

Abstract
The discovery of functionally biased and physiologically beneficial ligands directed toward G-protein coupled receptors (GPCRs) has provided the impetus to design dopamine D2 receptor (D2R) targeted molecules that may be therapeutically advantageous for the treatment of certain neuropsychiatric or basal ganglia related disorders. Here we describe the synthesis of a novel series of D2R agonists linking the D2R unbiased agonist sumanirole with privileged secondary molecular fragments. The resulting ligands demonstrate improved D2R affinity and selectivity over sumanirole. Extensive in vitro functional studies and bias factor analysis led to the identification of a novel class of highly potent Go-protein biased full D2R agonists with more than 10-fold and 1000-fold bias selectivity toward activation of specific G-protein subtypes and β-arrestin, respectively. Intracellular electrophysiological recordings from midbrain dopamine neurons demonstrated that Go-protein selective agonists can elicit prolonged ligand-induced GIRK activity via D2Rs, which may be beneficial in the treatment of dyskinesias associated with dopamine system dysfunction.
Keywords: G protein-coupled receptors, D2R biased agonism, structure−activity relationships, bioluminescence resonance energy transfer (BRET), bias factor analysis, brain slice electrophysiology
Introduction
Dopamine receptors are classified in two subfamilies: D1-like receptors (D1R and D5R) evoking their physiological effects via activation of stimulatory Gs/olf heterotrimeric proteins and the D2-like receptors (D2R, D3R, and D4R) inducing their inhibitory responses by coupling with Gi/o/z proteins and leading decreased cAMP accumulation.1 Dopamine (DA) is the endogenous catecholamine neurotransmitter, and several neuropsychiatric and neurologic disorders are associated with hyper- or hypo-activation of dopaminergic pathways in the central nervous system.2−4 Indeed, multiple pharmacotherapeutic approaches have targeted the D2R subtypes to ameliorate symptoms associated with these disorders.5−7 Canonical agonists and partial agonists preferentially activate the D2R and D3R subtypes by binding to the orthosteric binding site (OBS, the same binding site where DA binds), and have been used as pharmacological tools as well as candidates in clinical trials for the treatment of Parkinson’s disease (PD), schizophrenia, and restless legs syndrome (RLS), with varying degrees of success.8−13
In the past few years, there has been a growing interest in developing small molecules targeting the allosteric binding site (ABS) or a secondary binding pocket (SBP)—a site topographically distinct from the OBS, but able to modulate affinity and/or intrinsic activity of orthosteric ligands within specific G-protein coupled receptor (GPCR) subfamilies.14−17 More recently, bivalent or bitopic ligands, that is, molecules in which primary/orthosteric and secondary pharmacophores (SP) are linked together, have emerged as a new approach to discover high affinity and subtype selective drugs.14,18,19 By simultaneously engaging both the OBS and the SBP, these molecules combine increased selectivity resulting from the interaction of a SP with the SBP, and high affinity associated with structurally well-defined primary pharmacophores (PP) targeting the OBS.20 In particular, this approach has been extremely successful in creating novel generations of D3R selective antagonists with potencies and subtype selectivities never observed before.19,21−23
Recent investigations have provided data revealing how GPCRs can signal not only via the canonical G-protein mediated activation pathways, but also by engaging specific proteins, most prominently, β-arrestins, that can activate their own G-protein independent signaling cascades leading to unique physiological outcomes.24−27 Several bitopic ligands, targeting different GPCR families, have been identified to demonstrate biased signaling (functionally selective), defined as the ability to fully activate one specific signaling pathway while displaying limited activation of other pathway(s) for the same receptor.28,29 This might be a consequence of these ligands stabilizing specific receptor conformations, thereby facilitating or limiting the recruitment and activation of either G-proteins or β-arrestins.30
Over the past decade, several research groups have directed efforts toward the synthesis of novel selective D2R biased agonists. In particular, multiple lead molecules have been identified as selective full or partial agonists for D2R-mediated β-arrestin recruitment, potentially providing the tools to dissect the mechanistic role of β-arrestins in dopaminergic receptors involved in schizophrenia as well as PD.31−38 However, to achieve a more comprehensive understanding of D2R cellular pharmacology and signaling physiology, highly selective G-protein biased D2R agonists are needed for comparison with the β-arrestin biased agonists.39−41 Indeed, the focus of D2R drug development research, has recently expanded toward the identification of D2R agonists capable of inducing selective activation of the Gi/o proteins with consequent inhibition of cAMP accumulation, without recruiting β-arrestins.31,42−44 The development of such ligands would greatly facilitate our understanding of which pathways are responsible for positive therapeutic responses and which ones are associated with undesired “off target” side effects.45−47 Here we report a bitopic molecular approach to further explore D2R agonist SAR43,48 based on a lead compound, 1 (Figure 1) and the discovery of novel and highly G-protein biased agonists with physiological significance.
Figure 1.

Lead and reference compounds that inspired new SAR studies. Molecular bitopic approach used to design new D2 agonists or partial agonists: benzothiazole or 2-methylindoline secondary pharmacophores inspired by the positive allosteric modulator 2;16 sumanirole primary pharmacophore or its simplified pharmacophore; classic n-butylamide linker or its trans-cyclopropyl constrained analogue.
Results
Design and Synthesis of Novel D2R Bitopic Agonists
We previously identified 1(43) (Figure 1) as a novel G-protein biased D2R agonist, based on the functionally unbiased or balanced D2R agonist, sumanirole ((R)-5,6-dihydro-5-(methylamino)-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one). To further explore the structural requirements of G-protein bias and potentially improve our lead molecule we embarked on synthetic strategies to (i) replace the indole-2-carboxamide SP of 1 with a new set of secondary aromatic pharmacophores, inspired by a recently published D2-like positive allosteric modulator (PAM);16 (ii) simplify the sumanirole PP by removal of the imidazolinone ring to confirm the importance of the N-1 position for D2R selectivity and binding pose in OBS;48 and (iii) modify the classic n-butylamide linker chain by introducing a cyclopropyl ring conferring rigidity and chirality (Figure 1).49 The most promising novel compounds presented multiple chiral centers, requiring the preparation of their respective diastereoisomers or enantiomers, allowing a better understanding of the geometrical orientations necessary for optimal interaction with the D2R binding site.
The choice of PPs, linkers, and SPs of this new library of compounds was aimed to combine structural fragments inspired by previously characterized D2R ligands presenting unique pharmacological properties (Figure 1). Synthetic schemes, structural and purity characterization of all the new compounds are described in detail in the Supporting Information. In addition, sumanirole,43,48434 (β-arrestin D2R biased partial agonist), and 2(16) (D2R/D3R PAM) were synthesized following previously reported procedures and tested as references in our biological assays. Diastereomeric ratios, as well as diastereomeric purities, were determined for selected lead compounds by chiral high-performance liquid chromatography (HPLC) (chromatograms reported in supplementary Figures S1–S6; detailed HPLC analytical methods are described in the experimental section and Supporting Information).
Radioligand Binding Studies Identify D2R Agonists with High Affinity
The affinity (Ki) values of sumanirole and its novel analogues, as well as the D2-like PAM 2 were determined using the agonist [3H]-(R)-(+)-7-OH-DPAT (Table 1).43,48,50 We previously established that D2-like receptor agonists more readily compete against a radiolabeled agonist, allowing the accurate evaluation of the affinity for the receptors’ active state.48 Sumanirole (Ki = 80.6 nM) was moderately selective over the D3R (D3R/D2R = 10) in agreement with previous experimental data.43,48 Compound 2, being selective for the ABS, showed effectively no competition for the OBS labeled with [3H]-(R)-(+)-7-OH-DPAT (Ki = >100 000 nM and 14 400 nM for D2R and D3R, respectively). This lack of competition for the OBS, consistent with the previous results,16 was also confirmed via sumanirole dose–response affinity curves in the presence of 1 μM or 10 μM of 2. Indeed, when tested in combination with 2, sumanirole did not show any major difference in its affinity and selectivity profiles for either D2R or D3R. The lack of D2R or D3R specific radioligands for the ABS, precludes the use of competition binding assays for PAM affinities. Thus, detailed cell based functional studies were previously described.16
Table 1. Radioligand Competition Binding Data.



Equilibrium dissociation constants (Ki) were derived from IC50 values using the Cheng–Prusoff equation. Each Ki value represents the arithmetic mean ± SEM of at least three independent experiments, each performed in duplicate or triplicate.
