

SUMMARY:
Fragile X syndrome (FXS), the most common genetic form of intellectual disability in males, is caused by silencing of the FMR1 gene by hypermethylation of the CGG expansion mutation in the 5’UTR region of FMR1 in FXS patients. Here, we applied recently developed DNA methylation editing tools to reverse this hypermethylation event. Targeted demethylation of the CGG expansion by dCas9-Tet1/sgRNA switched the heterochromatin status of the upstream FMR1 promoter to an active chromatin state restoring a persistent expression of FMR1 in FXS iPSCs. Neurons derived from methylation edited FXS iPSCs rescued the electrophysiological abnormalities and restored a wild-type phenotype upon the mutant neurons. FMR1 expression in edited neurons was maintained in vivo after engrafting into the mouse brain. Finally, demethylation of the CGG repeats in post-mitotic FXS neurons also reactivated FMR1. Our data establish demethylation of the CGG expansion is sufficient for FMR1 reactivation, suggesting potential therapeutic strategies for FXS.
In Brief
Rescue of Fragile X syndrome neurons by CRISPR-mediated DNA methylation editing of the FMR1 gene
Graphical Abstract
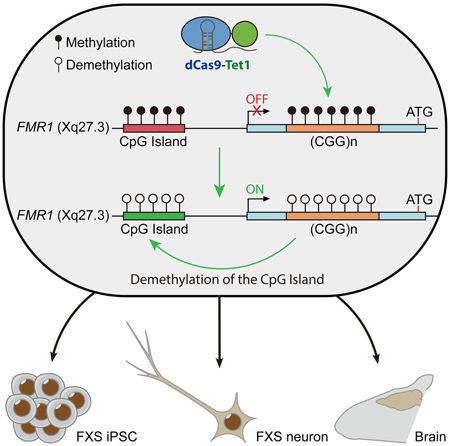
INTRODUCTION
Fragile X syndrome (FXS) is the most common genetic form of intellectual disability with an incidence of one in 3,600 males. Patients with FXS display a broad spectrum of autistic phenotypes such as intellectual, cognitive, and social deficits (Contractor et al., 2015; Pugin et al., 2017; Santoro et al., 2012; Sidorov et al., 2013). These deficits are attributed to the loss of the Fragile X Mental Retardation Protein (FMRP) encoded by the FMR1 gene during brain development. FMRP is a RNA binding protein in neurons and has been shown as a molecular brake for local protein synthesis at developing synapses and hence is essential for the maintenance of normal synaptic plasticity (Darnell et al., 2011; Sidorov et al., 2013; Smith et al., 2014). Indeed, loss of FMRP expression in patient-derived neurons leads to deregulated production and membrane insertion of neurotransmitter receptors and ion channels, causing synaptic hyper-excitability that affects proper function of various neural circuits in the central nervous system (Contractor et al., 2015; Irwin et al., 2000; Santoro et al., 2012). CGG trinucleotide repeat (>200) expansion mutation at the 5’ UTR region of FMR1, accompanied with DNA hypermethylation, was thought to result in a heterochromatin formation at the FMR1 promoter and subsequent silencing of FMR1 expression in FXS (Avitzour et al., 2014; Coffee et al., 1999; Colak et al., 2014; de Esch et al., 2014; Halevy et al., 2015; Urbach et al., 2010; Verkerk et al., 1991), but the molecular mechanism are not fully understood.
Currently, there is no cure or treatment for FXS likely due to the lack of mechanistic understanding of FXS pathophysiology at the molecular and cellular level and the enormous complexity of the FXS neuronal circuitry phenotype (Contractor et al., 2015; Santoro et al., 2012). Insertion of CGG repeats into the mouse Fmr1 locus did not result in DNA hypermethylation or repression of Fmr1 expression (Berman et al., 2014). While Fmr1 knockout (KO) mice partially recapitulate the neuronal hyper-excitability and excessive spinogenesis in the brain (1994; Bernardet and Crusio, 2006; Comery et al., 1997; Huber et al., 2002), the FXS mouse model neither harbors the accurate genetic context nor fully recapitulates the phenotypes of FXS patients. For example, the PAK1 inhibitor and negative allosteric modulator for mGluR5 have been found to ameliorate synaptic functions in the Fmr1 KO mouse model, but have variable efficacy in FXS patients (Dolan et al., 2013; Dolen et al., 2007; Erickson et al., 2017; Hagerman et al., 2009; Hayashi et al., 2007; Michalon et al., 2012). Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) derived from FXS patients have been reported to properly model the hypermethylation of CGG repeats and the silencing of FMR1 (Eiges et al., 2007; Urbach et al., 2010), representing a useful and complementary tool to study FXS.
The CRISPR/Cas9 system, developed for gene editing in the mammalian genome (Cong et al., 2013; Gilbert et al., 2014; Jinek et al., 2012; Komor et al., 2017; Mali et al., 2013; Wang et al., 2013), has been used for correcting disease-causing mutations (Huai et al., 2017; Long et al., 2016; Nelson et al., 2016; Park et al., 2015b; Tabebordbar et al., 2016). Similar gene editing approaches have been used to shorten the CGG trinucleotide repeats at the 5’ UTR to partially restore FMR1 expression and normalize the physiological function of FXS patient derived cells in the dish (Park et al., 2015a; Xie et al., 2016), providing a proof of principle for targeted therapies that involve FMR1 reactivation. Also, 5-Aza-2’-deoxycytidine treatment, inhibition of polycomb repressive complexes, and other molecular intervention for transcriptional regulation have shown to partially reactivate FMR1 in patient fibroblast cells (Crunkhorn, 2017; Erickson et al., 2017; Hagerman et al., 2009; Kumari and Usdin, 2016; Tabolacci et al., 2016), suggesting the epigenetic regulation plays a major role for FMR1 silencing in FXS.
To dissect the functional significance of DNA methylation events in the human genome (Lister et al., 2013; Lister et al., 2009; Schultz et al., 2015), we and others have designed DNA methylation editing tools by fusion of a catalytically inactivate Cas9 with the DNA methylation modification enzymes Dnmt or Tet (dCas9-Dnmt/Tet) allowing for targeted modification of DNA methylation in the mammalian genome in vitro and in vivo (Choudhury et al., 2016; Liu et al., 2016; Vojta et al., 2016; Xu et al., 2016). In the present study, we applied this tool to reverse the hypermethylation of CGG repeats at the FMR1 locus in multiple FXS patient derived iPSCs. We designed a single sgRNA to guide the dCas9-Tet1 to efficiently demethylate the CGG repeats in the pathological FMR1 locus. Complete demethylation of the CGG expansion induced hypomethylation of the CpG island, increased H3K27 acetylation and H3K4 trimethylation, decreased H3K9 trimethylation at the FMR1 promoter, and unlocked the epigenetic silencing of the FMR1 gene restoring FMRP expression in FXS iPSCs and neurons with no significant off-targeting effect. Expression of FMR1 and demethylation of its promoter in edited FXS cells were maintained for at least two weeks after inhibition of dCas9-Tet1 by a bacteriophage protein AcrIIA4. Epigenetic editing rescued the electrophysiological abnormalities of FXS neurons and the reactivation of FMR1 was maintained in edited neurons in vivo following transplantation into the mouse brain. We also demonstrated that demethylation of the CGG repeats in post-mitotic FXS neurons reactivated FMR1 and reversed the spontaneous hyperactivity associated with FXS neurons. Our study provides a proof of concept that reversion of gene inactivation by epigenome editing may be a valid therapeutic strategy for disorders that involve epigenetic silencing.
