

Abstract
Both circulating adiponectin (APN) and cardiac APN exert cardioprotective effects and improve insulin sensitivity and mitochondrial function. Low circulating APN serves as a biomarker for cardiovascular risk. Ablation of adiponectin receptor 1 (AdipoRl) causes myocardial mitochondrial dysfunction. Although high salt intake is a contributor to cardiovascular disease, how it modulates the expression of APN or AdipoRl in cardiomyocytes is not known. We report that APN mRNA expression was attenuated in a dose-dependent manner in mouse cardiomyocyte cell line HL-1 exposed to salt concentrations ranging from 0.75% to 1.5% for 12 h. High-salt exposure (0.88% and 1.25% for 12 h) also suppressed APN and AdipoRl protein expression significantly in rat cardiac muscle H9c2 cells. Co-immunostaining for AdipoR1 and mitochondrial complex 1 indicated that AdipoR1 may be co-localized with mitochondria. These data show for the first time that high salt is an important suppressor of cardiovascular protective APN and AdipoR1.
Keywords: adiponectin, adiponectin receptor type 1, salt, cardiovascular disease
Résumé :
L’adiponectine (APN) en circulation comme l’APN cardiaque ont des effets cardioprotecteurs et améliorent la sensibilité à l’insuline ainsi que le fonctionnement des mitochondries. Les faibles taux d’APN en circulation servent de biomarqueurs pour le risque cardiovasculaire. L’ablation des récepteurs 1 de l’APN (AdipoR1) entraîne un dysfonctionnement des mitochondries dans le myocarde. Bien qu’un apport élevé en sel contribue à la maladie cardiovasculaire, on ne sait pas comment ce facteur assure la modulation de l’expression de l’APN ou de l’AdipoR1 dans les cardiomyocytes. Nous rapportons que l’expression de l’ARNm de l’APN était atténuée de manière proportionnelle à la dose dans des lignées HL-1 de cardiomyocytes murins exposées à des concentrations de sel se situant entre 0,75 % et 1,5 % pendant 12 h. L’exposition à des taux de sel élevés (à 0,88 % et 1,25 % pendant 12 h) a aussi entraîné une inhibition importante de l’expression de l’APN et de la protéine AdipoR1 dans des cellules de muscle cardiaque H9c2 de rat. La co-immunocoloration de l’AdipoR1 et du complexe mitochondrial 1 a montré que l’AdipoR1 pourrait être colocalisée avec la mito-chondrie. Ces données montrent pour la première fois qu’un taux de sel élevé est un important facteur de suppression des effets protecteurs de l’APN et de l’AdipoR1 sur le système cardiovasculaire. [Traduit par la Rédaction]
Keywords: adiponectine, récepteur de l’adiponectine de type 1, sel, maladie cardiovasculaire
Introduction
Adiponectin (APN) is an anti-inflammatory, anti-apoptotic, and anti-atherogenic adipokine that improves insulin sensitivity and renders cardiovascular protection (Shibata et al. 2004; van Stijn et al. 2014). Accumulating clinical evidence indicates that circulating APN levels are inversely correlated to obesity, diabetes, and hypertension (Davis et al. 2015; Lee et al. 2016; Mantovani et al. 2016). Thus, low circulating APN levels are considered a risk factor for these metabolic diseases. Moreover, genetic polymorphisms in the APN gene modulate blood pressure responses to dietary sodium and potassium intake (Chu et al. 2016). However, in conditions of chronic heart failure, APN resistance is observed because, in such conditions, circulating APN levels are increased with no effect on insulin resistance (Sente et al. 2016). It is unclear what sources contribute to this increase in circulating APN in chronic heart failure. Although adipose tissue is the main source of APN, recent studies have identified localized production of APN in skeletal muscle cells and cardiomyocytes (Amin et al. 2010; Skurk et al. 2008). The exact roles of these local APN systems are yet to be elucidated.
