

Abstract
A marked decrease in malaria-related deaths worldwide has been attributed to the administration of effective antimalarials against Plasmodium falciparum, in particular, artemisinin-based combination therapies (ACTs). Increasingly, ACTs are also used to treat Plasmodium vivax, the second major human malaria parasite. However, resistance to frontline artemisinins and partner drugs is now causing the failure of P. falciparum ACTs in southeast Asia. In this Review, we discuss our current knowledge of markers and mechanisms of resistance to artemisinins and ACTs. In particular, we describe the identification of mutations in the propeller domains of Kelch 13 as the primary marker for artemisinin resistance in P. falciparum and explore two major mechanisms of resistance that have been independently proposed: the activation of the unfolded protein response and proteostatic dysregulation of parasite phosphatidylinositol 3- kinase. We emphasize the continuing challenges and the imminent need to understand mechanisms of resistance to improve parasite detection strategies, develop new combinations to eliminate resistant parasites and prevent their global spread.
Although malaria has taken a staggering toll on human health in the past, the 21st century seems poised to consider its elimination and eradication. Plasmodium falciparum causes the most virulent malaria infections in humans and is responsible for most of the malaria- related deaths. From 2010 to 2015, through the control of the mosquito vector and fast-acting artemisinin-based combination therapies (ACTs), the numbers of new cases of P. falciparum malaria and of malaria-related deaths in the world decreased by ~20% and ~30%, respectively1. However, a substantial global burden remains, with over 400,000 deaths and 200 million new cases reported in 2016 (REF. 2). In addition, Plasmodium vivax, which can infect humans and causes relapsing malaria, is geographically widespread3.
An infection with P. falciparum begins when sporozoites are injected into the bloodstream of the vertebrate host through a mosquito bite and invade hepatocytes (the liver stage). In liver cells, sporozoites form merozoites, which are then released into the bloodstream and infect erythrocytes (the blood stage) (FIG. 1a). The parasites develop into the ring form and subsequently develop into proliferative (and morphologically distinct) trophozoite and schizont stages. At the end of the 48 h asexual life cycle, the schizonts rupture and release daughter merozoites into the plasma to initiate infection of new erythrocytes. Merozoite release coincides with periodic fever every 2 days, which is associated with P. falciparum malaria. In addition to the symptoms of acute infection, blood-stage parasites are also responsible for the severe and often fatal disease pathologies of malaria caused by parasite adhesion to the host endothelium, as the parasites are sequestered and concentrated in tissues, which leads to organ dysfunction4,5. Thus, all antimalarials that are currently used target the proliferative trophozoites and schizonts. Artemisinins also block the ring stage and the sexual parasite stages that lead to transmission from the blood to the mosquito6 when the vector takes a blood meal (FIG. 1a).
Figure 1 |. Life cycle of Plasmodium falciparum and epidemiology of antimalarial drug resistance.

a | Infection begins when a mosquito bite releases sporozoites into the bloodstream of the vertebrate host. Sporozoites subsequently infect hepatocytes, in which they proliferate and develop into merozoites. Merozoites are released into blood, where they invade erythrocytes. The parasites develop into the ring form and subsequently develop into proliferative (and morphologically distinct) trophozoite and schizont stages. When the schizont lyses, new merozoites are released, which initiate a new asexual cycle in the blood. A small proportion of ring-form parasites develop into gametocytes (sexual stages), which are taken up by the mosquito during a blood meal. In the mosquito midgut, male and female gametes emerge and fuse to form zygotes, which further differentiate into oocysts. Each oocyst divides to produce and release thousands of haploid sporozoites into the mosquito body cavity. These sporozoites travel and invade the mosquito salivary glands, from where they are injected into the human host. Trophozoite and schizont stages are sequestered in host tissue to cause severe disease. All antimalarial classes shown (and comprehensively reviewed in TABLE 1) target the asexual trophozoite and schizont stages. Antifolates, primaquine (8-aminoquinoline) and atovaquone (naphthoquinone) also target liver-stage parasites. Endoperoxides (artemisinins) target all asexual and early sexual blood-stage parasites. Primaquine is the only drug that targets latency in the liver to prevent the relapsing infection characteristic of Plasmodium vivax. b | Detailed maps showing the distribution of P. falciparum resistance to chloroquine, sulfadoxine-pyrimethamine and artemisinin in Africa and southeast Asia18,19,21,61,62,74,119–125. Artemisinin resistance is based on two criteria: clearance and PfKelch13-associated mutation. Although the A578S mutation has been reported115, no pfkelch13 mutations associated with resistance have been reported yet in Bangladesh (resistance is indicated at the Bangladesh-Myanmar border). Resistance to chloroquine and sulfadoxine- pyrimethamine (but not artemisinin) has emerged in the Amazon basin of South America126 (not shown). Resistance of P. vivax to chloroquine is emerging127 (not shown). Each dot represents a region of emergence of drug resistance. DRC, Democratic Republic of the Congo. Part a adapted from REF. 128.
Before the use of ACTs, chloroquine and subsequently sulfadoxine-pyrimethamine were effective, inexpensive, safe and widely used to treat acute infections with P. falciparum7,8. Mefloquine, quinine and lumefantrine are commonly used antimalarials in endemic regions; however, mefloquine has substantially greater side effects than chloroquine and sulfadoxine- pyrimethamine9. Clinical failure and spread of resistance to chloroquine and sulfadoxine-pyrimethamine10–15 led to the introduction of ACTs in Africa. In ACTs, artemisinins confer rapid and potent effectiveness, while their high rates of clearance from plasma (half-life <1 h (REF. 16)) are offset by a second, longer-lived antimalarial (such as lumefantrine, mefloquine, piperaquine (PPQ), amodiaquine or sulfadoxine-pyrimethamine) as a partner drug (Supplementary information S1 (table)). The underlying rationale is that artemisinins rapidly eliminate the majority of the parasites within days by mechanisms that are distinct from those of the partner drug, which eliminates residual parasites over weeks, raising expectations that parasites that emerge resistant to the artemisinin drug would still be eliminated by the partner drug.
Resistance has emerged to all known antimalarials (FIG. 1b; TABLE 1), and therefore, tracking drug sensitivities of clinical infections is critical to inform treatment strategy as well as strategies of mass drug administration that aim to eliminate malaria. The detection of drug-resistant parasites and their spread was greatly facilitated by the discovery of molecular resistance markers17–21. Genome-wide association studies (GWAS) of parasites obtained directly from infected individuals can be powerful in identifying chromosomal regions and their associated genes under high selective pressure20,22,23. However, to establish a causal marker requires the ability to reproduce the proliferation of resistant pathogens in the laboratory and in genetic studies. This was readily achieved using standard assays for parasites that are resistant to all antimalarials24, except for artemisinins, for which establishing resistance in the laboratory took almost 5 years from their first identification in the field (BOX 1).
Table 1|.
Current antimalarial drugs and associated resistance markers
| Chemical class | Common name | Targeted parasite stage | Genetic marker for drug resistance |
|
|---|---|---|---|---|
| Plasmodium falciparum | Plasmodium vivax | |||
| Sesquiterpene lactone endoperoxides | Artemisinin* | All parasite stages | pfkelch13 (REF. 18) | Unknown |
| Artesunate*‡ | All parasite stages | pfkelch13 (REF. 18) | Unknown | |
| Artemether*‡ | All parasite stages | pfkelch13 (REF. 18) | Unknown | |
| Dihydroartemisinin*‡ | All parasite stages | pfkelch13 (REF. 18) | Unknown | |
| 4-Aminoquinolines | Chloroquineठ| Blood stages (trophozoite and schizont) | pfcrt17,26 | pvmdr1 (REF. 81) |
| Amodiaquine*‡ | Blood stages (trophozoite and schizont) | pfcrt, pfmdr1 (REF. 129) | Unknown | |
| Piperaquine*‡ | Blood stages (trophozoite and schizont) | pfplm2 (REFS 37,38), pfcrt55,130 | Unknown | |
| Pyronaridine | Blood stages (ring, trophozoite and schizont) | pfcrt131 | Unknown | |
| Naphthoquine* | Blood stages (trophozoite and schizont) | Unknown | Unknown | |
| Amino alcohols | Quinine§ | Blood stages (trophozoite and stages I to III gametocytes) | pfcrt, pfmdr1 (REFS 51,132) | Unknown |
| Mefloquine* | Blood stages (trophozoite and schizont) | pfmdr1 (REFS 133–135) | pvmdr1 (REFS 81,82) | |
| Lumefantrine*‡ | Blood stages (trophozoite and schizont) | pfcrt, pfmdr1 (REFS 132,136) | Unknown | |
| Halofantrine§ | Blood stages (trophozoite and schizont) | pfcrt, pfmdr1 (REF. 133) | Unknown | |
| 8-Aminoquinoline | Primaquine*‡ | Blood (gametocyte) and liver (schizont) forms | Unknown | Unknown |
| Antifolates | Pyrimethamine* | Blood and liver schizont and mosquito stage (oocysts) | pfdhfr137 | pvdhfr83 |
| Sulfadoxine* | Blood and liver schizont | pfdhps137 | pvdhps83,138 | |
| Proguanil* | Blood stages (schizont and gametocyte) and liver schizont | pfdhfr139 | Unknown | |
| Naphthoquinone | Atovaquone§ | Blood stages (schizont and gametocyte) and liver schizont | pfcytb140 | Unknown |
| Antibiotics | Clindamycin§ | Blood stages | Apicoplast target57 | Unknown |
| Doxycycline§ | Blood stages | Apicoplast target57 | Unknown | |
| Tetracycline§ | Blood stages | Apicoplast target57 | Unknown | |
crt, chloroquine-resistance transporter; cytb, cytochrome b; dhfr, dihydrofolate reductase; dhps, dihydropteroate synthase; mdr1, multidrug resistance protein; pf, Plasmodium falciparum gene; pv, Plasmodium vivax gene; pfkelch13, P. falciparum Kelch 13; plm2, plasmepsin 2.