RESULTS
Demethylation of the CGG repeats to reactivate FMR1 in FXS iPSCs
To test whether demethylation of the hypermethylated CGG repeats can reactivate FMR1, we infected FX52 iPSCs, a previously described FXS iPSC line (Urbach et al., 2010) containing about 450 CGG repeats in the 5’ UTR region of FMR1, with lentiviruses expressing dCas9-Tet1-P2A-tBFP (dC-T) and a mCherry-expressing sgRNA targeting the “GGCGGCGGCGGCGGCGGCGGNGG” sequence (CGG sgRNA), and collected the cells expressing both vectors by FACS (Figure 1A). As shown in Figure 1B, the expression level of FMR1 mRNA in the cells with dC-T/CGG sgRNA was restored to 90% of the one in wild-type WIBR1 hESCs, whereas the cells expressing a catalytically inactive Tet1 (dC-dT) and the same sgRNA did not reactivate FMR1. Further, FMRP protein expression was restored in dC-T/CGG sgRNA expressing FX52 iPSCs to 73% of the wild-type level in WIBR1 cells as shown by immunofluorescence staining and western blotting in Figure 1C and1D. Methylation analysis of the CGG repeats showed a significant reduction of methylation from 100% in mock FX52 iPSCs to 4% in dC-T/CGG sgRNA expressing FX52 iPSCs (Figure 1E) and bisulfite sequencing (BS-seq) of the FMR1 promoter showed a robust demethylation after infection with lentiviral dC-T/CGG sgRNA vectors (Figure 1F and1G). These data indicate that dCas9-Tet1 targeted by the CGG sgRNA can efficiently demethylate the CGG repeats in FX52 iPSCs resulting in subsequent demethylation of the FMR1 promoter and reactivation of FMR1 both in the transcriptional and translational level. Two additional FXS iPSC lines with more than 450 CGG trinucleotide repeats (Brick et al., 2014; Xie et al., 2016) were also used to test the reactivation of FMR1 by dCas9-Tet1/CGG sgRNA. As shown in Figure S1A and S1B, FMR1 was reactivated after lentiviral transduction of dCas9-Tet1 and CGG sgRNA in both cell lines. FMR1 was reactivated to ~60% and ~30% of the level seen in WIBR1 wild-type cells respectively. The different levels of FMR1 expression are likely due to variable vector expressions in these particular experiments (see Figure 2A). We conclude that targeted demethylation of the CGG repeat expansion by dCas9-Tet1/CGG sgRNA can reactivate FMR1 in multiple FXS patient derived iPSCs. FX52 iPSC line was used for further studies.
Figure 1. Reactivation of FMR1 by dCas9-Tet1 in FXS iPSCs.

(A) Schematic representation of targeting the CGG repeats of FMR1 by dCas9-Tet1 with a CGG sgRNA to erase methylation and activate FMR1 expression. (B) A previously reported FXS iPSC line (FX52) was infected with lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) with a mCherry-expressing sgRNA targeting the CGG repeats “GGCGGCGGCGGCGGCGGCGGNGG” (CGG sgRNA) or lentiviruses expressing dCas9 fused with a catalytically dead Tet1 (dC-dT) with the same sgRNA. Double positive (BFP+; mCherry+) cells were isolated by FACS after infection. The expression level of FMR1 were quantified by qPCR analysis, and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of three biological replicates. (C) Cells in B were cultured on feeder MEFs and subject to immunofluorescence staining. Scale bar: 100 um. Stained in green for FMRP, red for Cas9, and blue for DNA. (D) Cells in B were subject to western blot analysis. The protein level of FMRP was quantified by ImageJ and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of two biological replicates. (E) Methylation levels of the CGG repeats in the FMR1 locus. Shown is the mean percentage of two biological replicates. (F) Bisulfite sequencing of cells described in B. (G) Methylation levels of individual CpGs in the FMR1 promoter region. Shown is the mean percentage ± SD of two biological replicates. See also Figure S1.
Figure 2. Off-target effects of dCas9-Tet1/CGG sgRNA.

(A) RT-qPCR analysis of three FX52 iPSC lines with different expression levels of dCas9-Tet1 and different restoration levels of FMR1 normalized to the wild-type WIBR1 hESCs. (B) A Manhattan plot depicting genome-wide binding sites of dCas9-Tet1/CGG sgRNA identified in cells described in A. All peaks with p value < 10−15 are shown. (C) BS-seq and Pyro-seq of the 6 top off-target candidate gene loci that overlapped with methylated promoter regions according to a human ESC/iPSC methylome study previously reported (Lister et al., 2009) and showed highest binding affinity of dCas9-Tet1 in B. CGG sgRNA target site (GGCGGCGGCGGCGGCGGCGGNGG) is illustrated with a red hexagon. Mismatch target site is illustrated with a yellow dot. CpGs are indicated by vertical bars. Shown is the mean percentage ± SD of two biological replicates. (D) Gene expression analyses of the 6 top off-target candidate genes by qPCR. Shown is the mean percentage ± SD of two biological replicates. (E) Anti-Cas9 ChIP-BS-seq of FX52 iPSC expressing dCas9-Tet1/CGG sgRNA (line #1 in A) or dCas9-dTet1/CGG sgRNA. 28 binding sites with change of methylation larger than 10% are labeled in red; FMR1 is labeled in green. The diameter of a circle is in proportion to the number of CGG sgRNA target sites; the binding sites with mismatched target site are indicated by the smallest circles. Blue lines of circles indicate the binding sites overlapping with a promoter region. The dash lines mark the 10% methylation difference between samples. (F) RNA-seq of mock and dCas9-Tet1/CGG sgRNA expressing FX52 iPSC line #1 described in A. Red dots highlight the 28 genes with a change of methylation larger than 10% identified in E. FMR1 is labeled with a green dot. The dashed red lines mark the 4-fold difference between the samples. See also Figure S1.
Off-target effect of dCas9-Tet1 is minimal
The presence of the CGG sgRNA targeting sequence “GGCGGCGGCGGCGGCGGCGGNGG” in other genomic loci raises concerns regarding off-target effects of the dCas9-Tet1/sgRNA system used. We examined the genome-wide binding sites for dCas9-Tet1 with CGG sgRNA by an anti-Cas9 ChIP-seq experiment using three FX52 iPSC lines expressing different levels of dCas9-Tet1 generated by lentiviral transduction with different viral titers. We observed that the restoration level of FMR1 decreased from 90% to 20% when the expression level of dCas9-Tet1 was reduced from 100% to 21% (Figure 2A), whereas reduction of CGG sgRNA did not affect the level of FMR1 reactivation (Figure S1C), suggesting dCas9-Tet1 rather than sgRNA was the limiting factor. The number of genome-wide dCas9-Tet1 binding sites was also decreased when the expression level of dCas9-Tet1 was reduced (Figure 2B). To examine possible off-target effects in more detail we chose the FX52 iPSC line #1 for further analysis as it has the highest number of dCas9-Tet1 binding sites among the three lines in which 90% of the FMR1 mRNA level was restored (Figure 2A). Among these binding sites, we first analyzed the top 6 candidate loci overlapping with methylated promoter regions according to a human ESC/iPSC methylome study previously reported (Lister et al., 2009) and showing the highest binding affinity of dCas9-Tet1 in ChIP-seq (Figure 2B). BS-seq showed a 20% and 30% reduction of methylation levels for SHCBP1L with one CGG sgRNA targeting site and RGPD1 with 6 targeting sites respectively but no detectable methylation changes for the other four genes (Figure 2C). The level of demethylation in SHCBP1L and RGPD1 loci likely correlate with the number of dCas9-Tet1/CGG sgRNA targeting sites. Gene expression analysis by qPCR showed either undetectable or minor changes with a 2.2-fold upregulation of SHCBP1L as the highest level of change (Figure 2D). To evaluate the methylation editing at a genome-wide level, we performed an anti-Cas9 ChIP-BS-seq experiment using FX52 iPSCs expressing dCas9-Tet1/CGG sgRNA (line #1 in Figure 2A) or dCas9-dTet1/CGG sgRNA. The result of this experiment allowed comparison of methylation levels at 1,018 dCas9-Tet1 binding sites between these two lines (Figure 2E). 29 loci showed a change of methylation larger than 10% with the FMR1 locus displaying the most significant methylation decrease (85%) in edited cells. Importantly, RNA-seq analysis of mock and methylation edited FX52 cells showed no significant change in overall gene expression (a correlation coefficient as 0.99) in Figure 2F. In contrast to the 1,500 fold upregulation of FMR1 expression, either no change or a minor change (maximal 4-fold) of expression was detected for the 28 genes with a change of methylation larger than 10% identified by ChIP-BS-seq. These data suggest that the off-target effect of dCas9-Tet1/CGG sgRNA on the DNA methylation and transcription levels is minimal and could be further minimized by titration of the expression level of dCas9-Tet1.
