

Abstract
The combination of next generation sequencing (NGS) and automated liquid handling platforms has led to a revolution in single-cell genomic studies. However, many molecules that are critical to understanding the functional roles of cells in a complex tissue or organ, are not directly encoded in the genome, and therefore cannot be profiled with NGS. Lipids, for example, play a critical role in many metabolic processes but cannot be detected by sequencing. Recent developments in quantitative imaging, particularly coherent Raman scattering (CRS) techniques, have produced a suite of tools for studying lipid content in single cells. This article reviews CRS imaging and computational image processing techniques for non-destructive profiling of dynamic changes in lipid composition and spatial distribution at the single-cell level. As quantitative CRS imaging progresses synergistically with microfluidic and microscopic platforms for single-cell genomic analysis, we anticipate that these techniques will bring researchers closer towards combined lipidomic and genomic analysis.
Graphical Abstract
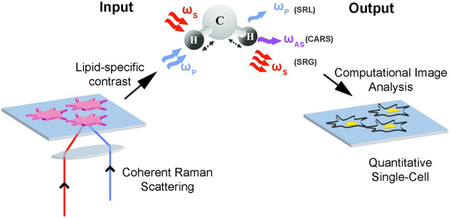
Non-destructive spatial characterization of lipid droplets using coherent Raman scattering microscopy and computational image analysis algorithms at the single-cell level
1. Introduction
While each cell in an organism has essentially the same genome, variation in gene regulation gives rise to vast cellular heterogeneity. This heterogeneity is present in tissues and even among populations of cells of the same type. Cellular heterogeneity plays an important role in many biological processes, including cell fate determination,1,2 cancer development and relapse,3,4 and drug resistance.5 Investigations at the single-cell level are therefore critical for uncovering this heterogeneity which otherwise is masked by ensemble measurements. Many single-cell analysis techniques have been enabled by next generation sequencing (NGS). Reverse transcription of mRNA followed by high-throughput sequencing of cDNA (RNA-sequencing) allows transcriptome-wide gene expression profiling. Recently, microfluidic platforms have made it possible to isolate mRNA from hundreds to thousands of single cells in a single experiment, thereby enabling comprehensive mapping of cellular composition of biological tissues and organs. These technological advances have paved the way towards constructing a human cell atlas.6 The Human Cell Atlas project7 aims to leverage high-throughput single-cell RNA sequencing (scRNA-seq) along with other single-cell measurements, to quantitatively characterize cellular identity throughout the human body, tracking both developmental and disease states, within their contextual niche. Recent work has already composed transcriptional catalogues of both mouse and human organs, including the brain,8,9 the thymus,10 the pancreas,11 and two recent reports of a comprehensive mouse atlas.12,13
While scRNA-seq has proven to be a robust tool for quantifying cellular identity, there are many molecules, which play critical roles in cellular function, that are not directly encoded in the genome and therefore cannot be detected with measurements that are based on sequencing. Metabolites and lipids are examples of such molecules that cannot be profiled using NGS but are important for regulation of cellular function. Lipids are predominantly involved in energy storage in cells and provide structural integrity to biological membranes.14,15 Lipids also participate in signaling pathways14,16 and interact with proteins to regulate their functions.17,18 In cells, neutral lipids and phospholipids are stored in organelles called lipid droplets (LDs).For a long time, LDs’ only role was perceived as storage of lipids.19 It is now well-accepted that LDs are dynamic organelles with functions in energy production,20 protein degradation,21 and lipid metabolism homeostasis.22 The dysregulation of lipid metabolism has been linked to many human diseases such as cancer, obesity, and diabetes. For example, imbalance in the number of intracellular LDs has been reported to be associated with multiple cancers23 and has been shown to promote cancer progression.24 Obesity is associated with eventual accumulation of lipids in nonadipose tissues25,26 which subsequently interferes with local insulin signaling and plays a key role in the development of type II diabetes.27 Consequently, quantifying changes in lipid metabolism is critical for understanding disease pathways and screening for targeted therapeutics.28,29
There are many different species of cellular lipids with structural variations in their hydrophobic and hydrophilic regions. Intracellular LDs store a wide distribution of lipid molecules and lipid metabolism directly alters the lipid composition of LDs.30,31 The field of lipidomics aims to study changes in lipid metabolism in response to physiological, pathological, and environmental conditions by characterizing this compositional distribution of all cellular lipids.
Established techniques for lipidomic analysis include gas or liquid chromatography-mass spectrometry (GC/LC-MS)32-34 and shotgun mass spectrometry.35,36 GC/LC-MS and shotgun techniques allow for targeted and untargeted detection of lipid molecules, respectively, when implemented on biological extracts from a population of cells (Fig. 1A). Recent advancements in sample preparation and ionization techniques have further enabled researchers to profile the lipidome at the single-cell level based on microarray for MS (MAMS), single-cell matrix assisted laser desorption/ionization-MS (MALDI-MS), and subcellular content aspiration-based MS techniques;37-40 Imaging mass spectrometry (IMS) is an imaging method that allows for visualization and quantification of spatial distribution of lipids in intact biological systems.41-45 Implementation of IMS techniques requires extensive sample preparation46 with spatial resolution ranging from submicron to hundreds of microns depending on the ion source.47 Typically, sensitivity of MS-based techniques lies in the picomolar range with detection specificity of hundreds of lipid species simultaneously.48 Such high sensitivity and specificity of MS-based techniques comes at the cost of destructive measurements.
Figure 1.

Pipeline of mass spectrometry (MS) and microscopic quantitative imaging for lipidomic analysis (a) In MS-based techniques, lipid is extracted from bulk cells. Extracted lipid can be separated using a gas/liquid chromatographic column before mass spectrometric detection, or directly infused in mass spectrometer for untargeted detection. (b) In quantitative imaging-based techniques, multiple live cells in the field of view are first imaged non-destructively to generate a lipid-specific contrast. The image is then computationally analyzed to segment cells and quantify properties of subcellular lipid droplets on the single-cell level.
