

Abstract
Human melanocortin-4 receptor (hMC4R) mutations have been implicated as the cause for about 6–8% of all severe obesity cases. Drug-like molecules that are able to rescue the functional activity of mutated receptors are highly desirable to combat genetic obesity among this population of patients. One such molecule is the selective MC4R agonist RM-493 (setmelanotide). While this molecule has been shown to activate mutated receptors with 20-fold higher potency over the endogenous agonist, little is known about its binding mode and how it effectively interacts with hMC4R despite the presence of mutations. In this study, a MC4R homology model was constructed based on the X-ray crystal structure of the adenosine A2A receptor in the active state. Four MC4R mutations commonly found in genetically obese patients and known to effect ligand binding in vitro were introduced into the constructed model. RM-493 was then docked into the wild-type and mutated models in order to better elucidate the possible binding modes for this promising drug candidate and assess how it may be interacting with MC4R to effectively activate receptor polymorphisms. The results reflected the orthosteric interactions of both the endogenous and synthetic ligands with the MC4R, which is supported by the site-directed mutagenesis studies. Meanwhile it helped explain the decremental affinity and potency of these ligands with the receptor polymorphisms. More significantly, our findings indicated that the structural characteristics of RM-493 may allow for enhanced receptor-ligand interactions, particularly through those with the putative allosteric binding sites, which facilitated the ligand to stabilize the active state of native and mutant MC4Rs to maintain reasonably high affinity and potency.
Keywords: obesity, MC4R agonist, mutations, homology model, docking, allosteric binding modes
Graphical Abstract
1. INTRODUCTION
G-protein coupled receptors (GPCRs) are a superfamily of membrane proteins that exist in most organ systems throughout the body and are involved in numerous physiological processes as well as numerous disease pathogeneses. Due to their many important roles, GPCRs have been heavily exploited for drug discovery and development efforts. It is estimated that approximately 35% of the drugs on the market today target GPCRs.1 Even with the success of GPCR ligands in drug design and development, it has been difficult to attain ligands that are highly specific among receptor subtypes. Efforts to design ligands that target GPCRs have primarily focused on the orthosteric binding site, simulating the endogenous substrate. While this approach has afforded a multitude of drug-like compounds, the orthosteric binding site is highly conserved across GPCR subtypes, making selectivity a challenge. Therefore, several drug discovery efforts have sought to explore allosteric binding sites.2–5 These are alternative, druggable sites on GPCRs separate from the orthosteric binding site. These sites are less conserved among GPCR subtypes, thus making them an appealing target for the design of selective GPCR ligands. There are generally three categories of ligands that target the allosteric sites. These are positive allosteric modulators (PAMs), which enhance the receptor response to the orthosteric ligand, negative allosteric modulators (NAMs), which decrease the receptor response to the orthosteric ligand, and silent allosteric modulators (SAMs), which bind to the allosteric site but have no effect on receptor response to the orthosteric ligand.2–5 These ligands are highly desirable as they are generally more selective and are devoid of the negative side effects that accompany orthosteric ligands.6–11 However, due to their ulterior binding site, allosteric ligands tend to have a lower affinity than orthosteric ligands and are typically only able to exert their function in the presence of the orthosteric ligand. Recently a newer class of ligands that simultaneously target both the orthosteric and allosteric binding sites has emerged in research. These ligands are termed “bitopic” ligands.12–16 Traditionally these ligands are comprised of an orthosteric ligand connected to an allosteric ligand by a linker. The aim in developing these ligands is producing compounds with both high affinity (via orthosteric sites) and high selectivity (via allosteric sites).
Despite our increased understanding of GPCR binding sites, both orthosteric and allosteric, the dynamic nature of GPCRs and their physiological role in membranes has made it difficult to solve their 3D structures. This has proven to be a limiting factor in the discovery of novel orthosteric, allosteric, and recently bitopic ligands. Therefore, construction of homology models of GPCRs with therapeutic potential is an important tool to better understand their putative 3D structure and exploit these results in the design of novel molecules. In this report, the construction of such homology models and molecular modeling operations were pursued for an increasingly popular antiobesity therapeutic target, the melanocortin-4 receptor (MC4R).
The melanocortin-4 receptor is a 332 amino acid GPCR that serves as the key regulator in the appetite-controlling pathway within the hypothalamus.17 When hunger signals such as ghrelin come from the gut, they act on orexigenic neurons by binding to their respective receptor.18 This stimulates the release of the melanocortin-4 receptor inverse agonist, agouti-related protein (AgRP). AgRP lessens MC4R activation, thus preventing satiety signals, and induces the perception of hunger. When satiety signals such as insulin from the pancreas or leptin from adipose tissues bind to their respective receptors on anorexigenic neurons, the MC4R agonist α-melanocyte stimulating hormone (α-MSH) is released, after which it binds to and activates MC4R, producing satiety signaling.17,18 Dysregulation or mutation of this receptor has been implicated as the cause of about 6–8% of all obesity cases.19,20 These mutations can decrease the receptor affinity for the endogenous agonist, decrease surface expression levels of the receptor, or result in impaired signal transduction. Therefore, melanocortin-4 has become an important receptor target to study for the treatment of obesity.19,20
At present, no crystal structure for this receptor is available; therefore, molecular modeling and homology model construction represent the most accessible approaches to understand and visualize receptor conformation and ligand binding for the cases of both wild-type (WT) and mutated receptors. Various homology models have been constructed and previously reported in the literature.21–23 These models were constructed using either the rhodopsin or β–2 adrenergic receptors as the template. With the number of new and diverse GPCR crystal structures available increasing constantly, new and more suitable templates will be beneficial to better simulate the active conformation of the MC4R.
In this study, a homology model of the MC4R was constructed using the adenosine A2A receptor crystal structure (PDB 3QAK) as the construction template. This homology model was validated by docking each putative model with the tetrapeptide pharmacophore of the endogenous MC4R agonist α-MSH (Figure 1). The α-MSH tetrapeptide sequence has a very well established binding profile, and has been verified by several site-directed mutagenesis studies and molecular modeling studies.21–27 The chosen model was further validated using the well-known MC4R small molecule agonist THIQ (Figure 1). In order to better understand MC4R mutations and their effect on ligand binding and subsequent receptor activation and satiety signaling, a novel MC4R agonist and clinical candidate RM-493 (setmelanotide, Figure 1) was studied for its molecular mechanism to activate wild-type and mutant receptors with a significantly higher potency as compared to the endogenous substrate α-MSH. RM-493 is a potent and selective MC4R agonist that entered Phase 3 clinical trials in May 2017 for the treatment of obesity caused by genetic variations in the MC4R appetite-controlling pathway.27–31 Herein, four mutations commonly found in obese adults and children (Ser127Leu, Ser58Cys, Ile102Thr, and Gly252Ser), which are known to directly impact α-MSH potency in vitro, were reproduced in the homology model. RM-493 was then docked into the wild-type homology model as well as the four mutated models to understand its possible binding modes and mechanism of action as an antiobesity agent.