Activation of the heterochromatic FMR1 promoter
As shown previously, the FMR1 promoter region in FXS patient cells is in a heterochromatic conformation with DNA hypermethylation, decreased histone acetylation and H3K4 trimethylation and increased histone H3K9 trimethylation (Coffee et al., 2002; Coffee et al., 1999). To dissect the molecular mechanism of FMR1 reactivation by dCas9-Tet1/CGG sgRNA mediated demethylation of the CGG repeats, we examined the epigenetic state of the FMR1 promoter by ChIP-seq assays with antibodies against RNA polymerase II (RNAPII), H3K4me3, H3K27Ac, H3K27me3, and H3K9me3. As shown in Figure 3A, RNAPII was recruited to the FMR1 promoter in the methylation edited cells but not in the cells expressing dCas9-dTet1/CGG sgRNA. The active chromatin marks H3K4me3 and H3K27Ac were localized to the promoter regions in the edited cells with the repressive marker H3K9me3 being reduced, suggesting a switch of the heterochromatin state of the FMR1 promoter to an active chromatin conformation after demethylation of the CGG repeats. Two control loci including POU5F1 and MYOD1 showed no detectable change for these marks in edited cells. Genome-wide analysis of RNAPII occupancy showed that FMR1 is the most upregulated gene in the edited cells while the 28 genes with a change of methylation larger than 10% showed either no change or a minor change of RNAPII occupancy (less than 3-fold) except GSE1 with a 5-fold change (Figure 3B). Nevertheless the expression level of GSE1 did not change in the edited cells (Table S3). These data confirm a selective activation of FMR1 by dCas9-Tet1/CGG sgRNA. Genome-wide analysis of histone H3K4me3 distribution again highlighted the effect at the FMR1 locus with either no change or a minor change (less than 3-fold) for all the 28 genes (Figure 3C). Our observations support the role of DNA methylation of the CGG repeats as the major epigenetic block in silencing of FMR1, and argue that demethylation of these repeats is sufficient to rebuild an active chromatin status for the FMR1 promoter allowing for its reactivation.
Figure 3. Chromatin conformation of the reactivated FMR1 promoter.

(A) Anti-RNA polymerase II (RNAPII), anti-histone H3K4me3, anti-H3K27Ac, anti-H3K27me, and anti-H3K9me3 ChIP-seq signals at the FMR1, POU5F1, and MYOD1 loci in mock and edited FX52 iPSCs. (B-C) Genome-wide RNAPII occupancy (B) and histone H3K4me3 occupancy (C) of mock and edited FX52 iPSCs. Red dots highlight the 28 genes associated with a change of methylation larger than 10% as described in Figure 2E. FMR1 is labeled with a green dot. Promoters with a 3-fold or more change in factor occupancy were plotted as blue circles.
Kinetics and persistence of FMR1 reactivation
To gain insight into the kinetics of FMR1 reactivation by methylation editing, we performed a time course experiment to monitor the expression of FMR1. As shown in Figure 4A, the expression of FMR1 was first detected at 9-day time point after infection with dCas9-Tet1/CGG sgRNA lentiviruses and peaked around 3-week accompanied by demethylation of the FMR1 promoter (Figure 4D). To examine the persistence of methylation editing, we used AcrIIA4, a previously described inhibitor of Cas9/dCas9 (Rauch et al., 2017), to inactivate dCas9-Tet1. Expression of AcrIIA4 blocked the binding of dCas9-Tet1 to the FMR1 locus as shown in Figure 4B. We observed that FMR1 expression and demethylation of its promoter were maintained in the presence of AcrIIA4 for at least 2 weeks (Figure 4C and4D), suggesting constitutive presence of dCas9-Tet1 at the CGG repeats may not be required to sustain FMR1 reactivation through cell division.
Figure 4. The kinetics and persistence of methylation editing.

(A) FX52 iPSCs were infected with lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) and a CGG sgRNA, and were harvested for qPCR analysis of FMR1 expression at various time points. Shown is the mean expression level relative to that in a CGG deletion FX52 iPSC line (± SD of two biological replicates). This CGG deletion FX52 iPSC line was previously reported to restore FMR1 expression (Park et al., 2015a). (B) 293T cells were transfected with dC-T alone, with dC-T/CGG sgRNA, or with dC-T/CGG sgRNA and AcrIIA4. The cells were subject to an anti-Cas9 ChIP experiment and the enrichment of dC-T at the FMR1 and BDNF loci was examined by qPCR analysis. Shown is the mean of relative binding normalized to the input ± SD from three biological replicates. (C) Cells on Day-23 (labeled as Meth edited) in A were infected with lentivirus expressing AcrIIA4 and harvested for qPCR analysis at various time points. Shown is the mean FMR1 (red bar) and AcrIIA4 (blue bar) expression levels relative to the levels in the CGG deletion FX52 iPSC line (CGGΔ) and the cells on Day-3 after AcrIIA4 infection (D3), respectively (± SD of two biological replicates). (D) Pyro-seq of cells in A and C. Shown is the mean of percentage ± SD of two biological replicates.
Rescue of FXS phenotype in edited neurons
To evaluate the effect of FMR1 reactivation on the rescue of FXS-related cellular phenotypes, post-mitotic neurons were derived from the methylation edited FX52 iPSCs as shown in Figure 5A with a well-established differentiation protocol (Chambers et al., 2009). Gene expression analysis of lineage-specific markers suggested comparable differentiation states between wild type and mutant neural cultures (Figure 5B). The expression level of FMR1 in neurons expressing dC-T/CGG sgRNA was 82% of the one in wild type neurons and FMR1 remained silent in the neurons expressing dC-dT/CGG sgRNA (Figure 5C), suggesting that differentiation of edited FXS iPSCs did not affect the FMR1 reactivation. Immunohistochemistry confirmed that FMRP protein remained expressed in the edited neurons (Figure 5D). Genome-wide methylation analysis by ChIP-BS-seq using neurons derived from FX52 iPSCs expressing dC-T/CGG sgRNA or dC-dT/CGG sgRNA allowed for comparison of methylation levels at 670 binding sites, and identified 43 sites with a change of methylation larger than 10% including a 38% decrease of methylation at the FMR1 locus (Table S2). Nevertheless, transcriptome analysis of FX52 mock neurons and edited neurons outlined FMR1 as the most upregulated gene (481-fold) and showed either no change or a minor change (4-fold) of expression for the 41 genes and 9-fold change for RGPD1 gene associated with a larger than 10% methylation change highlighted with red dots in Figure 5E, suggesting a specific reactivation of FMR1. To examine the electrophysiological properties of methylation edited FXS neurons, multi-electrode array (MEA) assay was performed with wild type WIBR1 neurons and FX52 mock, dC-T/CGG sgRNA, and dC-dT/CGG sgRNA expressing neurons. As shown in Figure 5F, the significantly higher firing rate of FX52 neurons was reduced to the levels of wild-type neurons in dC-T/CGG sgRNA expressing neurons but not in neurons expressing dC-dT/CGG sgRNA. These results suggest that reactivation of FMR1 reversed the spontaneous hyperactive phenotype of FXS neurons.
Figure 5. Phenotypical rescue of FXS-related cellular deficits.

(A) Methylation edited FX52 iPSCs were differentiated into neuronal processor cells (NPCs) and post-mitotic neurons. Scale bar: 500 um. (B) Expression levels of linage-specific markers in neurons described in A measured by qPCR. Shown is the mean percentage ± SD of two biological replicates. (C) FMR1 expression level of neurons in A measured by qPCR. Shown is the mean percentage ± SD of two biological replicates. (D) Neurons in A were subject to immunofluorescence staining. Scale bar: 100 um. Stained in green for FMRP, red for Cas9, blue for DNA, and grey for MAP2. (E) RNA-seq of mock and dCas9-Tet1/CGG sgRNA expressing FX52 neurons described in A. Red circles highlight 43 genes with a change of methylation larger than 10% in dCas9-Tet1/CGG sgRNA expressing neurons (Table S2). FMR1 is labeled with a green dot. The dashed red lines mark a 4-fold difference between the samples. (F) Multi-electrode array (MEA) of wild-type WIBR1 neurons and FX52 mock, dCas9-Tet1/CGG sgRNA or dCas9-dTet1/CGG sgRNA expressing neurons. Shown is the mean ± SD of biological triplicates for each type of neurons. (G) Schematic representation of engraftment of 1:1 mixed cells containing mock FX52 NPCs labeled with GFP and methylation edited FX52 NPCs labeled with RFP into the P1 mouse brain and harvested for analysis one month post-engraftment. (H) Representative confocal micrographs of cells engrafted in the mouse brain at one month post-transplantation. Scale bar: 50 μm. Stained in red for RFP, green for GFP, magenta for FMRP, and blue for DNA. A total of 496 RFP-positive neurons and 149 GFP-positive neurons from three mice after one month were counted. 56% ± 9% of RFP neurons were FMRP-positive, whereas all GFP positive neurons were FMRP-negative. A total of 203 RFP-positive neurons and 117 GFP-positive neurons from two mice after three months were counted. 57% ± 3% of RFP neurons were FMRP-positive, whereas all GFP positive neurons were FMRP-negative.