Quantitative microscopic imaging techniques are complementary to MS-based technology and allow for non-destructive spatial characterization of LDs in live cells but with less lipid specificity. The non-destructive nature of optical microscopy allows researchers to perform time-resolved imaging to investigate dynamic cellular behavior. Furthermore, live-cell imaging can be coupled with subsequent molecular measurements such as sequencing or mass-spectrometry. Also, when combined with image processing algorithms, microscopy enables researchers to gather subcellular information such as LD morphology, or LD composition. This circumvents the need for physical isolation of single cells, thereby increasing the speed of data acquisition (Fig. 1B).
Amongst quantitative microscopic imaging techniques, fluorescence imaging allows for quantification down to a single molecule level. Fluorescence imaging with lipid-soluble dyes, lipid-binding probes, or fluorophore-conjugated lipids, has been used to study the composition and morphology of LDs.49,50 In some cases, the process of labeling can alter the distribution of cellular lipids. For example, Yen et al. showed that staining based on both Nile red and BODIPY does not correlate with fat stores for the model organism C. elegans.51 Complementary to fluorescence imaging are label-free optical techniques such as phase contrast,52 differential interference contrast,53 quantitative phase-imaging,54 and third harmonic generation microscopy55 that have been used to visualize LDs. In order to extend the capabilities of label-free imaging techniques for lipid profiling and quantification, magnetic resonance imaging (MRI) and coherent Raman scattering (CRS) techniques have been implemented to provide a lipid-specific contrast. MRI is an imaging technique based on nuclear magnetic resonance that has been implemented for quantification of total fat content and lipid accumulation.56-58 The high penetration depth achieved from near-IR imaging allows researchers to implement MRI techniques in vivo. For in vitro and in vivo label-free mapping of LD composition, CRS imaging techniques are used. CRS techniques include coherent anti-Stokes Raman scattering (CARS) imaging and stimulated Raman scattering (SRS) imaging, both of which have been widely used to quantify LDs at the single-cell level with high spatial and temporal resolution. In this review, we will highlight applications of CRS techniques for quantifying LDs. We will also discuss object recognition algorithms for identification of LD and cellular boundaries in an image. Such segmentation analysis is necessary for microscopy to be used for quantitative single-cell analysis. We will conclude by discussing the implications of non-destructive CRS techniques towards promises of multi-omic analysis at the single-cell level.
2. Coherent Raman Scattering (CRS) Microscopy
CRS microscopy provides a label-free approach for profiling the chemical composition of biological specimens by probing the characteristic vibrational modes of molecular bonds. Because of the strong vibrational modes associated with CH2, CRS is particularly powerful for imaging intracellular lipids. For selective imaging of lipids, the asymmetric-stretching vibrational mode of the carbon–hydrogen bond is probed at 2,845 cm−1 (Fig. 2A). CRS is induced by simultaneously illuminating the specimen with two photons at frequencies ωp (pump) and ωs (Stokes). When the difference in frequency between the two photons equals a vibrational frequency that is characteristic of the target molecule (Ω = ωp-ωs), the Raman scattering cross-section is resonantly enhanced giving rise to a strong CRS signal. Coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) are two imaging modalities that operate on this principle. In CARS, a signal is detected at the anti-Stokes frequency, ωAS, given by ωAS = 2ωp - ωs. CARS relies on homodyne detection, as ωAS can be separated from both the incoming frequencies ωp and ωs using a dichroic mirror or optical filters.59 In SRS, one of the two incoming photons, ωs or ωp, is amplitude modulated and the signal is detected as a loss or gain in the intensity of the pump or Stokes photon respectively (Fig. 2B). Therefore, SRS techniques utilize heterodyne detection schemes and require a lock-in amplifier to amplify the modulated stimulated Raman loss or gain.60 As CARS and SRS are nonlinear optical processes, signal is only generated at the focal plane of the objective, enabling intrinsic three-dimensional sectioning by scanning in the x, y, and z axes. Long-term live cell imaging is also possible as CRS contrast is not limited by photobleaching. CARS and SRS are diffraction limited techniques and therefore offer quantification at a subcellular level with resolution as low as hundreds of nanometers. The CARS signal is quadratic with respect to the concentration of resonant chemical bonds and the SRS signal is linear. SRS also has a higher signal to noise ratio (SNR) as compared to CARS because there is no non-resonant background. However, heterodyne detection in SRS requires additional instrumentation (lock-in amplifier) which is bypassed in CARS by using appropriate filters for homodyne detection.
Figure 2.

Vibrational imaging of lipids using coherent Raman scattering. (A) Spontaneous Raman spectra of oleic acid. The red solid line indicates asymmetric stretching vibrational mode of the carbon–hydrogen bond at 2,845 cm-1. (B) Schematic of excitation and detection for coherent Raman scattering. For both coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS)imaging, a characteristic vibrational mode of the CH2 bond in lipids is excited with two incoming photons at the pump (ωp) and Stokes (ωs) frequency. Stimulated Raman loss (SRL) is detected as a loss in the pump intensity and stimulated Raman gain (SRG) is detected as a gain in the Stokes intensity. CARS is detected at the anti-Stokes frequency, ωAS.
Multiphoton excitation techniques like CRS imaging employ ultrashort pulsed lasers to obtain high concentrations of laser power inside the sample, which is necessary for efficient excitation of the targeted vibrational mode. A possible consequence of this elevated laser irradiance is photodamage to cells and tissues. Schonle and Hell developed a model for investigating the effects of optical absorption (in near-IR, by water in biological specimens) on focal heating during multiphoton excitation microscopy.61 Their results showed an increase in focal temperature by not more than 3K for an average laser power of 100 mW at the focal plane, suggesting that heating through linear absorption does not play a destructive role. However, the required peak laser power, to maintain an average laser power of 100 mW, may lead to nonlinear photodamage. Other studies have shown that maintaining laser power below 10 mW at the focal plane is considered to be a safe range for sample integrity.62,63 Some applications of CRS imaging may require higher laser power for fast and efficient excitation of the resonant mode.64,65 For such purposes, optimizing the average and peak laser power should be the first step towards maintaining a strong signal while minimizing photodamage to the sample.66 Work has been done by several research groups to identify and define criterias for characterization of photodamage induced by nonlinear imaging.67-70
In this section, we discuss investigations using CRS techniques for quantifying LDs. In section 3, we will then discuss object recognition algorithms applicable for cell and LD boundary determination. Section 4 will focus on biological investigations using CRS techniques coupled with segmentation algorithms for quantitative single-cell and single-lipid droplet analysis.