Compounds synthesized following the previously reported procedures34 and tested using the binding protocols describe in the experimental section.
Data previously reported.43
For comparison, we also synthesized 4 and 5, previously described D2R β-arrestin biased agonists.34 Given their bitopic structures, the presence of the canonical 2,3-dichlorophenyl piperazine as the PP, and of the 5-substituted benzothiazole (same synthon of 2, but different in its regiochemistry) as the SP, it was of interest to test them in parallel with our compounds and study their binding profiles. Also, as both 4 and 5 were previously tested in competition with the antagonist [3H]-N-methylspiperone, we sought to further assess their binding affinities using the agonist [3H]-(R)-(+)-7-OH-DPAT. In agreement with the original literature34 neither of the two compounds were D2R selective, with 5 presenting a slightly longer butyl chain and showing ∼2-fold higher affinity for D2R (D3R/D2R = 4.85) compared to 4, consistent with our observations of optimal linker length for D2R affinity and selectivity.
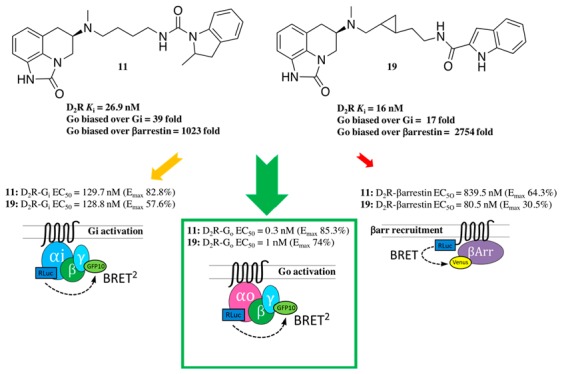
The parent molecule, 1, for this series of compounds is included in Table 1 for comparison. Replacing the terminal indole amide of 1 with the regioisomeric benzothiazoles from 2 results in 8 and 9, the affinities of which at D2R and D3R, were comparably decreased (∼2–5-fold) compared to 1, likely due to the indole amide being a privileged structure.14,21,43 When the racemic 2-methylindoline amide from 2 was inserted as the SP (10) via a slightly shorter butylamide linker, lower D2R and D3R affinities of 262 nM and 3340 nM resulted. However, by increasing the chain length via a butyl-urea-like linker (11), D2R affinity was rescued (D2R Ki = 26.9 nM). As 11 is a racemate, we synthesized its enantiomers. The (R)- configuration of the sumanirole ring is known to be essential for its affinity; however, much less is known about spatial requirements necessary for the SP. Chiral resolution of the racemic 2-methylindoline scaffold showed the R-methyl enantiomer of the 2-methylindoline ((R,R)-13) had higher D2R affinity compared to (R,S)-12, (D2R Ki = 21.6 and 51.4 nM, respectively). Moreover (R,R)-13 showed the highest D2R selectivity over D3R among this series (D3R/D2R = 14). Of note, the finding that the 2R-methylindoline scaffold was the higher affinity enantiomer agreed with the previous findings showing that the (R)-enantiomer of 2 is the eutomer for its positive allosteric modulation properties.16
Deconstructing the tricyclic scaffold of sumanirole by removing the imidazolinone ring to give the 1,2,3,4-tetrahydroquinoline PP (14, 15, and 16) maintained similar D3R binding with respect to their sumanirole bitopic counterparts (21, 9, and 11, respectively), but lost D2R affinities, with Ki values ranging from 232 to 466 nM. The decrease in D2R affinity is consistent with the previous observation that the sumanirole imidazolinone N-1 (Figure 1) interaction with Ser5.42 in D2R OBS is important to achieve the most favorable binding pose, leading to increased affinity and subtype selectivity.48 Thus, the removal of the imidazolinone ring negatively impacted binding of these simplified analogues. The only exceptions were compounds 17 and 18. The former included the n-propyl substituent on the same nitrogen as the 2-indolebutylamide linker, and the latter was dialkylated with two 2-benzothiazolebutylamide linkers at both available nitrogens. Compound 17 showed affinities overlapping with its homologue 22 and preferential D3R selectivity (Ki = 9.37 nM and 1.34 nM for D2R and D3R, respectively). In the case of 18, the secondary benzothiazole pharmacophore appended in position N-1 of the tetrahydroquinoline ring is responsible for the similar or higher D2R affinity (Ki = 45.7 nM) when compared to its related monosubstituted bitopic analogue 15.
Finally, in the last series, the two trans-cyclopropyl analogues, 19 and 20, were synthesized as inspired by the D3R selective antagonist 3 (Figure 1)22 in addition to sharing the same 2-carboxylindoleamide SP with 1. Compound 19, bearing the trans-cyclopropyl ring one methylene unit distant from the PP and two from the SP demonstrated the highest D2R affinity among this new series of compounds (D2R Ki = 16 nM), and its D2R Ki value was about 5-fold lower than that of sumanirole.
Sumanirole and selected compounds, 1, 11, (R,S)-12, (R,R)-13, 19, and 20 were also tested for their binding affinities on D4R (Table S3), D1R, and serotonin 5-HT1A, 5-HT2A, and 5-HT2C receptors (Tables S4–S6). Although some off-target activities were observed, all of these analogues were D2R preferential.
Cellular Functional Assays Using BRET Reveal G-Protein Bias
With the aim of identifying a new generation of potent G-protein biased agonists, compounds with promising binding profiles were further evaluated with four different D2R bioluminescence resonance energy transfer (BRET) functional assays to comprehensively evaluate activation of specific G-protein signaling pathways: (i) D2SR Gi1-protein activation, (ii) D2SR GoA-protein activation, (iii) cAMP accumulation inhibition by D2SR, and (iv) D2SR β-arrestin recruitment. To further validate our experimental observations, bias factors40,51−54 were calculated to integrate both potencies and efficacies in evaluating the bias for G-protein/cAMP signaling versus β-arrestin recruitment, and for Gi versus Go protein activation. The evaluation of bias factors among G-protein subfamilies, despite being observed before for other GPCRs,55,56 has not been fully explored for the D2-like receptors. On the basis of the binding results, N-5 sumanirole compounds with D2R affinities <50 nM and D3R/D2R selectivity >2 were selected for functional testing. Two major subtypes of Gi-like proteins (Gi1 and GoA)57,58 were tested for D2R-mediated G protein activation. To correlate their function at the immediate downstream, inhibition of adenylyl cyclase, thus inhibition of cAMP production, was measured using CAMYEL biosensor.59 Finally, a BRET-based β-arrestin 2 recruitment assay was used to detect ligand-induced D2R- β-arrestin 2 interaction. All the assays were measured at 10 and 30 min after the ligand addition. We have focused on interpretation of 30 min results across the four assays since the β-arrestin 2 recruitment assay does not yield quantifiable signals until 30 min even with GRK2 coexpression.43 All dose response plots were fitted with monophasic sigmoidal curves (supplementary Figure S7). Comparing results at 30 min allows the temporal interpretation to be consistent among different assays and avoids confounds resulting from data collected at variable time points.51
Quinpirole was used as a reference agonist for D2R activity since it confers efficacy and potency as balanced as dopamine in cAMP inhibition and β-arrestin 2 recruitment assays without non-D2R off-target contribution (e.g., adrenaline receptors).43,50 Sumanirole, displayed similar efficacy levels in the four assays tested, albeit with potency levels slightly lower than those of quinpirole. Compound 2,16 as expected, demonstrated low efficacy when tested alone in the Gi1 activation and β-arrestin 2 recruitment assays. As described in the chemistry section, 2 comprises two moieties (i.e., benzothiazole and methylindoline). The position 5-substituted benzthiazole analogue 8 was fully efficacious and more potent than sumanirole, with an EC50 = 143.9 nM in the Gi activation assay, whereas benzothiazole substitution at position 2 (9) resulted in a somewhat lower EC50 = 300.6 nM, but approximately 3-times more potent than sumanirole (Table 2). Methylindoline substitution with a butylamide linker at N-5 gave the diastereomeric mixture 11 and its enantiomers (R,S)-12 and (R,R)-13. All three compounds showed markedly lowered EC50 values for activation of both Gi1 and GoA proteins while maintaining a similar level of EC50 for β-arrestin 2 recruitment compared to sumanirole. Compared to 1, the cyclopropyl linked analogue 19 showed a much higher EC50 for Gi1 activation while maintaining similar profiles for the other assays. Compound 20, on the other hand, showed higher EC50 values for both Gi1 and GoA activation as well as cAMP inhibition.