Drug used in artemisinin-based combination therapy.
Antimalarial drug used alone or in combination for the treatment of P. vivax malaria.
Antimalarial drug used alone or in combination with molecules other than artemisinin derivatives.
Box 1 |. Conversion of clinical phenotypes into molecular and laboratory readouts of artemisinin resistance.
Genome-wide association studies (GWAS) identified genetic variants that are associated with clinical artemisinin-resistant phenotypes and implicated two regions on chromosome 13 under high selection pressure in parasites from areas of southeast Asia22,58 (see the figure, part a). The gene encoding Plasmodium falciparum Kelch 13 (PfKelch13) was identified as a single genetic determinant of resistance; an African parasite strain, selected under high artemisinin pressure for several years, was shown to survive drug exposure in a newly developed resistant ring-stage survival assay (RSA). Whole-genome sequencing revealed the M476I mutation in the β-propeller domain of PfKelch13 (which is encoded by PF3D7_1343700). The major C580Y mutation of the PfKelch13 β-propeller was associated with ~80% of resistant strains in southeast Asia, with R539T and I543T mutations showing second and third place prevalence, respectively. Polymorphisms in pfkelch13 were rapidly mapped throughout southeast Asia and Africa (and, to a very limited degree, in Bangladesh and India)19,21,61,64,65,115. GWAS23 also established the presence of several artemisinin-resistant founder populations in Cambodia and Vietnam. The C580Y mutation arose independently in three different Cambodian founders along with polymorphisms in ferredoxin (pffd), apicoplast ribosomal protein S10 (pfarps10), multidrug resistance protein 2 (pfmdr2) and chloroquine-resistance transporter (pfcrt), which suggests that unexpected genetic interactions affect levels of resistance, parasite fitness and/or potential for transmission to mosquitoes. Artemisinin resistance in P. falciparum was first established in the clinic as delayed clearance of ring-stage parasites in patients (see the figure, part b, left panel)69,70. The graph shows increased delay in parasite clearance as resistance emerged from 2004 to 2015. The RSA (see the figure, part b, middle panel) exposes rings to short pulses of pharmacologically relevant plasma concentrations (700 nM) of dihydroartemisinin (DHA; the active metabolite of all artemisinins) for 6 h, thereby mimicking characteristically short drug exposure in patients86. After removal of DHA, parasites are cultured for another 66 h and assessed for survival. Early young ring-stage parasites (after 0–3 h) manifest the highest levels of resistance and are therefore used in the RSA. The assay provided the first robust correlation between reduced clearance of clinically resistant parasites and reduced killing by DHA in vitro. Determination of the median percentage of viable parasites at 72 h from culture-adapted isolates from the artemisinin-resistant strain from the Pailin province of western Cambodia revealed significantly higher survival in the RSA for this strain than for artemisinin-sensitive parasites from the Ratanakiri province of eastern Cambodia86,116. The RSA can be modified to test the efficacy of new drugs that are being developed in the antimalarial pipeline against diverse artemisinin-resistant parasites at different drug concentrations (see the figure, part b, right panel). Part a from REF. 23, Macmillan Publishers Limited. Part b left panel adapted from REF. 79. Part b right panel adapted from REF. 112.

Although the identification of genetic resistance markers advances detection, understanding the mechanisms of resistance enables the development of rational strategies for containment and treatment to eliminate malaria. Amplification of and/or SNPs in target catalytic enzymes or efflux pumps are known to confer resistance to many antimalarials25–28. The inhibitory activity of artemisinins depends on the cleavage of their endoperoxide bridge, which yields free radicals, suggesting that these drugs alkylate key target proteins to kill parasites. However, recent proteomic studies reveal that there are probably hundreds of targets, and thus, killing may be due to more generalized degeneration of the protein milieu (proteopathy)20,30. The gene encoding P. falciparum Kelch 13 (PfKelch13), which is located on chromosome 13, was shown to be the major causative artemisinin-resistance marker18,31,32. PfKelch13 is predicted to be a regulator of protein quality control. It belongs to a eukaryotic evolutionary gene family of ~60 members33. Mammalian orthologues of PfKelch13 confer resistance to cancer drugs that kill tumours by inducing proteopathy34. This supports the idea that PfKelch13-mediated atemisinin resistance may also have a role in restoring complex systems of protein functions (so-called proteostasis) in the parasite to promote its survival from artemisinin-induced proteopathy. Mechanistically, this is distinct from amplification or mutation of a single target enzyme or pump causing resistance.
Two major mechanisms of artemisinin resistance that have been independently proposed are the activation of the unfolded protein response (UPR)35 and proteostatic dysregulation of P. falciparum phosphatidylinositol 3-kinase (PfPI3K), which leads to increased levels of the lipid product phosphatidylinositol-3-phosphate (PtdIns3P)36. Whether those mechanisms function in one or parallel effector pathways of resistance remains unknown. Simultaneous resistance to partner drugs has accelerated clinical failure of ACTs37,38, emphasizing the need to better manage existing ACTs and other antimalarials as well as to develop potent new drug combinations through a discovery pipeline informed by mechanisms of resistance to ACTs. Recent studies also suggested that host immunity contributes to resistance39, which seems to be a unique characteristic of artemisinin resistance that may affect the elimination of malaria.
In this Review, we integrate key lessons learned from clinical, genetic and molecular analyses of P. falciparum and P. vivax drug treatment failure over the past 5 years, with a particular focus on artemisinin resistance. We discuss resistance markers and drug targets that have been identified, focusing on the two major effector pathways of artemisinin resistance: the UPR and PfPI3K. We emphasize how understanding mechanisms of drug resistance is critical in eliminating the parasites, which places high demands on detection, risk assessment, therapy and drug development to treat both symptomatic and asymptomatic infections with heterogeneous parasite burdens in high-transmission and low-transmission areas, all of which are dynamic frontiers on the path to the end game of malaria elimination.
Antimalarial drugs and resistance
Drug targets and resistance markers.
To date, not all molecular targets of known antimalarials that are currently in use are defined. Drug resistance can be mediated by a direct catalytic mechanism, or it can be due to amplification of the gene encoding the target enzyme or transporter that pumps the drug out of the parasite. Additionally, resistance can be mediated by processes that mitigate toxicity induced by the drug.
Sulfadoxine and pyrimethamine inhibit the P. falciparum enzymes dihydropteroate synthase (PfDhps) and dihydrofolate reductase (PfDhfr), respectively, which function in the folate pathway. Resistance to these antimalarials is conferred by dominant mutations in catalytic sites and/or amplification of the pfdhps and pfdhfr genes15,40–42. Multiple mutations in pfdhps and pfdhfr confer resistance to sulfadoxine-pyrimethamine in endemic regions, and parasites with quintuple mutations (pfdhfrN51I,C59R,S108N and pfdhpsA437G,K540E) have been linked to sulfadoxine-pyrimethamine resistance in Africa43. Sulfadoxine-pyrimethamine has been partnered with the artemisinin-based compound artesunate (Supplementary information S1 (table)). Double, triple and quadruple mutations in pfdhfr and pfdhps genes led to replacement of this drug combination with a second ACT composed of the artemisinin-based compound artemether and the partner drug lumefantrine in North East India44.
Atovaquone (of the naphthoquinone drug class) is partnered with proguanil in the drug Malarone (GlaxoSmithKline). Owing to its high cost as well as the emergence of resistance to atovaquone, Malarone is mainly used by travellers rather than resident populations of endemic countries. P. falciparum cytochrome b (PfCytB), which is a mitochondrial electron donor, is the target of atovaquone. Mutations in pfcytb that lead to changes in its catalytic activity render resistance to atovaquone28. In combination, atovaquone and proguanil (in its inactive prodrug form) dissipate mitochondrial membrane potential45. This synergistic action is lost in parasites with mutated pfcytb46; in particular, clinical failure of atovaquone is associated with pfcytbY268S/C/N. Notably, parasites with mutations in pfcytb (albeit not Y268S/C/N) show reduced transmission to mosquitoes. This has led to the suggestion that although atovaquone resistance may arise, it will not easily spread, and thus, atovaquone-proguanil may be useful for prophylaxis in elimination strategies (where it is important to block transmission to vectors)47.
Chloroquine targets the polymerization of free haem within the food vacuole of the parasite. In the food vacuole, haemoglobin that has been taken up from the host is digested into amino acids, which are used for parasite protein synthesis, and into Fe2+- containing haem48. Fe2+-containing haem is oxidized to Fe3+-containing protoporphyrin IX (FPIX), which is toxic to the parasite and therefore is converted to the polymer haemozoin (the black pigment of malaria). Chloroquine disrupts haemozoin formation. The major chloroquine-resistance mechanism is drug efflux via the P. falciparum chloroquine-resistance transporter (encoded by pfcrt) located at the food vacuole. An SNP, K76T, in pfcrt was universally associated with chloroquine resistance in Africa17 (TABLE 1), and globally, K76T and other additional mutations in pfcrt are associated with the development of chloroquine resistance26,49–52.