APN receptors include adiponectin receptor 1 (AdipoRl) that is primarily expressed by skeletal muscle and AdiopoR2 that is mainly expressed by liver (Silva et al. 2014; Singh et al. 2017). The predominant APN receptor expressed in cardiomyocytes is AdipoR1 AdipoR1 deficiency in the heart causes myocardial mitochondrial dysfunction in mice and highlights the critical role of APN and its receptors in mitochondrial function (Koentges et al. 2015). Previous studies have shown that APN and its receptors are expressed in adult ventricular cardiomyocytes (Ding et al. 2007). In diabetic rats, cardiac expression of APN and its receptors is suppressed. ngiotensin II (Ang II) type 1 receptor (AT1R) inhibitor telmisar-tan, and activation of peroxisome proliferator-activated receptor gamma by rosiglitazone, elevate cardiac expression of APN and its receptors in diabetic rats (Guo et al. 2012a, 2012b).
Although Ang II infusion and activation of renin-angiotensin-aldosterone system suppress circulating APN, Ang II has an opposing effect on cardiac APN. In neonatal rat ventricular myocytes, Ang II induces elevation of APN mRNA and this effect is mediated via the Ang II type 2 receptor (AT2R)/nitric oxide/cGMP/protein kinase G signaling pathway (Guo et al. 2011). Because AT2R/nitric oxide/cGMP/protein kinase G signaling pathway attenuates Ang II-induced hypertension, increase in APN via this pathway in cardiomyocytes could be a protective mechanism. Both Ang II and high-salt diet are major contributors to cardiovascular diseases including left ventricular hypertrophy and chronic heart failure (He et al. 2011). Interestingly, high-salt-induced dysregulation of glucose homeostasis could be reversed by activation of PPARδ/ APN-mediated inhibition of SGLT2 (Zhao et al. 2016). To date, there are no studies that show the effect of high salt on the expression of cardiac APN or APNR1. This study was undertaken to examine the effect of high salt on the expression of APN and APNR1 under in vitro conditions using atrial and ventricular cardiac muscle cell lines of rat (H9c2) and mouse (HL-1).
Materials and methods
Cell culture and treatments
The cardiac ventricular cell line H92C was grown in Dulbeco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (both from ATCC) and 1% penicillin/streptomycin (Thermo Fisher). Mouse atrial cardiomyocyte HL-1 cells were a gift from William Claycomb at Louisiana State University Medical Center and cultured as described previously (Arnold et al. 2014; Mahmood and Pulakat 2015). For Ang II and salt treatments (12 h), cells were maintained in serum-free medium supplemented with Ang II (100 nmol/L), or salt (NaCl) concentrations adjusted to be between 0.75% and 1.5% of salt.
RNA isolation and quantitative real-time RT-PCR
Isolation of mRNA from HL-1 cells was performed using mir-Vana miRNA isolation kit (Ambion) following the manufacturer’s protocol. The mRNA was quantified using NanoDrop (Thermo Scientific) and stored at −80 °C until further processing. c-DNA synthesis for mRNA and qRT-PCR was performed as described previously (Arnold et al. 2014). Taqman primers for mouse AdipoQ and 18S RNA (gene expression assays) from Applied Biosystems Life Technologies were used. Experiments were performed in triplicates for each biological sample with Taqman Fast Universal PCR Master Mix 2X (Applied Biosystems Life Technologies). 18S RNA was used as internal control. Relative quantification values were obtained by determining ΔCT values followed by determining ΔΔCT values and then relative quantification values via the equation 2(-ΔΔCT).
Immunofluorescence
Immunofluorescence analysis of the expression of APN1 and AdipoRl in H9c2 cells in response to treatments with salt or Ang II or their combination was performed using anti-APN and anti-AdipoRl antibodies (Abcam Inc., 1:50 dilution) and Alexa Fluor 488 or Alexa Fluor 594 goat anti-rabbit (Abcam Inc.) (1:200 dilution) as described previously (Arnold et al. 2014; Mahmood and Pulakat 2015). Analysis of co-localization of AdipoR1 with mitochondria was performed by double immunostaining using anti-AdipoR1 and anti-mitochondrial complex 4 subunit 1 (MTCO1) antibodies. All experiments were done in at least triplicates.
Statistics
Statistical analysis was performed using the SPSS 20 software package. Results were expressed as mean ± SEM (standard error of mean). Differences among groups were tested by using one-way ANOVA followed up with Tukey’s test or Student’s t test, as appropriate, and two-tailed p values are reported. A p value of 0.05 was considered statistically significant.