Table 2. D2-Mediated Gi and Go Activation, cAMP Inhibition, and β-Arrestin2 Recruitmenta.
| quinpirole | sumanirole | 1 | 2 | 8 | 9 | 11 | 12 | 13 | 19 | 20 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gi1 Activation | |||||||||||
| Emax (%) | 100.0 ± 8.4 | 95.6 ± 7.7 | 51.0 ± 4.4 | 8.1 ± 8.2 | 107.6 ± 7.3 | 85.1 ± 8.6 | 82.8 ± 6.9 | 79.0 ± 5.9 | 71.6 ± 7.9 | 57.6 ± 4.9 | 72.4 ± 4.6 |
| EC50 (nM) | 111.4 ± 48.6 | 922.6 ± 332.4 | 8.1 ± 4.1 | 39.7 ± 39.7 | 143.9 ± 52.6 | 300.6 ± 147.1 | 129.7 ± 55.8 | 303.4 ± 118.0 | 95.7 ± 50.8 | 128.8 ± 56.3 | 184.1 ± 63.6 |
| GoA Activation | |||||||||||
| Emax (%) | 100.0 ± 5.8 | 97.1 ± 4.5 | 71.0 ± 4.5 | 31.9 ± 7.0 | 92.1 ± 7.2 | 75.5 ± 7.8 | 85.3 ± 5.3 | 72.2 ± 6.0 | 78.9 ± 6.7 | 74.0 ± 5.4 | 80.3 ± 3.8 |
| EC50 (nM) | 34.3 ± 12.7 | 89.7 ± 24.6 | 0.8 ± 0.4 | 61.1 ± 48.8 | 7.3 ± 3.6 | 117.0 ± 59.0 | 0.3 ± 0.1 | 0.7 ± 0.4 | 0.9 ± 0.5 | 1.0 ± 0.5 | 102.8 ± 28.2 |
| cAMP Inhibition | |||||||||||
| Emax (%) | 100.0 ± 5.6 | 87.9 ± 1.4 | 70.9 ± 2.8 | 58.5 ± 30.2 | 95.4 ± 4.7 | 84.0 ± 6.2 | 77.1 ± 7.5 | 92.8 ± 6.5 | 86.1 ± 6.0 | 60.5 ± 1.5 | 59.8 ± 1.5 |
| EC50 (nM) | 14.4 ± 5.2 | 35.1 ± 4.2 | 6.8 ± 1.9 | 22.9 ± 19.0 | 5.2 ± 1.8 | 79.1 ± 31.7 | 61.7 ± 31.1 | 72.4 ± 28.3 | 40.1 ± 16.8 | 6.0 ± 1.1 | 81.1 ± 13.1 |
| βArrestin2 Recruitment | |||||||||||
| Emax (%) | 100.0 ± 23.2 | 91.0 ± 3.8b | 66.4 ± 1.8b | NA | 76.4 ± 20.1 | 64.2 ± 25.6 | 64.3 ± 24.6 | 65.8 ± 19.5 | 70.2 ± 21.3 | 30.5 ± 2.6 | 26.0 ± 2.1 |
| EC50 (nM) | 245.4 ± 193.6 | 501.2 ± 148.5b | 154.5 ± 30.7b | NA | 208.4 ± 172.9 | 794.3 ± 711.4 | 839.5 ± 742.1 | 599.8 ± 497.3 | 674.5 ± 558.7 | 80.5 ± 35.7 | 207.5 ± 85.8 |
Potency (expressed as EC50) and efficacy values (%, normalized to quinpirole Emax) at 30 min for hD2R expressed in HEK293 cells. The values represent the arithmetic mean ± SEM of at least three independent experiments, each performed in triplicate.
Data previously reported.43
Bias Factor Comparison between cAMP and β-Arrestin
Next, pairwise analysis was made among different assays by employing the operational model of bias factor calculation.51−53 First, cAMP inhibition, the major signaling event of Gi-coupled D2R, was compared to the recruitment of β-arrestin-2 (Table 3). While all of the N-5 sumanirole bitopic analogues showed a modest bias toward cAMP inhibition (>0.5 in log scale), 1, 8, 19, and 20 showed a particularly pronounced bias toward cAMP inhibition (>1.0 in log scale).
Table 3. Bias Factors in Logarithmic Scale for D2-Mediated Gi vs Go Activation, cAMP Inhibition vs β-Arrestin2 Recruitment, and Go Activation vs β-Arrestin2 Recruitmenta.
| compared parameters | quinpirole | sumanirole | 1 | 8 | 9 | 11 | 12 | 13 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|---|---|
| Gi1–GoA activation | 0.00 | –0.54 | –0.98 | –0.44 | 0.43 | -1.59 | -1.18 | -1.13 | -1.24 | 0.24 |
| cAMP−βarrestin | 0.00 | –0.10 | 2.04 | 1.05 | 0.56 | 0.60 | 0.73 | 0.66 | 1.93 | 1.39 |
| GoA−βarrestin | 0.00 | 0.15 | 1.51 | 1.21 | 0.55 | 3.01 | 2.01 | 2.15 | 3.44 | 2.50 |
Bias Factor Comparison between Gi1 and GoA
As cAMP inhibition likely reflects the activation of all the Gi-like proteins, we specifically compared the activations of the two most expressed Gi subtypes in the brain, Gi1 and GoA. Notably, most of the N-5 analogues including sumanirole showed modest bias (<−0.5 in log scale) toward GoA over Gi1 activation. In particular, 11, 12, 13, and 19 showed a marked (<−1.0 in log scale) GoA bias suggesting the SP’s added effect toward GoA activation.
Bias Factor Comparison between GoA and β-Arrestin
From a receptor conformation stand point, structural mechanisms for ligand-induced G protein activation vs β-arrestin recruitment bias have been intensively investigated.38,60 D2R-mediated GoA activation has been implicated in GIRK activation due to the extensive colocalization of GoA and D2R expression in relevant brain areas.58,61 Therefore, we next compared GoA activation and β-arrestin 2 recruitment and analyzed the bias factors between the two. In this analysis, most of the N-5 analogues showed a marked (>1.0 in log scale) GoA activation bias over β-arrestin 2 recruitment. These bias factors are higher than those of cAMP inhibition vs β-arrestin 2 recruitment, likely because the bias factors incorporate Gi1 vs GoA. Remarkably, 11, 12, 13, 19, and 20 exhibit a strong bias (>2.0 in log scale) with 11 and 19, showing an unprecedented >1000-fold bias for Go versus β-arrestin, significantly departing from the balanced or unbiased functional activity of quinpirole and sumanirole.
D2R G-Protein Biased Agonists Are Physiologically Distinct from Sumanirole
In particular, we studied GoA vs β-arrestin 2 bias in functional response in D2R-expressing neurons. In an attempt to correlate biased effects with neuronal activity, the G protein biased compounds (11, 12, 13, 19) were characterized using electrophysiological measurements in neurons in brain slices, and this was compared to those of sumanirole. In D2R-expressing dopamine neurons in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc), the activation of G protein coupled inward-rectifying K+ (GIRK) channels is linked to G protein activity via βγ-subunit coupling.61 Meanwhile, β-arrestin 2-mediated receptor desensitization can counteract G protein activity including its downstream GIRK channel activation.61 Therefore, G protein-biased ligands may confer prolonged GIRK activity in the D2R-expressing neurons compared to nonbiased ligands due to lower levels of receptor desensitization via β-arrestin-2 coactivation.
We conducted ex vivo slice electrophysiology to study a time course response of ligand-induced GIRK activity in D2R-expressing dopaminergic neurons (Figure 2A).
Figure 2.

Average traces of ligand-induced current change (pA) with perfusion of corresponding drugs from 10 to 15 min: (A) gray/black 3/30 μM dopamine, (B) gray/black 3/30 μM sumanirole, (C) light purple/purple 3/30 μM 19, (D) light blue/blue 3/30 μM 12, (E) light orange/orange 3/30 μM 13, (F) light red/red 3/30 μM 11. Current peaks are indicated by arrows. Numbers of animals/neurons recorded are reported in the methods section.