Mutations and amplification in broad, xenobiotic efflux pumps (such as P. falciparum multidrug resistance protein (pfmdrl), which encodes a protein that is also located in the membrane of the food vacuole) confer measurable levels of resistance to many antimalarials (TABLE 1). Mutations in and/or amplification of pfmdr1 confer resistance to partner drugs such as mefloquine and lumefantrine and thus limit effective treatment with ACTs53. Indeed, widespread amplification of pfmdr1 in southeast Asia led to replacement of artesunate and mefloquine by dihydroartemisinin (DHA; the active metabolite of all artemisinins) and PPQ. Although PPQ is structurally related to chloroquine, it is effective against parasites that harbour some resistance mutations in pfcrt and is therefore of value even in the presence of chloroquine resistance52,54. Notably, the introduction of the pfcrtC101F mutation into chloroquine-resistant P. falciparum parasites reduced chloroquine transport and thus rendered them chloroquine sensitive but PPQ resistant55. Together, these studies provide molecular evidence that different alleles of pfcrt differently affect resistance to chloroquine and PPQ.
Artemether-lumefantrine and artesunate- amodiaquine also interact with pfcrt and pfmdr1, but each ACT selects different resistance alleles in these genes56, suggesting that artesunate-amodiaquine is effective in treating parasites resistant to a partner drug such as lumefantrine. Parasite genetic profiles responsive to PPQ selection suggest that ACTs containing PPQ are also effective against resistance to lumefantrine (reviewed in REFS 56,57).
Risk factors for artemisinin resistance.
From 2008 to 2013, many parasite determinants were shown to be risk factors for artemisinin resistance, largely through population-transcriptomic and population-genetic studies22,58–60. These population-based tools were combined with extended laboratory drug selections (carried out over 5 years) to reveal mutations in the β-propeller domains of PfKelch13 (PfKelch13-propeller mutations) as the primary marker for artemisinin resistance in P. falciparum malaria18. Identification of PfKelch13 galvanized mapping the spread of resistance to these frontline antimalarials worldwide19,21,61 (FIG. 1b).
PfKelch13 is neither a transporter nor an enzyme. Rather, its structure reveals an amino-terminal BTB domain (common to all Broad-complex, Tramtrack and Bric-a-brac domain (BTB) family proteins; also known as the poxvirus and zinc-finger (POZ) domain) with six Kelch domains (from which it derives its name) (FIG. 2). The BTB domain promotes dimerization, while Kelch domains enable binding to specific substrates to accelerate their ubiquitylation and proteasomal degradation. Mutations in Kelch domains decrease substrate binding, ubiquitylation and targeting to proteasomes: the net effect is increased levels of substrate accumulation (and relocalization), which are frequently associated with mammalian genetic disorders33. Resistance mutations in pfkelch13 (FIG. 2a,b) have been detected throughout southeast Asia and Myanmar21 and have emerged by parasite spread and independently20,23. Notably, the major mutations in Myanmar differ from those in Cambodia21,62,63. Low-abundance Cambodian polymorphisms in pfkelch13 have been reported in India64,65 but were not associated with treatment failure. Nonetheless, the predicted path of artemisinin resistance from southeast Asia to Myanmar, through Bangladesh and India, seems to be similar to that by which chloroquine resistance became pervasive in Africa66,67.
Figure 2 |. Schematic and structural representation of Plasmodium falciparum Kelch 13 and its hypothesized function as a substrate adapter for a cullin E3 ligase.

a | Plasmodium falciparum Kelch 13 (PfKelchl3) contains a single BTB domain (common to all Broad-complex, Tramtrack and Bric-a-brac domain (BTB) family proteins). The BTB domain is a homodimerization domain at the amino terminus of proteins that contain multiple copies of Kelch repeats. PfKelch13 contains six β-propeller domains, also called Kelch domains, characteristic of families of Kelch proteins conserved in eukaryotes. The amino acid sequences of the PfKelch13 β-propeller domains are shown, with cysteine residues indicated in red (Cys447, Cys469, Cys473, Cys532, Cys542 and Cys696). C580Y (shown in orange) is the most prevalent resistance mutation in Cambodia. Cysteine mutations identified in Cambodia that have been validated in the laboratory (R539T and I543T32) and a major mutation (F446I) found in Myanmar62,63 as well as the M476I18 mutation (which confers high-level resistance in the laboratory) are indicated in yellow. Mutations that are not associated (A578S)61 or are less associated (Y493T) with resistance or that have been identified in clinical isolates in minor populations (R561H, Y493T and P574L) are indicated in green19. b | A 3D model of PfKelch13 amino acid residues 338–726 generated by pyMOL from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB; ID: 4YY8) shows R539T, I543T, F446I and M476I proximal to mutations in cysteine residues primarily in β strands and A578S and Y493T mutations in loops. c | The single BTB domain of PfKelch13 is expected to bind the cullin-really interesting new gene (RING)-E3 ubiquitin ligase complex, whereas the β-propeller Kelch domain binds substrate to facilitate ubiquitin transfer from the E2 ligase, which is also present in the complex, for degradation by the 26S proteasome. Mutation in (β-propeller) Kelch domains reduces the affinity of PfKelch13 for its substrates, their ubiquitylation and proteasomal degradation, thereby increasing substrate half-life in the cytoplasm. The major southeast Asia artemisinin-resistance mutation, PfKelch13-C580Y, has been shown to decrease PfKelch13 binding and ubiquitylation of P. falciparum phosphatidylinositol 3-kinase (PfPI3K), leading to increased levels of the kinase itself and of its lipid product phosphatidylinositol-3-phosphate (PtdIns3P), which confers artemisinin resistance36. The PfKelch13-R539T mutation was also found to increase PtdIns3P levels36.
By 2010, in all countries where malaria was endemic, ACTs were the recommended first-line therapy for uncomplicated P. falciparum malaria68 (Supplementary information S1 (table)). Unfortunately, partial resistance to artemisinins had emerged even earlier (as early as 2008)69,70, likely owing to historic use of artemisinin monotherapy in southeast Asia. ACTs substantially reduced parasite burdens in Africa71,72, but considerable levels of malaria remain. If extensive resistance to ACTs developed in Africa, it would have potential for seeding epidemics there. By contrast, malaria burdens are relatively low in Asia (and sometimes are reliably detected only by ultrasensitive PCR), but a large fraction of these parasites in asymptomatic individuals is resistant to artemisinins73. As transmission of these parasites could spread resistance, mass drug administration was recently undertaken with the ACT DHA-PPQ37,38. This combination was effective in clearing parasites from a majority of asymptomatic individuals, but it did not result in complete elimination and yielded parasite strains dually resistant to both DHA and PPQ, thereby providing a compelling rationale for developing a triple artemisinin combination therapy74.
Parasites from southeast Asia that are resistant to both DHA and PPQ show simultaneous polymorphisms in pfkelch13 and amplification of parasite proteases plasmepsin 2 and plasmepsin 3 (REFS 37,38). In the presence of resistance to the artemisinin-based drug alone, the partner drug functions as a monotherapy, which results in the rapid emergence of resistance to the partner as well, thereby generating dually resistant parasites. Moreover, ‘fit’ Cambodian PfKelch13-C580Y mutations of artemisinin resistance that spread rapidly may accelerate resistance to partner drugs such as PPQ75. Plasmepsin 2 and plasmepsin 3 are involved in haemoglobin digestion (in the food vacuole), and the acceleration of this process in resistant parasites may increase the release of free amino acids to the nutrient pool of the parasite. This may stimulate new protein synthesis to overcome inhibition of haemoglobin catabolism and haem detoxification by PPQ and support proteostasis mediated by PfKelch13-based artemisinin resistance. The link between PPQ resistance and amplification of plasmepsins suggests that PPQ targets early steps of haemoglobin digestion (in addition to terminal steps of haem polymerization). Consequently, resistance to drugs that target intermediate determinants of the haemoglobin degradation pathway may also contribute to multidrug resistance. As pfmdrl and pfcrt affect the activity of partner drugs53,76 (BOX 1), both determinants have an important role in multidrug resistance and ACT resistance (possibly even in ring stages that are now reported to degrade haemoglobin to release Fe2+-containing haem for endoperoxide cleavage and artemisinin action77, even though they lack a prominent food vacuole in which haemoglobin is degraded in later trophozoite and schizont stages). The prevalence of background mutations to partner drugs also renders ACT a monotherapy, thereby increasing the likelihood of the emergence of resistance to artemisinins. In sum, mechanisms of resistance to different individual drugs in an ACT may iteratively modulate multidrug resistance to emerging combination therapies.
A recent study reports on clinical artemisinin resistance (as detected by increased parasite clearance times in patients administered with drugs) in the absence of mutation in pfkelch13 (REF. 78). Studies on longitudinal genomic surveillance of artemisinin resistance suggest that after PfKelch13-C580Y, the strongest temporal changes are seen at SNPs in phosphatidylinositol 4-kinase and Sec14-like cytosolic factor or phosphatidylinositol-phosphatidylcholine transfer protein (a phosphatidylinositol-phosphatidylcholine exchange protein)79. Laboratory findings provide independent evidence that PtdIns3P alone is sufficient to confer resistance in the absence of pfkelch13 mutation36. Together, these studies suggest that polymorphisms and/or amplification of genes in the phosphoinositide pathways of the parasite are particularly insightful in tracking both PfKelch13-dependent and PfKelch13-independent resistance.