Figure 1.

Chemical structures of known MC4R agonists, α-MSH tetrapeptide His6-Phe7-Arg8-Trp9 pharmacophore, the small molecule THIQ, and the clinical candidate RM-493 (setmelanotide).
2. RESULTS AND DISCUSSION
2.1. Template Selection.
One of the first few steps in generating a homology model is choosing an appropriate template, ideally one that is at least reasonably homologous to the target protein, MC4R.32–34 To achieve this, a tier based screening approach was adopted to determine the optimal template for the construction of the models. The alignment search tool (BLAST) was applied to compare the primary sequence of MC4R to a database of various protein sequences for which crystal structures are available in PDB.33–39 The primary sequence of MC4R protein was retrieved from UniProtKB/Swiss-Prot database (primary accession number P32245) and inserted into the query. From all the crystal structures of GPCRs that were available at the time of this study, the human A2A adenosine receptor (AA2AR) showed the highest overall score with a sequence identity of 28% and a sequence similarity of 46%. Due to the high variability in the loop regions among GPCRs, further analysis was done to compare the homology of the individual transmembrane domains of the AA2AR template sequence to those of MC4R. Pair alignment of the two sequences was performed, and % homology in each region was derived from ratios of residues that have identity or similarity divided by the total number of residues per transmembrane domain. The results suggest that the AA2AR is a reasonable choice based upon the percentage of homology to MC4R in the transmembrane regions (58%). Additionally, the A2A adenosine receptor is one of MC4Rs closest neighbors on the GPCR phylogenetic tree, making the crystal structure of the A2A adenosine receptor a viable choice as the template for homology modeling.37,40 There are 25 crystal structures of the adenosine A2A receptor in the PDB. Therefore, a second-tier of screening was adopted to find an active state crystal structure that best resembled the protein of interest. There are 6 agonist bound crystal structures of the adenosine A2A receptor in the PDB. Of these, 5 are bound to small molecules. These structures may not be optimal for the generation of homology models because the endogenous ligand, α-MSH, for MC4R is a large peptide. One AA2AR structure (PDB 3QAK) was cocrystallized with a large molecule, UKA (C40H47N11O6, MW = 777.87) that better resembles the size of the MC4R tetrapeptide (C32H42N10O5, MW = 646.33) to be used in docking studies. Therefore, 3QAK was chosen as the template for the generation of MC4R homology models.
2.2. Sequence Alignment and Homology Model Construction.
The sequence alignment (Figure 2) was adjusted by removing the cocrystallized lysozyme of the template protein and truncating the N-terminus of the target protein (MC4R) to better match the length of the template. Additionally, there was a gap present within TM3 of the template protein. This gap, 7 residues in length, was retained in the ECL1 domain to ensure proper construction of the transmembrane helices TM2 and TM3 for the MC4R target protein, as these helices are imperative for binding and GPCR structure. Unlike most GPCRs, the MC4R has a very short extracellular loop 2 (ECL2) and a very long extracellular loop 3 (ECL3). This makes it very difficult to model these loops using the AA2AR template, which has a relatively longer ECL2 and shorter ECL3. To resolve this, all loops in the template were omitted first in the alignment file, and the transmembrane helices were aligned. The omitted loops were inserted as gaps in the alignment file. The modeling software was allowed to reconstruct these regions using the loop residues of MC4R later on.41 MODELLER then used the adjusted alignment file to generate 100 homology models with differing geometric conformations.
Figure 2.

Multisequence alignment of MC4R with representative GPCR sequences. ADRB2, human β2-adrenergic receptor; ADRB1, human β1-adrenergic receptor; CXCR4, human chemokine receptor CXCR4; OPSD, bovine rhodopsin; AA2AR, human A2A-adenosine receptor. The most conserved residues among GPCR superfamily members are marked in red.
2.3. Model Screening, Evaluation, and Validation.
The tetrapeptide sequence of the endogenous agonist α-MSH (NH2-His6-Phe7-Arg8-Trp9-Ac) was chosen for validation of the constructed models. This tetrapeptide sequence contains the essential pharmacophore features for melanocortin receptor recognition and is the minimal peptide sequence necessary for MC4R activation.42,43 The tetrapeptide pharmacophore, rather than the full endogenous peptide, was also chosen to lessen the computation power needed to dock such a large molecule into 100 models. The α-MSH tetrapeptide was docked into all 100 models and evaluated by GOLD scores.24 Based on the generated GOLD scores, the top pose was chosen, and it showed favorable interactions between residues Asp126 and Asp122 of TM3 of the protein and both Arg8 and the backbone nitrogen of His7 of the tetrapeptide, consistent with mutagenesis data.25,26 Additionally, potential interactions were observed between the side chains of Phe7 and Trp9 of the tetrapeptide and the hydrophobic residues on TM6, Phe261 and Trp258, also consistent with mutagenesis data and previously reported modeling studies (Figure 3A).21–27 Therefore, it was concluded that this model was acceptable for further application.
Figure 3.

Highest GOLD scored mode of (A) α-MSH tetrapeptide (cyan, sticks) and (B) THIQ (magenta, sticks). (C) RM-493 (green, sticks) in generated MC4R homology model (gray, cartoon). Amino acids involved in interactions are shown as gray sticks. Dashed lines indicated potential hydrogen bonds.