In contrast to ligand concentrations used in experiments with isolated cells for which receptor access is not limited, higher ligand concentrations are necessary in brain slices to rapidly reach receptor equilibrium because of the diffusion barrier presented by tissue thickness.62 Therefore, consistent with previous studies using DA,63,64 ligand concentrations of 3 and 30 μM were chosen for bath perfusion application in the slice experiments (Figure 2). In whole cell voltage-clamp recordings, dopamine and sumanirole produced an increase in the holding current (e.g., outward current) needed to maintain the membrane potential at −60 mV. The currents reached peak levels between 10 and 15 min after beginning drug onset (indicated by arrows in Figure 2), and this was taken to indicate equilibration of these compounds at receptor sites in the tissue. In contrast, all of the N-5 bitopic analogues showed slower peak kinetics, often reaching peaks after the 15 min time point when ligand perfusion ended. Decay kinetics were also slower for the N-5 bitopic analogues, compared to sumanirole and dopamine, where peak currents were maintained for 15–20 min after drug application was terminated. The slower kinetics are more clearly demonstrated in Figure 3, where peak currents at drug wash in (measured at min 10–min 15 after drug onset; gray bars), and average currents during wash out (measured at min 30–min 35; black bars) are plotted (Figure 3B). Finally, the extent of washout of GIRK current activity, was analyzed by calculating ratios between average currents during washout (from min 30 to min 35) and peak currents (measured from min 10 to min 15) at either the 3 μM (Figure 3C) or the 30 μM (Figure 3D) concentrations of all drugs. Here, it can be seen that all of the N-5 bitopic analogues exhibited significantly higher ratios than sumanirole or dopamine. As the 20 min washout volume (3.6 mL/min × 20 min) exceeds the several rounds of bath exchange (1.3 mL/exchange), the elevated current in the 30–35 min window is unlikely from residual compounds leaching out of the tissue section (i.e., tissue reservoir). The D2R-selective antagonist sulpiride inhibited the change in the holding current induced by either sumanirole or 11, confirming that the effects of these compounds reflect D2R activation (Figure 4). Thus, we demonstrate the ability of N-5 bitopic compounds to promote sustained D2R-mediated currents in midbrain DA neurons
Figure 3.

Analysis of prolonged current increase by bivalent ligands: (A) Schematic timeline of electrophysiological recording with perfusion of corresponding drugs from 10 to 15 min, (B) peak current (pA) between 10 and 15 min (gray bars) and average current (pA) between 30 and 35 min (black bars), (C) percentage ratio between average 30–35 min and peak 10–15 min for 3 μM ligands (same color scheme as Figure S7), (D) percentage ratio between average 30–35 min and peak 10–15 min for 30 μM ligands (same color scheme as Figure S7). Values were statistically analyzed by one-way analysis of variance (ANOVA) repeated measure followed by Newman–Keuls post hoc test. p-Values are as indicated: **p < 0.01 or ***p < 0.001. The error bars represent SEM. Numbers of animals/neurons recorded are reported in the methods section.
Figure 4.

Average time course of ligand-induced current change (pA) following perfusion of agonists from 10 to 20 min and the D2 antagonist sulpiride from 15 to 20 min: (A) 30 μM sumanirole and 100 μM sulpiride, (B) 30 μM 11 and 100 μM sulpiride. The insets show peak holding current change prior to and following sulpiride application for each individual cell. Values were statistically analyzed by paired t test. p-Values are as indicated: *p < 0.05. The error bars represent SEM. Numbers of animals/neurons recorded are reported in the methods section.
Discussion
One of the greatest challenges in drug discovery is to develop highly selective agonists that are able to discriminate among closely related receptor subtypes, the protein structures of which are highly homologous, particularly in their OBS.19,65−67 This promiscuity in ligand–receptor interactions may be one of the multiple reasons associated with side effects observed with the current D2-like drugs approved for clinical use. Conversely, “biased magic bullets” have also been proposed as a viable new drug discovery approach wherein functional bias may be more important than subtype selectivity.38 At this stage, it is likely too early to determine which strategy will yield more effective medications. However, accumulating data support the development of new molecular tools with which to investigate the potential of functionally biased drug discovery approaches.46
Sumanirole is one the most well characterized D2R full agonists.48,68,69 In addition to showing moderate affinity and selectivity for the D2R over the D3R, it has been pharmacologically studied in multiple in vivo models and evaluated in clinical trials for the treatment of RLS and PD.68−73 Although never receiving FDA approval for clinical use in the U.S.A., it remains a valuable tool for drug discovery and lead optimization.43,48
Previous drug design and structure–activity relationship studies led to the discovery of 1, the first sumanirole-based D2R G-protein biased full agonist.43
To date, reports on selective allosteric modulators for the D2-like receptors have been sparse;16,74−77 however, the recently characterized PAM 2(16) confirmed that an ABS in D2R can be targeted in order to enhance potencies and affinities of classic orthosteric agonists (i.e., dopamine). Synthons inspired by 2 (i.e., 2-methylindoline and benzothiazole aromatic moieties (Figure 1)) were covalently inserted in the N-5 position of sumanirole or its deconstructed analogue 1,2,3,4-tetrahydroquinoline, via an n-butylamide linker to give ligands that might simultaneously target the OBS and SBP. This structural hybridization strategy to make bitopic ligands with novel secondary pharmacophores led to the discovery of the first Go protein ≫ Gi protein > β-arrestin agonist 11 and its enantiomers (R,R)-13 and (R,S)-12.
Moreover, in addition to identifying a new class of bitopic molecules that confer G-protein bias, we further demonstrate that the nature of the linker is critical not only to separate the PP from the SP by an optimal distance, but also to induce specific orientations and binding poses, to create their own key interactions within the receptor. Specifically, we recently observed how the highly D3R selective antagonist 3 (Figure 1), presenting a trans-cyclopropyl ring in the linker chain, showed additional D3R noncompetitive antagonism/negative allosteric modulation (NAM), depending exclusively on the chirality of the cyclopropyl ring and its relative distance from the PP and SP.22 Inspired by this observation, 1 was modified by inserting the trans-cyclopropyl ring in its linker, in both the relative distance combinations, to investigate the linker’s role in D2R binding and functional bias (Figure 1). Indeed, 19 and 20 showed significantly higher (>1000-fold) Go-protein bias over βarrestin as compared to ∼30-fold for 1. To our knowledge, biased profiles among G-protein subtypes have not been described for D2-like receptors, and thus these leads can be exploited for their unique functional potential.
Moreover, extensive in vitro BRET functional studies and biased factor calculations validated the specificity of 11, (R,R)-13, (R,S)-12, and 19 toward D2R G-protein activation pathways. G-protein activation bias for these key bitopic compounds was further characterized by in vitro electrophysiology, allowing for direct physiological observation of the biased agonism at the neuronal level, in comparison to sumanirole. The electrophysiology experiments showed that each of our novel G-protein biased D2R agonists activated GIRK, and that this persisted over a longer time course than DA or sumanirole, consistent with reduced β-arrestin recruitment and reduced receptor desensitization. Future biophysical methodologies may elucidate how the specific spatial orientations of the secondary pharmacophore, and the trans-cyclopropyl linker are responsible for the increased affinities, potencies, and Go-biased activation with respect to the canonical orthosteric agonist sumanirole. Moreover, the preferential targeting and activation of Go-protein, the most predominant in the central nervous system, will inspire further in vivo studies with these novel tools in animal models of basal ganglia related disorders, including Parkinson’s disease, with the aim to unravel the specific physiological effects associated with D2R G-protein biased agonism. In summary, as the transmission of biased signaling is far more complex than a simple binary function,29 the development of small molecule tools evaluated in genetic models47 provide a potentially superior therapeutic approach over current medications available to treat disorders associated with dopaminergic dysfunction.