P. vivax infection is limited to reticulocytes, which are precursors to erythrocytes and present in very low numbers in blood. Therefore, P. vivax parasitaemia is not as high as that of P. falciparum, but this species causes substantial disease, including severe malaria80. There is widespread and increasing resistance of P. vivax to chloroquine and sulfadoxine-pyrimethamine (TABLE 1; Supplementary information S1 (table))81–84. ACTs are now used to treat malaria caused by P vivax (Supplementary information S1 (table))85. An orthologue of PfKelch13 exists in P. vivax (PvKelch13), but there are no reports yet of P. vivax resistance to artemisinins. The major limitation for the treatment of P vivax infections with ACTs is that latent liver-stage parasites are not targeted, and, therefore, relapse is not blocked. Latent infection with P. vivax in the liver can be targeted only by primaquine, the use of which is recommended in combination with blood-stage antimalarials (Supplementary information S1 (table)).
Challenges in identifying artemisinin-resistance markers.
The identification of PfKelch13 as a resistance marker presented unique challenges (BOX 1). To begin with, when clinical isolates with delayed in vivo clearance (characteristic of artemisinin resistance) were adapted to culture, the parasites failed to show concomitant drug-resistant survival in conventional in vitro half-maximal inhibitory concentration (IC50) assays. The ring-stage survival assay (RSA) (BOX 1) presented an important advance in the development of an in vitro correlate of clinical resistance that mimics the short exposure of ring parasites to the high plasma concentration of artemisinins (700 nM) seen in patients86. In addition, it took several years to select parasites in the laboratory that survived concentrations of DHA that were 1,000-fold higher than the IC50 of this drug. Both were needed to reveal mutation in pfkelch13 as a critical resistance event18. Exquisitely precise genome editing technologies31,32 provided genetic evidence that major polymorphisms in pfkelch13 that were associated with resistant clinical strains were indeed causal for artemisinin resistance as detected by the RSA across parasite genetic strains (BOX 2). Population studies established PfKelch13-C580Y as the major mutation (accounting for 80% of resistant cases) responsible for clinical artemisinin resistance in southeast Asia19. This mutation can result in high levels of resistance, although this varies depending on the genetic background of the parasite31,32. One explanation for this observed strain-specific variation in resistance is that although PfKelch13 is linked to resistance, additional genes (whose expression varies in different genetic strains) modify PfKelch13 function in artemisinin resistance. The mutation F446I found in parasites in Myanmar is associated with moderate clinical resistance62,63.
Box 2 |. Genome editing techniques to validate artemisinin resistance.
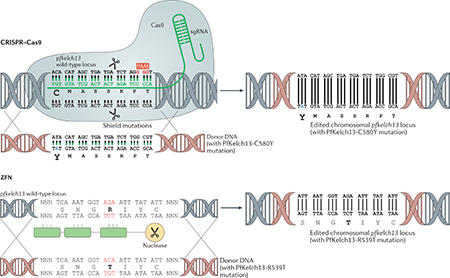
Genome editing in Plasmodium falciparum enables engineering at the chromosomal locus by use of CRISPR-Cas9 or zinc-finger nucleases (ZFNs). CRISPR-Cas9 technology is based on bacterial defence mechanisms whereby the Cas9 nuclease recognizes and eliminates foreign DNA from invading bacteriophages117. The site-specificity of the Cas9 endonuclease is provided by the single guide RNA (sgRNA), which enables complementary base pairing with the gene region of interest (for example, the P. falciparum Kelch 13 (PfKelch13) C580 position for the C580Y mutation) and directs the nuclease to DNA double-stranded breaks (DSBs; indicated by the scissors) (see the figure). DSBs are repaired by homologous recombination by use of donor DNA, as P. falciparum is deficient in error-prone non-homologous end-joining. In addition, the activity of Cas9 requires a short 2–5 nucleotide NGG sequence known as the protospacer-associated motif (PAM) in the vicinity. ‘Shield’ mutations make the edited site resistant to re-cleavage by Cas9. A recent study used CRISPR-Cas9 to engineer a single point mutation in the genome of the sensitive African strain PfNF54 to yield a PfNF54 strain with pfkelch13C580Y and proved that the pfkelch13 mutation was causal for artemisinin resistance31. ZFNs were the first site-specific genome editing nucleases used in P. falciparum118. ZFNs consist of an array of DNA-binding zinc-finger domains, with each finger recognizing a specific nucleotide triplet. The number of fingers can accordingly be custom-designed to target a specific region of the DNA and induce a DSB by the nuclease domain (scissors). The nucleases in ZFNs function as dimers, so the DNA-binding domains are engineered to recognize upstream and downstream regions for any sequence of interest to mediate a DSB. One study used ZFN technology to rapidly introduce or remove pfkelch13 mutations in P. falciparum reference strains and clinical isolates from Cambodia to prove that different mutations cause different levels of resistance (as measured by the RSA)32. They also showed that the major mutation C580Y induced different levels of resistance in different parasite genetic backgrounds.
Challenges remain in current assays for artemisinin and ACT resistance. For example, survival as expressed by the RSA describes only two parasite categories: those with an RSA value <1 (which are sensitive) and those with an RSA value >1 (which are resistant). Importantly, we do not understand why in a parasite population that is genetically homogenous in the resistance mutation C580Y (induced by an SNP in the genome of a sensitive strain), only a fraction of parasites survive drug exposure in the RSA (for example, 12–14% survival in the engineered strain PfNF54 with PfKelch13-C580Y compared with <1% survival in the wild-type PfNF54 strain). In other words, how do PfKelch13 mutants become heterogeneous in their phenotypic survival response to DHA even when they are genetically identical? There is urgent need to understand the mechanism and whether it accelerates acquisition of simultaneous resistance to a second partner drug.
Molecular mechanisms of artemisinin resistance
Plasmodium falciparum Kelch 13 structure and function.
The best studied mammalian orthologue of PfKelch13 is Kelch-like ECH-associated protein 1 (KEAP1), which functions by binding and ubiquitylating nuclear factor erythroid 2-related factor 2 (NFE2L2), a nuclear transcription factor, to control its protein levels87. Mutation of KEAP1 results in increased protein levels of NFE2L2 and its translocation to the nucleus. Therefore, in prior reviews of artemisinin resistance, emphasis had been given to the idea that PfKelch13 may also regulate an analogous transcriptional factor in the parasite88,89. However, in eukaryotes, the Kelch family comprises over 60 proteins with diverse substrates33. PfKelch13 belongs to the KBTBD subfamily, members of which contain a BTB domain (see above) that functions as an adaptor between E3 ubiquitin ligases and Kelch domains (FIG. 2c) to form active ubiquitylation complexes that regulate turnover of a wide range of substrates with distinct cellular functions and location90–92.
The structure of PfKelch13 does suggest a redox function owing to the presence of seven cysteine residues in the β-propeller domain (FIG. 2a,b) (the corresponding KEAP1 region contains eight cysteines). When cellular thiol levels become depleted and oxidative stress increases, cysteine residues in a redox sensor protein, or residues close to them, mutate and render a conformational change to compensate for the oxidative damage. When this occurs in a β-propeller domain, its target substrate is released, accumulates and may also change location to trigger a global antioxidative response (like a transcriptional signal to increase cellular thiol levels). In PfKelch13, mutations in the β-propeller domain are predicted to induce a conformational change and decrease the affinity of PfKelch13 for its target protein, which can potentially change both the levels and location of the target protein or target proteins. The C580Y mutation (the major artemisinin-resistance mutation in southeast Asia) shows reduced binding to and ubiquity- lation of PfPI3K, increasing the levels of the kinase itself and of its lipid product PtdIns3P36. The effects of other PfKelch13 mutations on PfPI3K binding are not known, but R539T also increases PtdIns3P36. In addition, R539T and another prevalent Cambodian mutation, I543T, and major Myanmar mutations F446I and M476I (a highly resistant laboratory mutation) are located within 1–3 amino acids of a cysteine residue, whereas lesser to minor mutations Y493T, R561H and P574L or polymorphisms not associated with resistance (A578S) are located in distal loop regions (FIG. 2a,b). It is possible that the extent of redox sensing by PfKelch13 may have a role in the level and/or prevalence of mutations in southeast Asia (and other areas globally). Mutations in the Kelch domain are expected to increase substrate levels: thus, redox sensing by PfKelch13 is also expected to increase substrate levels and their associated mechanisms of resistance (FIG. 2c).
Major effector pathways of artemisinin resistance: the unfolded protein response and Plasmodium falciparum phosphatidylinositol 3-kinase.
The activity of artemisinin against a parasite is dependent on cleavage of the endoperoxide bridge in the drug, and recent studies suggest that the resulting free radicals do not have a specific target but rather insert indiscriminately into proteins throughout the parasite. Thus, the killing mechanisms may be due to proteopathy (see above). Recovery from proteopathy requires multiple cellular functions that include removal of misfolded, aggregated toxic proteins and their replacement through new protein translation, translocation, folding and vesicular export, which together contribute to proteostasis.