The MC4R homology model was overlapped and compared to the AA2AR crystal structure. The overall weighted root mean square distance (RMSD) between them was 7.405 Å and the backbone RMSD between only the transmembrane regions was 4.620 Å. The model of MC4R in the active state mostly resembled the structure of AA2AR, including the important proline kinks in TM6 (Pro260) and TM7 (Pro299), part of the (N/D)PxxY motif conserved in the rhodopsin superfamily of GPCRs.44–48 However, as expected, the homology model and the AA2AR template differ significantly in the loop region, yielding the larger overall standard deviation, due to the low homology between MC4R and the template in these regions. The extracellular loops of the protein are largely outward facing, consistent with the active state of the protein being capable of accommodating large peptides.21,23,49 Additional observations included the shortened N-terminal end of TM1, the shift of the extracellular part of TM7 toward TM1, and the significant movement of the extracellular portion of TM6 toward TM7, all consistent with the proposed activation mechanism of MC4R.22,50–52
This model was further analyzed using Molprobity and ProSa (see Supporting Information). The Ramachandran plot from Molprobity showed that almost all amino acid residues within the model had reasonable bond lengths and bond angles. As gathered from the plot, 93.2% (261/280) of all residues were in favored (98%) regions, and 98.6% (276/280) of all residues were in allowed (>99.8%) regions. The 4 residues identified as having unfavorable φ and ψ angles were Gln115, Ile195, Cys196, and Cys318. The first three were located on the extracellular loops and should not affect ligand binding. Similarly, Cys318 should not impact ligand binding either, as it is located on the C-terminus of the protein. In the ProSA validation study, the model had a calculated z score of –2.56, which falls into the range of scores typically found for native proteins of a similar size.
2.4. THIQ Docking Studies.
A small molecule, THIQ (Merck), was chosen as the validation ligand because it has been well studied as an MC4R agonist. It has been used previously in numerous modeling studies, and as a reference ligand for the design of several small molecule α-MSH peptidomimetics.23,53–55 It also has very well established site-directed mutagenesis data supporting its interactions with several similar residues to those that bind to the tetrapeptide such as Asp122, Asp126, and Phe261. However, it was also reported to interact with additional residues not implicated in the binding of α-MSH such as Asn123, which makes it an ideal ligand to use to substantiate the constructed homology models.53 THIQ was docked into the chosen model with 100 GA runs and clustered according to pose similarity. Fifteen different clusters were generated. All were analyzed for their interactions with the model to see which cluster best matched the interactions implicated by site-directed mutagenesis data.44,56 In the top scoring pose, the nitrogen atom of the 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (TIC) group showed possible interactions with the carboxylate groups of Asp122 and Asp126 of the orthosteric site, common to α-MSH. Additional π–π interactions could be observed between its p-chlorophenyl and Phe261 and between its triazole ring and Trp258, also in the common orthosteric binding site (Figure 3B). Additionally, THIQ may interact with Asn123, located in a putative allosteric site, providing THIQ with greater specificity for MC4R as compared to α-MSH over other melanocortin receptor subtypes.53 Altogether, this model was able to accommodate both the endogenous tetrapeptide and the small molecule THIQ and match site-directed mutagenesis data for both compounds. Therefore, it was concluded that this model would be suitable to model the binding of the clinical candidate RM-493 and subsequent genetic protein mutations.
2.5. RM-493 in the Wild-Type Model.
RM-493 (setmelanotide) is a potent MC4R agonist in development by Rhythm Pharmaceuticals for the treatment of obesity caused by genetic mutations, and it recently entered Phase III clinical trials.28 It was shown to possess a 37-fold higher affinity than α-MSH in wild-type (WT) binding assays (Ki = 0.71 nM) and was 20-fold more potent in WT activation assays (EC50 = 1.5 nM). In in vivo studies, RM-493 was shown to reduce food intake and weight gain in normal rats, and reduce fatty liver disease and hyperinsulinemia in diet-induced obese mice when administered peripherally.27,29 When RM-493 was docked into the wild-type model, the top scoring clustered pose showed possible interactions between RM-493 and several residues implicated to be critical for α-MSH binding by site-directed mutagenesis studies (Figure 3C). Arg1 and Arg6 of RM-493 showed interactions with Asp122 and Asp126, respectively. Additional possible π–π interactions could be seen between Trp7 and both Trp258 and Phe261 of MC4R, also similar to the tetrapeptide-binding mode. However, our docking results also suggested several additional interactions between RM-493 and hMC4R that were not seen for α-MSH. These included polar interactions at the top of TM3 between Arg1 and Leu106 and Asp111 and His4 and Asn123. This suggests the ability for RM-493 to access the putative allosteric binding site at the top of TM3 as predicted by studies involving THIQ. Additional conceivable hydrophobic interactions were observed between Phe5 and both Tyr268 and Tyr276, also not implicated in α-MSH binding. These residues, however, have been implicated in the binding of other large cyclic peptides for MC4R such as MT-II, insinuating an alternative allosteric binding site at the top of TM6 that may provide greater specificity for large cyclic ligands.55 As predicted by previous NMR and modeling studies, α-MSH was thought to bind in a hairpin loop conformation, placing the tetrapeptide messaging sequence into the activation site of the receptor, and the rest of the peptide pointing out of the binding pocket toward the extracellular space.57 On the other hand, from our docking studies, the entire rigid and cyclic structural skeleton of RM-493 was accommodated within the whole binding pocket. The presence of these extra amino acid residues allowed for several additional interactions, potentially within multiple allosteric binding sites (Table 1) and may be the cause of increased affinity and potency as compared to α-MSH in the WT protein.
Table 1.
Amino Acids Residues Involved in Wild-Type Protein Interacting with MC4R Ligandsa
| α-MSH tetrapeptide | RM-493 | |
|---|---|---|
| residues | Glu100, Asp122, Asp126, Phe261, Trp258, His264 | Glu100, Asp122, Asp126, Phe261, Trp258, His264, Asn123, Leu106, Asp111, Tyr276, Tyr268 |
Amino acid residues potentially involved in allosteric binding contributing to agonist specificity and potency in bold.
2.6. Point Mutations.
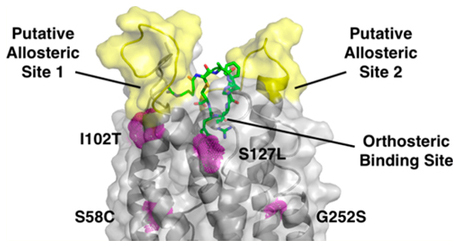
The MC4R point mutations were chosen among the various categories found in obese patients, based on their available binding and functional activity data for the clinical candidate RM-493 binding.27 The 4 chosen point mutations (Ser127Leu, Ser58Leu, Ile102Thr, Gly252Ser) are from class 2B and 2C of MC4R mutations and result in reduced potency of the endogenous ligand α-MSH.27 The mutations known to directly impact ligand potency would provide the most direct comparison between α-MSH and RM-493 binding through molecular modeling. Each point mutation was located in a distinct region of the receptor; however, they were all located within or near the orthosteric-binding pocket of the receptor (Figure 4A). After energy minimization, short-term dynamics studies (1000 fs) were performed to note any conformational changes. Larger scale molecular dynamics simulations for refinement in a full water-membrane environment were not utilized due to the extreme large time spans necessary to evaluate active state proteins. When the mutated minimized proteins were overlapped with the wild-type (Figure 4B,C,D,E), it was observed that the mutant Ile102Thr had the greatest conformational change among residues located near the mutation.