FMR1 reactivation in edited FXS neurons is sustained after engrafting into mouse brains
To test whether the reactivation of FMR1 in the methylation edited FXS cells is sustainable in vivo, FX52 mock or methylation edited neuronal precursor cells (NPCs) were labeled with GFP or RFP lentiviruses respectively, and then the mixture of these two types of NPCs was injected into the P1 mouse brain for subsequent analysis one- or three-month post transplantation (Figure 5G). Immunofluorescence staining of the mouse brain sections showed that 56% and 57% of the edited FX52 neurons (RFP-positive) in oneand three-month post transplanted mice, respectively, expressed FMRP whereas FX52 mock neurons (GFP-positive) were negative for FMRP expression (Figure 5H), suggesting that FMR1 reactivation can be maintained in vivo after transplantation.
Deletion of CGG repeats results in a similar phenotypic rescue as epigenetic editing
To evaluate the rescue effects observed in methylation edited FXS cells compared with cells lacking the CGG expansion mutation, we characterized a pair of FXS iPSC lines including an isogenic line with CGG deletion by CRISPR/Cas9 technology (Xie et al., 2016). Both FMR1 mRNA and FMRP protein were restored in the CGG deletion line (Figure 6A–C). The methylation level of the FMR1 promoter was reduced to 39% in the CGG deletion line (Figure 6D and6E). Importantly, the spontaneous hyperactivity associated with FXS neurons was significantly reduced in the CGG deletion FXS neurons as shown in Figure 6F and6G. Thus, the decrease of the FMR1 promoter methylation and the reversal of the hyperactivity in the CGG deletion FXS cells are consistent with our observations on the methylation edited FXS cells, suggesting a functional rescue of FXS-related cellular phenotypes by the demethylation of the CGG expansion.
Figure 6. Deletion of CGG repeats to rescue FXS phenotypes.

(A) Schematic representation of deletion of the CGG repeats in the FMR1 locus by CRISPR/Cas9 to activate FMR1 expression. (B) FMR1 mRNA level measured by RT-qPCR in a control iPSC line, a FXS iPSC line with more than 450 CGG repeats and a CGG deletion FXS iPSC line generated by CRISPR/Cas9 gene editing (Xie et al., 2016). Shown is the mean ± SD of 3 biological replicates. (C) FMRP expression in neurons derived from iPSCs in B. Stained in blue for DAPI, green for MAP2, and red for FMRP. Scale bar: 100 um. (D) Summary of the methylation levels of the CGG repeats and the FMR1 promoter for the cells in B. (E) Methylation levels of CpGs in the FMR1 promoter of the cells in B. (F) MEA assay of the neurons derived from the iPSCs in B with the Ngn2 method (Zhang et al., 2013). Shown is the mean ± SD of 6 biological replicates. (G) Quantification of neural firing rate at 22 days post plating on the MEA plate. Shown is the mean ± SD of 6 biological replicates.
Direct reactivation of FMR1 in FXS neurons
As neurons are the most disease-relevant cell type in FXS patients, we tested whether FMR1 could be reactivated with dCas9-Tet1/CGG sgRNA in post-mitotic neurons derived from FXS iPSCs. After infection with lentiviruses expressing dC-T or dC-dT with CGG sgRNA, the mRNA level of FMR1 in dC-T/CGG sgRNA expressing FXS neurons was restored to 45% of the one in wild type neurons but dC-dT/CGG sgRNA did not reactivate FMR1 (Figure 7B). FMRP proteins were only detected in the neurons expressing dC-T/CGG sgRNA but not dC-dT/CGG sgRNA (Figure 7A). These results suggest that reactivation of FMR1 in post-mitotic FXS neurons is achievable by dCas9-Tet1/CGG sgRNA but less efficiently as compared to FXS iPSCs shown in Figure 1B and1C. Analysis of the CGG methylation status showed 30% reduction in the edited neurons compared to FXS mock neurons (Figure 7C). BS-seq of the FMR1 promoter in these neurons showed a 20% decrease of the methylation level in the edited FXS neurons as compared to FXS mock neurons (Figure 7D). The demethylation of the CGG repeats and subsequent demethylation of the FMR1 promoter and the reactivation of the FMR1 gene were less robust as compared to that seen in edited iPSCs (Figure 1). This is likely due to our inability to isolate double vector-infected neurons by FACS and the different DNA demethylation mechanism in post-mitotic neurons compared to dividing iPSCs (Wu and Zhang, 2014). Nevertheless, MEA assays revealed a rescue of the electrophysiological abnormalities in the edited FXS neurons (Figure 7E) suggesting the spontaneous hyperactivity associated with FXS neurons was reversed after the reactivation of FMR1 in these neurons.
Figure 7. Direct reactivation of FMR1 in the FXS neurons.

(A) FX52 neurons were infected with lentiviruses expressing dCas9-Tet1 (dC-T) and the CGG sgRNA or dCas9-dTet1 (dC-dT) with the same sgRNA. Infected neurons were subject to immunofluorescence staining. Scale bar: 50 um. Stained in green for FMRP, red for Cas9, blue for DNA, and grey for MAP2. (B) The expression level of FMR1 in A was quantified by qPCR analysis, and shown as the mean relative to the level in WIBR1 hESCs ± SD of two biological replicates. (C) Methylation levels of the CGG repeats in the FMR1 locus. Shown is the mean percentage ± SD of two biological replicates. (D) Bisulfite sequencing of the FMR1 promoter in cells described in A. Shown is the mean percentage ± SD of two biological replicates. (E) MEA assay of the neurons in A. Shown is the mean percentage ± SD of two biological replicates.
DISCUSSION
In this study, we applied recently developed DNA methylation editing tools to reverse the hypermethylation of the CGG repeats in the FMR1 locus. An iPSC-based FXS model was chosen as it recapitulates the hypermethylation of CGG repeat expansion and epigenetic silencing of FMR1. Our results demonstrate that targeted demethylation of the CGG repeats reactivated FMR1 in multiple FXS iPSCs as well as in in vitro derived FXS neurons. Demethylation of the CGG repeats resulted in conversion of the heterochromatic state to an active state of the non-targeted upstream FMR1 promoter. Thus, our data provide the first direct evidence that demethylation of the CGG repeats is sufficient to reactivate FMR1. Importantly, methylation editing reversed the abnormal electrophysiological phenotype of FXS neurons, and the expression of FMRP in edited neurons was maintained in vivo after transplantation into the mouse brain. To address potential off-target effects associated with the dCas9-Tet1/CGG sgRNA system, we performed a genome-wide survey to identify binding sites of dCas9-Tet1 with CGG sgRNA and examined the methylation levels of these sites. ChIP-BS-seq revealed a small set of sites with changes in methylation larger than 10% and RNA-seq analysis showed no major off-target effects on transcriptional levels of these genes suggesting a highly specific effect by targeted methylation editing.
Trinucleotide repeat expansions play a pathological role in several neurological, neurodegenerative, and neuromuscular disorders such as FXS, Huntington’s disease, spinocerebellar ataxia, and myotonic dystrophy (Nelson et al., 2013; Pearson et al., 2005). Repeats can elicit toxicity through a series of overlapping pathogenic cascades, including gain-of-function or loss-of-function at the protein and/or RNA level(s). Silencing of FMR1 with more than 200 CGG repeats is considered as the cause for FXS. However the carrier patients with 55–200 CGG repeats have overproduction of FMR1 mRNA but reduction of FMRP protein and develop Fragile X-Associated Tremor and Ataxia syndrome (FXTAS) with some features of the FXS phenotype including progressive cerebellar tremor and ataxia, cognitive impairment, mild parkinsonian symptoms and brain atrophy (Hagerman et al., 2001). The repeat-associated non-AUG (RAN) translation of a transfected CGG repeat-containing construct has been shown to generate toxic neuronal proteins (Kearse et al., 2016; Todd et al., 2013). Also, RNA inclusions were found in a small subgroup (6–11%) of FXTAS nuclei (Tassone et al., 2004; Wojciechowska and Krzyzosiak, 2011). Restoration of FMRP protein and rescue of FXS cellular phenotypes after reactivation of FMR1 argue that the cellular toxicity assumed for CGG repeat-containing FMR1 RNA is likely to be minimal in the methylation edited FXS cells within the short period of culture time. It remains an open question whether the reactivated FMR1 in FXS cells results in RAN and/or forms intranuclear RNA inclusions, but our tool provides a unique experimental system to further investigate the toxicity of CGG repeats in neurons.
