < 0.05. (g) Number of tumor spheres in integrin β3 knockdown MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. *p < 0.05.
FSTL1 is targeted by mir-137
miRNAs regulate gene expression by binding their target mRNA 3ʹ-UTR regions, thereby causing direct changes in cellular functions. The target prediction algorithm TargetScan identified miR-137 as a potential regulator of FSTL1 (Figure 5a). Substantiating this prediction, past research has determined that miR-137 regulates breast cancer cell chemoresistance.28 To assess whether FSTL1 is targeted by miR-137, we generated wild-type and mutant luciferase report plasmids containing the FSTL1 3ʹ-UTR (Figure 5b). Luciferase activity was notably reduced after the cells were transfected with the wild-type plasmid, suggesting that miR-137 targets the FSTL1 3ʹ-UTR (Figure 5c). To further demonstrate the effects of miR-137 on FSTL1 expression, we first increased miR-137 levels in MDA-MB-231, MDA-MB-468 and HCC38 (Figure 5d). qRT-PCR and western blot assays then showed that miR-137 did indeed significantly decrease FSTL1 expression (Figure 5e and f). Overall, these results indicate that miR-137 targets FSTL1 and reduces its mRNA and protein levels.
Figure 5.
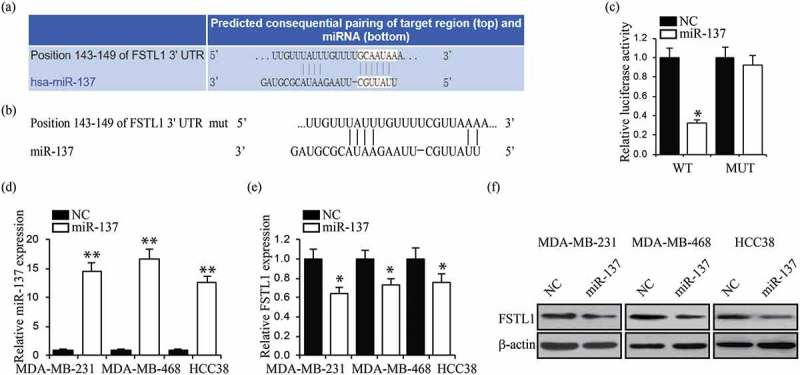
FSTL1 is targeted by miR-137. (a) Predicted binding site in the 3ʹUTR of the FSTL1 gene with miR-137. (b) Mutant of FSTL1 3ʹUTR reporter plasmid was constructed. (c) Luciferase reporter assays were performed in 293T cells with co-transfection of indicated wild-type or mutant 3ʹUTR FSTL1 constructs and miR-137 or NC (negative control). *p < 0.05. (d) Relative expressions of miR-137 in MDA-MB-231, MDA-MB-468 and HCC38 cells with miR-137 or NC transfection. **p < 0.01. (e, f) RT-PCR (e) and western blot (f) analyses of FSTL1 expression in MDA-MB-231, MDA-MB-468 and HCC38 cells with miR-137 or NC transfection. **p < 0.01.
FSTL1 is required for mir-137-regulated wnt/β-catenin signaling
To investigate whether miR-137 regulates Wnt/β-catenin signaling in breast cancer cell through FSTL1, we examined expression levels of downstream genes, including β-catenin, myc, and cyclin D1, after FSTL1 overexpression in miR-137 transfected breast cancer cells (Figure 6a). Western blot assays showed that FSTL1 overexpression rescued miR-137-induced decreases of Wnt/β-catenin signaling target gene expression (Figure 6b). Luciferase activity assays further demonstrated that FSTL1 prevents miR-137 inhibition of Wnt/β-catenin signaling (Figure 6c). The totality of these findings reveals the function of the miR-137/FSTL1/integrin β3/Wnt/β-catenin signaling axis in breast cancer cells.
Figure 6.