Two major mechanisms of artemisinin resistance in P. falciparum have been proposed35,36. Population-based transcriptomic studies35 examined resistant clinical isolates of ring-stage parasites and provided evidence for the transcriptional increase of two major chaperone complexes, the reactive oxidative stress complex (ROSC) linked to the UPR and the T-complex protein 1 (TCP1) ring complex (TRiC). The studies suggested that the increased expression of those chaperone complexes mitigates toxic protein aggregates in the endoplasmic reticulum (ER) and cytoplasm by accelerating their removal through the proteasome (via the ROSC) or by restoring proper folding (via the TRiC). Other studies examined ubiquitylation-enhanced parasite stress and altered patterns of development in resistant para- sites93. A link between ubiquitylation and the UPR was not described, but proteasomal inhibitors were found to synergize with artemisinins in parasite killing93, a property that is also shared by selective inhibitors of the plasmodial proteasome in both drug-sensitive and drug-resistant parasites94. Proteasomal inhibitors also seem to synergize with other antimalarial compounds95. The function of the UPR (principally through the ROSC) is to re-establish ER homeostasis by overcoming imbalanced protein-folding capacity and more importantly by triggering a transcriptional response to stimulate gene expression and restore redox conditions throughout the cell. The UPR is one of multiple proteostasis mechanisms used to maintain quality control of protein folding in eukaryotes. We discuss four major relevant pathways and their potential pertinence for mechanisms of artemisinin resistance in P. falciparum malaria (FIG. 3).
Figure 3 |. Model of protein quality control in the endoplasmic reticulum lumen and cytoplasm of eukaryotes illustrating mechanisms of artemisinin resistance in Plasmodium falciparum.

In eukaryotes, proteostasis pathways function in protein quality control in the endoplasmic reticulum (ER) and the cytoplasm and stimulate protein translation, translocation, vesicular export and additional chaperone functions to restore proper folding and function of proteins. They have been proposed to rescue Plasmodium falciparum cells from artemisinin-induced protein alkylation, aggregation and toxicity (or proteopathy) and death. There are four major pathways that maintain quality control of proteins in the ER and cytoplasm of eukaryotes, as indicated in the figure. In the ER-associated degradation (ERAD) pathway (I), misfolded proteins are unfolded by binding to the ER chaperone immunoglobulin heavy chain-binding protein (BiP), translocated from the ER to the cytoplasm, ubiquitylated and targeted for proteasomal degradation by the ubiquitin-26S proteasome pathway (II). Misfolded proteins in the ER are initially removed by ERAD, but when this pathway gets saturated, BiP continues to bind to misfolded proteins in the ER lumen. BiP is a key component of the reactive oxygen stress complex (ROSC) pathway (III) in the ER lumen, which is regulated by the unfolded protein response (UPR). Under equilibrium conditions, BiP binds to and keeps inactive transmembrane ER stress-response proteins activating cAMP-dependent transcription factor (ATF6), inositol-requiring protein 1 (IRE1) and protein-kinase R (PKR)-like ER kinase (PERK; which targets eukaryotic translation initiation factor 2A, eIF2A, to suppress translation). Because BiP has a high preference for unfolded proteins, accumulation of these proteins in the ER lumen decreases the binding of BiP to transmembrane stress-response proteins, leading to their activation. In clinical isolates of artemisinin-resistant P. falciparum, increased transcript levels of the gene encoding BiP have been detected, possibly indicating a mechanism of artemisinin resistance to remove misfolded, aggregated toxic proteins and to activate putative transmembrane ER stress-response proteins. Because P. falciparum orthologues of PERK and eIF2A exist, activation of PERK might yield a UPR mechanism of translational repression and explain why artemisinin-resistant ring parasites may temporarily slow down. However, P. falciparum does not encode for orthologues of transcription factors of ATF6 and IRE1, which are the main drivers of UPR in eukaryotes. Therefore, these major UPR pathways of transcriptional activation of redox survival (needed for resistance) have yet to be established in P. falciparum. Thus, how the parasite induces the UPR-induced transcriptional increase of ROSC and the compensatory global antioxidant responses needed for resistance is unclear. Cytoplasmic unfolded and misfolded proteins may also be removed by autophagy (IV), including by macroautophagy, which involves phosphatidylinositol-3-phosphate (PtdIns3P)-dependent expansion of the ER. However, macroautophagy has not been established in P. falciparum. Chaperone-mediated autophagy is a second type of autophagy that also assists in the removal of misfolded proteins via interactions with cytoplasmic heat shock protein 70 (HSC70) and HSC90. T-Complex protein 1 (TCP1) ring complex (TRiC) chaperones enable misfolded proteins to become properly folded in the cytoplasm (and hence, TRiC substrates do not undergo chaperone-mediated autophagy). Resistant clinical isolates have been shown to have increased transcript levels of genes encoding TRiC chaperones. Thus, a model has been proposed whereby the transcriptional increase of two major chaperone complexes, the ROSC linked to the UPR and the TRiC, might mitigate toxic protein aggregates in the ER and cytoplasm by accelerating their removal through the proteasome (via the ROSC) or by restoring proper folding (via the TRiC). PfKelch13 mutations C580Y and R539T increase levels of the P. falciparum phosphatidylinositol 3-kinase (PfPI3K) and its lipid product phosphatidylinositol-3-phosphate (PtdIns3P). Thus, stimulation of macroautophagy by elevation of PtdIns3P suggests a second mechanism to remove toxic protein aggregates from the parasite.
The ER-associated degradation (ERAD) pathway is a steady-state mechanism by which misfolded proteins in the ER are unfolded, translocated back to the cytoplasm and ubiquitylated for subsequent degradation by the proteasome (FIG. 3). For our model, proteasomal degradation is limited to the 26S proteasome, as PfKelch13 mediates ubiquitylation (the 20S proteasome involved in ubiquitin-independent degradation is not considered here). When the ERAD pathway gets saturated, immunoglobulin heavy chain-binding protein (BiP; also known as HSPA5), which is the major ER chaperone and a key component of the ROSC, binds to luminal misfolded proteins, resulting in its disassociation from and activation of ER transmembrane sensors (to which BiP is usually bound). P. falciparum encodes the ER transmembrane sensor protein-kinase R (PKR)- like ER kinase (PERK; also known as eIF2AK3), which phosphorylates elongation initiation factor 2A (eIF2A), suggesting a mechanism of translational repression as an adaptive response to misfolded proteins that transiently delays maturation of mutant parasites96 (and also explains the morphologies of apparent arrest)97. Of note, P. falciparum lacks orthologues of ER transmembrane stress-response receptors such as activating transcription factor 6 (ATF6) and inositol-requiring protein 1 (IRE1) as well as ATF4, and thus, the transcriptional pathways leading to the increased expression of UPR components remain unknown. Initial analyses of PfKelch13 mutants provided evidence that BiP protein levels are not increased during even the peak of the ring stages of artemisinin-resistant parasites (that is, after 0–3 h)36. It is therefore possible that the pfkelch13 mutation alone is insufficient to induce the global transcriptional effect of the UPR. In clinical isolates of drug-resistant P. falciparum, there is an increase in transcription of BiP, TRiC proteins and ROSC components (other than BiP). This may also be a response to drug-induced formation of toxic protein aggregates. Parasite BiP has been reported as a high confidence protein, alkylated and aggregated after exposure to artemisinins29, which confirms that it can be a drug target. However, in the absence of transcription factor orthologues of ATF6, IRE1 or ATF4, the UPR alone seems limited to the parasite ER and to a cytoplasmic chaperone mechanism (through the TRiC), rather than global transcriptional reprogramming to activate redox-protective functions, and thus may be insufficient to rescue widespread artemisinin-induced proteopathy (as well as nucleic acid and lipid damage).
A second mechanism of resistance independently emerged from studies of PfPI3K, a parasite lipid kinase expressed during the blood stages36. Phylogenetic analyses revealed that PfPI3K is an orthologue of yeast phosphatidylinositol 3-kinase Vps34, which is a class III kinase with a single lipid product, PtdIns3P, that localizes in cytoplasmic orientation in the cell98,99. PfPI3K was found to be potently inhibited by DHA, which suggested that the kinase is a substrate for PfKelch13. Indeed, PfPI3K was shown to be isolated in a complex with PfKelch13, ubiquitylated and targeted for proteasomal degradation36. Moreover, evidence suggested that PfKelch13 resistance mutations abrogate binding to and ubiquitylation of PfPI3K and lead to increased levels of the kinase and its lipid product PtdIns3P (FIGS 2c,3). Evidence was also presented that overexpression of PtdIns3P induced resistance independent of mutation of PfKelch13. In yeast (which, like P. falciparum, contains only a single PI3K), Vps34 regulates autophagy (FIG. 3), which can remove misfolded proteins in the cytoplasm. Similarly, phosphatidylinositol 4,5-bisphosphate 3-kinase (PIK3), a mammalian orthologue of Vps34, is central to PtdIns3P-dependent expansion of the autophagosome, which leads to macroautophagy100. However, little is known about macroautophagy in P. falciparum. In P. falciparum, PtdIns3P has been detected at the apicoplast, food vacuole and ER101–103. Most of these studies used fluorescent reporter systems, and it will be important to localize endogenous PtdIns3P in the parasite. Malaria parasites contain a subset of autophagy- related genes (ATG), including autophagy-associated protein Atg8 (atg8), which is required for the formation of autophagosomal membranes and has been shown to be expressed in blood-stage parasites104,105. Atg8 has been localized to the apicoplast as well as to other vesicles in the parasite, but macroautophagy was not yet shown to occur at the apicoplast. Macroautophagy could in principle represent a vesicle-dependent mechanism to remove toxic aggregates and misfolded proteins that have been damaged by artemisinins at any cellular location. As membrane vesicles are derived from the ER, they may also be coupled to increased translation and other proteostasis mechanisms (including nuclear processes) to restore cellular homeostasis and parasite survival and, in principle, to confer resistance to any drug that functions by alkylation of cellular targets.