Accurate and efficient targeting with a controllable off-target effect is the ultimate goal for gene therapy and epigenome editing towards therapeutic intervention. To examine the potential off-target effect of dCas9-Tet1/CGG sgRNA, we performed a series of genome-wide analyses of the methylation edited FXS cells. Compared to the robust reduction on the methylation of the FMR1 locus (84% decrease), a global methylation analysis of FXS cells expressing dCas9-Tet1/CGG sgRNA or dCas9-dTet1/CGG sgRNA by ChIP-BS-seq revealed that only a small set of binding loci (28 among 1018 sites in edited FXS iPSC, and 42 among 670 sites in edited FXS neurons) changed the level of methylation by more than 10%. The high methylation editing specificity and efficiency of dCas9-Tet1/CGG sgRNA are likely due to the repetitive nature of the CGG repeats present in the FMR1 locus in FXS cells, which allows for an enrichment of the dCas9-Tet1 machinery on the condensed CGG repeats for efficient methylation editing. Importantly, RNA-seq revealed either no change or minor changes of expression for the genes associated with these sites, suggesting a highly specific effect on FMR1 reactivation by methylation editing. Nevertheless, off-target binding of dCas9-Tet1 is a concern. We found that the level of dCas9-Tet1 vector expression was positively correlated with both, the expression level of FMR1 and the number of off-target binding sites of dCas9-Tet1 suggesting that the dCas9-Tet1/CGG sgRNA is a tunable methylation editing tool that allows for restoration of FMR1 at different expression levels with a potentially controllable off-target effect to safely rescue FXS phenotypes. Furthermore, we observed that reactivation of FMR1 and demethylation of its promoter did not require a constitutive editing by dCas9-Tet1 and was maintained for at least two weeks after inactivation of dCas9-Tet1. Another strategy to minimize off-target binding would be to target the dCas9-Tet1 to the FMR1 promoter instead of the CGG repeats as targeting the promoter would allow for designing unique sgRNAs and reducing potential off-target binding of dCas9-Tet1.
Whether the FXS phenotype can be reversed postnatally is unknown. It has been shown that re-expression of FMRP by AAV-based gene delivery could partially rescue the neuronal phenotype in Fmr1 mutant mice suggesting that the functional deficits may be at least partially reversible (Gholizadeh et al., 2014; Zeier et al., 2009). We have demonstrated that restoration of FMR1 can be achieved in in vitro derived FXS neurons by dCas9-Tet1/CGG sgRNA-mediated methylation editing. Electrophysiological analysis of the edited neurons with 45% restoration of FMR1 showed a similar behavior as the wild-type neurons, indicating that the FXS cellular phenotype is likely reversible in neurons and full restoration of FMR1 may not be necessary for a functional rescue opening a potential therapeutic window for the treatment of FXS.
In summary, our study demonstrates that demethylation of the CGG repeats is sufficient to reactivate FMR1. Thus, methylation editing is a valid strategy to reactivate FMR1 and to rescue the FXS-related cellular phenotypes, providing a proof-of-concept paradigm to study disease-associated abnormal DNA methylation events. This method for epigenome editing can be easily applied to examine the causality of disease-associated DNA methylation events and evaluate the consequence after targeted reversal of DNA methylation status holding great potentials for the future research for novel therapies.
Methods and Resources
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Rudolf Jaenisch (jaenisch@wi.mit.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines and breeding strategies
An immuno-compromised mouse line (NOD-scid IL2Rgnull) was used for the transplantation experiment. Mice were housed in the animal facility at Whitehead Institute with sterilized water, food, and cages. Caretakers wear PPE in the mouse room. Male and female mice at the P1 stage were used for transplantation experiments, and no sex differences were observed. Mice were handled in accordance with institutional guidelines and approved by the Committee on Animal Care (CAC) and Department of Comparative Medicine (DCM) of Massachusetts Institute of Technology.
METHOD DETAILS
Plasmid design and construction
PCR amplified Tet1 catalytic domain from pJFA344C7 (Addgene plasmid: 49236), Tet1 inactive catalytic domain from MLM3739 (Addgene plasmid: 49959), and tagBFP (synthesized gene block) were cloned into FUW vector (Addgene plasmid: 14882) with AscI, EcoRI and PfIMI to package lentiviruses. The CGG sgRNA expression plasmids were cloned by inserting annealed oligos into modified pgRNA plasmid (Addgene plasmid: 44248) with AarI site. A synthetic gBlock encoding the bacteriophage AcrIIA4 purchased from IDT was cloned into a modified FUW vector with AscI and EcoRI to package lentiviruses. All constructs were sequenced before transfection. Primer information for sgRNA design and construction is listed in Supplemental Table S4. Related plasmids have been deposited into Addgene plasmid database.
Cell culture and lentivirus production
All the FXS iPSC lines were derived from male patients and reported previously (see KEY RESOURCES TABLE). The CGG repeat expansion mutations were verified by Claritas Genomics Inc with Asuragen AmplideX® mPCR approach and the mycoplasma test was negative. FXS iPSCs were cultured either with mTeSR™1 medium (STEMCELL, #85850) or on irradiated mouse embryonic fibroblasts (MEFs) with standard hESCs medium: [DMEM/F12 (Invitrogen) supplemented with 15% fetal bovine serum (Gibco HI FBS, 10082–147), 5% KnockOut Serum Replacement (Invitrogen), 2 mM L-glutamine (MPBio), 1% nonessential amino acids (Invitrogen), 1% penicillin-streptomycin (Lonza), 0.1 mM β-mercaptoethanol (Sigma) and 4 ng/ml FGF2 (R&D systems)]. Lentiviruses expressing dCas9-Tet1-P2A-BFP, sgRNAs, and AcrIIA4 were produced by transfecting HEK293T cells with FUW constructs or pgRNA constructs together with standard packaging vectors (pCMV-dR8.74 and pCMV-VSVG) followed by ultra-centrifugation-based concentration. Virus titer (T) was calculated based on the infection efficiency for 293T cells, where T = (P*N) /(V), T = titer (TU/ul), P = % of infection positive cells according to the fluorescence marker, N = number of cells at the time of transduction, V = total volume of virus used. Note TU stands for transduction unit. Lentiviruses labeling NPCs (EF1A-GFP and EF1A-RFP) were purchased from Cellomics Technology.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Cas9 (IF staining) | Active Motif | Cat#61577 (7A9-3A3) |
| Mouse monoclonal anti-Cas9 (ChIP) | Active Motif | Cat#61757 (8C1-F10) |
| Chicken polyclonal anti-GFP | Aves Labs | Cat#GFP-1020 |
| Chicken polyclonal anti-MAP2 | Encor Biotech | Cat# CPCA-MAP2 |
| Rabbit polyclonal anti-FMRP (WB, ICC, IHC) | Cell Signaling | Cat#4317 |
| Mouse monoclonal anti-FMRP (ICC, IHC) | Biolegend | Cat#834701 |
| Mouse monoclonal anti-FMR1PolyG | EMD Millipore | Cat#MABN784 |
| Goat polyclonal anti-mCherry | SICGEN | Cat#AB0040-200 |
| Mouse monoclonal anti-RNA Polymerase II | Abcam | Cat#ab817 |
| Rabbit polyclonal anti-H3K27Ac | Abcam | Cat#ab4729 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898 |
| Mouse monoclonal anti-H3K27me3 | Abcam | Cat#ab6002 |
| Rabbit polyclonal anti-H3K4me3 | Millipore | Cat#07-473 |
| Chemicals | ||
| Doxycycline hyclate | Sigma Aldrich | Cat#D9891-100G |
| Critical Commercial Assays | ||
| EpiTect Bisulfite Kit | Qiagen | Cat#59104 |
| EpiNext™ High-Sensitivity Bisulfite-Seq kit | EPIGENTEK | Cat#P-1056 |
| EpiNext™ NGS Barcode Set-12 | EPIGENTEK | Cat#P-1060 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| Zymoclean Gel DNA Recovery Kit | Zymo Research | Cat#D4002 |
| DNA Clean & Concentrator-5 | Zymo Research | Cat#D4013 |
| X-tremeGENE™ 9 DNA Transfection Reagent | Sigma Aldrich | Cat#6365809001 |
| Direct-zol RNA Miniprep | Zymo Research | Cat#R2050 |
| SuperScript III First-Strand Synthesis SuperMix | Life Technologies | Cat#18080400 |
| Fast SYBR Green Master Mix | Life Technologies | Cat#4385618 |
| Deposited Data | ||
| Raw data files for sequencing | NCBI Gene Expression Omnibus | Database: GSE102655, GSE102684, GSE108171, GSE108498, GSE108577 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD-scid IL2Rgnull, | Jackson Laboratories | Stock No: 005557 |
| Experimental Models: Cell Lines | ||
| FX52 iPSCs | (Urbach et al., 2010) | N/A |
| FXS iPSC (135.3) | (Brick et al., 2014) | N/A |
| FXS iPSC (FXS_SW) | (Xie et al., 2016) | N/A |
| Recombinant DNA | ||
| Fuw-dCas9-Tet1-P2A-BFP | This paper | N/A |
| pgRNA | This paper | N/A |
| Fuw-AcrIIA4-P2A-GFP | This paper | N/A |
| See “Plasmid design and construction” in METHOD DETAILS section | ||
| Sequence-Based Reagents | ||
| See Table S4, S5, S6 for primer sequences | This paper | N/A |
| Software and Algorithms | ||
| MACS (ChIP-seq algorithms) | Wu et al., 2014b | http://liulab.dfci.harvard.edu/MACS/ |
| ImageJ (Fiji) | NIH | http://imagej.net/Fiji |
Multi-electrode array recording
Two- or four-week-old differentiating neuronal cultures were dissociated using Accutase and 5 × 105 cells were plated on each single well in the PEI-coated Axion Biosystems # M768-GL1–30Pt200 arrays. Recordings of spontaneous activities during a 5-minute period were performed on days indicated. Biological triplicates for each type of neurons were included.