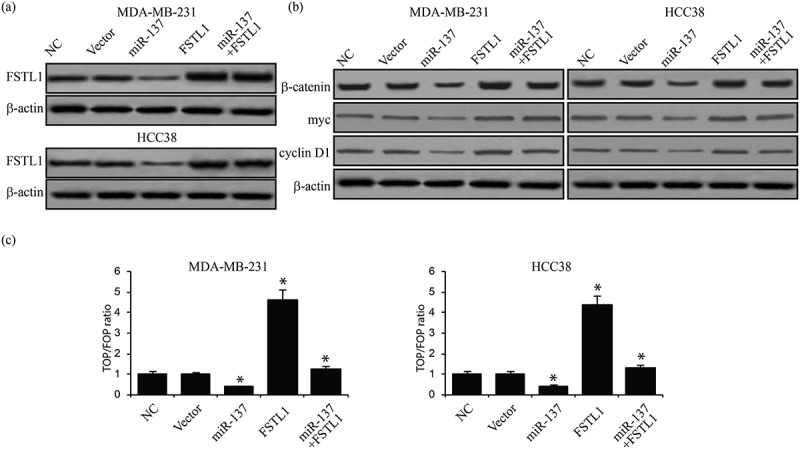
FSTL1 is required for miR-137-regulated Wnt/β-catenin signaling. (a) Western blot of FSTL1 expression in miR-137 transfected MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. (b) Western blot assays of Wnt/β-catenin signaling target gene expression in miR-137 transfected MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. *p < 0.05. (c) TOP/FOP ratios in miR-137 transfected MDA-MB-231 and HCC38 cells with or without FSTL1 overexpression. *p < 0.05.
Discussion
In this study, we have revealed a critical role for FSTL1 in breast cancer cell stemness and chemoresistance. We have also identified miR-137 as a regulator of the FSTL1/integrin β3/Wnt/β-catenin signaling axis. Using human TNBC specimens, cells, and drug-resistant TNBC cells, we began by demonstrating with RT-PCR, western blotting, and IHC that FSTL1 is overexpressed in TNBC specimens as compared to non-TNBC specimens and in breast cancer cell lines as compared to normal mammary epithelial cells. After then finding that FSTL1 is required for significant multiple drug resistance and breast cancer cell stemness traits, we used CDDP- and DOX-resistant breast cancer cell lines, TOP/FOP flash assays, tumor sphere formation assays, and western blotting to demonstrate that FSTL1 promotes oncogenic phenotypes through integrin β3-mediated Wnt/β-catenin signaling. Finally, we found that miR-137 regulates Wnt/β-catenin signaling in breast cancer cell through FSTL1.
Integrin β3 is a member of the integrin family of cell surface receptors. Like other integrins, it regulates molecular interactions across cell membranes, primarily between the cell cytoskeleton and extracellular matrix.29 These functions are critical for cell adhesion, mobility, and proliferation, among other cell processes crucial to oncogenesis.30 Integrin β3 recently became of interest for its role in regulating vasculature in the context of a variety of cancers, including breast, cervical, and pancreatic.31 It has also been shown to promote breast cancer cell invasion, migration, growth factor release, and the epithelial-mesenchymal transition (EMT).32-34 The present study builds on prior work into the complex relationship between integrin β3 and breast cancer oncogenesis by establishing that FSTL1 activates the Wnt/β-catenin signaling pathway through integrin β3. This finding is highly compelling because it opens the possibility for targeting FSTL1 or integrin β3 in a coordinated manner to suppress breast cancer cell oncogenesis.
The Wnt family is comprised of secreted glycoproteins that act as signaling molecules to control numerous cell developmental processes, including cell proliferation, migration, differentiation, and polarity.35 The Wnt/β-catenin signaling pathway acts through cell surface receptors like integrin β3 to effect intracellular transduction. When dysregulated, Wnt/β-catenin signaling can cause developmental defects and oncogenesis.36 In our previous studies, we demonstrated that FSTL1 and Wnt/β-catenin signaling interact in lung development25,26 and that Wnt/β-catenin signaling regulates breast cancer stem cell development and chemoresistance.27 In this study, we tested our hypothesis that FSTL1 promotes breast cancer cell stemness and chemoresistance by promoting Wnt/β-catenin signaling. Not only did we find that this hypothesis was correct, but we also determined that miR-137 inhibits this FSTL1/integrin β3/Wnt/β-catenin signaling axis.