Figure 4.

(A) Location of the mutated residues (magenta, spheres) within the protein (gray, cartoon) and labeled putative allosteric sites. (B-E) Overlap of wild-type (gray) and mutated proteins (magenta). (B) Mutation of Ser58 to Cys58 causes disruption of the H-bond network stabilizing TM6. Hydrogen bonds with distances in the wild-type protein shown as dashed lines (black). Distances increase between N294 and S295 from 2.9 to 4.2 Å, between N294 and S58 from 3.0 to 3.2 Å, and between D90 and N62 from 3.3 to 5.3 Å. Distances between residues upon mutation not shown for clarity. (C) Mutation of Ile102 to Thr102 disrupts the TM2-TM3 interface imperative for receptor activation. (D) Mutation of S127 to Leu127 causes a shift in Asp122 and Asp126, critical residues for ligand recognition. (E) Mutation of Gly252 to Ser252 interrupts helix-helix interactions necessary for stabilization of TM6.
2.7. RM-493 and α-MSH Tetrapeptide in Mutated Receptor Models.
When RM-493 was docked into the four missense mutation models, it was observed that none of the mutated residues were directly involved in its binding; however, all of the mutations are located proximal to residues implicated as important for MC4R activation in the orthosteric binding site, which may explain their in vitro effects on ligand binding affinity and potency (Table 2).
Table 2.
Comparison of Inhibition Constants (Ki) and 50% Maximum Concentration (EC50) Values for α-MSH and RM-493 in Radioligand Binding and Intracellular cAMP Production Assays27
| mutation | Ki (nM) | EC50 (nM) | ||
|---|---|---|---|---|
| α-MSH | RM-493 | α-MSH | RM-493 | |
| WT | 26 ± 7.0 | 0.71 ± 0.16 | 30 ± 7.5 | 1.5 ± 0.38 |
| SerS8Cys | 53 ± 12 | 1.7 ± 4.3 | 670 ± 90 | 46 ± 8.0 |
| Ile102Thr | 210 ± 94 | 2.2 ± 0.21 | 5800 ± 850 | 150 ± 57 |
| Ser127Leu | 160 ± 40 | 3.3 ± 0.13 | 640 ± 190 | 17 ± 6.0 |
| Gly252Ser | 160 ± 66 | 2.3 ± 0.30 | 210 ± 45 | 13 ± 2.0 |
Ile102Thr and Ser127Leu are both located close to the MC4R orthosteric ligand-binding pocket and may cause a shift in the residues Glu100, Asp122, and Asp126, which are necessary for receptor recognition and activation by the endogenous agonist α-MSH.19,21,22,27,51,55,58,59 Ser58Cys and Gly252Ser are located further from the binding pocket and are likely involved in ligand-induced conformational changes important for MC4R activation.
2.7.1. Ile102Thr.
Ile102 is located on TM2 close to the TM2-TM3 interface site. This site is composed of Glu100, Ile104, Asp122, and Ile125 and is believed to be critical for receptor activation mediated by ionic interactions with the ligand.39 Aside from ligand recognition, this site may initiate the movement of TM3 in receptor activation, similar to other predicted GPCR activation mechanisms.51,60 From short-term dynamics studies without bound ligand, it was observed that the Ile102Thr mutation produced the largest movement among its surrounding residues (Figure 4C). These residues include Ile104, Glu100, Asp122, and Ile125, indicating substantial perturbation of the TM2-TM3 interface by this mutation. As shown in Figure 4C, mutation of the Ile102 residue to a smaller Thr102 residue may cause the rotation of Glu100 downward and away from the TM2-TM3 interface by 1.4 Å as well as Ile104 downward and away from the interface by 1.2 Å (Figure 4C). The movement of these residues away from the interface may disrupt the rotation of TM3 and therefore impair receptor activation. This disrupted interface, which includes the critical residue Asp122 for binding, may explain the decreased affinity and potency seen for both α-MSH and RM-493 upon mutation as supported by binding data (Table 2). However, in the case of RM-493, additional interactions with residues at the top of TM3 in a putative allosteric binding site, such as Leu106 and Asp111, may compensate such disruption and help stabilize TM3 in the active state resulting in a higher affinity and potency of RM-493 in the mutated protein as compared to α-MSH (Figure 5B). The ability for these allosteric interactions to help stabilize the TM3 indicates that RM-493 may be acting as a positive allosteric modulator (PAM) by maintaining the spatial and structural conformation needed for receptor activation.
Figure 5.
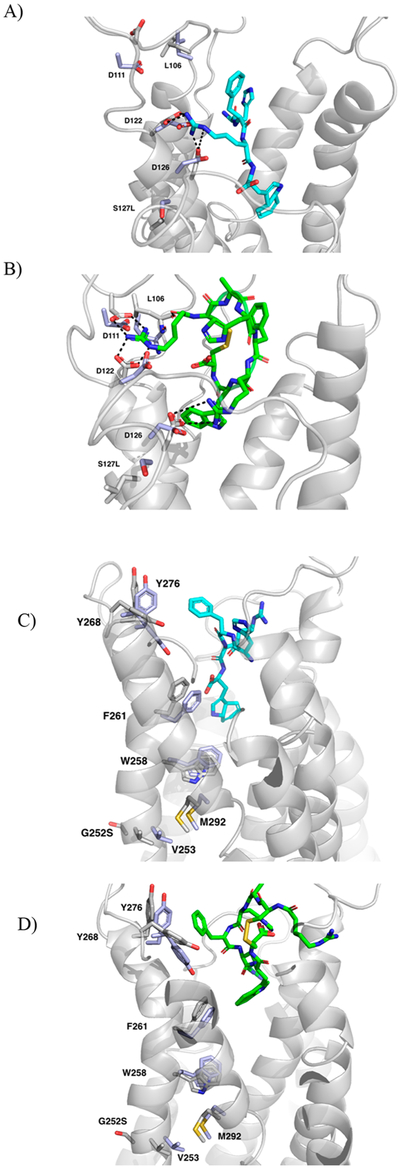
(A) Ser127Leu mutant receptor with bound α-MSH tetrapeptide. (B) Ser127Leu mutant receptor with bound RM-493. (C) Gly252Ser mutant receptor bound with α-MSH tetrapeptide. (D) Gly252Ser mutant receptor bound with RM-493. Mutant receptors are shown as gray cartoon with the bound RM-493 as green sticks. Interacting residues are indicated by white sticks and overlapped with the corresponding residues in the wild-type receptor (purple, sticks). Potential hydrogen bonds denoted as dashed lines (black).