2.1. CARS and SRS
As CARS signal is quadratic with molecular concentration of the resonant bond, quantification using CARS requires processing of signal intensity. For example, Chen et al. derived a formula to calibrate CARS intensity to accurately report the number of lipid molecules in the scattering volume.71 In this study, they developed an automated image analysis algorithm for quantification of lipid content in single cells. Rinia et al. adopted another strategy where they implemented spectral-analysis tools in conjunction with multiplex CARS for retrieval of spontaneous Raman-like spectra which is linear with the number of vibrating molecules.72 In this study, they analyzed the retrieved spontaneous Raman-like spectra to map the acyl chain unsaturation and acyl chain order within individual LDs in adipocytes, which were incubated with exogenous free fatty acids (FFA) of varying compositions (Fig. 3). They found heterogeneity in lipid composition and packing in individual LDs and demonstrated that this heterogeneity was dependent on the FFA composition of incubation mixture. In contrast to CARS, SRS signal is linear with the number of vibrating molecules, thereby making quantification more straightforward. Freudiger et al. demonstrated SRS as a contrast mechanism for imaging biological specimens.60 They monitored the uptake and metabolism of unsaturated FFA by imaging at 3015 cm−1 wavenumber specific to the =C-H bond in unsaturated fatty acids. Wang et al. used SRS microscopy combined with RNA interference screening to determine lipid storage regulatory genes in C. elegans.73 Lipid storage capacity was quantified based on mean SRS intensity. Using this technique, they were able to screen for 272 genes and found 8 new regulatory genes for fat storage. Besides quantifying LDs, CRS techniques have been critical towards visualizing LD growth and formation thereby revealing new lipid functions in cellular environment.74,75 Nan et al. demonstrated vibrational imaging of LDs using CARS and monitored LD formation during differentiation of 3T3-L1 fibroblast cells into adipocytes.76 They found that after adding adipogenic differentiation media, there was an initial clearance of LDs at the early stage of differentiation followed by formation of large LDs (Fig.4). Le and Cheng combined CARS microscopy with fluorescence imaging and flow cytometry to investigate heterogeneity in rates of LD formation in differentiating 3T3-L1 cells.77 They found that phenotypic variability among differentiating 3T3-L1 cells was dependent on the kinetics of an insulin signaling cascade.
Figure 3.

Multiplex coherent anti-Stokes Raman scattering (CARS) imaging of 3T3-L1-derived adipocyte to map the composition and packing of individual lipid droplets. Cells were incubated in a 1:3 mix of unsaturated : saturated fatty acid (A) Brightfield image of an adipocyte. Spontaneous Raman-like spectra in the (B) CC-stretch and (C) CH-stretch regions for locations indicated (in D). Retrieved spectra was then analyzed for mapping the (D) lipid concentration, (E) acyl chain unsaturation and (F) acyl chain order on the same adipocyte. Reprinted from Biophysical Journal, Volume 95, Issue 10, H. A. Rinia, K. N. J. Burger, M. Bonn and M. Müller, Quantitative Label-Free Imaging of Lipid Composition and Packing of Individual Cellular Lipid Droplets Using Multiplex CARS Microscopy, Pages 4908-4914, Copyright (2018), with permission from Elsevier.
Figure 4.

Monitoring lipid droplet formation during differentiation of 3T3-L1 cells using CARS at 2845 cm-1. Images were taken at different times after adding differentiation induction media: (A) 0 h, (B) 24 h, (C) 48 h, (D) 60 h, (E) 96 h, and (F) 192 h. Republished with permission of American Soc for Biochemistry & Molecular Biology, from Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy, X. Nan, J.-X. Cheng and X. S. Xie, volume 44, edition 11, Copyright (2018); permission conveyed through Copyright Clearance Center, Inc.
2.2. Vibrational Raman tags
Imaging at a single frequency is insufficient for monitoring the uptake of saturated fatty acids because all vibrational markers of saturated fatty acids are shared by unsaturated fatty acids. However, no endogenous molecular species, including lipids, vibrate in the range from 1800 cm −1 to 2800 cm −1, known as the “Raman-silent region” in cells. Raman tags are biorthogonal vibrational labels that consist of chemical bonds having a unique Raman shift in the cell’s silent region. Fatty acids have been conjugated with Raman tags for tracking their uptake dynamics. Stable isotope substitution using 2H 78,79 or conjugation with alkyne tags80,81 are the two major strategies employed with CRS techniques. Wei et al. demonstrated metabolic incorporation of saturated FFA into triglycerides and its storage in LDs using alkyne tagging together with SRS.82 Li and Cheng demonstrated direct visualization and quantification of glucose metabolism in single cells using SRS microscopy coupled with isotope labeling (glucose-d7).83 They demonstrated up-regulation of de novo lipogenesis in pancreatic and prostate cancer cell lines as compared to healthy cell lines. They also showed that compared to pancreatic cancer cells, prostate cancer cells have lower level of de novo lipogenesis but higher level of dietary lipid uptake. On the other hand, Hu et al. monitored glucose uptake activity in live cells using a glucose analogue labeled with an alkyne tag (3-O-propargyl-d-glucose, 3-OPG).84 In their study, they found that glioblastoma cells have a higher level of de novo lipogenesis as compared to cervical cancer cells. These studies demonstrated that cancer cells with differing metabolic activities can be distinguished using Raman tagging strategies. It will be interesting to see if the reported results can be validated for prostate and pancreatic cells using alkyne tagging and for cervical and glioblastoma cells using isotope labeling.