miRNAs are a class of small (approximately 20–24 nucleotides) noncoding RNAs that post-transcriptionally regulate mRNA activity.37 binding the 3ʹUTRs of target mRNAs, they cause translational repression or message cleavage, thereby acting as the endogenous equivalents of short interfering RNAs (siRNAs).38 While miRNAs were initially recognized for their evolutionarily conserved roles in development and other crucial cell processes like stress responses,39 increasing evidence suggests their involvement in pathological processes, including oncogenesis40 and drug resistance.41 miRNAs, especially when dysregulated, have been implicated in the etiology of a variety of cancers, including breast cancer.42 They can even serve as diagnostic, prognostic, and treatment biomarkers.43 In the present study, we have determined for the first time that miR-137 negatively regulates FSTL1 activity, thereby suppressing the Wnt/β-catenin signaling pathway, breast cancer cell stemness, and multiple drug chemoresistance.
Prior to this study, miR-137 has been shown to play a role in a variety of cancers, largely as a negative regulator of oncogenesis. In 2012, Yuanyin Zhao et al. found that miR-137 reduces the breast cancer nuclear receptor estrogen-related receptor α (ERRα) and negatively regulates breast cancer cell proliferation and migration.44 Apart from its purported role in breast cancer, miR-137 induces differentiation of human glioblastoma-multiform derived stem cells and cell cycle arrest,45 targets Rho GTPase family member Cdc42 to inhibit colorectal cancer cell proliferation, invasion, and cell cycle progression,46 and suppresses lung cancer cell proliferation by targeting Cdc42 and Cdk6.47 In this study, we used the mRNA target prediction algorithm TargetScan to determine that miR-137 binds the 3ʹUTR of FSTL1. We then used luciferase assays with plasmids containing either the wild-type or mutated 3ʹUTR of FSTL1 to confirm that miR-137 reduces FSTL1 mRNA and protein levels. Our discovery of the miR-137/FSTL1/integrin β3/Wnt/β-catenin signaling axis in breast cancer cells has the potential to advance individualized therapies for aggressive breast cancers that currently lack targeted treatment regimens.
Funding Statement
The study was supported by Natural Science Foundation of Heilongjiang Province (ZD2016018) and National Natural Science Foundation of China (81673006).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A.. Global cancer statistics, 2012. CA: a Cancer Journal for Clinicians. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Lacey JV Jr., Devesa SS, Brinton LA. Recent trends in breast cancer incidence and mortality. Environ Mol Mutagen. 39;2002:82–88. [DOI] [PubMed] [Google Scholar]
- 3.Troxell ML, Long T, Hornick JL, Ambaye AB, Jensen KC. Comparison of estrogen and progesterone receptor antibody reagents using proficiency testing data. Arch Pathol Lab Med. 2017;141:1402–1412. doi: 10.5858/arpa.2016-0497-OA. [DOI] [PubMed] [Google Scholar]
- 4.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 5.Mancini P, Angeloni A, Risi E, Orsi E, Mezi S. Standard of care and promising new agents for triple negative metastatic breast cancer. Cancers (Basel). 2014;6:2187–2223. doi: 10.3390/cancers6042187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C, Kar S, Lai X, Cai W, Arfuso F, Sethi G, Lobie PE, Goh BC, Lim LHK, Hartman M, et al. Triple negative breast cancer in Asia: an insider’s view. Cancer Treat Rev. 2018;62:29–38. doi: 10.1016/j.ctrv.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 7.O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MHZ, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell M, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clinical. 2015;3:257–275. doi: 10.1016/j.bbacli.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An J, Wang L, Zhao Y, Hao Q, Zhang Y, Zhang J, Yang C, Liu L, Wang W, Fang D, et al. Effects of FSTL1 on cell proliferation in breast cancer cell line MDA‑MB‑231 and its brain metastatic variant MDA‑MB‑231‑BR. Oncology Reports. 