Methods
Radioligand Binding Studies
Radioligand binding assays were conducted similarly to previously described.43,48,50 HEK293 cells stably expressing human D2LR or D3R or D4.4 were grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using premixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL of hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 14 500 rpm (∼25 000 g) for 30 min at 4 °C. The pellet was then resuspended in fresh binding buffer. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration. For [3H]-(R)-(+)-7-OH-DPAT binding studies, membranes were harvested fresh; the binding buffer was made from 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 7.4. On test day, each test compound was diluted into half-log serial dilutions using 30% DMSO vehicle. To assist the solubilization of free-base compounds at the desired stock concentration, 10 μL of glacial acetic acid was added along with the DMSO. Membranes were diluted in fresh binding buffer. Radioligand competition experiments were conducted in 96-well plates containing 300 μL of fresh binding buffer, 50 μL of diluted test compound, 100 μL of membranes (40–80 μg/well, 20–40 μg/well, and 30–60 μg/well total protein for hD2LR, hD3R, and hD4.4R, respectively), and 50 μL of radioligand diluted in binding buffer ([3H]-(R)-(+)-7-OH-DPAT: 1.5 nM final concentration for hD2L, 0.5 nM final concentration for hD3, and 3 nM final concentration for hD4.4 ARC, Saint Louis, MO). Aliquots of [3H]-(R)-(+)-7-OH-DPAT solution were also quantified accurately to determine how much radioactivity was added, taking in account the experimentally determined counter efficiency. Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in duplicate or triplicate, and the reaction was incubated for 90 min at room temperature. The reaction was terminated by filtration through a PerkinElmer Uni-Filter-96 GF/B, presoaked for 90 min in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL (3 × 1 mL/well) of ice-cold binding buffer; 65 μL of PerkinElmer MicroScint 20 Scintillation Cocktail was added to each well and filters were counted using a PerkinElmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose–response curves, and Ki values were calculated using the Cheng–Prusoff equation;78Kd values for [3H]-(R)-(+)-7-OH-DPAT were determined via separate homologous competitive binding experiments. These analyses were performed using GraphPad Prism version 6.00 for Macintosh (GraphPad Software, San Diego, CA). Ki values were determined from at least three independent experiments and are reported as mean ± SEM.
Bioluminescence Resonance Energy Transfer (BRET) Studies
Variations of bioluminescence resonance energy transfer (BRET) assay were performed to detect receptor ligand-induced events. A constant amount of plasmid cDNA (15 μg) was transfected into human embryonic kidney cells 293 T (HEK-293T) using polyethylenimine (PEI; Sigma) in a 1:2 weight ratio in 10 cm plates. Cells were maintained in culture with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Atlanta), 2 mM l-glutamine (Gibco), and 1% penicillin streptomycin (Gibco) and kept in an incubator at 37 °C and 5% CO2. The transfected amount and ratio among the receptor and heterotrimeric G proteins were tested for the optimized dynamic range in drug-induced BRET. Experiments were performed approximately 48 h post-transfection. As reported previously,50 cells were collected, washed, and resuspended in phosphate-buffered saline (PBS). Approximately 200 000 cells/well were distributed in 96-well plates, and 5 μM coelenterazine H (luciferase substrate, BRET1) or 5 μM coelenterazine 400a (luciferase substrate, BRET2) was added to each well. One minute after the addition of coelenterazine, D2R ligands were added to each well. Four different configurations of BRET were used: (i) Gαi1-γ2 protein activation, (ii) GαoA-γ2 protein activation, (iii) cAMP inhibition, and (iv) β-arrestin-2 recruitment. (i) Gαi1-γ2 protein activation assay uses Gαi1-Rluc-γ2-GFP10 for a resonance energy transfer (RET) pair. D2R and untagged Gβ1 constructs were cotransfected; (ii) GαoA-γ2 protein activation assay uses GαoA-Rluc-γ2-GFP10 for a RET pair. D2R and untagged Gβ1 constructs were cotransfected; (iii) cAMP production assay uses a CAMYEL biosensor construct that contains Rluc and YFP allowing detection of intracellular cAMP change59 in conjunction with receptor coexpression. D2R-Gi/o activation was studied by agonist-induced inhibition of cAMP production. Cells were prestimulated with 10 μM forskolin (Sigma) 10 min prior to agonist treatment. (iv) β-Arrestin-2 recruitment uses D2R-Rluc-β-arrestin-2-Venus for a RET pair. GRK2 was cotransfected to assist an enhanced phosphorylation required for the β-arrestin-2 recruitment. The donor luminescence as well as the acceptor fluorescence was always quantified for consistent expression levels across different experiments such that no significant expression differences between Gαi1-Rluc-γ2-GFP10 and GαoA-Rluc-γ2-GFP10 pairs were observed, for instance. For BRET1, venus was excited at 500 nm and measured at an emission wavelength of 530 nm. For BRET2, GFP10 was excited at 405 nm and measured at an emission wavelength of 515 nm. Both fluorophores were measured over 1 s of recording, using a Mithras LB940 microplate reader (Berthold Technologies, Bad Wildbad, Germany). The BRET1 signal from the same batch of cells was calculated as the ratio of the light emitted by Venus (530 nm) over that emitted by coelenterazine H (485 nm), and the BRET2 signal from the same batch of cells was calculated as the ratio of the light emitted by GFP10 (515 nm) over that emitted by coelenterazine 400a (400 nm). BRET change was defined as BRET ratio for the corresponding drug minus BRET ratio in the absence of the drug. Emax values are expressed as the basal subtracted BRET change and in the dose–response graphs. Data and statistical analysis were performed with Prism 7 (GraphPad Software).
Animals
Drd2-EGFP reporter BAC mice (S118Gsat/Mmnc, RRID:MMRRC_000230_UNC) backcrossed onto a C57BL/6J background were obtained from Mutant Mouse Resource and Research Center (MMRRC). Male Drd2-EGFP mice were used for slice electrophysiology experiments. Animals were housed with food and water available ad libitum in temperature-controlled and humidity-controlled rooms and were maintained on a 12 h light/dark cycle. They were experimentally naive at the start of the study and were maintained under the approved protocol of the Institutional Care and Use Committee of the Intramural Research Program, National Institute on Drug Abuse.
Brain Slice Electrophysiology
Experiments were performed based on a previous report with modifications.79 Horizontal slices (220 μm) were prepared from male adult Drd2-eGFP mouse brain using a vibrating tissue slicer (VT-1200S, Leica). Prior to decapitation, mice were anesthetized and perfused with modified artificial cerebral spinal fluid (maCSF) containing (in mM): 92 NMDG, 20 HEPES, 25 glucose, 30 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 10 MgSO4, 0.5 CaCl2, 300–310 mOsm, at pH 7.3–7.4. The brains were sectioned in cold maCSF, saturated with 95% O2 and 5% CO2 (carbogen), and then placed in the same buffer, maintained at 32 °C, for 10 min. The slices were then transferred to a holding chamber filled with carbogen saturated aCSF (holding aCSF) containing, in mM: 92 NaCl, 20 HEPES, 25 glucose, 30 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 1 MgSO4, 2 CaCl2, 300–310 mOsm, at pH 7.3–7.4. During electrophysiological recordings, slices were continuously perfused at 2 mL/min with carbogen-saturated aCSF containing (in mM): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 26 NaHCO3, 11 glucose, 2.4 CaCl2, 300–310 mOsm, at pH 7.3–7.4, supplemented with 200 μM sodium bisulfite and 100 nM tetrodotoxin. The temperature of the recording chamber was maintained at 31–32 °C. Electrodes (3–5 MΩ) were backfilled with an internal solution containing (in mM): 120 mM K gluconate, 20 KCl, 0.05 EGTA, 10 HEPES, 1.5 MgCl2, 2.18 Na2 ATP, 0.38 Na GTP, 10.19 Na phosphocreatine, 280–285 mOsm, and pH 7.3–7.4. Cells were visualized on an upright microscope using infrared differential interference contrast video microscopy. Whole-cell current-clamp recordings were made using a MultiClamp 700B amplifier (2 kHz low-pass Bessel filter and 10 kHz digitization) with pClamp 10.5 software (Molecular Devices). Presumed dopaminergic neurons in the ventral tegmental area and substantia nigra pars compacta were identified by green fluorescence, membrane resistance, and morphology in the Drd2-EGFP mouse brain slices. Series resistance (10–25 MΩ) was monitored using a 5 mV hyperpolarizing pulse (50 ms) given every 20 s, and only recordings that remained stable (monitored by series resistance) over the period of data collection were used. On breaking into neurons, the resting membrane potentials were between −45 and −65 mV. Holding current was recorded in voltage clamp mode at a corresponding resting membrane potential. The following numbers of cells were recorded for the drug perfusion conditions listed: 3, 30 μM dopamine (13 cells/5 animals, 16 cells/7 animals); 3, 30 μM sumanirole (10 cells/5 animals, 10 neurons/6 animals); 3, 30 μM 19 (9 cells/3 animals, 11 neurons/4 animals); 3, 30 μM 12 (6 cells/4 animals, 9 neurons/4 animals); 3, 30 μM 13 (5 cells/3 animals, 10 neurons/7 animals); 3, 30 μM 11 (11 cells/4 animals, 11 neurons/8 animals). The following numbers of cells were recorded for the antagonist inhibition: 30 μM sumanirole and 100 μM sulpiride (7 neurons/3 animals) and 30 μM 11 and 100 μM sulpiride (6 neurons/3 animals). All data are reported as mean ± SEM. Data were analyzed in Clampex and statistically analyzed with Prism 7 (GraphPad Software) by one-way ANOVA followed by Newman–Keuls post hoc test.