2.3. Hyperspectral SRS
Single-channel imaging of deuterated or alkyne-tagged lipids has been demonstrated as a useful tool for tracking uptake dynamics of a targeted lipid molecule. For unbiased profiling of the distribution of cellular lipids in response to changes in cellular metabolic states, hyperspectral SRS (hSRS) imaging is implemented. hSRS imaging enables researchers to separately quantify lipid molecules with overlapping Raman spectra by utilizing subtle differences in the spectral intensity across a range of wavenumbers.85,86 hSRS techniques are often used in conjunction with spectral-analysis tools to retrieve the Raman spectra of different molecules from the convoluted SRS spectra. The retrieved spectra can be used to reconstruct the compositional distribution images for each lipid species (Fig.5).87,88 Li et al. employed hSRS imaging to quantitatively analyze the composition of intracellular lipids inside single ovarian cancer and non-cancer stem cells and reported higher levels of unsaturated lipids in cancer cells based on the ratio of intensities at 3002 cm−1 and 2900 cm−1 wavenumber.89 Alfonso-García et al. used hSRS coupled with unsupervised vertex component spectral analysis to study the metabolism and storage of deuterated cholesterol (D38-cholesterol).90 They utilized the spectral differences in the CH fingerprint region between D38-cholesterol and natural cholesterol to map the distribution of esterified and unesterified cholesterol in LDs. They found that subpopulations of LDs exist each with a predominant storage of esterified or free cholesterol. They also found that steroidogenic Y1 cells store triacylglycerol (TAG) and cholesteryl esters (CE) in different LDs. It is known that steroidogenic cells and macrophages primarily accumulate CE in LDs and liver cells primarily accumulate TAG in LDs.91,92 This study observed accumulation of TAG in steroidogenic cells but didn’t perform any investigation in macrophages or liver cells.90 In contrast, Fu et al. detected only CE containing LDs in macrophages and only TAG containing LDs in hepatocytes.93 In this study, spectral differences between TAG and CE were utilized to quantitatively profile the two classes of neutral lipids. Based on these observations, it will be interesting to see whether lipid sorting occurs in macrophages, liver cells and other cell types using Alfonso-García’s methodology. Fu et al. also characterized lipid compositional changes associated with metabolic disorders and further extended hSRS coupled with isotope labeling to simultaneously trace saturated and unsaturated fatty acids.93
Figure 5.

Hyperspectral stimulated Raman scattering (hSRS) imaging for mapping three types of polymer beads with overlapping but distinct Raman spectra (A) Spontaneous Raman spectra of the three polymer beads. The black solid line indicates overlapping Raman spectra at 3028 cm-1 (B) stimulated Raman scattering (SRS) imaging of the three polymer beads at 3028 cm-1 with different color arrows pointing out corresponding beads (C) SRS spectra for the three polymer beads pointed out by the arrows (in B). (D) Color-code distribution of the three polymer beads generated using hSRS imaging coupled with spectral decomposition. PMMA: Poly (methyl methacrylate). Reprinted with permission from D. Fu, G. Holtom, C. Freudiger, X. Zhang and X. S. Xie, J. Phys. Chem. B, 2013, 117, 4634–4640 Copyright (2018) American Chemical Society.
3. Object Recognition Algorithms
While optical microscopy has the spatial resolution necessary to be an inherently single-cell measurement, interpretation of micrographs in the single-cell paradigm is not always straightforward. Historically, microscopy has been used in low volume, manual, and generally qualitative, descriptions of biological samples. Such an approach, in addition to being susceptible to interpretation bias, is now increasingly impractical as image data has become larger and more complex. Furthermore, with the push in the life sciences towards generating results with greater statistical power, there is more demand for quantitative analyses of images, which all but necessitates computation. Image analysis algorithms have been under development since the pre-digital age, and the past two decades have seen many improvements in their application to biological datasets.
One of the most basic, and arguably most important, questions that can be asked about an image is where are the boundaries between objects? When quantifying metabolic composition of cells, it is important to have an objective methodology for defining objects. In tissue this amounts to cell boundaries, in individual cells, the subcellular structures and organelles such as LDs. Traditional techniques for answering this question often start with contrast enhancement and gradient-based edge detection methods. The simplest approach is thresholding, with automatic threshold determination by algorithms such as Otsu’s method,94 or balanced histogram thresholding.95 Thresholding tends to separate objects and is also often employed to aid in background correction. Convolution with operators such as the Sobel,96 Canny,97 or other gradient operators can provide information on sharp line boundaries. For more general shape extraction, the Hough transform has been a popular choice in a wide variety of fields. First patented in 1962 for line identification,98 it was then generalized to arbitrary shapes.99 It is well-suited to identifying regularly shaped features which can vary in dimension across an image.
These gradient or edge detection techniques are then frequently combined with a watershed-based algorithm,100 which imagines filling basins from minima in the images and draws boundaries where the watersheds meet. Implementations of these techniques can be found in all major programming languages, and are also included in many widely available image analysis software suites, like Fiji.101 They have therefore been applied, in a number of combinations and variations, for analysis of LD size and number distribution.102,103
More recently, the field of computer vision has shifted focus to machine learning approaches for everything from automatic feature extraction to image classification. This has been driven in large part by the success of convolutional neural networks (CNN), and their rapid development in the past decade. First introduced over 20 years ago,104,105 initial adoption was slow, but the list of current variations and applications is now constantly growing. CNNs work similarly to conventional, or ‘fully-connected,’ neural networks but reduce the number of parameters that need to be learned by using convolutions rather than transformation matrices that relate every point in the image to every point in the output. This is in some ways analogous to some traditional methods listed above, but instead of pre-selecting, e.g. a gradient filter, the filter is learned by the network, and there are many filtering steps. While generally more computationally intensive, fully-connected neural networks have also found use in image analysis.