Results
Regulation of APN by salt and Ang II in mouse atrial cardiac muscle cell line HL-1
As shown in Fig. 1A, exposure of HL-1 cells to salt (0.75%−1.5%) significantly suppressed APN (Adipoq) mRNA expression in a dose-dependent manner. In contrast, exposure to Ang II (100 nmol/L) increased APN (Adipoq) mRNA levels and this is consistent with previous reports (Guo et al. 2011). To our knowledge, this is the first report that shows high salt suppresses endogenous expression of APN in cardiac cells.
Fig. 1.
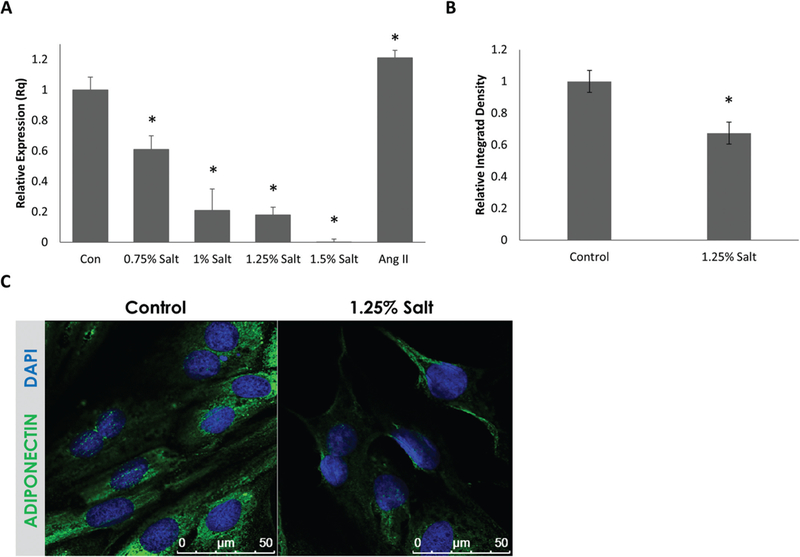
Adiponectin (APN) (AdipoQ) mRNA levels are suppressed by high-salt conditions in a dose-dependent manner in mouse atrial HL-1 cells and APN protein expression is suppressed by high-salt conditions in rat ventricular cardiac muscle cell line H9c2. (A) qRT-PCR data on the expression of APN (AdipoQ) mRNA in response to high-salt conditions in mouse HL-1 cells. APN mRNA expression was suppressed in a dose-dependent manner by high-salt conditions (n = 3 biological replicates). (B) Immunofluorescent intensity quantification (by integrated density) of rat H9c2 probed for APN. Expression of APN was significantly suppressed in cells treated with 1.25% NaCl containing medium. (C) Representative immunofluorescent images showing APN expression in H9c2 cells (n > 15). Values are means ± SEM. DAPI, 4’,6-diamidino-2-phenylindole, fluorescent nuclear stain. *, p < 0.05 vs. Control.
Regulation of APN by salt in rat ventricular cardiac muscle cell line H9c2
To further validate that high salt suppresses APN expression in cardiac cells, we tested the effect of 1.25% salt on APN protein expression in rat cardiac muscle cell line H9c2. As shown in Figs. 1B and 1C, immunofluorescence analysis indicated that APN protein expression was suppressed by 33% in H9c2 cells in response to 1.25% salt.
AdiopoR1 is also affected by high-salt conditions, while Ang II can partially mitigate this effect
To further investigate the impact of high-salt exposure on the APN signaling in cardiac muscle, we investigated how it modulates AdipoR1 protein expression in H9c2 cells. As shown in Fig. 2, AdipoR1 protein expression was significantly suppressed in H9c2 cells exposed to 1.25% salt (50%) and 0.88% salt (21%). Ang II (100 nmol/L) did not significantly affect AdipoR1 protein expression under these experimental conditions. Addition of Ang II with 1.25% salt did not rescue salt-induced loss of AdipoR1 protein expression. However, addition of Ang II with 0.88% salt could restore AdipoR1 protein expression to levels comparable with control. Thus, Ang II could partially mitigate loss of AdipoR1 expression in response to salt when salt concentration was not very high. To our knowledge, this is the first report that shows high-salt exposure suppresses AdipoR1 expression.
Fig. 2.