Acknowledgments
This project was supported by the National Institute on Drug Abuse (NIDA-IRP) Z1A DA0000609. The authors thank Dr. Sergi Ferré and Dr. Ning-Sheng Cai for sharing instrumentation for tissue culture and BRET studies, Dr. Antonello Bonci for helpful discussions about electrophysiology experiments, Dr. Aaron Janowsky for off-target binding screening, Dr. Ludovic Muller from the Structural Biology Core for high resolution mass spectrometry analyses, Francisco O. Battiti and Dr. Anver Basha Shaik for helpful discussions about chemistry experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.8b00060.
Chemistry methods, NMR data and synthetic schemes; microanalyses, high resolution mass spectroscopy, and HPLC-DAD data; D2-Mediated Gi and Go activation, cAMP inhibition and β-arrestin2 recruitment; off-target binding data; HPLC chromatograms; BRET dose-response curves; web of bias plot; references (PDF)
Author Contributions
# A.B. and H.Y. contributed equally to this work. A.H.N., A.B. and H.Y. designed the project and wrote the manuscript with input of all authors; A.B., H.Y., A.H.N., L.S, C.R.L and A.F.H. designed and/or supervised the experiments and data analyses. A.B., H.Y., A.M.G. and V.K. performed experiments.
The authors declare no competing financial interest.
Supplementary Material
References
- Beaulieu J. M.; Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63 (1), 182–217. 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Kehne J. H.; Andree T. H.; Heinrich J. N. (2008) D2 receptor partial agonists: treatment of CNS disorders of dopamine function. Curr. Top. Med. Chem. 8 (12), 1068–88. 10.2174/156802608785161394. [DOI] [PubMed] [Google Scholar]
- Joyce J. N.; Millan M. J. (2007) Dopamine D3 receptor agonists for protection and repair in Parkinson’s disease. Curr. Opin. Pharmacol. 7 (1), 100–5. 10.1016/j.coph.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Ferrari-Toninelli G.; Bonini S. A.; Cenini G.; Maccarinelli G.; Grilli M.; Uberti D.; Memo M. (2008) Dopamine receptor agonists for protection and repair in Parkinson’s disease. Curr. Top. Med. Chem. 8 (12), 1089–99. 10.2174/156802608785161402. [DOI] [PubMed] [Google Scholar]
- Madras B. K. (2013) History of the discovery of the antipsychotic dopamine D2 receptor: a basis for the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 22 (1), 62–78. 10.1080/0964704X.2012.678199. [DOI] [PubMed] [Google Scholar]
- Citrome L. (2014) Unmet needs in the treatment of schizophrenia: new targets to help different symptom domains. J. Clin. Psychiatry 75, 21–6. 10.4088/JCP.13049su1c.04. [DOI] [PubMed] [Google Scholar]
- Lako I. M.; Liemburg E. J.; Van den Heuvel E. R.; Knegtering H.; Bruggeman R.; Taxis K. (2014) Estimating dopamine D(2) receptor occupancy for doses of 8 antipsychotics: a meta-analysis: a reply. J. Clin. Psychopharmacol. 34 (4), 532–3. 10.1097/JCP.0000000000000172. [DOI] [PubMed] [Google Scholar]
- Olanow C. W. (2002) The role of dopamine agonists in the treatment of early Parkinson’s disease. Neurology 58, S33–41. 10.1212/WNL.58.suppl_1.S33. [DOI] [PubMed] [Google Scholar]
- Bonuccelli U.; Del Dotto P.; Rascol O. (2009) Role of dopamine receptor agonists in the treatment of early Parkinson’s disease. Parkinsonism Relat. Disord. 15, S44–53. 10.1016/S1353-8020(09)70835-1. [DOI] [PubMed] [Google Scholar]
- Luo D.; Sharma H.; Yedlapudi D.; Antonio T.; Reith M. E.; Dutta A. K. (2016) Novel multifunctional dopamine D2/D3 receptors agonists with potential neuroprotection and anti-alpha synuclein protein aggregation properties. Bioorg. Med. Chem. 24 (21), 5088–5102. 10.1016/j.bmc.2016.08.021. [DOI] [PubMed] [Google Scholar]
- Watson D. J. G.; King M. V.; Gyertyan I.; Kiss B.; Adham N.; Fone K. C. F. (2016) The dopamine D(3)-preferring D(2)/D(3) dopamine receptor partial agonist, cariprazine, reverses behavioural changes in a rat neurodevelopmental model for schizophrenia. Eur. Neuropsychopharmacol. 26 (2), 208–224. 10.1016/j.euroneuro.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Manconi M.; Ferri R.; Zucconi M.; Clemens S.; Giarolli L.; Bottasini V.; Ferini-Strambi L. (2011) Preferential D2 or preferential D3 dopamine agonists in restless legs syndrome. Neurology 77 (2), 110–7. 10.1212/WNL.0b013e3182242d91. [DOI] [PubMed] [Google Scholar]
- Seeman P. (2006) Targeting the dopamine D2 receptor in schizophrenia. Expert Opin. Ther. Targets 10 (4), 515–31. 10.1517/14728222.10.4.515. [DOI] [PubMed] [Google Scholar]
- Mistry S. N.; Shonberg J.; Draper-Joyce C. J.; Klein Herenbrink C.; Michino M.; Shi L.; Christopoulos A.; Capuano B.; Scammells P. J.; Lane J. R. (2015) Discovery of a Novel Class of Negative Allosteric Modulator of the Dopamine D2 Receptor Through Fragmentation of a Bitopic Ligand. J. Med. Chem. 58 (17), 6819–43. 10.1021/acs.jmedchem.5b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolbeare K.; Pontoriero G. F.; Gupta S. K.; Mishra R. K.; Johnson R. L. (2003) Iso-lactam and reduced amide analogues of the peptidomimetic dopamine receptor modulator 3(R)-[(2(S)-pyrrolidinylcarbonyl)amino]-2-oxo-1-pyrrolidineacetamide. Bioorg. Med. Chem. 11 (18), 4103–12. 10.1016/S0968-0896(03)00396-1. [DOI] [PubMed] [Google Scholar]
- Wood M.; Ates A.; Andre V. M.; Michel A.; Barnaby R.; Gillard M. (2016) Vitro and In Vivo Identification of Novel Positive Allosteric Modulators of the Human Dopamine D2 and D3 Receptor. Mol. Pharmacol. 89 (2), 303–12. 10.1124/mol.115.100172. [DOI] [PubMed] [Google Scholar]
- Huynh T.; Valant C.; Crosby I. T.; Sexton P. M.; Christopoulos A.; Capuano B. (2015) Synthesis and pharmacological evaluation of M4 muscarinic receptor positive allosteric modulators derived from VU10004. ACS Chem. Neurosci. 6 (6), 838–44. 10.1021/acschemneuro.5b00035. [DOI] [PubMed] [Google Scholar]
- Bonifazi A.; Yano H.; Del Bello F.; Farande A.; Quaglia W.; Petrelli R.; Matucci R.; Nesi M.; Vistoli G.; Ferre S.; Piergentili A. (2014) Synthesis and biological evaluation of a novel series of heterobivalent muscarinic ligands based on xanomeline and 1-[3-(4-butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28–1). J. Med. Chem. 57 (21), 9065–77. 10.1021/jm501173q. [DOI] [PubMed] [Google Scholar]
- Keck T. M.; Burzynski C.; Shi L.; Newman A. H. (2014) Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol. 69, 267–300. 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J. R.; May L. T.; Parton R. G.; Sexton P. M.; Christopoulos A. (2017) A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 13 (9), 929–937. 10.1038/nchembio.2431. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Bonifazi A.; Ellenberger M. P.; Keck T. M.; Pommier E.; Rais R.; Slusher B. S.; Gardner E.; You Z. B.; Xi Z. X.; Newman A. H. (2016) Highly Selective Dopamine D3 Receptor (D3R) Antagonists and Partial Agonists Based on Eticlopride and the D3R Crystal Structure: New Leads for Opioid Dependence Treatment. J. Med. Chem. 59 (16), 7634–50. 10.1021/acs.jmedchem.6b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Moritz A. E.; Keck T. M.; Bonifazi A.; Ellenberger M. P.; Sibley C. D.; Free R. B.; Shi L.; Lane J. R.; Sibley D. R.; Newman A. H. (2017) Synthesis and Pharmacological Characterization of Novel trans-Cyclopropylmethyl-Linked Bivalent Ligands That Exhibit Selectivity and Allosteric Pharmacology at the Dopamine D3 Receptor (D3R). J. Med. Chem. 60 (4), 1478–1494. 