The major drawback for using CNNs or deep learning architectures generally is the need for training data. This has slowed adoption in the field of lipidomics, although CNNs have been successfully applied to numerous types of microscopy data. Medical imaging has been a recent adopter, with hundreds of successful demonstrations in the last three years.106 Importantly, these demonstrations span a wide-variety of disciplines but utilize similar network architectures. Many are straightforward modifications of well-known networks, and often rely on already trained networks as starting points, suggesting a similar strategy may be effective for lipidomics. Single-cell segmentation, cell cycle progression and disease state identification, have been recently demonstrated using CNNs on fluorescent images.107 Chen et al. also recently showed algal cell classification based on lipid content, using time-stretch quantitative phase imaging and deep neural networks.108
A final consideration, is that many of the imaging techniques used for lipid characterization contain additional information beyond the purely morphological. Most of the analysis algorithms discussed thus far have focused on segmentation and object identification. This makes them generalizable to all types of images, but also makes them blind to the additional information that can be encoded in some microscopy datasets. In some cases, it is therefore advantageous to utilize more specialized algorithms for analysis, hyperspectral coherent Raman imaging being a prime example. Fu and Xie demonstrated the ability to segment subcellular structures, including lipid droplets, from a hSRS dataset using a spectral phasor method adapted from the fluorescence lifetime imaging field.109 Di Napoli et al. were also able to monitor uptake of different lipid components using hyperspectral CARS,110 using an unsupervised retrieval algorithm.111
4. Quantitative CRS for single-cell and single-LD analysis
High signal to noise ratio (SNR) associated with concentrated CH2 bonds in lipids allows researchers to monitor the dynamics of LDs in a straightforward fashion using CRS techniques coupled with LD recognition and trajectory tracking packages. Jungst et al. demonstrated tracking of LDs using fast, long-term three-dimensional CARS imaging at 2850 cm−1 in order to investigate the dynamics of LD fusion in living adipocytes undergoing differentiation.112 They used the Imaris software package (http://bitplane.com) for detection and tracking of LDs. In Imaris, thresholding is performed for automated segmentation of LDs. Morphological characterization of identified LDs is then performed including radius and volume rendering. Detected LDs are then tracked by selecting for appropriate three-dimensional tracking algorithm. Based on the lipid transfer rates obtained, researchers suggested a model in which lipid transfer is driven by the pressure difference between participating LDs through a putative fusion pore, whose size depends on the size of the donor LD.
Zhang et al. used SRS microscopy to study the dynamics of LDs using three-dimensional SRS imaging at 2850 cm−1.113 They implemented a feature point tracking algorithm, as developed for the Particle Tracker software, 114 for monitoring LD movements. In this software, feature points are localized by finding local intensity maxima in the filtered image. The retrieved positions are then refined to reduce the standard deviation of the position measurement, which takes into consideration a user-provided threshold. Once point location matrices have been defined for each frame in the time-resolved image, a cost function is minimized to find a set of associations for tracking each point. Using this software, researchers demonstrated that the dynamics of LDs, quantified using maximum displacement and speed as the parameters, can be used to differentiate changes in lipid metabolism in living cells. They studied changes in lipid metabolism upon glucose starvation and refeeding and showed that their methodology could predict increase in lipolysis upon starvation as expected.
Medyukhina et al. developed an image processing approach for detection of nuclear and cellular boundaries from co-registered two-photon excited fluorescence (TPEF) and CARS images respectively.115 For nuclei boundary determination, they first used the local gray-scale minimum from denoised TPEF images for localization of nuclei centers. The gradient maxima from each nucleus location was used to detect the nuclear boundary. Once nuclei locations and boundaries were validated, they subsequently used TPEF images to delineate the cellular boundaries in the denoised CARS images. They assumed that the cellular boundary corresponds to the first local gradient minimum behind the nuclear boundary. Finally, they demonstrated the implementation of this approach for automated segmentation of cells and nuclei in brain tumor samples.
In order to reveal single-cell heterogeneity, data has to be acquired from multiple single cells for statistically significant conclusions. Cao et al. characterized the mechanisms of LD growth and formation upon lipid accumulation, as induced by exogenous FFA, at the single-cell level using SRS microscopy.116 LD growth and formation was monitored by tracking the number, average size, and average SRS intensity of LDs in a single cell under various concentrations of FFA. To increase throughput and therefore statistical power, all experiments were performed on a microfluidic platform capable of delivering controlled concentration of FFA to uniquely addressable nanoliter cell culture colonies. Images were obtained at 2850 cm−1 to identify LDs (Fig. 6A). A second set of images were taken at the protein-rich CH3 stretching vibration at 2950 cm−1 to extract boundaries of single cells. Thresholding was performed to generate a LD and cell mask. The position and morphology of each LD was then recorded and assigned to an individual cell (Fig. 6B). In this investigation, researchers found that lipid accumulation in nonadipocyte cells is mainly reflected in the increase of LD number, as opposed to an increase in their size or lipid concentration.
Figure 6.

Stimulated Raman scattering (SRS) image processing pipeline for determining cellular boundaries and characterizing lipid droplets in single cells. (A) three-dimensional lipid-specific images were acquired at 2850 cm-1. The signal was processed to generate a lipid droplet mask. The lipid droplet mask was analyzed for three-dimensional morphological characterization (B) three-dimensional protein-specific images were acquired at 2950 cm-1 for cell boundary segmentation and cell mask generation. The position of each LD was then recorded and assigned to an individual cell. Reprinted with permission from C. Cao, D. Zhou, T. Chen, A. M. Streets and Y. Huang, Anal. Chem., 2016, 88, 4931–4939 Copyright (2018) American Chemical Society.
5. From lipidomic to multiomic analysis
Highly-multiplexed barcoding strategies and automated fluid handling has now made it possible to profile the transcriptome from thousands of single cells in one experiment. However, in order to understand the correlation between gene expression and metabolic states at the single-cell level, multiple measurements must be made on the same single cell. Because CRS imaging in non-destructive, cells can be sequenced directly downstream of lipidomic analysis, thereby making implementation of multi-omic approaches possible. In this section, we will discuss the applicability of utilizing the developed microfluidic and microscopic platforms for combined single-cell genomic and lipidomic analysis.
5.1. Microfluidic Platforms
Microfluidic technology has proven critical for increasing the throughput of NGS techniques permitting profiling of genome-wide features from a large number of single cells. Implementation of single-cell sequencing requires single-cell isolation. In microfluidic platforms, this is typically achieved by valve-based compartmentalization,117,118 droplet encapsulation,119,120 and microwell separation.121,122 After single-cell isolation, downstream library preparation reactions are implemented. Another advantage of microfluidic devices is the optical transparency of the polymer used for chip fabrication, polydimethylsiloxane (PDMS), which enables researchers to visualize sequencing protocols in real-time using a microscope. Because of the optical transparent nature of microfluidic devices and the necessity to physically isolate single cells, lipidomic and genomic analysis can be performed on the same single cell by acquiring images upstream of library preparation reactions (Fig. 7A).
Figure 7.