2.7.2. Ser127Leu.
Ser127Leu is located on TM3 and is close to Asp122 and Asp126, the critical residues for endogenous ligand binding. From the model, when Ser127 is mutated to Leu127, the side chain of Asp122 rotates 2.5 Å downward toward TM4, and the side chain of Asp126 shifts downward 1.4 Å (Figure 4D). As both Asp122 and Asp126 are located in the orthosteric site and are critical for ligand recognition and receptor activation, any movement among these residues could impair ligand affinity, as seen from the data in Table 2. Additionally, as previously discussed, Asp122 is thought to be involved in an important interaction site with Glu100, Ile104, and Ile125 at the interface between TM2 and TM3. Upon mutation, the movement of Asp122 away from TM3 and toward TM4 could play a role in interfering with the interactions in this region of the protein. Interference in the orthosteric binding site may explain the decreased affinity and potency seen with both ligands in the Ser127Leu mutated protein as compared to the wild-type (Table 2). Even so, RM-493 is still able to activate the mutated MC4R effectively with a 38-fold higher potency as compared to α-MSH.27 This again could be due to additional potential interactions shown by RM-493 with residues surrounding Ile102Thr and Ser127Leu, such as Leu106 and Asp111 in an allosteric binding site, that were not observed in α-MSH binding (Figure 5A,B). Again, the capacity for the allosteric interacting portion of RM-493 to stabilize the receptor and maintain adequate affinity and potency despite mutations in the orthosteric pocket supports the ability of this compound to act as a PAM. Another conceivable explanation is the additional arginine residue on RM-493. The presence of an additional arginine provides two points of interaction for Asp122 and Asp126 rather than just the one (Arg8) in α-MSH. This may provide stronger interactions regardless of the orientation and movement of the Asp122 and Asp126 residues upon mutation.
2.7.3. Ser58Cys.
Ser58Cys is located at the interface between TM1 and TM7 and has been implicated as an integral part of the hydrogen bond network that stabilizes the active state of MC4R.19,22,61 This network is understood to be formed by several polar residues including Ser58 and Asn62 from TM1; Tyr80, Asp90, Ser94, Asn97, and Glu100 from TM2; Ser132, Ser136, Asp146, and Arg147 from TM3; and Asn294, Ser295, Asp298, and Tyr302 from TM7.22 From the model, hydrogen-bonding interactions were observed between Ser58 and Asn294 on TM7. Upon mutation to cysteine, this polar interaction is weakened. While the sulfur atom could also engage in hydrogen bonding, it is bulkier and generally regarded as a poor hydrogen bond acceptor. This weakened interaction may result in a destabilized hydrogen bond network (Figure 4B) and explain the decrease in affinity and potency observed for both α-MSH and RM-493 in binding and functional assays (Table 2). However, despite mutation, RM-493 maintains a 30-fold higher affinity and a 15-fold higher potency as compared to α-MSH.16 As this mutation is not in the binding pocket, it is likely that the increased affinity and potency of RM-493 in this mutation is compensated largely by the additional positively modulating allosteric ligand-receptor interactions of RM-493 with Leu106 and Asp111.
2.7.4. Gly252Ser.
Gly252Ser is located on TM6 next to Val253, a critical residue for supporting helix-helix interactions and controlling the conformation of TM6 through hydrophobic interactions with Met292 (Figure 4E).19,22,62,63 It is known that TM6 plays a very important role in GPCR activation, and these hydrophobic interactions between TM6 and TM7 may help to mediate helical interactions involved in activation. Upon activation, it is predicted that TM6 undergoes conformational change in the helical region below the conserved proline-kink residue (Pro260), which includes Gly252 and Val253.22 Due to the change in size and electronic nature caused by mutating a glycine to a serine, it is possible that these important helix-helix interactions at the interface might be disrupted, thus impairing receptor activation. It can be seen that upon mutation of the glycine to serine, Val253 and Met292 both shift downward and away from the TM6–7 interface by 1.2 and 1.4 Å, respectively (Figure 4E). The shift of these residues increases the distance between them from 3.9 Å in the wild-type to 4.8 Å in the mutant (Figure 4E). This may lead to weakened hydrophobic interactions that could help to destabilize the helix-helix interactions and produce a decrease in potency for both RM-493 and α-MSH. However, consistent with the previously discussed mutations, RM-493 remained 16-fold more potent than α-MSH. One possible explanation for such higher affinity and potency for RM-493 as compared to α-MSH may be the additional ligand-receptor interactions, particularly with the residues located at the top of TM6 and TM7 such as Tyr268 and Tyr276, that were not implicated in α-MSH binding. (Figure 5C,D). In addition to hydrophobic interactions with Phe261 and Trp258 in the orthosteric site, also common to α-MSH, the additional hydrophobic interactions with Tyr268 and Tyr276 may help stabilize the conformational change needed for activation despite the disrupted helical interface and could represent an alternative positive allosteric site.27
3. SUMMARY AND CONCLUSIONS
A homology model of MC4R was constructed based on the crystal structure of agonist-bound adenosine A2A receptor. The model was optimized and validated by adopting two MC4R ligands, the endogenous ligand α-MSH tetrapeptide pharmacophore and the small molecule agonist THIQ. The ligands were docked into the constructed homology models and the ligand-protein complexes were analyzed using several criteria including fitness scores and available site-directed mutagenesis data. Based on the validated docking results, it seemed that both of the ligands bound into a common upper-transmembrane domain of the MC4R model, consistent with previously constructed models and mutagenesis data. The point mutation studies indicate that the polymorphisms used in this study may cause binding disruptions both in the orthosteric binding pocket and in several transmembrane regions essential for the conformational changes that occur during receptor activation. The docking studies of both the α-MSH tetrapeptide and the clinical candidate RM-493 within the mutated models show that the disruptions in the orthosteric binding site and the transmembrane regions may be the cause of the decremental affinity and potency of these ligands with the receptor polymorphisms. The docking studies also suggest that the cyclic structure of RM-493 and its ability to interact more extensively with MC4R may help activate and stabilize the receptor, even in the presence of polymorphisms. These additional interactions may be with putatively positive allosteric binding sites near the extracellular loop region of TM3 and TM6.64 The capacity for RM-493 to bind to the orthosteric binding site with high affinity and simultaneously modulate receptor activation despite mutation via interactions with putative allosteric sites suggest the possibility that it may act as both a positive allosteric modulator and an orthosteric ligand, making it a “nontraditional” bitopic ligand. Typically, bitopic ligands are orthosteric ligands connected to allosteric ligands by a linker; however, RM-493 is a cyclic peptide large enough to simultaneously interact with both sites without the need for the traditional orthosteric/allosteric building blocks and a linker. With the absence of crystallographic data, the molecular modeling work presented here may serve as a platform to enable the design of future selective and potent ligands capable of interacting simultaneously with the orthosteric and allosteric binding sites. This “bitopic” binding mode may allow such ligands to retain potency and efficacy despite the presence of genetic variants.5,13,14 Such ligands would be invaluable for the future treatment of obesity and associated endocrine disorders in patients with MC4R polymorphisms.