10.1021/acs.jmedchem.6b01688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng C. A.; Bakare O. M.; Zhan J.; Banala A. K.; Burzynski C.; Pommier E.; Keck T. M.; Donthamsetti P.; Javitch J. A.; Rais R.; Slusher B. S.; Xi Z. X.; Newman A. H. (2015) High Affinity Dopamine D3 Receptor (D3R)-Selective Antagonists Attenuate Heroin Self-Administration in Wild-Type but not D3R Knockout Mice. J. Med. Chem. 58 (15), 6195–213. 10.1021/acs.jmedchem.5b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman R. B. (2007) GPCR functional selectivity has therapeutic impact. Trends Pharmacol. Sci. 28 (8), 390–6. 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J. D.; Clarke W. P.; von Zastrow M.; Nichols D. E.; Kobilka B.; Weinstein H.; Javitch J. A.; Roth B. L.; Christopoulos A.; Sexton P. M.; Miller K. J.; Spedding M.; Mailman R. B. (2006) Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320 (1), 1–13. 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Marti-Solano M.; Babu M. M.; Sexton P. M. (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19 (10), 638–653. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- Wang W.; Qiao Y.; Li Z. (2018) New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 39 (4), 367–386. 10.1016/j.tips.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Schrage R.; Kostenis E. (2017) Functional selectivity and dualsteric/bitopic GPCR targeting. Curr. Opin. Pharmacol. 32, 85–90. 10.1016/j.coph.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. (2018) Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discovery 17 (4), 243–260. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S.; Provasi D.; Filizola M. (2016) How Oliceridine (TRV-130) Binds and Stabilizes a mu-Opioid Receptor Conformational State That Selectively Triggers G Protein Signaling Pathways. Biochemistry 55 (46), 6456–6466. 10.1021/acs.biochem.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller D.; Banerjee A.; Uzuneser T. C.; Skultety M.; Huth T.; Plouffe B.; Hubner H.; Alzheimer C.; Friedland K.; Muller C. P.; Bouvier M.; Gmeiner P. (2017) Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1,5-a]pyridine Substructure. J. Med. Chem. 60 (7), 2908–2929. 10.1021/acs.jmedchem.6b01857. [DOI] [PubMed] [Google Scholar]
- Mannel B.; Dengler D.; Shonberg J.; Hubner H.; Moller D.; Gmeiner P. (2017) Hydroxy-Substituted Heteroarylpiperazines: Novel Scaffolds for β-Arrestin-Biased D2R Agonists. J. Med. Chem. 60 (11), 4693–4713. 10.1021/acs.jmedchem.7b00363. [DOI] [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U. S. A. 108 (45), 18488–93. 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Sassano M. F.; Zheng L.; Setola V.; Chen M.; Bai X.; Frye S. V.; Wetsel W. C.; Roth B. L.; Jin J. (2012) Structure-functional selectivity relationship studies of β-arrestin-biased dopamine D(2) receptor agonists. J. Med. Chem. 55 (16), 7141–53. 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs N. M.; Peterson S. M.; Caron M. G. (2017) New Concepts in Dopamine D2 Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biol. Psychiatry 81 (1), 78–85. 10.1016/j.biopsych.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs N. M.; Gee S. M.; Pack T. F.; McCorvy J. D.; Evron T.; Snyder J. C.; Yang X.; Rodriguiz R. M.; Borrelli E.; Wetsel W. C.; Jin J.; Roth B. L.; O’Donnell P.; Caron M. G. (2016) Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U. S. A. 113 (50), E8178–e8186. 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K.; Gomez-Soler M.; Valle-Leon M.; Lopez-Cano M.; Taura J. J.; Ciruela F.; Fernandez-Duenas V. (2018) Antipsychotic-Like Efficacy of Dopamine D2 Receptor-Biased Ligands is Dependent on Adenosine A2A Receptor Expression. Mol. Neurobiol. 55 (6), 4952–4958. 10.1007/s12035-017-0696-y. [DOI] [PubMed] [Google Scholar]
- McCorvy J. D.; Butler K. V.; Kelly B.; Rechsteiner K.; Karpiak J.; Betz R. M.; Kormos B. L.; Shoichet B. K.; Dror R. O.; Jin J.; Roth B. L. (2017) Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 14 (2), 126–134. 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S. M.; Pack T. F.; Caron M. G. (2015) Receptor, Ligand and Transducer Contributions to Dopamine D2 Receptor Functional Selectivity. PLoS One 10 (10), e0141637. 10.1371/journal.pone.0141637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free R. B.; Chun L. S.; Moritz A. E.; Miller B. N.; Doyle T. B.; Conroy J. L.; Padron A.; Meade J. A.; Xiao J.; Hu X.; Dulcey A. E.; Han Y.; Duan L.; Titus S.; Bryant-Genevier M.; Barnaeva E.; Ferrer M.; Javitch J. A.; Beuming T.; Shi L.; Southall N. T.; Marugan J. J.; Sibley D. R. (2014) Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 86 (1), 96–105. 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun L. S.; Vekariya R. H.; Free R. B.; Li Y.; Lin D. T.; Su P.; Liu F.; Namkung Y.; Laporte S. A.; Moritz A. E.; Aube J.; Frankowski K. J.; Sibley D. R. (2018) Structure-Activity Investigation of a G Protein-Biased Agonist Reveals Molecular Determinants for Biased Signaling of the D2 Dopamine Receptor. Front. Synaptic Neurosci. 10, 2. 10.3389/fnsyn.2018.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; McCorvy J. D.; Fischer M. G.; Butler K. V.; Shen Y.; Roth B. L.; Jin J. (2016) Discovery of G Protein-Biased D2 Dopamine Receptor Partial Agonists. J. Med. Chem. 59 (23), 10601–10618. 10.1021/acs.jmedchem.6b01208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi A.; Yano H.; Ellenberger M. P.; Muller L.; Kumar V.; Zou M. F.; Cai N. S.; Guerrero A. M.; Woods A. S.; Shi L.; Newman A. H. (2017) Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J. Med. Chem. 60 (7), 2890–2907. 10.1021/acs.jmedchem.6b01875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo M.; Klein Herenbrink C.; Christopoulos A.; Lane J. R.; Capuano B. (2014) Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D(2) receptor. J. Med. Chem. 57 (11), 4924–39. 10.1021/jm500457x. [DOI] [PubMed] [Google Scholar]
- Siuda E. R.; Carr R. 3rd; Rominger D. H.; Violin J. D. (2017) Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr. Opin. Pharmacol. 32, 77–84. 10.1016/j.coph.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Michel M. C.; Charlton S. J. (2018) Biased Agonism in Drug Discovery-Is It Too Soon to Choose a Path?. Mol. Pharmacol. 93 (4), 259–265. 10.1124/mol.117.110890. [DOI] [PubMed] [Google Scholar]
- Rose S. J.; Pack T. F.; Peterson S. M.; Payne K.; Borrelli E.; Caron M. G. (2018) Engineered D2R Variants Reveal the Balanced and Biased Contributions of G-Protein and β-Arrestin to Dopamine-Dependent Functions. Neuropsychopharmacology 43 (5), 1164–1173. 10.1038/npp.2017.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M. F.; Keck T. M.; Kumar V.; Donthamsetti P.; Michino M.; Burzynski C.; Schweppe C.; Bonifazi A.; Free R. B.; Sibley D. R.; Janowsky A.; Shi L.; Javitch J. A.; Newman A. H. (2016) Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J. Med. Chem. 59 (7), 2973–88. 10.1021/acs.jmedchem.5b01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele T. T. (2016) The ″Cyclopropyl Fragment″ is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 59 (19), 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- Sanchez-Soto M.; Bonifazi A.; Cai N. S.; Ellenberger M. P.; Newman A. H.; Ferre S.; Yano H. (2016) Evidence for Noncanonical Neurotransmitter Activation: Norepinephrine as a Dopamine D2-Like Receptor Agonist. Mol. Pharmacol. 89 (4), 457–66. 10.1124/mol.115.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C.; Sykes D. A.; Donthamsetti P.; Canals M.; Coudrat T.; Shonberg J.; Scammells P. J.; Capuano B.; Sexton P. M.; Charlton S. J.; Javitch J. A.; Christopoulos A.; Lane J. R. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842. 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. W.; Leff P. (1983) Operational models of pharmacological agonism. Proceedings of the Royal Society of London. Series B, Biological Sciences 220 (1219), 141–62. 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Watson C.; Muniz-Medina V.; Christopoulos A.; Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3 (3), 193–203. 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott L. A.; Hall D. A.; Holliday N. D. (2016) Unravelling intrinsic efficacy and ligand bias at G protein coupled receptors: A practical guide to assessing functional data. Biochem. Pharmacol. 101, 1–12. 10.1016/j.bcp.2015.10.011. [DOI] [PubMed] [Google Scholar]