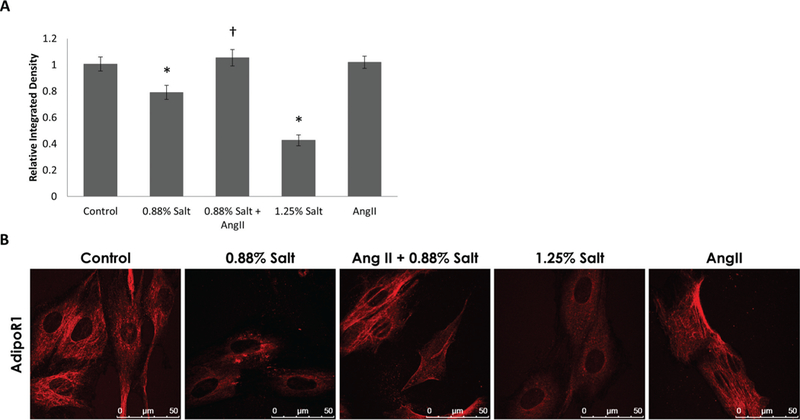
Adiponectin receptor (AdipoRl) protein expression is suppressed by high-salt conditions in H9c2 cells and Ang II could partially restore AdipoRl protein expression. (A) Graph shows immunofluorescence intensity quantification (by integrated density) for rat H9c2 probed for AdipoRl. Suppression of AdipoRl was observed in response to 0.88% and 1.25% salt treatment and this effect was reversed by 0.88% Salt + Ang II (n = 3 for each group, average of 40 cells measured per group). (B) Representative images of H9c2 cells stained with anti-AdipoRl (scale bars = 50 μm). Values are means ± SEM. *, p < 0.05 vs. Control; †, p < 0.05 vs. 0.88% Salt.
AdipoR1 may be co-localized with mitochondria
AdipoR1 is a 7-transmembrane domain receptor that is structurally and functionally distinct from classical G-protein coupled receptors, because it has an inverted membrane topology with acytoplasmic amino terminus and a short extracellular carboxyl terminus. AdipoR1 deficiency in myocardium is reported to induce mitochondrial dysfunction (Koentges et al. 2015). Because this observation highlights the crucial role of AdipoR1 in mitochondrial structure and function, we investigated whether AdipoR1 is present on mitochondria. As shown in Fig. 3, co-immunostaining with anti-AdipoR1 and anti-MTCO1 (mitochondria marker) showed that AdipoR1 seems to be present on mitochondria, in addition to the cell membrane.
Fig. 3.
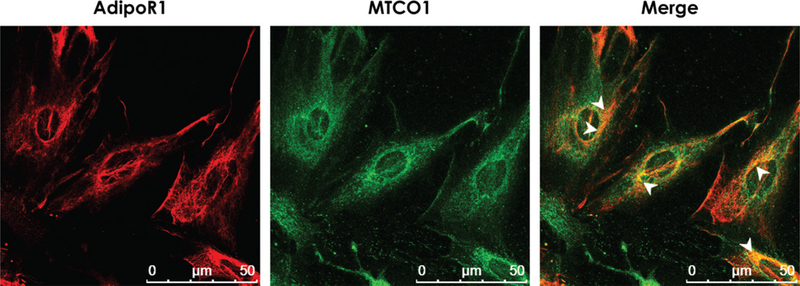
Adiponectin receptor 1(AdipoR1)may be present on mitochondria.Representative images generated by co-staining of H9c2 cell for AdipoR1 and mitochondrial complex 4 subnit 1 (MTCO1:as a mitochondrial marker) shows a pattern of co-localization between the 2 proteins,suggesting the presence of the AdipoR1 on the mitochondrial surface. Orange color reprents overlap of the fluorescent signals. Areas of significant overlap are indicated by white arrowheads.
Discussion
Accumulating clinical evidence show that a low circulating level of APN is an independent risk factor for cardiovascular disease, whereas patients with high circulating APN have a significantly reduced risk of myocardial infarction even after adjustment for body mass index, history of diabetes, and hypertension (Koentges et al. 2015; Lee et al. 2016; Mantovani et al. 2016). Mice lacking APN exhibit enhanced concentric cardiac hypertrophy following pressure overload and increased myocardial infarct size following ligation of the left anterior descending coronary artery, and these pathologies could be ameliorated by administration of recombinant APN (Shibata et al. 2004, 2005). APN is synthesized and secreted by human and murine cardiomyocytes and endogenously produced APN protects cardiomyocytes from hypertrophy induced by a high-fat diet (Amin et al. 2010). Additionally, loss of AdipoR1 induced myocardial mitochondrial dysfunction whereas loss of AdipoR2 did not have such effect. Thus, APN-AdipoR1 signaling has an important role in cardiomyocyte survival and mitochondrial function.