Transplantation of FXS NPCs into mouse brain
Cultured human neural precursor cells (NPC) were dissociated using Accutase (Life Technologies) and resuspended in phosphate buffer saline without calcium and magnesium prior to injection, at a concentration of 6×104 cells/μL. NOD-scid IL2Rgnull mouse pups of either sex were manually injected within a day of birth with a total of 300,000 hNPC dispersed over five injection sites, as described previously (Windrem et al., 2014). The mice were perfused with 4% paraformaldehyde for immunohistochemistry at 1 month of age (n = 3 mice) and 3 months of age (n = 2 mice), and 100 μm-thick brain sections were prepared for immunofluorescence analyses with the antibodies indicated.
Immunocytochemistry, immunohistochemistry, microscopy, and image analysis
FXS iPSCs and neurons were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. Cells were permeablized with PBST (1 × PBS solution with 0.1% Triton X-100) before blocking with 10% Normal Donkey Serum (NDS) in PBST. Cells were then incubated with appropriately diluted primary antibodies in PBST with 5% NDS for 1 hours at room temperature or 12 hours at 4 °C, washed with PBST for 3 times at room temperature and then incubated with desired secondary antibodies in TBST with 5% NDS and DAPI to counter stain the nuclei. Mouse brain slices were incubated for an hour in PBS with 0.5% Triton and 3% normal serum. Slices were then incubated with desired primary antibodies in the same solution for 72 hours at 4 °C, washed with PBS for 3 times at room temperature and then incubated with secondary antibodies in PBS for 24 hours away from light at room temperature. Staining of the nuclei with DAPI was then performed. Sections were washed 3 times with PBS before slide mounting. The following antibodies were used in this study: Chicken anti-GFP (1:1000, Aves Labs), Rabbit anti-FMRP (1:50, Cell Signaling), Chicken anti-MAP2 (1:1000, Encor Biotech), Goat anti-mCherry (1:1000, SICGEN). Images were captured on a Zeiss LSM710 confocal microscope and processed with Zen software, ImageJ/Fiji, and Adobe Photoshop. For imaging-based quantification, unless otherwise specified, 3–5 representative images were quantified and data were plotted as mean ± SD with Excel or Graphpad Prism.
FACS analysis
To isolate the infection-positive cell after lentiviral transduction, the treated cells were dissociated with trypsin and single-cell suspensions were prepared in growth medium subject to a BD FACSAria cell sorter according to the manufacture’s protocol at the Whitehead Institute Flow Cytometry Core. Data were analyzed with FlowJo software.
Western blot
Cells were lysed by RIPA buffer with proteinase inhibitor (Invitrogen), and subject to standard immunoblotting analysis. Mouse anti-Cas9 (1:1000, Active Motif), mouse α-Tubulin (1:1000, Sigma), mouse anti-FMR1polyG (1:1000, EMD Millipore), rabbit anti-FMRP (1:100, Cell Signaling) antibodies were used.
RT-qPCR
Cells were harvested using Trizol followed by Direct-zol (Zymo Research), according to manufacturer’s instructions. RNA was converted to cDNA using First-strand cDNA synthesis (Invitrogen SuperScript III). Quantitative PCR reactions were prepared with SYBR Green (Invitrogen), and performed in 7900HT Fast ABI instrument. Primer information for RT-qPCR is listed in Supplemental Table S5.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described in (Lee et al., 2006) with a few adaptations. Cells were crosslinked for 15 minutes at room temperature by the addition of one-tenth volume of fresh 11% formaldehyde solution (11% formaldehyde, 50 mM HEPES pH 7.3, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0) to the growth media followed by 5 min quenching with 125 mM glycine. Cells were rinsed twice with 1X PBS and harvested using a silicon scraper and flash frozen in liquid nitrogen. Frozen crosslinked cells were stored at −80°C. For immunoprecipitabon of lysate from 100 million cells, 50 μl of Protein G Dynabeads (Life Technologies #10009D) and 5 μg of antibody were prepared as follows. Dynabeads were washed 3X for 5 minutes with 0.5% BSA (w/v) in PBS. Magnetic beads were bound with the antibody overnight at 4°C, and then washed 3X with 0.5% BSA (w/v) in PBS.
Cells were prepared for ChIP as follows. All buffers contained freshly prepared 1× cOmplete protease inhibitors (Roche, 11873580001). Frozen crosslinked cells were thawed on ice and then resuspended in lysis buffer I (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1× protease inhibitors) and rotated for 10 minutes at 4°C, then spun at 1350 rcf for 5 minutes at 4°C. The pellet was resuspended in lysis buffer II (10 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1× protease inhibitors) and rotated for 10 minutes at 4°C and spun at 1350 rcf for 5 minutes at 4°C. The pellet was resuspend in sonication buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.1% SDS, and 1% Triton X-100, 1× protease inhibitors) and then sonicated on a Misonix 3000 sonicator for 10 cycles at 30 seconds each on ice (18–21 W) with 60 seconds on ice between cycles. Sonicated lysates were cleared once by centrifugation at 16,000 rcf for 10 minutes at 4°C. 50 uL was reserved for input, and then the remainder was incubated overnight at 4°C with magnetic beads bound with antibody to enrich for DNA fragments bound by the indicated factor. Beads were washed twice with each of the following buffers: wash buffer A (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), wash buffer B (50 mM HEPES-KOH pH 7.9, 500 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), wash buffer C (20 mM Tris-HCl pH8.0, 250 mM LiCl, 1 mM EDTA pH 8.0, 0.5% Na-Deoxycholate, 0.5% IGEPAL C-630 0.1% SDS), wash buffer D (TE with 0.2% Triton X-100), and TE buffer. DNA was eluted off the beads by incubation at 65°C for 1 hour with intermittent vortexing in 200 uL elution buffer (50 mM Tris-HCL pH 8.0, 10 mM EDTA, 1% SDS). Cross-links were reversed overnight at 65°C. To purify eluted DNA, 200 uL TE was added and then RNA was degraded by the addition of 2.5 μL of 33 mg/mL RNase A (Sigma, R4642) and incubation at 37°C for 2 hours. Protein was degraded by the addition of 10 μL of 20 mg/mL proteinase K (Invitrogen, 25530049) and incubation at 55°C for 2 hours. A phenol:chloroform:isoamyl alcohol extraction was performed followed by an ethanol precipitation. The DNA was then resuspended in 50 uL TE and used for sequencing. Purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries. Libraries for Illumina sequencing were prepared following the Illumina TruSeq DNA Sample Preparation v2 kit. Amplified libraries were size-selected using a 2% gel cassette in the Pippin Prep system from Sage Science set to capture fragments between 200 and 400 bp. Libraries were quantified by qPCR using the KAPA Biosystems Illumina Library Quantification kit according to kit protocols. Libraries were sequenced on the Illumina HiSeq 2500 for 40 bases in single read mode.