Whether the increases in the levels of PtdIns3P (through proteostatic control of PfPI3K; see below) and the ROSC indicate common and/or related mechanisms of resistance has sparked discussion88,89. Mutations in pfkelch13 are expected to lead to increased levels of cytoplasmic protein substrates such as PfPI3K. Mutations in pfkelch13 may also increase levels of cytoplasmic chaperones such as those in the TRiC and in the ROSC in the ER lumen (but the mechanisms of increase remain unknown). Notably, protein misfolding is thermodynamically variable, and the degree of protein folding (and the functional activity of the protein) varies; therefore, the amplitude of resistance may vary among individual parasites (FIG. 4). Mutants with a higher amplitude of resistance may be better equipped to remove misfolded or aggregated proteins induced by artemisinins and survive (FIG. 4), possibly explaining why isogenic pfkelch13 mutations show heterogeneity in survival in the RSA (BOX 1). Further, in addition to PfKelch13, a wide range of mechanisms of parasite protein quality control may potentially confer artemisinin resistance (FIG. 3).
Figure 4 |. A working model for heterogeneity in levels of artemisinin resistance.

In wild-type parasites, in addition to properly folded proteins, misfolded or poorly folded proteins are also made. These ill-folded proteins are bound by Plasmodium falciparum Kelch 13 (PfKelch13), ubiquitylated and targeted to the proteasome for degradation, which keeps their levels low in the parasite. In PfKelch13 mutants, in the absence of ubiquitylation and protein quality control, PfKelch13 substrates (blue) accumulate with varying levels of folding and activity (shown by numbers of loops and intensity of blue colour, respectively). PfKelch13 substrates such as P. falciparum phosphatidylinositol 3-kinase (PfPI3K) may function in pathways that restore protein folding or remove aggregates to mitigate the toxicity induced by the drug. Therefore, genotypically identical parasites have heterogeneous resistance mechanisms to remove the toxic misfolded aggregates induced by artemisinins. As such, following artemisinin-induced toxicity due to protein alkylation and oxidation, only parasites with sufficient resistance mechanisms (which allow clearance of toxic intermediates and restoring of protein folding) survive and proliferate.
Therapy and risk assessment
The identification of resistance markers and mechanisms of resistance guides treatment strategies. Of the ACTs available, PPQ in combination with artemisinin derivatives is expected to be recommended as the first- line treatment or as preventive treatment for infants or pregnant women in Africa. The combined analyses of pfkelch13 polymorphisms and plasmepsin 2 copy number (along with pfmdr1 amplification) provide a molecular prediction kit to enable continued treatment with ACT that is based on risk analyses in the population. A gold standard of artemisinin resistance is delayed clearance of parasites as directly measured in a patient in the clinic, and thus, this metric of drug resistance is expected to be influenced by the host immune response39. Higher levels of immunity are expected to accelerate parasite clearance and mask resistance. Haemoglobinopathies, which alter the redox properties of host erythrocytes, may also influence the effectiveness of artemisinins owing to the increased levels of oxidative stress in infected blood cells106,107. This suggests that both the moderately high parasite burdens still seen in regions of Africa, which elicit a protective immune response, and the health status of the host modulate the spread of artemisinin resistance. Because of this, it will be important to monitor clinical polymorphisms that emerge using RSAs (and other laboratory assays that do not take host innate and adaptive immunity into account). This will flag the risk of potential clinical resistance that may emerge as both parasite burden and corresponding immunity wane. In southeast Asia, mefloquine will be added to DHA-PPQ and amodiaquine will be added to artemether-lumefantrine to generate triple ACTs as an elimination strategy74. This needs careful monitoring because mefloquine can affect heart rhythms and has neurological side effects. Mass drug administration in southeast Asia is further challenged by the emergence of PfKelch13-independent artemisinin resistance that may also occur together with resistance to a second drug (such as PPQ) and that is not invariably linked to amplification and/or mutation of the multidrug resistance transporter PfMdrl (REF. 78).
Outlook
The antimalarial discovery pipeline (reviewed in REF. 108) aims to develop next-generation single-dose therapies that contain both fast-acting (to replace artemisinins) and long-acting (to replace partners) drugs. Of the fast-acting synthetic endoperoxides, OZ277 (which has moved to registration) shows cross-resistance to the pfkelch13 mutations C580Y, R539T and I543T, which are associated with artemisinins, whereas other compounds show cross-resistance with less prevalent pfkelch13 mutations (R539T and I543T)109,110. This suggests that mechanisms of resistance to synthetic endoperoxides and artemisinin are similar but that some endoperoxides in the antimalarial pipeline slow the spread of the maj or artemisinin-resistance mutation, PfKelch13-C580Y. Lead compounds that are not as fast-acting111,112 may be suitable for triple combinations with the caveat that they may be less effective if they are effluxed out of parasites owing to amplification of pfmdrl, which is often found in multidrug-resistant parasites53. As increased levels of PtdIns3P have been shown to induce resistance, PFPI3K presents a potentially important target, but new inhibitor series have not yet been described. Mechanistic studies suggest that resistance to artemisinins can affect many cellular targets. Therefore, it will be important to develop new drugs with known targets to strengthen discovery strategies for compounds that are fast-acting and long-acting against blood-stage parasites113 as well as to develop drugs that target transmission to the mosquito vector114.
As elimination strategies are focused in local or regional efforts, the distribution of malaria has become highly heterogeneous worldwide. It will be important to comprehensively sample parasite populations. Measurement of clinical parasite clearance demands infrastructure for appropriate patient monitoring, which may be impossible to achieve in severely resource-restricted areas (which is also where malaria burdens remain the highest). The RSA requires adapting clinical samples to culture, which is difficult to achieve at the low parasitaemia seen in southeast Asia. PfKelch13-independent mechanisms cannot yet be detected by genome sequencing. This argues for future validation through functional assays that capture the entire resistance pathway and that can be executed without culture and with small amounts of blood to reduce the resources needed in high-transmission areas and to justify engagement of asymptomatic populations in low-transmission settings. Eliminating a major microbial pathogen with drugs alone in individuals with very low parasite burdens that do not elicit a substantial protective immune response may also yield hitherto unknown biological paradigms of resistance and detection.
Supplementary Material
Sporozoites.
A malaria parasite stage that is injected by the mosquito and that infects liver cells.
Merozoites.
A malaria parasite stage that infects red blood cells (also known as erythrocytes).
Endoperoxide.
A peroxide group (O-O) that bridges two atoms of a larger molecule and whose cleavage gives rise to reactive free radicals that can oxidize and aggregate proteins (as well as DNA and lipids).
Proteopathy.
A disease state where proteins become structurally abnormal and disrupt cellular function.
Proteostasis.
A network process or system that integrates translation, signalling pathways, molecular chaperones and protein degradation to enable cells to control the abundance and folding of the proteome.
Parasitaemia.
The number of infected red cells per total number of red cells.
Autophagosome.
A double-membraned vesicle containing cellular material slated to be degraded by autophagy.
Macroautophagy.
A process by which a membrane or phagophore forms near cargo and then expands until it encloses the cargo, which is subsequently degraded by autophagy. Macroautophagy depends on phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) and its lipid product phosphatidylinositol-3- phosphate (PtdIns3P).
Apicoplast.
An organelle that is a remnant of a non-photosynthetic plastid found in many apicomplexan parasites, including Plasmodium falciparum.
Haemoglobinopathies.
Single-gene disorders that result in abnormal structure of the haemoglobin molecule.
Acknowledgements.
The authors apologize to colleagues whose work could not be cited owing to the broad scope of the Review and space limitation. They thank members of the Haldar laboratory for insightful discussion. Work in the authors’ laboratories was supported by the US National Institutes of Health (R01 HL069630 and HL130330) and India Government Department of Science and Technology (ECR/2015/000387) and Department of Biotechnology Ramalingaswami Re-entry Fellowship (BT/HRD/35/02/2006).
Footnotes
Competing interests statement
The authors declare no competing financial interest.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Microbiology thanks Didier Menard, and the other anonymous reviewer(s), for their contribution to the peer review of this work.