Despite extensive epidemiological evidence that suggests a high-salt diet is an independent predictor of heart disease along with heart failure, the exact mechanisms by which high salt increases this susceptibility is not clear. Given the role of APN-AdipoR1 signaling in cardiomyocyte survival, we hypothesized that high salt may suppress this cardioprotective signaling. Data presented here show that APN expression can be suppressed in cardiac muscle cell lines derived from mouse atrium (HL-1) and rat ventricle (H9c2) by high-salt conditions. Although these cell lines do not exhibit all properties of primary cardiomyocytes, they are widely used as surrogates to study cardiac signaling mechanisms. We found that APN (AdipoQ) mRNA expression was suppressed in a dose-dependent manner by high-salt conditions and, at a salt concentration of 1.5%, there was almost no expression of AdipoQ mRNA in HL-1 cells as detected by quantitative RT-PCR (Fig. 1A). The fact that 18S mRNA expression in the HL-1 cells treated with 1.5% salt was similar to that of untreated HL-1 cells validated the severe suppression of AdipoQ mRNA expression by 1.5% salt. Previous studies have shown that Ang II elevates expression of APN in neonatal cardiomyocytes (Guo et al. 2011). Consistent with this, we also observed an elevation of AdipoQ mRNA in HL-1 cells in response to Ang II treatment.
The suppression of APN protein by high-salt conditions in H9c2 cells further confirmed the fact that salt is a negative regulator of APN expression (Fig. 1B). In H9c2 cells, Ang II treatment did not significantly affect AdipoR1 expression. However, Ang II treatment could rescue 0.88%-salt-induced suppression of AdipoR1 protein expression (Fig. 2). These observations indicate a novel cross talk between salt-induced signaling and Ang II that is mediated via APN. However, Ang II treatment was ineffective in restoring the AdipoR1 protein expression in the presence of 1.25% salt (Supplementary Fig. S12). Therefore, the protective effect of Ang II in improving AdipoR1 expression in the presence of high salt is dependent on the extent of salt in the medium.
The heart tissues of mice lacking AdipoRl gene expression showed significant mitochondrial structural and functional damage (Koentges et al. 2015). Thus, AdipoR1 has a critical role in mitochondrial survival. However, it is not known whether AdipoR1 is present on the mitochondrial membranes. Data presented here suggest that MTCO1 seems to co-localize with AdipoR1. Therefore, it is tempting to speculate that endogenous APN in cardiac muscle cells may directly regulate AdipoR1 signaling in an intracrine manner and protect mitochondrial structure and function.
Collectively, our data shows for the first time, to our knowledge, a novel pathway by which high-salt conditions can lead to suppression of cardioprotective APN and its receptor AdipoR1 directly within cardiomyocytes (Fig. 4). Considering that high-salt diet is associated with cardiac hypertrophy, diastolic dysfunction, and heart failure, the role of suppression of cardioprotective APN and AdipoR1 by high-salt diet warrants further investigation.
Fig. 4.
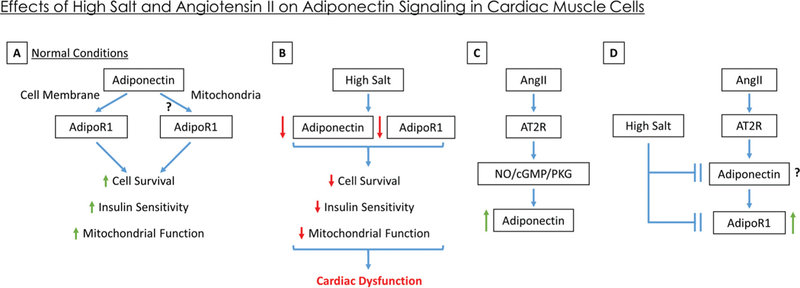
Schematic representation of the effect of high salt and Angiotensin II (Ang II) on cardiac adiponectin signaling. (A) In normal conditions, it is known that adiponectin acts through the adiponectin receptor (AdipoR1) present on the cell surface to improve cardiac muscle survival, insulin sensitivity, and mitochondrial function. Our data suggest presence of AdipoR1 on the mitochondrial surface and imply a potential intracrine signaling for adiponectin via mitochondrial AdipoR1. (B) In conditions of high salt, both adiponectin and AdipoR1 expression are suppressed, which decreases cell survival, insulin sensitivity, and mitochondrial function, ultimately leading to cardiac dysfunction. (C) Treatment of cardiac cells with Ang II increases adiponection expression via Ang II Type 2 (AT2R)-mediated activation of the nitric oxide/cGMP/Protein Kinase G pathway (as shown in literature; Guo et al. 2011). (D) Co-treatement of cardiac cells in high salt with Ang II abolishes the suppressive effects of high salt on AdipoR1 expression (returning it to normal levels) while their effect on cardiac adiponectin needs to be studied. [Color online.]