Cas9 ChIP-seq peak calling method
Cas9 ChIP-seq data was analyzed as follows. Reads are de-multiplexed and mapped to human genome (hg19) using STAR (Dobin et al., 2013), requiring unique mapping and perfect match. Peaks are called using MACS (Zhang et al., 2008) with equal number of collapsed reads sampled to match sequencing depth. For Figure 2B, all IP samples were compared to the same input sample with a peak p value cut off of 10−15, and peaks are listed in Table S1. Raw data can be accessed with GSE102655 at NCBI Gene Expression Omnibus.
ChIP-seq Scatter plot method
For analysis of factor occupancy at gene promoters in Figure 3, we defined promoters as two kilobase regions centered on the transcription start sites of all RefSeq annotated transcripts (GRCh38/hg19, RefSeq Genes). To quantify the occupancy of each factor at promoters, we first quantified the ChIP-seq signal at each promoter region in reads per million mapped reads (rpm). To do this, we extended ChIP-seq reads to 200 bp, calculated the number of ChIP-seq reads that aligned to each promoter using bamToGFF (https://github.com/BradnerLab/pipeline), and divided by the number of million mapped reads in the dataset. For each promoter, we then subtracted the signal in the matching input control samples, which were also quantified in rpm as described above, to produce an input normalized ChIP-seq signal. In cases where the input normalized ChIP-seq signal was less than zero, the value was set to zero. To visualize differences in factor occupancy at promoters in different samples, we plotted the input normalized ChIP-seq signals for each promoter as a scatter plot. This was performed by adding a pseudocount of 0.1 to each input normalized ChIP-seq signal value and plotting the log2 transformed values. Promoters that showed a 3 fold or more change in factor occupancy were plotted as blue circles, and 28 loci with a change of methylation level larger than 10% were plotted as red dots. Raw data can be accessed with GSE102684 at NCBI Gene Expression Omnibus.
ChIP-BS-seq
Anti-Cas9 ChIP experiment was performed as described above. The BS conversion and sequencing library preparation were performed according to the instructions by EpiNext High-Sensitivity Bisulfite-Seq Kit (EPIGENTEK, #P-1056A) and EpiNext NGS Barcode (EPIGENTEK, #P-1060). To analyze the raw data, the adapter sequences in the illumina reads identified with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) were removed with Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). BS-Seq aligner Bismark (Krueger and Andrews, 2011) was used for assigning reads to human genome hg19 and calling methylation with bismark_methylation_extractor. To increase the number of uniquely mapped reads, after the first bismark alignment, 5 bases from the 5’ and one base from the 3’ of the unmapped reads were trimmed based on FastQC analysis. The resulting trimmed reads were then aligned to genome with Bismark. In both cases, bismark was ran with the options “--non_directional -un --ambiguous --bowtie2 -N 1 -p 4 --score_min L,−6,−0.3 --solexa1.3-quals”. To compare the methylation levels of dCas9-Tet1 binding sites between dC-T and dC-dT samples, only the anti-Cas9 ChIP-seq peaks that included at least 20 CpG sites in which each CpG was covered with at least 10 reads in iPSCs and 5 reads in neurons by ChIP-BS-seq were selected to calculate the methylation levels. The number of binding sites in iPSC cells is 1018 and 670 in neurons. Sites with at least 10% changes of methylation level (average CpG methylation level of each site) were highlighted in red and the genes whose transcript start sites were no more than 100 bp away from closest off-target sites were circled in blue in Figure 2E, Figure 2F and Figure 5E. The scan_for_matches was utilized to search for the GGCGGCGGCGGCGGCGGCGGNGG motif in the sequences derived from those binding sites. R scripts were written for generating graphs. Peaks with calculated methylation levels are listed in Table S2. Raw data along with methylation levels from Bismark can be accessed with GSE108171 at NCBI Gene Expression Omnibus.
Bisulfite Conversion, PCR and Sequencing
Bisulfite conversion of DNA was established using the EpiTect Bisulfite Kit (Qiagen) following the manufacturer’s instructions. The resulting modified DNA was amplified by first round of nested PCR, following a second round using loci specific PCR primers (Supplemental Table S6). The first round of nested PCR was done as follows: 94 °C for 4 min; 55 °C for 2 min; 72 °C for 2 min; Repeat steps 1–3 1 X; 94 °C for 1 min; 55 °C for 2 min; 72 °C for 2 min; Repeat steps 5–7 35X; 72 °C for 5 min; Hold 12 °C. The second round of PCR was as follows: 95 °C for 4 min; 94 °C for 1 min; 55 °C for 2 min; 72 °C for 2 min; Repeat steps 2–4 35 X; 72 °C for 5 min; Hold 12°C. The resulting amplified products were gel-purified, sub-cloned into a pCR2.1-TOPO-TA cloning vector (Life technologies), and sequenced. Primer information for bisulfite sequencing is listed in Supplemental Table S6.
DNA Methylation analysis
Pyro-seq of all bisulfite converted genomic DNA samples were performed with PyroMark Q48 Autoprep (Qiagen) according to the manufacturer’s instructions. Primers information for pyro-seq is listed in Supplemental Table S6. Methylation analysis of CGG trinucleotide repeats: Methylation status off CGG repeats were analyzed by Claritas Genomics Inc. with Asuragen AmplideX® mPCR approach.
RNA-seq and analysis
The 40 bp single-end reads from Illumina had good quality by checking with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped to hg19 using TopHat v2.1.1 (Kim et al., 2013) with options as “solexa1.3-quals” mode and “no-novel-juncs”. The gene model for TopHat was created by merging knownGene in gtf format with kgXref table. Both knownGene and kgXref were downloaded from UCSC table browser in hg19 assembly. The read counts were obtained using featureCounts (Liao et al., 2014) function from Subread package (Liao et al., 2013) with strandness option as −r 2. Reads were normalized with DESeq2 (Love et al., 2014). The biological replicates in the neuron samples, processed at different time periods, have batch effect as suggested by principle component analysis. Consequently, Combat was used for reducing this batch effect. The log2 values of the normalized reads were plotted in the scatterplots in Figure 2F, and 5E. To avoid log0 as undefined value, the number of reads for each gene was increased by one. Raw data along with gene expression levels were deposited to NCBI Gene Expression Omnibus GSE108498.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n and measures (mean ± SD) and statistical significance are reported in the Figures and the Figure Legends. Data is judged to be statistically significant when p < 0.05 by two-tailed Student’s T-Test or 2-way ANOVA, where appropriate.
DATA AND SOFTWARE AVAILABILITY
Raw data files for sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GSE102655, GSE102684, GSE108171, GSE108498. These SubSeries are linked to one SuperSeries GSE108577.
Supplementary Material
Figure S1. Reactivation of FMR1 by dCas9-Tet1 with CGG sgRNA in multiple FXS iPSC lines (A) One previously reported FXS iPSC line (135.3) with more than 450 CGG repeats was infected with lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) with a mCherry expressing sgRNA targeting the CGG repeats “GGCGGCGGCGGCGGCGGCGGNGG” (CCG sgRNA) or lentiviruses expressing dCas9 fused with a catalytically dead Tet1 (dC-dT) with the same sgRNA. Double positive (BFP+; mCherry+) cells were isolated by FACS after infection. The expression level of FMR1 were quantified by qPCR analysis and compared to an isogenic line with shortened CGG repeat expansion, and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of two biological replicates. (B) Another previously reported FXS iPSC line (SW) with more than 450 CGG repeats was used to examine FMR1 expression as did in B. The expression level of FMR1 were quantified by qPCR analysis and compared to an isogenic line with shortened CGG repeat expansion, and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of two biological replicates. (C) FX52 iPSCs were infected with different ratios of lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) and a CGG sgRNA. Infected cells were harvested for qPCR analysis of FMR1 expression at various time points. Shown is the mean of relative percentages to the one in a CGG deletion FX52 iPSC line ± SD of two biological replicates.
Table S1_anti-Cas9 ChIP-seq (related to Figure 2B)
Table S2_anti-Cas9 ChIP-BS-seq (related to Figure 2E)
Table S4_Primer sequences to construct sgRNAs (related to Methods and Resources)
Table S5_qPCR primers (related to Methods and Resources)
Table S6_Bisulfite sequencing and pyrosequencing primers (related to Methods and Resources)
Highlights.