Combining lipidomic and genomic analysis at the single-cell level. (A)Lipidomic and genomic analysis using microfluidic single-cell isolation. A single cell is physically isolated in a small chamber using valve-based compartmentalization. While the cell is trapped, images are acquired in a non-destructive fashion using coherent Raman scattering (CRS) imaging for lipidomic analysis. The cell is then pushed downstream for library preparation and finally sequenced using next generation sequencing (NGS) techniques. (B) Lipidomic and genomic analysis using microscopy and computational cell-segmentation. Multiple live cells are imaged on a coverglass using CRS. Individual cells are then computationally isolated using object recognition algorithms and images are analyzed for lipidomic analysis at the single-cell level. The transcriptome of the same cells is then profiled using in-situ sequencing techniques.
Streets et al. developed a microfluidic platform for whole-transcriptome profiling of single cells.118 In this device, cells were isolated in nanoliter-scale trapping chambers using a valve-based strategy. CRS imaging can be performed while cells are trapped thereby allowing researchers to perform combined lipidomic and genomic analysis on the same cell. Lane et al. integrated epifluorescence microscopy with scRNA-seq on a commercial microfluidic platform, Fluidigm C1.123 They used this approach to measure both the dynamics of activation for a specific transcription factor and the global transcriptional response in the same individual cell. Instead of fluorescence microscopy, label-free CRS imaging can be implemented on this platform for combined lipidomic and genomic analysis on the same cell. Gierahn et al4124 and Bose et al.125 developed platforms for massively parallel scRNA-seq based on gravitational settling of single cells in subnanoliter and picoliter-scale microwells respectively. As cells are stationary while isolated in microwells, this solid-phase capture can be utilized for high-resolution CRS imaging upstream of library preparation reactions. Zhang et al. developed a flow cytometer based on Raman scattering for fast, high-throughput single-cell analysis.126 They developed a multiplex stimulated Raman scattering flow cytometry (SRS-FC) technique for measuring chemical contents of single cells. This technique can be extended for quantifying lipid content in single cells. These cells can then be isolated using droplet encapsulation platforms119,120 for scRNA-seq. Thus coupling SRS-FC with droplet encapsulation-based microfluidic platforms will allow researchers to perform combined lipidomic and genomic analysis on the same cell. Such coupled datasets will transform the way we understand single-cell biology by enabling researchers to study the correlation between single-cell phenotype and gene expression profile.
5.1.1. High-Speed CRS Imaging
Microfluidic devices have enabled researchers to perform single-cell analysis in a high-throughput fashion. In the previous paragraph, we discussed the applicability of microfluidic platforms for retrieving lipidomic (CRS imaging) as well as transcriptomic (scRNA-seq) information from the same single cell, thereby advancing towards multi-omic approaches. However, implementation of such coupled experiments on hundreds to thousands of single cells will require application of high-speed CRS imaging techniques for fast single-cell lipidome profiling. As discussed previously, hyperspectral imaging techniques are essential for profiling the distribution of multiple cellular lipids simultaneously. Thus, it becomes critical to employ hyperspectral CRS techniques capable of rapid spectral acquisition at microsecond scale. Recent developments in CARS and SRS instrumentation have been influential in accelerating the spectral acquisition rate. For example, Liao et al. demonstrated parallel acquisition of SRS signal over 180 cm−1 bandwidth (~20 spectral data points) with 42 μs pixel dwell time using spectrally focused laser pulses and a homebuilt microsecond optical delay-line tuner.127 He et al. integrated a galvanometer mirror-based rapid-scanning optical delay line with spectrally focused laser pulses to acquire a spectrum with 20 data points in 40 μs.128 Liao et al. built an array of tuned amplifiers for lock-in free parallel acquisition of SRS signal over 180 cm−1 bandwidth (~20 spectral data points) with 32 μs pixel dwell time using multiplexed SRS.129,130 Alshaykh et al. integrated a rapid acoustooptic delay line with spectrally focused laser pulses to achieve parallel acquisition of SRS signal over 180 cm−1 bandwidth (~20 spectral data points) with 12.8 μs pixel dwell time.131 Hashimoto et al. coupled a rapid-scanning retro-reflective optical path length scanner with a Fourier-transform CARS (FT-CARS) system to accomplish spectral acquisition rate of 20,000 spectra/second over 1300 cm−1 bandwidth (~130 spectral data points).132 Tamamitsu et al. updated this system to incorporate a more rapidly scanning optical delay line thereby achieving spectral acquisition rate of 50,000 spectra/sec (~500 spectral data points).133 Recently, Coluccelli et al. demonstrated parallel detection of CARS signal with Raman shifts of ~ 3000 cm−1 using FT-CARS. The system was based on a single high-power Yb-fiber laser source coupled to a FT interferometer with pixel dwell time of 160 μs (~675 spectral data points).134 Thus, such studies focused on development of rapid CRS imaging techniques demonstrate the promise of coupling high-content spectral imaging with high-throughput single-cell analysis.
5.2. Microscopic Platforms
An alternative to physical isolation for single-cell genomic analysis is to employ techniques that turn the genomic information into optical information in situ. Fluorescence in situ hybridization (FISH) is a technique that uses fluorescent probes that bind specifically to complementary nucleic acid sequences. Thus, researchers can obtain spatial information about the distribution and subcellular localization of specific DNA or RNA molecules. In-situ sequencing leverages FISH to extract sequence information from tens to hundreds of targeted transcripts for large scale gene expression profiling in single cells.135-137 Such methods preserve the microenvironment of the biological sample allowing single molecule RNA sequencing and localization without removing cells from their original context. These emerging technologies are enabling a new-wave of spatial transcriptomic studies, which link single-cell gene expression to cellular niche in a tissue or organ. Since FISH techniques are fundamentally based on imaging, quantitative CRS techniques for lipidomic analysis can be combined with in situ sequencing for multi-omic single-cell analysis. Figure 7B illustrates how single-cell transcriptomics might be combined with CRS-based single-cell lipidomics.