- Quoyer J.; Janz J. M.; Luo J.; Ren Y.; Armando S.; Lukashova V.; Benovic J. L.; Carlson K. E.; Hunt S. W. 3rd; Bouvier M. (2013) Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc. Natl. Acad. Sci. U. S. A. 110 (52), E5088–97. 10.1073/pnas.1312515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnelli M.; Sauliere A.; Manning M.; Bouvier M.; Gales C.; Chini B. (2012) Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J. Biol. Chem. 287 (6), 3617–29. 10.1074/jbc.M111.277178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M.; Bajpayee N. S. (2009) Molecular mechanisms of go signaling. Neurosignals 17 (1), 23–41. 10.1159/000186688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M.; Spicher K.; Boulay G.; Wang Y.; Birnbaumer L. (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc. Natl. Acad. Sci. U. S. A. 98 (6), 3577–82. 10.1073/pnas.051632598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L. I.; Collins J.; Davis R.; Lin K. M.; DeCamp D.; Roach T.; Hsueh R.; Rebres R. A.; Ross E. M.; Taussig R.; Fraser I.; Sternweis P. C. (2007) Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282 (14), 10576–84. 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Stevens R. C.; Roth B. L. (2017) How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170 (3), 414–427. 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C.; Slesinger P. A. (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11 (5), 301–15. 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredell J. A.; Turnquist P. A.; Maciver M. B.; Pearce R. A. (2004) Determination of diffusion and partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br. J. Anaesth. 93 (6), 810–7. 10.1093/bja/aeh272. [DOI] [PubMed] [Google Scholar]
- Lacey M. G.; Mercuri N. B.; North R. A. (1988) On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J. Physiol. 401, 437–53. 10.1113/jphysiol.1988.sp017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin R. A.; Huyghe D.; Li X.; Parakala M.; Aisenberg E.; Moss S. J.; Slesinger P. A. (2018) GIRK currents in VTA dopamine neurons control the sensitivity of mice to cocaine-induced locomotor sensitization. Proc. Natl. Acad. Sci. U. S. A. 115 (40), E9479–E9488. 10.1073/pnas.1807788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A. E.; Free R. B.; Sibley D. R. (2018) Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell. Signalling 41, 75–81. 10.1016/j.cellsig.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330 (6007), 1091–5. 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggio G. M.; Bucolo C.; Platania C. B.; Salomone S.; Drago F. (2016) Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther. 165, 164–77. 10.1016/j.pharmthera.2016.06.007. [DOI] [PubMed] [Google Scholar]
- McCall R. B.; Lookingland K. J.; Bedard P. J.; Huff R. M. (2005) Sumanirole, a highly dopamine D2-selective receptor agonist: in vitro and in vivo pharmacological characterization and efficacy in animal models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 314 (3), 1248–56. 10.1124/jpet.105.084202. [DOI] [PubMed] [Google Scholar]
- Heier R. F.; Dolak L. A.; Duncan J. N.; Hyslop D. K.; Lipton M. F.; Martin I. J.; Mauragis M. A.; Piercey M. F.; Nichols N. F.; Schreur P. J.; Smith M. W.; Moon M. W. (1997) Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo[4,5,1-ij]quinolin-5-amine and its metabolites. J. Med. Chem. 40 (5), 639–46. 10.1021/jm960360q. [DOI] [PubMed] [Google Scholar]
- Sethy V. H.; Ellerbrock B. R.; Wu H. U. (1997) 95666E: a potential anti-parkinsonian drug with anxiolytic activity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 21 (5), 873–83. 10.1016/S0278-5846(97)00086-9. [DOI] [PubMed] [Google Scholar]
- Barone P.; Lamb J.; Ellis A.; Clarke Z. (2007) Sumanirole versus placebo or ropinirole for the adjunctive treatment of patients with advanced Parkinson’s disease. Mov. Disord. 22 (4), 483–9. 10.1002/mds.21191. [DOI] [PubMed] [Google Scholar]
- Garcia-Borreguero D.; Winkelman J.; Adams A.; Ellis A.; Morris M.; Lamb J.; Layton G.; Versavel M. (2007) Efficacy and tolerability of sumanirole in restless legs syndrome: a phase II, randomized, double-blind, placebo-controlled, dose-response study. Sleep Med. 8 (2), 119–27. 10.1016/j.sleep.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Zintzaras E.; Kitsios G. D.; Papathanasiou A. A.; Konitsiotis S.; Miligkos M.; Rodopoulou P.; Hadjigeorgiou G. M. (2010) Randomized trials of dopamine agonists in restless legs syndrome: a systematic review, quality assessment, and meta-analysis. Clin. Ther. 32 (2), 221–37. 10.1016/j.clinthera.2010.01.028. [DOI] [PubMed] [Google Scholar]
- Basu D.; Tian Y.; Bhandari J.; Jiang J. R.; Hui P.; Johnson R. L.; Mishra R. K. (2013) Effects of the dopamine D2 allosteric modulator, PAOPA, on the expression of GRK2, arrestin-3, ERK1/2, and on receptor internalization. PLoS One 8 (8), e70736. 10.1371/journal.pone.0070736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaert M. G.; Daya R. P.; Dyck B. A.; Johnson R. L.; Mishra R. K. (2013) PAOPA, a potent dopamine D2 receptor allosteric modulator, prevents and reverses behavioral and biochemical abnormalities in an amphetamine-sensitized preclinical animal model of schizophrenia. Eur. Neuropsychopharmacol. 23 (3), 253–62. 10.1016/j.euroneuro.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Rossi M.; Fasciani I.; Marampon F.; Maggio R.; Scarselli M. (2017) The First Negative Allosteric Modulator for Dopamine D2 and D3 Receptors, SB269652 May Lead to a New Generation of Antipsychotic Drugs. Mol. Pharmacol. 91 (6), 586–594. 10.1124/mol.116.107607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinathan A.; Draper-Joyce C.; Szabo M.; Christopoulos A.; Scammells P. J.; Lane J. R.; Capuano B. (2019) Subtle Modifications to the Indole-2-carboxamide Motif of the Negative Allosteric Modulator N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1 H)-yl)ethyl)cyclohexyl)-1 H-indole-2-carboxamide (SB269652) Yield Dramatic Changes in Pharmacological Activity at the Dopamine D2 Receptor. J. Med. Chem. 62, 371. 10.1021/acs.jmedchem.8b00192. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22 (23), 3099–108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Yano H.; Cai N. S.; Xu M.; Verma R. K.; Rea W.; Hoffman A. F.; Shi L.; Javitch J. A.; Bonci A.; Ferre S. (2018) Gs- versus Golf-dependent functional selectivity mediated by the dopamine D1 receptor. Nat. Commun. 9 (1), 486. 10.1038/s41467-017-02606-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.