Targeted demethylation of CGG repeats by dCas9-Tet1 reactivates FMR1 in FXS cells
Demethylation of CGG repeats induces an active chromatin status for FMR1 promoter
Methylation edited FXS neurons behave similarly as wild type neurons
FMR1 reactivation by dCas9-Tet1 is sustainable in a human/mouse chimeric model
ACKNOWLEDGMENTS
We thank Patti Wisniewski, Patrick Autissier and Hanna Aharonov for FACS sorting, and thank Maisam Mitalipova, Dongdong Fu, Robert Plasschaert, Malkiel Cohen, Carrie Ng, and Vivian Villegas for reagents, technical assistance and inputs on the manuscript. We thank Steve Warren in Emory University for providing FXS_SW isogenic iPSC lines. This study was supported by NIH grants HD045022, MH104610, R01NS088538 and R01GM123511. X.S.L. is supported by a Damon Runyon Cancer Foundation Postdoctoral Fellowship and funding from Rett Syndrome Research Trust, H.W. was supported by a NARSAD Young Investigator Fellowship, X.W. is supported by a Helen Hay Whitney Foundation Postdoctoral Fellowship. R.J. is co-founder of Fate Therapeutics, Fulcrum Therapeutics and Omega Therapeutics and R.A.Y is a co-founder of Syros Pharmaceuticals, Marauder Therapeutics and Omega Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
REFERENCES:
- (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33. [PubMed] [Google Scholar]
- Avitzour M, Mor-Shaked H, Yanovsky-Dagan S, Aharoni S, Altarescu G, Renbaum P, Eldar-Geva T, Schonberger O, Levy-Lahad E, Epsztejn-Litman S, et al. (2014). FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 3, 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RF, Buijsen RA, Usdin K, Pintado E, Kooy F, Pretto D, Pessah IN, Nelson DL, Zalewski Z, Charlet-Bergeurand N, et al. (2014). Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J Neurodev Disord 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardet M, and Crusio WE (2006). Fmr1 KO mice as a possible model of autistic features. Scientific World Journal 6, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick DJ, Nethercott HE, Montesano S, Banuelos MG, Stover AE, Schutte SS, O’Dowd DK, Hagerman RJ, Ono M, Hessl DR, et al. (2014). The Autism Spectrum Disorders Stem Cell Resource at Children’s Hospital of Orange County: Implications for Disease Modeling and Drug Discovery. Stem cells translational medicine 3, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury SR, Cui Y, Lubecka K, Stefanska B, and Irudayaraj J (2016). CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7, 46545–46556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Zhang F, Ceman S, Warren ST, and Reines D (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am J Hum Genet 71, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Zhang F, Warren ST, and Reines D (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22, 98–101. [DOI] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, and Jaffrey SR (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, and Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proceedings of the National Academy of Sciences of the United States of America 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, and Portera-Cailliau C (2015). Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunkhorn S (2017). Genetic disorders: Repurposing metformin in FXS. Nat Rev Drug Discov 16, 456. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Esch CE, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST, van der Wal E, Douben H, Gunhanlar N, Kushner SA, et al. (2014). Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Reports 3, 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, and Tonegawa S (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proceedings of the National Academy of Sciences of the United States of America 110, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, et al. (2007). Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1, 568–577. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati EV, Sweeney JA, Fitzpatrick SE, Brown WT, Budimirovic D, Hagerman RJ, et al. (2017). Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholizadeh S, Arsenault J, Xuan IC, Pacey LK, and Hampson DR (2014). Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 39, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, and Hagerman PJ (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130. [DOI] [PubMed] [Google Scholar]
- Halevy T, Czech C, and Benvenisty N (2015). Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports 4, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, and Tonegawa S (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America 104, 11489–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai C, Jia C, Sun R, Xu P, Min T, Wang Q, Zheng C, Chen H, and Lu D (2017). CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice. Hum Genet 136, 875–883. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences of the United States of America 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, and Greenough WT (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10, 1038–1044. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, and Todd PK (2016). CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell 62, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2017). CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari D, and Usdin K (2016). Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum Mol Genet 25, 3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 341, 629-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, and Jaenisch R (2016). Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247 e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, and Olson EN (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, and Lindemann L (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, Orr HT, and Warren ST (2013). The unstable repeats--three evolving faces of neurological disease. Neuron 77, 825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, and Kim DW (2015a). Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep 13, 234–241. [DOI] [PubMed] [Google Scholar]
- Park CY, Kim DH, Son JS, Sung JJ, Lee J, Bae S, Kim JH, Kim DW, and Kim JS (2015b). Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 17, 213–220. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, and Cleary JD (2005). Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6, 729–742. [DOI] [PubMed] [Google Scholar]
- Pugin A, Faundes V, Santa Maria L, Curotto B, Aliaga S, Salas I, Soto P, Bravo P, Pena MI, and Alliende MA (2017). Clinical, molecular, and pharmacological aspects of FMR1 related disorders. Neurologia 32, 241–252. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, and Warren ST (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 7, 219–245. [DOI] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorov MS, Auerbach BD, and Bear MF (2013). Fragile X mental retardation protein and synaptic plasticity. Mol Brain 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LN, Jedynak JP, Fontenot MR, Hale CF, Dietz KC, Taniguchi M, Thomas FS, Zirlin BC, Birnbaum SG, Huber KM, et al. (2014). Fragile X mental retardation protein regulates synaptic and behavioral plasticity to repeated cocaine administration. Neuron 82, 645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabolacci E, Palumbo F, Nobile V, and Neri G (2016). Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome. Genes (Basel) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, and Hagerman PJ (2004). FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol 1, 103–105. [DOI] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, and Benvenisty N (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. [DOI] [PubMed] [Google Scholar]
- Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, Klasic M, and Zoldos V (2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res 44, 5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Morrow C, Munir J, Chandler-Militello D, Wang S, and Goldman SA (2014). A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J Neurosci 34, 16153–16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowska M, and Krzyzosiak WJ (2011). Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20, 3811–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Zhang Y (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N, Gong H, Suhl JA, Chopra P, Wang T, and Warren ST (2016). Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS One 11, e0165499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, Ruan K, Wang F, Xu GL, and Hu R (2016). A CRISPR-based approach for targeted DNA demethylation. Cell discovery 2, 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeier Z, Kumar A, Bodhinathan K, Feller JA, Foster TC, and Bloom DC (2009). Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther 16, 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reactivation of FMR1 by dCas9-Tet1 with CGG sgRNA in multiple FXS iPSC lines (A) One previously reported FXS iPSC line (135.3) with more than 450 CGG repeats was infected with lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) with a mCherry expressing sgRNA targeting the CGG repeats “GGCGGCGGCGGCGGCGGCGGNGG” (CCG sgRNA) or lentiviruses expressing dCas9 fused with a catalytically dead Tet1 (dC-dT) with the same sgRNA. Double positive (BFP+; mCherry+) cells were isolated by FACS after infection. The expression level of FMR1 were quantified by qPCR analysis and compared to an isogenic line with shortened CGG repeat expansion, and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of two biological replicates. (B) Another previously reported FXS iPSC line (SW) with more than 450 CGG repeats was used to examine FMR1 expression as did in B. The expression level of FMR1 were quantified by qPCR analysis and compared to an isogenic line with shortened CGG repeat expansion, and shown as the mean of relative percentages to the one in WIBR1 hESCs ± SD of two biological replicates. (C) FX52 iPSCs were infected with different ratios of lentiviruses expressing dCas9-Tet1-P2A-BFP (dC-T) and a CGG sgRNA. Infected cells were harvested for qPCR analysis of FMR1 expression at various time points. Shown is the mean of relative percentages to the one in a CGG deletion FX52 iPSC line ± SD of two biological replicates.
Table S1_anti-Cas9 ChIP-seq (related to Figure 2B)
Table S2_anti-Cas9 ChIP-BS-seq (related to Figure 2E)
Table S4_Primer sequences to construct sgRNAs (related to Methods and Resources)
Table S5_qPCR primers (related to Methods and Resources)
Table S6_Bisulfite sequencing and pyrosequencing primers (related to Methods and Resources)
Data Availability Statement
Raw data files for sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GSE102655, GSE102684, GSE108171, GSE108498. These SubSeries are linked to one SuperSeries GSE108577.