6. Conclusion
Coherent Raman scattering (CRS) techniques have become an essential tool for profiling LDs in single-cells by enabling researchers to quantify intracellular lipids in a non-destructive and time-resolved fashion. As the development of CRS instrumentation progresses towards higher specificity, sensitivity, and faster hyperspectral imaging, and next generation sequencing techniques advance towards higher throughput single-cell genomic analysis with lesser bias, coupling these techniques will lead to a more acute understanding of the regulation of metabolic pathways. For example, adipocytes display a wide range of functions and phenotypes, from energy storage in large unilocular LDs (white adipocytes) to thermogenic lipolysis of small LDs (brown adipocytes). Adult humans were thought to only have white adipose tissue with brown adipose tissue being essentially absent after infancy. 138,139 In the early 2000s, observations in the field of nuclear medicine started challenging this notion.140,141 Multiple studies performing positron emission tomography (PET) with [18F]-fluorodeoxyglucose (FDG) for staging of cancer observed increased uptake of glucose in tumor-unrelated areas.140,141 These areas were found in the neck and shoulder region and presented itself with features of adipose tissue. It was hypothesized that this FDG uptake could represent activated brown adipose tissue in adult humans and this was finally demonstrated by three independent studies in 2009.142-144 Now, the existence of brown adipose tissue in adult humans is a well-accepted fact in the research community. Rodents also have a third kind of adipocyte called beige adipocyte, which has a different developmental origin from brown adipocytes.145 This fact naturally raises the question of whether humans also possess beige adipocytes. Interestingly, recent investigations of human brown adipocytes have reported the mixed presence of presumed beige adipocytes.146,147 These claims have been reported based on the upregulation of beige adipocyte markers as identified in rodents. Consequently, it is clear that we are only just beginning to understand and appreciate the vast cellular diversity of human adipose tissue. These data raise some critical questions about the composition of human adipose tissue that might only be addressed with single-cell measurements. Technology that couples CRS for lipid profiling and RNA-sequencing for gene expression analysis in single cells could greatly advance our understanding of adipocyte heterogeneity. We anticipate that imaging and sequencing single cells will be the next wave of multi-omic single-cell analysis.
Acknowledgements
This publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM124916. AMS is a Chan-Zuckerberg Biohub Investigator.
References
- 1.Losick R and Desplan C, Science, 2008, 320, 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND and Robson P, Dev. Cell, 2010, 18, 675–685. [DOI] [PubMed] [Google Scholar]
- 3.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J and Wigler M, Nature, 2011, 472, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian D, Zabala M, Bueno J, Neff NF, Wang J, Shelton AA, Visser B, Hisamori S, Shimono Y, Van De Wetering M, Clevers H, Clarke MF and Quake SR, Nat. Biotechnol, 2011, 29, 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altschuler SJ and Wu LF, Cell, 2010, 141, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nat. Methods, 2018, 15, 299. [Google Scholar]
- 7.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, Clevers H, Deplancke B, Dunham I, Eberwine J, Eils R, Enard W, Farmer A, Fugger L, Göttgens B, Hacohen N, Haniffa M, Hemberg M, Kim S, Klenerman P, Kriegstein A, Lein E, Linnarsson S, Lundberg E, Lundeberg J, Majumder P, Marioni JC, Merad M, Mhlanga M, Nawijn M, Netea M, Nolan G, Pe’er D, Phillipakis A, Ponting CP, Quake S, Reik W, Rozenblatt-Rosen O, Sanes J, Satija R, Schumacher TN, Shalek A, Shapiro E, Sharma P, Shin JW, Stegle O, Stratton M, Stubbington MJT, Theis FJ, Uhlen M, Van Oudenaarden A, Wagner A, Watt F, Weissman J, Wold B, Xavier R and Yosef N, Elife, , DOI: 10.7554/eLife.27041. [DOI] [Google Scholar]
- 8.Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA and Quake SR, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 7285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeisel A, Moz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J and Linnarsson S, Science, 2015, 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- 10.Kernfeld EM, Genga RMJ, Neherin K, Magaletta ME, Xu P and Maehr R, Immunity, 2018, 48, 1258–1270.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJP and van Oudenaarden A, Cell Syst, 2016, 3, 385–394.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quake SR, The TMC, Wyss-Coray T and Darmanis S, bioRxiv, 2018, 237446. [Google Scholar]
- 13.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, Huang D, Xu Y, Huang W, Jiang M, Jiang X, Mao J, Chen Y, Lu C, Xie J, Fang Q, Wang Y, Yue R, Li T, Huang H, Orkin SH, Yuan G-C, Chen M and Guo G, Cell, 2018, 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- 14.Fernandis AZ and Wenk MR, Curr. Opin. Lipidol, 2007, 18, 121–128. [DOI] [PubMed] [Google Scholar]
- 15.Sunshine H and Iruela-Arispe ML, Curr. Opin. Lipidol, 2017, 28, 408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higdon A, Diers AR, Oh JY, Landar A and Darley-Usmar VM, Biochem. J, 2012, 442, 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saliba A-E, Vonkova I and Gavin A-C, Nat. Rev. Mol. Cell Biol, 2015, 16, 753–761. [DOI] [PubMed] [Google Scholar]
- 18.Ayyappan JP, Paul A and Goo YH, Mol. Med. Rep, 2016, 13, 4527–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beller M, Thiel K, Thul PJ and Jäckle H, FEBS Lett, 2010, 584, 2176–2182. [DOI] [PubMed] [Google Scholar]
- 20.Welte MA, Curr. Biol, 2015, 25, R470–R481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bersuker K and Olzmann JA, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids, 2017, 1862, 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaushik S and Cuervo AM, Nat. Cell Biol, 2015, 17, 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL and Cheng JX, Cell Metab, 2014, 19, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW and Cravatt BF, Cell, 2010, 140, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, Cushman SW and Periwal V, PLoS Comput. Biol, 2009, 5, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carobbio S, Rodriguez-Cuenca S and Vidal-Puig A, Curr. Opin. Clin. Nutr. Metab. Care, 2011, 14, 520–526. [DOI] [PubMed] [Google Scholar]