Supplementary Material
Acknowledgements
This work was supported, in part, by the Life Science Mission Enhancement Fund from UM-Columbia (to L.P.), as well as research grant NIH NHLBI1R01HL118376–01 (to L.P.). This work was also supported by resources, including the use of facilities, from Research Services, Harry S. Truman Memorial Veterans’ Affairs Hospital.
Footnotes
This Brief Report is part of a Special Issue of selected papers from the 3rd Cardiovascular Forum for Promoting Centers of Excellence and Young Investigators held in Omaha, Nebraska, USA, on 10–12 September 2015.
Copyright remains with the author(s) or their institution(s). Permission for reuse (free in most cases) can be obtained from RightsLink.
A correction was made to the e-First version of this paper on 4 January 2017 prior to the final issue publication. The current online and print versions are identical and both contain the correction.
Contributor Information
Nicholas Arnold, Department of Medicine, University of Missouri, Columbia, MO, USA; Harry S. Truman Memorial Veterans’ Affairs Hospital, Columbia, MO, USA..
Abuzar Mahmood, Department of Medicine, University of Missouri, Columbia, MO, USA; Harry S. Truman Memorial Veterans’ Affairs Hospital, Columbia, MO, USA..
Maya Ramdas, Department of Biological Sciences, Mississippi State University, Starkville, MS, USA..
Paul P. Ehlinger, Department of Medicine, University of Missouri, Columbia, MO, USA; Harry S. Truman Memorial Veterans’ Affairs Hospital, Columbia, MO, USA.
Lakshmi Pulakat, Department of Medicine, University of Missouri, Columbia, MO, USA; Harry S. Truman Memorial Veterans’ Affairs Hospital, Columbia, MO, USA; Department of Nutrition and Exercise Physiology, University of Missouri, Columbia, MO, USA..
References
- Amin RH, Mathews ST, Alli A, and Leff T 2010. Endogenously produced adiponectin protects cardiomyocytes from hypertrophy by a PPARgamma-dependent autocrine mechanism. Am. J. Physiol. Heart Circ. Physiol. 299(3): H690–H698. doi: 10.1152/ajpheart.01032.2009. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold N, Koppula PR, Gul R, Luck C, and Pulakat L 2014. Regulation of cardiac expression of the diabetic marker microRNA miR-29. PLoS One, 9(7): e103284. doi: 10.1371/journal.pone.0103284. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Wang Y, Ren K. y., Yan D. y., Guo T. s., Zheng W. l., et al. 2016. Genetic variants in adiponectin and blood pressure responses to dietary sodium or potassium interventions: a family-based association study. J. Hum. Hypertens. 30: 563–570. doi: 10.1038/jhh.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SK, Gebreab SY, Xu R, Riestra P, Khan RJ, Sumner AE, et al. 2015. Association of adiponectin with type 2 diabetes and hypertension in African American men and women: the Jackson Heart Study. BMC Cardiovasc. Disord. 15:13. doi: 10.1186/s12872-015-0005-5. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding G, Qin Q, He N, Francis-David SC, Hou J, Liu J, et al. 2007. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J. Mol. Cell. Cardiol. 43(1): 73–84. doi: 10.1016/j.yjmcc.2007.04.014. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Li Y, Han R, Zhou H, and Wang M 2011. Angiotensin II upregulation of cardiomyocyte adiponectin production is nitric oxide/cyclic GMP dependent. Am. J. Med. Sci. 341(5): 350–355. doi: 10.1097/MAJ.0b013e318203abd5. PMID:. [DOI] [PubMed] [Google Scholar]