4. METHODS
4.1. Sequence Alignment and Homology Modeling.
All molecular modeling studies were performed using the SYBYL-X 2.1.1 package (Tripos LP, St. Louis, MO) on dual-core AMD Opteron 2.4 GHz processors. The amino acid sequence of the melanocortin-4 receptor (MC4R) was obtained from UniProtKB/Swiss-Prot (P32245). The amino acid sequences of all the currently available GPCR crystal structures were collected and compared to the sequence of MC4R using the Basic Local Alignment Search Tool (BLAST). Both overall homology levels and homology between individual regions were used to select the most appropriate template. The active adenosine A2A receptor (AA2AR) was eventually chosen as the template structure (PDB code 3QAK, 2.71 Å). The AA2AR sequence was aligned to that of the MC4R using ClustalX 2.1.65 Within ClustalX, manual adjustment of the sequence alignment was performed to eliminate gaps found within the transmembrane helices, remove the cocrystallized lysozyme, and truncate the N-terminus. Due to a relatively lower homology level between the template and MC4R in the extracellular loop regions, residues 32–42 in ICL1, 67–77 in ECL1, 105–117 in ICL2, 149–166 in ECL2, 207–221 in ICL3, and 257–268 in ECL3 were manually removed from the template first to allow the sequence alignment to be optimized. These loop regions were later reconstructed following the MC4R complete sequence upon construction of 100 homology models through MODELLER.
4.2. Ligand Construction.
The three ligands chosen for the docking studies include the MC4R endogenous peptide α-MSH tetrapeptide pharmacophore (sequence His6-Phe7-Arg8-Trp9), the small molecule MC4R agonist THIQ, and the selective peptide RM-493. The ligands were constructed in SYBYL-X 2.1.1 with standard bond lengths and angles. They were assigned Gasteige-Hückel charges and energy minimized with the Tripos Force Field (TAFF) also available in SYBYL-X 2.1.1. The nonbonded interaction cut off was 8 Å, dielectric constant was 1.00, and the energy minimization was run until the number of iterations exceeded 100 000 or when the energy gradient fell below 0.05 kcal/(mol × A).
4.3. Ligand Docking.
The α-MSH tetrapeptide was first docked into the 100 generated homology models using the genetic algorithm docking software GOLD 5.4 (Cambridge Crystallographic Data Centre, CCDC, Cambridge, UK). The putative docking site was defined by 10 Å around Asp126, consistent with site-directed mutagenesis data found in the literature. Ten poses were generated for the ligand in each of the 100 models and given a score from a fitness function (GOLD score) using generic GOLD docking parameters.66 The GOLD scores for each of the 10 poses within all 100 models were analyzed by merging the poses into the MC4R model using SYBYL-X 2.1.1. Out of the top 10 GOLD scoring models, the model showing the highest docking scores overall and the most favorable interactions matching previously reported site-directed mutagenesis data was chosen for further analysis. The subsequent receptor-ligand complex was energy minimized by applying the Tripos Force Field with Gasteiger-Hückel charges, a nonbonded interaction cut off of 8 Å, and a dielectric constant of 1.00. The minimization was run until the number of iterations exceeded 10 000 or when the energy gradient fell below 0.05 kcal/(mol × A). The stereochemical quality of the model was assessed using MolProbity and ProSA, and was aligned with the template structure 3QAK for comparison. Using the chosen model, both THIQ and RM-493 were subsequently docked using GOLD. The putative binding area was defined again using Asp126. Each ligand was docked into the model for 100 iterations using the generic GOLD parameters. The resulting docked solutions were clustered based on similarity of pose with the distance between clusters set as 4.0 Å. The docking poses for each ligand having the highest GOLD scores were merged into the MC4R model using SYBYLX 2.1.1 and energy minimized using the previously described parameters.
4.4. Point Mutations.
The point mutations were individually introduced in SYBYL-X 2.1.1, and then the entire mutated protein was energy minimized using the previously described parameters. After energy minimization, a short-term molecular dynamics (MD) simulation using SYBYL-X 2.1.1 was conducted. The simulation was run at a length of 1000 fs and a temperature of 300 K. The nonbonded cutoff was 8.00, and the dielectric constant was 1.00. Each step was defined as 1 fs, and snapshots were taken every 5 fs. The mutated proteins were then each overlapped with the wild-type receptor and aligned by homology using the biopolymer package in SYBYL-X 2.1.1. The mutated and wild-type proteins were extracted and overlapped, and then RMSD values were calculated for within 5 Å of the mutation to note any conformational changes that occurred.
4.5. Ligand Docking in Mutated Models.
The energy minimized RM-493-WT and α-MSH-WT complexes were each merged with the four previously constructed and energy minimized mutated models. Each mutated receptor was then aligned by homology to the WT receptor complex. After alignment, each ligand was extracted from the wild-type complex and remerged with each of the mutated receptor models. These new complexes were then energy minimized using the previously described minimization parameters. After minimization, a short-term molecular dynamics (MD) simulation using SYBYL-X 2.1.1 was conducted. The simulation was run at a length of 1000 fs and a temperature of 300 K. The nonbonded cutoff was 8.00, and the dielectric constant was 1.00. Each step was defined as 1 fs, and snapshots were taken every 5 fs. Similar MD procedures were also completed for the wild-type complexes.