2017;38:3001–3010. doi: 10.3892/or.2017.6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Mu T, Li T, Xie S, Zhou J, Liu M, Li D. Effects of FSTL1 on the proliferation and motility of breast cancer cells and vascular endothelial cells. Thoracic Cancer. 2017;8:606–612. doi: 10.1111/1759-7714.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anampa J, Makower D, Sparano JA. Progress in adjuvant chemotherapy for breast cancer: an overview. BMC Medicine. 2015;13:195. doi: 10.1186/s12916-015-0439-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. New England Journal of Medicine. 2009;360:790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- 12.Soliman H. Immunotherapy strategies in the treatment of breast cancer. Cancer Control. 2013;20:17–21. doi: 10.1177/107327481302000104. [DOI] [PubMed] [Google Scholar]
- 13.Fisher B, Anderson S, Bryant J, Margolese RG, Deutsch M, Fisher ER, Jeong JH, Wolmark N. Twenty-year follow-up of a randomized trial comparing total mastectomy, lumpectomy, and lumpectomy plus irradiation for the treatment of invasive breast cancer. 2009. doi: 10.1056/NEJMoa022152. [DOI] [PubMed] [Google Scholar]
- 14.Colleoni M, Sun Z, Price KN, Karlsson P, Forbes JF, Thürlimann B, Gianni L, Castiglione M, Gelber RD, Coates AS, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the international breast cancer study group trials I to V. Journal of Clinical Oncology. 2016;34:.927–935. doi: 10.1200/JCO2015623504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Assad L, Poniecka A, Hennessy B, Green M, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. 2016. doi: 10.1200/JCO2007106823. [DOI] [PubMed] [Google Scholar]
- 16.Sirohi B, Arnedos M, Popat S, Ashley S, Nerurkar A, Walsh G, Johnston S, Smith IE. Platinum-based chemotherapy in triple-negative breast cancer. Annals of Oncology. 2008;19:1847–1852. doi: 10.1093/annonc/mdn395. [DOI] [PubMed] [Google Scholar]
- 17.Tabchy A, Valero V, Vidaurre T, Lluch A, Gomez HL, Martin M, Qi Y, Barajas-Figueroa LJ, Souchon E, Coutant C, et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clinical Cancer Research. 2010;16:5351–5361. doi: 10.1158/1078-0432.CCR-10-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gianni L, Eiermann W, Semiglazov V, Lluch A, Tjulandin S, Zambetti M, Moliterni A, Vazquez F, Byakhov MJ, Lichinitser M, etal. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. The Lancet. 2010;375:377–384. [DOI] [PubMed] [Google Scholar]
- 19.Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, Kuzma CS, Pluard TJ, Somlo G, Port ER, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage ii to iii triple-negative breast cancer: CALGB 40603 (Alliance). 2016. doi: 10.1200/JCO2014570572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibanuma M, Mashimo J, Mita A, Kuroki T, Nose K. Cloning from a mouse osteoblastic cell line of a set of transforming‐growth‐factor‐β1‐regulated genes, one of which seems to encode a follistatin‐related polypeptide. The FEBS Journal. 1993;217:13–19. [DOI] [PubMed] [Google Scholar]
- 21.Sylva M, Moorman AF. van den Hoff MJ. Follistatin-like 1 in vertebrate development. Birth Defects Res C Embryo Today. 2013;99:61–69. doi: 10.1002/bdrc.21030. [DOI] [PubMed] [Google Scholar]
- 22.Wei K, Serpooshan V, Hurtado C, Diez-Cuñado M, Zhao M, Maruyama S, Zhu W, Fajardo G, Noseda M, Nakamura K, et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature. 2015;525:479. doi: 10.1038/525S9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouchi N, Asaumi Y, Ohashi K, Higuchi A, Sono-Romanelli S, Oshima Y, Walsh K. DIP2A functions as a FSTL1 receptor. J Biolo Chem. 2010;285:7127–7134. doi: 10.1074/jbc.M109.069468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundaram GM, Common JEA, Gopal FE, Srikanta S, Lakshman K, Lunny DP, Lim TC, Tanavde V, Lane EB, Sampath P. ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing. Nature. 2013;495:103–106. doi: 10.1038/nature11890. [DOI] [PubMed] [Google Scholar]