DATABASES
RSCB Protein Data Bank: http://www.rcsb.org/pdb/home/home.do
SUPPLEMENTARY INFORMATION
See online article: S1 (table)
References
- 1.World Health Organization. World Malaria Report 2015 (WHO, 2015). [Google Scholar]
- 2.World Health Organization. World Malaria Report 2016 (WHO, 2016). [Google Scholar]
- 3.Guerra CA et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLOS Negl. Trop. Dis 4, e774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haldar K, Murphy SC, Milner DA & Taylor TE Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu. Rev. Pathol 2, 217–249 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Milner DA Jr. et al. Quantitative assessment of multiorgan sequestration of parasites in fatal pediatric cerebral Malaria. J. Infect. Dis 212, 1317–1321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okell LC, Drakeley CJ, Ghani AC, Bousema T & Sutherland CJ Reduction of transmission from malaria patients by artemisinin combination therapies: a pooled analysis of six randomized trials. Malar. J 7, 125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen M & Mehlhorn H Seventy-five years of Resochin in the fight against malaria. Parasitol. Res 105, 609–627 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Jong EC & Nothdurft HD Current drugs for antimalarial chemoprophylaxis: a review of efficacy and safety. J. Travel Med 8, S48–S56 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Mawson A Mefloquine use, psychosis, and violence: a retinoid toxicity hypothesis. Med. Sci. Monit 19, 579–583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdrager J Epidemiology of the emergence and spread of drug-resistant falciparum malaria in South-East Asia and Australasia. J. Trop. Med. Hyg 89, 277–289 (1986). [PubMed] [Google Scholar]
- 11.Verdrager J Localized permanent epidemics: the genesis of chloroquine resistance in Plasmodium falciparum. Southeast Asian J. Trop. Med. Public Health 26, 23–28 (1995). [PubMed] [Google Scholar]
- 12.Payne D Spread of chloroquine resistance in Plasmodium falciparum. Parasitol. Today 3, 241–246 (1987). [DOI] [PubMed] [Google Scholar]
- 13.Gesase S et al. High resistance of Plasmodium falciparum to sulphadoxine/pyrimethamine in Northern Tanzania and the emergence of dhps resistance mutation at codon 581. PLOS ONE 4, e4569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mixson-Hayden T et al. Evidence of selective sweeps in genes conferring resistance to chloroquine and pyrimethamine in Plasmodium falciparum isolates in India. Antimicrob. Agents Chemother 54, 997–1006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah NK et al. Antimalarial drug resistance of Plasmodium falciparum in India: changes over time and space. Lancet. Infect. Dis 11,57–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meshnick SR, Taylor TE & Kamchonwongpaisan S Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol. Rev 60, 301–315 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Djimde A et al. A molecular marker for chloroquine- resistant falciparum malaria. N. Engl. J. Med 344, 257–263 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Ariey F et al. A molecular marker of artemisinin resistant Plasmodium falciparum malaria. Nature 505, 50–55 (2014).This study presents the identification of the first marker of artemisinin resistance in P. falciparum malaria, which was shown to be causal in later genetic studies.
- 19.Ashley EA et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 371,411–423 (2014).This is the first of a series of clinical and epidemiological studies showing that delayed clearance time of artemisinin resistance in patients was associated with mutations in the β-propeller domain of PfKelch13 and with their spread in southeast Asia.
- 20.Takala-Harrison S et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis 211, 670–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tun KM et al. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect. Dis 15, 415–421(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheeseman IH et al. A major genome region underlying artemisinin resistance in malaria. Science 336, 79–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miotto O et al. Genetic architecture of artemisinin- resistant Plasmodium falciparum. Nat. Genet 47, 226–234 (2015).This GWAS identifies the population structure of clinically resistant parasites and the genomic complexity of artemisinin resistance in southeast Asia.
- 24.Fidock DA, Rosenthal PJ, Croft SL, Brun R & Nwaka S Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discov 3, 509–520 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Fidock AD et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861–871 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidhu ABS, Verdier-Pinard D & Fidock DA Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298, 210–213 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Triglia T, Wang P, Sims PF, Hyde JE & Cowman AF Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J. 17, 3807–3815 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korsinczky M et al. Mutations in Plasmodium falciparum Cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob. Agents Chemother 44, 2100–2108 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ismail HM et al. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl Acad. Sci 113, 2080–2085 (2016).This study, together with reference 30, suggests ‘proteopathic’ toxicity of artemisinins.
- 30.Wang J et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun 6, 10111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghorbal M et al. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol 32, 819–821 (2014).This is the first report of the use of CRISPR-Cas9 in P. falciparum to engineer a single point mutation in the genome of the sensitive African strain PfNF54 to yield a PfNF54 strain with the PfKelch13-C580Y mutation, which shows an RSA value of 13–14; this study also proves that the pfkelch13 mutation was causal for artemisinin resistance.
- 32.Straimer J et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347, 428–431 (2015).In this study, zinc-finger nuclease technology is used to rapidly introduce or remove PfKelch13 β-propeller mutations in P falciparum reference strains and clinical isolates from Cambodia to prove that different mutations cause different levels of resistance (as measured by the RSA), the extent of which is sensitive to the genetic backgrounds of the parasites.
- 33.Gupta VA & Beggs AH Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet. Muscle 4, 11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikesitch N & Ling SC Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol 69, 97–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mok S et al. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347, 431–435 (2015).This study presents transcriptomic signatures of 1,000 clinical strains to reveal that the induction of the parasite UPR is associated with artemisinin resistance.
- 36.Mbengue A et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520, 683–687 (2015).This study shows that mutations of PfKelch13 increase PfPI3K to elevate PtdIns3P and confer artemisinin resistance.
- 37.Amato R et al. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype- phenotype association study. Lancet Infect. Dis 17, 164–173 (2017).This study, together with reference 38, identifies the molecular markers associated with P falciparum that are dually resistant to ACT with artemisinins and PPQ.
- 38.Witkowski B et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect. Dis 17, 174–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ataide R et al. Host immunity to Plasmodium falciparum and the assessment of emerging artemisinin resistance in a multinational cohort. Proc. Natl Acad. Sci. USA 114, 3515–3520 (2017).This is the first study investigating the effect of immunity on the parasite clearance rates associated with artemisinin resistance.
- 40.Tumwebaze P et al. Changing antimalarial drug resistance patterns identified by surveillance at three sites in Uganda. J. Infect. Dis 215, 631–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costa GL et al. Assessment of copy number variation in genes related to drug resistance in Plasmodium vivax and Plasmodium falciparum isolates from the Brazilian Amazon and a systematic review of the literature. Malar. J 16, 152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson DS, Walliker D & Wellems TE Evidence that a point mutation in dihydrofolate reductase- thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc. Natl Acad. Sci. USA 85, 9114–9118 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregson A & Plowe CV Mechanisms of resistance of malaria parasites to antifolates. Pharmacol. Rev 57, 117–145 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Mishra N et al. Declining efficacy of artesunate plus sulphadoxine-pyrimethamine in northeastern India. Malar. J 13, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srivastava IK & Vaidya AB A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob. Agents Chemother 43, 1334–1339 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaidya AB in Treatment and Prevention of Malaria: Antimalarial Drug Chemistry, Action and Use (eds Henry Staines M & Krishna Sanjeev) 127–139 (Springer; Basel, 2012). [Google Scholar]
- 47.Goodman CD & Buchanan HD & McFadden GI Is the mitochondrion a good malaria drug target? Trends Parasitol. 33, 185–193 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Sigala PA & Goldberg DE The peculiarities and paradoxes of Plasmodium heme metabolism. Annu. Rev. Microbiol 68, 259–278 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Nkrumah LJ et al. Probing the multifactorial basis of Plasmodium falciparum quinine resistance: evidence for a strain-specific contribution of the sodium-proton exchanger PfNHE. Mol. Biochem. Parasitol 165, 122–131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper RA et al. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharmacol 61, 35–42 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Cooper RA et al. Mutations in transmembrane domains 1,4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol. Microbiol 63, 270–282 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Petersen I et al. Balancing drug resistance and growth rates via compensatory mutations in the Plasmodium falciparum chloroquine resistance transporter. Mol. Microbiol 97, 381–395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Veiga MI et al. Globally prevalent PfMDRl mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat. Commun 7, 11553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pascual A et al. In vitro piperaquine susceptibility is not associated with the Plasmodium falciparum chloroquine resistance transporter gene. Malar. J 12, 431 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhingra SK et al. A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio 8, e00303–00317 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkatesan M et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate- amodiaquine. Am. J. Trop. Med. Hyg 91,833–843 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blasco B, Leroy D & Fidock DA Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat. Med 23, 917–928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takala-Harrison S et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc. Natl Acad. Sci. USA 110, 240–245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miotto O et al. Multiple populations of artemisinin resistant Plasmodium falciparum in Cambodia. Nat. Genet 45, 648–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mok S et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics 12, 391 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ménard D et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N. Engl. J. Med 374, 2453–2464 (2016).This study provides a summary of the distribution of polymorphisms of pfkelch13 that are linked to artemisinin resistance (or not) on a global scale.