Supplementary Material
Acknowledgments
Funding
This research was partially supported by PHS/NIH DA024022 and DA044855 (Y.Z.).
Footnotes
The authors declare no competing financial interest.
Authors will release the atomic coordinates and experimental data upon article publication.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00346.
Model validation using Molprobity and ProSA and coordinates of homology model (PDF)
REFERENCES
- (1). Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). Lane RJ, Abdul-Ridha A, and Canals M (2013) Regulation of G-protein coupled receptors by allosteric ligands. ACS Chem. Neurosci 4, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Conn PJ, Christopoulos A, and Lindsley CW (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). Nickols HH, and Conn PJ (2014) Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis 61, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5). Feng Z, Hu G, Ma S, and Xie X-Q (2015) Computational advances for the development of allosteric modulators and bitopic ligands in G-protein coupled receptors. AAPS J. 17, 1080–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6). Foster DJ, and Conn JP (2017) Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 94, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7). Conn JP, Lindsley CW, Meiler J, and Niswender CM (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discovery 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Mutel V, and Bettler B (2007) The pros of not being competitive. Curr. Neuropharmacol 5, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). Wenthur CJ, Gentry PR, Mathews TP, and Lindsley CW (2014) Drugs for allosteric sidtes on receptors. Annu. Rev. Pharmacol. Toxicol 54, 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Grover AK (2013) Use of allosteric targets in the discovery of safer drugs. Med. Princ. Pract 22, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). Abdel-Magid AF (2015) Allosteric modulators: an emerging concept in drug discovery. ACS Med. Chem. Lett 6, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12). Valant C, Lane JR, Sexton PM, and Christopoulos A (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of G-protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 52, 153–178. [DOI] [PubMed] [Google Scholar]
- (13). Lane JR, Sexton PM, and Christopoulos A (2013) Bridging the gap: bitopic ligands of F-protein-coupled receptors. Trends Pharmacol. Sci 34, 59–66. [DOI] [PubMed] [Google Scholar]
- (14). Kamal M, and Jockers R (2009) Bitopic ligands: all-in-one orthosteric and allosteric. F1000 Biol. Rep 1, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15). Fronik P, Gaiser BI, and Sejer Pederson D (2017) Bitopic ligands and metastable binding sites: opportunities for G protein-coupled receptor (GPCR) medicinal chemistry. J. Med. Chem 60 (4126), 4134. [DOI] [PubMed] [Google Scholar]
- (16). Mohr K, Trankle C, Kostenis E, Barocelli E, De Amici M, and Holzgrabe U (2010) Rational design of dualsteric GPCR ligands: quests and promise. Br. J. Pharmacol 159, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Garfield AS, Li C, Madara JC, Shah BP, Webber E, Steger JS, Campbell JN, Gavrilova O, Lee CE, Olson DP, Elmquist JK, Tannous BA, Krashes MJ, and Lowell BB (2015) A neural basis for melanocortin-4 receptor regulated appetite. Nat. Neurosci 18, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Kim GW, Lin JE, Valentino MA, Colon-Gonzalez F, and Waldman SA (2011) Regulation of appetite to treat obesity. Expert Rev. Clin. Pharmacol 4, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Govaerts C, Srinivasan S, Shapiro A, Zhang S, Picard F, Clement K, Lubrano-Berthelier C, and Vaisse C (2005) Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides 26, 1909–1919. [DOI] [PubMed] [Google Scholar]
- (20). Santini F, Maffei M, Pelosini C, Salvetti G, Scartabelli G, and Pinchera A (2009) Melanocortin-4 receptor mutations in obesity. Adv. Clin. Chem 48, 95–109. [PubMed] [Google Scholar]
- (21). Chapman KL, Kinsella GK, Cox A, Donnelly D, and Findlay JBC (2010) Interactions of the Melanocortin-4 Receptor with the Peptide Agonist NDP-MSH. J. Mol. Biol 401, 433–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). Tan K, Pogozheva ID, Yeo GSH, Hadaschik D, Keogh JM, Haskell-Leuvano C, O’Rahilly S, Mosberg HI, and Farooqi IS (2009) Functional characterization and structural modeling of obesity associated mutations in the melanocortin 4 receptor. Endocrinology 150, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). Pogozheva ID, Chai B-X, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, and Mosberg HI (2005) Interactions of Human Melanocortin 4 Receptor with Non Peptide and Peptide Agonists. Biochemistry 44, 11329–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24). Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, and Hadley ME (1980) 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. U. S. A 77, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25). Hruby VJ, Cai M, Cain J, Nyberg J, and Trivedi D (2011) Design of novel melanocortin receptor ligands: multiple receptors, complex pharmacology, the challenge. Eur. J. Pharmacol 660, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Holder JR, Bauzo RM, Xiang Z, and Haskell-Luevano C (2002) Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors. 1. Modifications at the His Position. J. Med. Chem 45, 2801–2810. [DOI] [PubMed] [Google Scholar]
- (27). Roubert P, Dubern B, Plas P, Lubrano-Berthelier C, Alihi R, Auger F, Deoliveira DB, Dong JZ, Basdevant A, Thurieau C, and Clément K (2010) Novel pharmacological MC4R agonists can efficiently activate mutated MC4R from obese patient with impaired endogenous agonist response. J. Endocrinol 207, 177–183. [DOI] [PubMed] [Google Scholar]
- (28). http://www.rhythmtx.com/news-resources/press-releases/rhythm-initiates-two-phase-2-clinical-trials-of-setmelanotide-rm-493-in-rare-genetic-disorders-of-obesity-caused-by-mc4-pathway-deficiencies/ (accessed Aug 8, 2018).