- Guo Z, Zhang R, Li J, and Xu G 2012a. Effect of telmisartan on the expression of adiponectin receptors and nicotinamide adenine dinucleotide phosphate oxidase in the heart and aorta in type 2 diabetic rats. Cardiovasc. Diabetol. 11: 94. doi: 10.1186/1475-2840-11-94. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Qin Z, Zhang R, Li J, and Yin Y 2012b. Effect of rosiglitazone on the expression of cardiac adiponectin receptors and NADPH oxidase in type 2 diabetic rats. Eur. J. Pharmacol. 685(1–3): 116–125. doi: 10.1016/j.ejphar.2012.04.010. PMID:. [DOI] [PubMed] [Google Scholar]
- He FJ, Burnier M, and Macgregor GA 2011. Nutrition in cardiovascular disease: salt in hypertension and heart failure. Eur. Heart J. 32(24): 3073–3080. doi: 10.1093/eurheartj/ehr194. PMID:. [DOI] [PubMed] [Google Scholar]
- Koentges C, König A, Pfeil K, Hölscher ME, Schnick T, Wende AR, et al. 2015. Myocardial mitochondrial dysfunction in mice lacking adiponectin receptor 1. Basic Res. Cardiol. 110(4): 37. doi: 10.1007/s00395-015-0495-4. PMID: . [DOI] [PubMed] [Google Scholar]
- Lee JJ, Britton KA, Pedley A, Massaro JM, Speliotes EK, Murabito JM, et al. 2016. Adipose tissue depots and their cross-sectional associations with circulating biomarkers of metabolic regulation. J. Am. Heart Assoc. 5(5): e002936. doi: 10.1161/JAHA.115.002936. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood A, and Pulakat L 2015. Differential effects of β-blockers, angiotensin II receptor blockers, and a novel AT2R agonist NP-6A4 on stress response of nutrient-starved cardiovascular cells. PLoS One, 10(12): e0144824. doi: 10.1371/journal.pone.0144824. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani RM, Rocha NP, Magalhães DM, Barbosa IG, Teixeira AL, and Simões e Silva AC 2016. Early changes in adipokines from overweight to obesity in children and adolescents. Jornal de Pediatria, 92: 624–630. doi: 10.1016/j.jped.2016.02.015. PMID:. [DOI] [PubMed] [Google Scholar]
- Sente T, Van Berendoncks AM, Hoymans VY, and Vrints CJ 2016. Adiponectin resistance in skeletal muscle: pathophysiological implications in chronic heart failure. J. Cachexia Sarcopenia Muscle, 7(3): 261–274. doi: 10.1002/jcsm.12086. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. 2004. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 10(12): 1384–1389. doi: 10.1038/nm1137. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. 2005. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 11: 1096–1103. doi: 10.1038/nm1295. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TE, Colombo G, and Schiavon LL 2014. Adiponectin: a multitasking player in the field of liver diseases. Diabetes Metab. 40(2): 95–107. doi: 10.1016/j.diabet.2013.11.004. PMID:. [DOI] [PubMed] [Google Scholar]
- Singh AK, Shree S, Chattopadhyay S, Kumar S, Gurjar A, Kushwaha S, et al. 2017. Small molecule adiponectin receptor agonist GTDF protects against skeletal muscle atrophy. Mol. Cell Endocrinol. 439: 273–285. doi: 10.1016/j.mce.2016.09.013. PMID:. [DOI] [PubMed] [Google Scholar]
- Skurk C, Wittchen F, Suckau L, Witt H, Noutsias M, Fechner H, et al. 2008. Description of a local cardiac adiponectin system and its deregulation in dilated cardiomyopathy. Eur. Heart J. 29: 1168–1180. doi: 10.1093/eurheartj/ehn136. PMID:. [DOI] [PubMed] [Google Scholar]
- van Stijn CM, Kim J, Barish GD, Tietge UJ, and Tangirala RK 2014. Adiponectin expression protects against angiotensin II-mediated inflammation and accelerated atherosclerosis. PLoS One, 9(1): e86404. doi: 10.1371/journal.pone.0086404. PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Gao P, Sun F, Li Q, Chen J, Yu H, et al. 2016. Sodium intake regulates glucose homeostasis through the PPARS/adiponectin-mediated SGLT2 pathway. Cell Metab. 23(4): 699–711. doi: 10.1016/j.cmet.2016.02.019. PMID:. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.