- 25.Geng Y, Dong Y, Yu M, Zhang L, Yan X, Sun J, Qiao L, Geng H, Nakajima M, Furuichi T, et al. Follistatin-like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7058–7063. doi: 10.1073/pnas.1007293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Liu Y, Li X, Zhao J, Geng Y, Ning W. Follistatin like-1 (Fstl1) is required for the normal formation of lung airway and vascular smooth muscle at birth. PloS one. 2017;12:e0177899. doi: 10.1371/journal.pone.0177899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Wang H, Li C, Zhao Y, Wu L, Du X, Han Z. VEGF-A/neuropilin 1 pathway confers cancer stemness via activating wnt/beta-catenin axis in breast cancer cells. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2017;44:1251–1262. doi: 10.1159/000485455. [DOI] [PubMed] [Google Scholar]
- 28.Zhu X, Li Y, Shen H, Li H, Long L, Hui L, Xu W. miR-137 restoration sensitizes multidrug-resistant MCF-7/ADM cells to anticancer agents by targeting YB-1. Acta biochimica et biophysica Sinica. 2013;45:80–86. doi: 10.1093/abbs/gms099. [DOI] [PubMed] [Google Scholar]
- 29.Barczyk M, Carracedo S, Gullberg D. Integrins. Cell and Tissue Research. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 110;2002:673–687. [DOI] [PubMed] [Google Scholar]
- 31.Sheldrake HM, Patterson LH. Function and antagonism of beta3 integrins in the development of cancer therapy. Current Cancer Drug Targets. 9;2009:519–540. [DOI] [PubMed] [Google Scholar]
- 32.Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Research: BCR. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takayama S, Ishii S, Ikeda T, Masamura S, Doi M, Kitajima M. The relationship between bone metastasis from human breast cancer and integrin alpha(v)beta3 expression. Anticancer Research. 25;2005:79–83. [PubMed] [Google Scholar]
- 34.Wang R, Li ZQ, Han X, Li BL, Mi XY, Sun LM, Song M, Han YC, Zhao Y, Wang EH. Integrin beta3 and its ligand regulate the expression of uPA through p38 MAPK in breast cancer. APMIS: Acta Pathologica, Microbiologica, Et Immunologica Scandinavica. 2010;118:909–917. doi: 10.1111/j.1600-0463.2010.02687.x. [DOI] [PubMed] [Google Scholar]
- 35.Kikuchi A, Yamamoto H, Sato A, Matsumoto S. New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011;291:21–71. doi: 10.1016/B978-0-12-386035-4.00002-1. [DOI] [PubMed] [Google Scholar]
- 36.Miller JR, Hocking AM, Brown JD, Moon RT. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene. 1999;18:7860–7872. doi: 10.1038/sj.onc.1203245. [DOI] [PubMed] [Google Scholar]
- 37.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 38.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 113;2003:673–676. [DOI] [PubMed] [Google Scholar]
- 39.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jansson MD. Biotech research and innovation centre and centre for epigenetics UoC, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark, Lund AH, biotech research and innovation centre and centre for epigenetics UoC, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark. MicroRNA and Cancer. Molecular Oncology. 2012;6:590–610. doi: 10.1016/j.molonc.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL, Ji M-H, Hu Q, Luo Z, Wu J-Z, et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One. 2014;9:e95240. doi: 10.1371/journal.pone.0095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 43.Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010;101:2087–2092. doi: 10.1111/j.1349-7006.2010.01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Li Y, Lou G, Zhao L, Xu Z, Zhang Y, He F, Vanacker J-M. MiR-137 targets estrogen-related receptor alpha and impairs the proliferative and migratory capacity of breast cancer cells. PLoS One. 2012;7:e39102. doi: 10.1371/journal.pone.0039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M, Vandenberg SR, Ginzinger DG, James CD, Costello JF, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Medicine. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu M, Lang N, Qiu M, Xu F, Li Q, Tang Q, Chen J, Chen X, Zhang S, Liu Z, et al. miR-137 targets Cdc42 expression, induces cell cycle G1 arrest and inhibits invasion in colorectal cancer cells. Int J Cancer. 2011;128:1269–1279. doi: 10.1002/ijc.25452. [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Li Y, Shen H, Li H, Long L, Hui L, Xu W. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 2013;587:73–81. doi: 10.1016/j.febslet.2012.11.004. [DOI] [PubMed] [Google Scholar]