- (29). Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, and Butler AA (2009) Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides 30, 1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30). Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LHT, Reitman ML, and Skarulis MC (2015) RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J. Clin. Endocrinol. Metab 100, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Fani L, Bak S, Delhanty P, van Rossum EFC, and van den Akker ELT (2014) The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int. J. Obes 38, 163–169. [DOI] [PubMed] [Google Scholar]
- (32). Park PS (2012) Ensemble of G Protein-Coupled Receptor Active States. Curr. Med. Chem 19, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Fiser A (2010) Template-Based Protein Structure Modeling. Methods Mol. Biol 673, 73–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34). Mobarec JC, Sanchez R, and Filizola M (2009) Modern Homology Modeling of G-Protein Coupled Receptors: Which Structural Template to Use? J. Med. Chem 52, 5207–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, and Ernst OP (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502. [DOI] [PubMed] [Google Scholar]
- (36). Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, and Stevens RC (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37). Xu F, Wu H, Katritch V, Han GW, Jacobson KW, Gao ZG, Cherezov V, and Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38). Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, and Stevens RC (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, and Kobilka BK (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, and Wüthrich K (2013) The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discovery 12, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Zhang Y, Sham YY, Rajamani R, Gao J, and Portoghese P (2005) Homology modeling and molecular dynamics simulations of the mu opioid receptor in a membrane-aqueous system. Chem-BioChem 6, 853–859. [DOI] [PubMed] [Google Scholar]
- (42). Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, et al. (1989) Alpha-melanotropin: the minimal active sequence in the lizard skin bioassay. Gen. Comp. Endocrinol 73, 157–163. [DOI] [PubMed] [Google Scholar]
- (43). Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, et al. (1987) alpha-Melanotropin: the minimal active sequence in the frog skin bioassay. J. Med. Chem 30, 2126–2130. [DOI] [PubMed] [Google Scholar]
- (44). Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, and Ernst OP (2003) Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. U. S. A 100, 2290–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45). Yang Z, Huang Z-L, and Tao Y-X (2015) Functions of DPLIY Motif and Helix 8 of Human Melanocortin-3 Receptor. J. Mol. Endocrinol 55, 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Urizar E, Claeysen S, Deupí X, Govaerts C, Costagliola S, Vassart G, and Pardo L (2005) An Activation Switch in the Rhodopsin Family of G Protein-coupled Receptors the Thyrotropin Receptor. J. Biol. Chem 280, 17135–17141. [DOI] [PubMed] [Google Scholar]
- (47). Gilchrist A Molecular Mechanisms of GPCR Ligand Binding. GPCR Molecular Pharmacology and Drug Targeting: Shifting Paradigms and New Directions; Wiley: Hoboken, NJ, 2010; pp 35–40. [Google Scholar]
- (48). Tao YX (2009) Mutations in melanocortin-4 receptor and human obesity. Prog. Mol. Biol. Transl. Sci 88, 173–204. [DOI] [PubMed] [Google Scholar]
- (49). Hogan K, Peluso S, Gould S, Parsons I, Ryan D, Wu L, and Visiers I (2006) Mapping the Binding Site of Melanocortin 4 Receptor Agonists: A Hydrophobic Pocket Formed by I3.28(125), I3.32(129), and I7.42(291) Is Critical for Receptor Activation. J. Med. Chem 49, 911–922. [DOI] [PubMed] [Google Scholar]
- (50). Yang Y, Chen M, Dimmitt R, and Harmon CM (2014) Structural Insight into the MC4R Conformational Changes via Different Agonist-Mediated Receptor Signaling. Biochemistry 53, 7086–7092. [DOI] [PubMed] [Google Scholar]
- (51). Lagerström MC, Klovins J, Fredriksson R, Fridmanis D, Haitina T, Ling MK, Berglund MM, and Schiöth HB (2003) High Affinity Agonistic Metal Ion Binding Sites within the Melanocortin 4 Receptor Illustrate Conformational Change of Transmembrane Region 3. J. Biol. Chem 278, 51521–51526. [DOI] [PubMed] [Google Scholar]
- (52). Chen M, Cai M, McPherson D, Hruby V, Harmon CM, and Yang Y (2009) Contribution of the transmembrane domain 6 of melanocortin-4 receptor to peptide [Pro5, DNaI (2′)8]-γ-MSH selectivity. Biochem. Pharmacol 77, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53). Yang Y, Cai M, Chen M, Qu H, McPherson D, Hruby V, and Harmon CM (2009) Key amino acid residues in the melanocortin-4 receptor for nonpeptide THIQ specific binding and signaling. Regul. Pept 155, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54). Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DBR, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, van der Ploeg LHT, Patchett AA, and Nargund RP (2002) Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. J. Med. Chem 45, 4589–4593. [DOI] [PubMed] [Google Scholar]
- (55). Xiang Z, Pogozheva ID, Sorenson NB, Wilczynski AM, Holder JR, Litherland SA, Millard WJ, Mosberg HI, and Haskell-Luevano C (2007) Peptide and Small Molecules Rescue the Functional Activity and Agonist Potency of Dysfunctional Human Melanocortin-4 Receptor Polymorphisms. Biochemistry 46, 8273–8287. [DOI] [PubMed] [Google Scholar]
- (56). Oosterom J, Garner KM, den Dekker WK, Nijenhuis WA, Gispen WH, Burbach JP, Barsh GS, and Adan RA (2001) Common requirements for melanocortin-4 receptor selectivity of structurally unrelated melanocortin agonist and endogenous antagonist, Agouti-related protein. J. Biol. Chem 276, 931–936. [DOI] [PubMed] [Google Scholar]
- (57). Lee JH, Lim SK, Huh SH, Lee D, and Lee W (1998) Solution structures of the melanocyte-stimulating hormones by two-dimensional NMR spectroscopy and dynamical simulated-annealing calculations. Eur. J. Biochem 257, 31–40. [DOI] [PubMed] [Google Scholar]
- (58). Ersoy BA, Pardo L, Zhang S, Thompson DA, Millhauser G, Govaerts C, and Vaisse C (2012) Mechanism of N-terminal modulation of activity at the melanocortin-4 receptor GPCR. Nat. Chem. Biol 8, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59). Tao Y (2010) -X. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr. Rev 31, 506–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60). Kobilka BK (2007) G protein coupled receptor structure and activation. Biochim. Biophys. Acta, Biomembr 1768, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61). Rene P, Le Gouill C, Pogozheva ID, Lee G, Mosberg HI, Farooqi IS, Valenzano KJ, and Bouvier M (2010) Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J. Pharmacol. Exp. Ther 335, 520–532. [DOI] [PubMed] [Google Scholar]
- (62). Dalton JA, Lans I, and Giraldo J (2015) Quantifying conformational changes in GPCRs: glimpse of a common functional mechanism. BMC Bioinf 16, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63). Nargund RP, Strack AM, and Fong TM (2006) Melanocortin-4 Receptor (MC4R) Agonists for the Treatment of Obesity. J. Med. Chem 49, 4035–4043. [DOI] [PubMed] [Google Scholar]
- (64). Yang Y (2011) Structure, function and regulation of the melanocortin receptors. Eur. J. Pharmacol 660, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65). Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, and Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- (66). Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, and Taylor RD (2003) Improved protein-ligand docking using GOLD. Proteins: Struct., Funct., Genet 52, 609–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.