- 62.Huang F et al. A single mutation in K13 predominates in Southern China and is associated with delayed clearance of Plasmodium falciparum following artemisinin treatment. J. Infect. Dis 21 2, 1629–1635 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tun KM et al. Parasite clearance rates in Upper Myanmar indicate a distinctive artemisinin resistance phenotype: a therapeutic efficacy study. Malar. J 15, 185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mishra N et al. Surveillance for artemisinin resistance in Plasmodium falciparum in India using the kelch13 molecular marker. Antimicrob. Agents Chemother. 59, 2548–2553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mishra N et al. Emerging polymorphisms in falciparum Kelch 13 gene in Northeastern region of India. Malar. J 15, 583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korenromp EL, Williams BG, Gouws E, Dye C & Snow RW Measurement of trends in childhood malaria mortality in Africa: an assessment of progress toward targets based on verbal autopsy. Lancet Infect. Dis 3, 349–358 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Trape JF et al. Impact of chloroquine resistance on malaria mortality. C. R. Acad. Sci III 321,689–697 (1998). [DOI] [PubMed] [Google Scholar]
- 68.World Health Organization. Global report on antimalarial drug efficacy and drug resistance: 2000–2010. (WHO, 2010). [Google Scholar]
- 69.Noedl H et al. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med 359, 2619–2620 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Dondorp AM et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 361, 1714 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barnes KI et al. Effect of artemether-lumefantrine policy and improved vector control on malaria burden in KwaZulu-Natal, South Africa. PLOS Med. 2, e330 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhattarai A et al. Impact of artemisinin-based combination therapy and insecticide-treated nets on malaria burden in Zanzibar. PLOS Med. 4, e309 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phommasone K et al. Asymptomatic Plasmodium infections in 18 villages of southern Savannakhet Province, Lao PDR (Laos). Malar. J 15, 296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Worldwide Antimalarial Resistance Network. Tracking resistance to artemisinin collaboration II. WWARN; http://www.wwarn.org/Working-Together/Partner-Projects/Tracking-Resistance-Artemisinin-Collaboration-ii(2017). [Google Scholar]
- 75.Imwong M et al. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet. Infect. Dis 5, 491–497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Srimuang K et al. Analysis of anti-malarial resistance markers in pfmdr1 and pfcrt across Southeast Asia in the tracking resistance to artemisinin collaboration. Malar. J 15, 541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xie SC et al. Haemoglobin degradation underpins the sensitivity of early ring stage Plasmodium falciparum to artemisinins. J. Cell Sci 129, 406–416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mukherjee A et al. Artemisinin resistance without pfkelch13 mutations in Plasmodium falciparum isolates from Cambodia. Malar. J 16, 195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cerqueira GC et al. Longitudinal genomic surveillance of Plasmodium falciparum malaria parasites reveals complex genomic architecture of emerging artemisinin resistance. Genome Biol. 18, 78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price RN et al. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg 77, 79–87 (2007). [PMC free article] [PubMed] [Google Scholar]
- 81.Suwanarusk R et al. Amplification of pvmdr1 associated with multidrug-resistant Plasmodium vivax. J. Infect. Dis 198, 1558–1564 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Imwong M et al. Gene amplification of the multidrug resistance 1 gene of Plasmodium vivax isolates from Thailand, Laos, and Myanmar. Antimicrob. Agents Chemother 52, 2657–2659 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu F et al. Mutations in the antifolate-resistance- associated genes dihydrofolate reductase and dihydropteroate synthase in Plasmodium vivax isolates from malaria-endemic countries. Am. J. Trop. Med. Hyg 83, 474–479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Price RN et al. Global extent of chloroquine- resistant Plasmodium vivax: a systematic review and meta-analysis. Lancet. Infect. Dis 14, 982–991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Douglas NM, Anstey NM, Angus BJ, Nosten F & Price RN Artemisinin combination therapy for vivax malaria? Lancet Infect. Dis 10, 405–416 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Witkowski B et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet. Infect. Dis 13, 1043–1049 (2013).This study presents the development of an in vitro correlate of in vivo P. falciparum artemisinin- resistant malaria.
- 87.Taguchi K & Yamamoto M The KEAP1-NRF2 System in Cancer. Front. Oncol 7, 85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fairhurst RM & Dondorp AM Artemisinin- Resistant Plasmodium falciparum Malaria. Microbiol. Spectr 4, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paloque L, Ramadani AP, Mercereau-Puijalon O, Augereau JM & Benoit-Vical F Plasmodium falciparum: multifaceted resistance to artemisinins. Malar. J 15, 149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sambuughin N et al. KBTBD13 interacts with Cullin 3 to form a functional ubiquitin ligase. Biochem. Biophys. Res. Commun 421,743–749 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geyer R, Wee S, Anderson S, Yates J & Wolf DA BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol. Cell 12, 783–790 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Canning P et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem 288, 7803–7814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dogovski C et al. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLOS Biol. 13, e1002132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H et al. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530, 233–236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prasad R et al. Blocking Plasmodium falciparum development via dual inhibition of hemoglobin degradation and the ubiquitin proteasome system by MG132. PLOS One 8, e73530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang M et al. PK4, a eukaryotic initiation factor 2α (eIF2a) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl Acad. Sci 109, 3956–3961 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng Q, Kyle DE & Gatton ML Artemisinin resistance in Plasmodium falciparum: a process linked to dormancy? Int. J. Parasitol. Drugs Drug Resist 2, 249–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vanhaesebroeck B et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem 70, 535–602 (2001). [DOI] [PubMed] [Google Scholar]
- 99.Vaid A, Ranjan R, Smythe WA, Hoppe HC & Sharma P PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood 115, 2500–2507 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dall’Armi C, Devereaux, Kelly A & Di Paolo G The role of lipids in the control of autophagy. Curr. Biol 23, R33–R45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tawk L et al. Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryot. Cell 9, 1519–1530 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tawk L et al. Phosphatidylinositol 3-monophosphate is involved in toxoplasma apicoplast biogenesis. PLOS Pathog. 7, e1001286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bhattacharjee S, Stahelin RV, Speicher KD, Speicher DW & Haldar K Endoplasmic reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell 148, 201–212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cervantes S et al. The multifunctional autophagy pathway in the human malaria parasite. Plasmodium falciparum. Autophagy 10, 80–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitamura K et al. Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLOS ONE 7, e42977 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cyrklaff M et al. Oxidative insult can induce malaria- protective trait of sickle and fetal erythrocytes. Nat. Commun 7, 13401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kavishe RA, Koenderink JB & Alifrangis M Oxidative stress in malaria and artemisinin combination therapy: pros and cons. FEBS J. 284, 2579–2591 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Phillips MA et al. Malaria . Nat Rev. Dis. Primers 3, 17050 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Straimer J et al. Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. mBio 8, e00172–e00117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O’Neill PM et al. A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance. Nat. Commun 8, 15159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dembele L et al. The Plasmodium PI(4)K inhibitor KDU691 selectively inhibits dihydroartemisinin- pretreated Plasmodium falciparum ring-stage parasites. Sci. Rep 7, 2325 (2017).References 111–114 present several potential new therapeutics from the antimalaria discovery and development pipeline.
- 112.Le Bihan A et al. Characterization of novel antimalarial compound ACT-451840: preclinical assessment of activity and dose-efficacy modeling. PLOS Med. 13, e1002138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McCarthy JS et al. Safety, tolerability, pharmacokinetics, and activity of the novel long- acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet. Infect. Diseases 17, 626–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baragana B et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mohon A et al. Mutations in Plasmodium falciparum K13 propeller gene from Bangladesh (2009–2013). Malar. J 13, 431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Witkowski B et al. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob. Agents Chemother 57, 914–923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bhaya D, Davison M & Barrangou R CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet 45, 273–297 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Straimer J et al. Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat. Methods 9, 993–998 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Amaratunga C et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect. Dis 16, 357–365 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Phyo AP et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 379, 1960–1966 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thriemer K et al. Delayed parasite clearance after treatment with dihydroartemisinin-piperaquine in Plasmodium falciparum malaria patients in central Vietnam. Antimicrob. Agents Chemother 58, 7049–7055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Z et al. Prevalence of K13-propeller polymorphisms in Plasmodium falciparum from China-Myanmar border in 2007–2012. Malar. J 14, 168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ye R et al. Distinctive origin of artemisinin-resistant Plasmodium falciparum on the China-Myanmar border. Sci. Rep 6, 20100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thanh NV et al. Rapid decline in the susceptibility of Plasmodium falciparum to dihydroartemisinin- piperaquine in the south of Vietnam. Malar. J 16, 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boulle M et al. Artemisinin-resistant Plasmodium falciparum K13 mutant alleles, Thailand-Myanmar border. Emerg. Infect. Dis 22, 1503–1505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gama BE, Lacerda MV, Daniel-Ribeiro CT & Ferreira-da-Cruz Mde F Chemoresistance of Plasmodium falciparum and Plasmodium vivax parasites in Brazil: consequences on disease morbidity and control. Mem. Inst. Oswaldo Cruz 106, 159–166 (2011). [DOI] [PubMed] [Google Scholar]
- 127.Goncalves LA, Cravo P & Ferreira MU Emerging Plasmodium vivax resistance to chloroquine in South America: an overview. Mem. Inst. Oswaldo Cruz 109, 534–539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Delves M et al. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 9, e1001169 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sa JM et al. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc. Natl Acad. Sci. USA 106, 18883–18889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Agrawal S et al. Association of a novel mutation in the Plasmodium falciparum chloroquine resistance transporter with decreased piperaquine sensitivity. J. Infect. Dis 216, 468–476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Madamet M et al. The Plasmodium falciparum chloroquine resistance transporter is associated with the ex vivo P. falciparum African parasite response to pyronaridine. Parasit. Vectors 9, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sidhu ABS et al. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis 194, 528–535 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sisowath C et al. In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by artemether- lumefantrine (Coartem). J. Infect. Dis. 191, 1014–1017 (2005). [DOI] [PubMed] [Google Scholar]
- 134.Sidhu AB, Valderramos SG & Fidock DA pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol. Microbiol 57, 913–926 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Price RN et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364, 438–447 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sisowath C et al. The role of pfmdr1 in Plasmodium falciparum tolerance to artemether-lumefantrine in Africa. Trop. Med. Int. Health 12, 736–742 (2007). [DOI] [PubMed] [Google Scholar]
- 137.Uhlemann AC & Krishna S Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr. Top. Microbiol. Immunol 295, 39–53 (2005). [DOI] [PubMed] [Google Scholar]
- 138.Imwong M et al. Limited polymorphism in the dihydropteroate synthetase gene (dhps) of Plasmodium vivax isolates from Thailand. Antimicrob. Agents Chemother 49, 4393–4395 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Parzy D et al. Proguanil resistance in Plasmodium falciparum African isolates: assessment by mutation- specific polymerase chain reaction and in vitro susceptibility testing. Am. J. Trop. Med. Hyg 57, 646–650 (1997). [DOI] [PubMed] [Google Scholar]
- 140.Gil JP et al. Detection of atovaquone and Malarone resistance conferring mutations in Plasmodium falciparum cytochrome b gene (cytb). Mol. Cell. Probes 17, 85–89 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.