- 27.Levelt E, Pavlides M, Banerjee R, Mahmod M, Kelly C, Sellwood J, Ariga R, Thomas S, Francis J, Rodgers C, Clarke W, Sabharwal N, Antoniades C, Schneider J, Robson M, Clarke K, Karamitsos T, Rider O and Neubauer S, J. Am. Coll. Cardiol, 2016, 68, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith TK, Reynolds TB and Denny PW, Biochem. Res. Int, 2012, 2012, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun JW, Phytochemistry, 2010, 71, 1625–1641. [DOI] [PubMed] [Google Scholar]
- 30.Visram M, Radulovic M, Steiner S, Malanovic N, Eichmann TO, Wolinski H, Rechberger GN and Tehlivets O, J. Biol. Chem, 2018, 293, 5544–5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiappini F, Coilly A, Kadar H, Gual P, Tran A, Desterke C, Samuel D, Duclos-Vallée JC, Touboul D, Bertrand-Michel J, Brunelle A, Guettier C and Le Naour F, Sci. Rep, 2017, 7, 46658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Köfeler HC, Fauland A, Rechberger GN and Trötzmüller M, Metabolites, 2012, 2, 19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cajka T and Fiehn O, TrAC - Trends Anal. Chem, 2014, 61, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Han J, Wang Z, Liu J, Wei J, Xiong S and Zhao Z, Int. J. Mol. Sci, 2014, 15, 10492–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Wang C, Han RH and Han X, Prog. Lipid Res, 2016, 61, 83–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X and Gross RW, Mass Spectrom. Rev, 2005, 24, 367–412. [DOI] [PubMed] [Google Scholar]
- 37.Tsuyama N, Mizuno H and Masujima T, Biol. Pharm. Bull, 2012, 35, 1425–1431. [DOI] [PubMed] [Google Scholar]
- 38.Zenobi R, Science (80-. )., , DOI: 10.1126/science.1243259. [DOI] [Google Scholar]
- 39.Rubakhin SS, Lanni EJ and Sweedler JV, Curr. Opin. Biotechnol, 2013, 24, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L and Vertes A, Angew. Chemie - Int. Ed, 2018, 57, 4466–4477. [DOI] [PubMed] [Google Scholar]
- 41.Boggio KJ, Obasuyi E, Sugino K, Nelson SB, Agar NYR and Agar JN, Expert Rev. Proteomics, 2011, 8, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanni EJ, Rubakhin SS and Sweedler JV, J. Proteomics, 2012, 75, 5036–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goto-Inoue N, Hayasaka T, Zaima N and Setou M, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids, 2011, 1811, 961–969. [DOI] [PubMed] [Google Scholar]
- 44.Touboul D, Brunelle A and Laprévote O, Biochimie, 2011, 93, 113–119. [DOI] [PubMed] [Google Scholar]
- 45.Gode D and Volmer DA, Analyst, 2013, 138, 1289–1315. [DOI] [PubMed] [Google Scholar]
- 46.Römpp A and Spengler B, Histochem. Cell Biol, 2013, 139, 759–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pól J, Strohalm M, Havlíček V and Volný M, Histochem. Cell Biol, 2010, 134, 423–443. [DOI] [PubMed] [Google Scholar]
- 48.Blanksby SJ and Mitchell TW, Annu. Rev. Anal. Chem, 2010, 3, 433–465. [DOI] [PubMed] [Google Scholar]
- 49.Maekawa M and Fairn GD, J. Cell Sci, 2014, 127, 4801–4812. [DOI] [PubMed] [Google Scholar]
- 50.Daemen S, van Zandvoort MAMJ, Parekh SH and Hesselink MKC, Mol. Metab, 2016, 5, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yen K, Le TT, Bansal A, Narasimhan SD, Cheng JX and Tissenbaum HA, PLoS One, 2010, 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sims JK, Rohr B, Miller E and Lee K, Tissue Eng. Part C Methods, 2015, 21, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McPhee CI, Zoriniants G, Langbein W and Borri P, Biophys. J, 2013, 105, 1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim K, Lee S, Yoon J, Heo J, Choi C and Park Y, Sci. Rep, , DOI: 10.1038/srep36815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Débarre D, Supatto W, Pena A-M, Fabre A, Tordjmann T, Combettes L, Schanne-Klein M-C and Beaurepaire E, Nat. Methods, 2006, 3, 47–53. [DOI] [PubMed] [Google Scholar]
- 56.Bley TA, Wieben O, François CJ, Brittain JH and Reeder SB, J. Magn. Reson. Imaging, 2010, 31, 4–18. [DOI] [PubMed] [Google Scholar]
- 57.Hwang J-H and Choi CS, Exp. Mol. Med, 2015, 47, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurowski K, Kochan K, Walczak J, Barańska M, Piekoszewski W and Buszewski B, Crit. Rev. Anal. Chem, 2017, 47, 418–437. [DOI] [PubMed] [Google Scholar]
- 59.Cheng JX, Jia YK, Zheng G and Xie XS, Biophys. J, 2002, 83, 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freudiger CW, Min W, Saar BG, Lu S, Holtom GR, He C, Tsai JC, Kang JX and Xie XS, Science (80-. ), 2008, 322, 1857–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schönle a and Hell SW, Opt. Lett, 1998, 23, 325–327. [DOI] [PubMed] [Google Scholar]
- 62.Galli R, Uckermann O, Andresen EF, Geiger KD, Koch E, Schackert G, Steiner G and Kirsch M, PLoS One, 2014, 9, e110295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nan X, Potma EO and Xie XS, Biophys. J, 2006, 91, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao L, Zhou H, Thrall MJ, Li F, Yang Y, Wang Z, Luo P, Wong KK, Palapattu GS and Wong STC, Biomed. Opt. Express, 2011, 2, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu F-K, Basu S, Igras V, Hoang MP, Ji M, Fu D, Holtom GR, Neel VA, Freudiger CW, Fisher DE and Xie XS, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 11624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang D, Slipchenko MN and Cheng JX, J. Phys. Chem. Lett, 2011, 2, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fu Y, Wang H, Shi R and Cheng J-X, Opt. Express, 2006, 14, 3942. [DOI] [PubMed] [Google Scholar]
- 68.Hopt A and Neher E, Biophys. J, 2001, 80, 2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Konig K, Simon U and Halbhuber KJ, Cell. Mol. Biol, 1996, 42, 1181–1194. [PubMed] [Google Scholar]
- 70.König K, So PTC, Mantulin WW and Gratton E, Opt. Lett, 1997, 22, 135. [DOI] [PubMed] [Google Scholar]
- 71.Chen WW, Chien CH, Wang CL, Wang HH, Wang YL, Ding ST, Lee TS and Chang TC, Anal. Bioanal. Chem, 2013, 405, 8549–8559. [DOI] [PubMed] [Google Scholar]
- 72.Rinia HA, Burger KNJ, Bonn M and Müller M, Biophys. J, 2008, 95, 4908–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang MC, Min W, Freudiger CW, Ruvkun G and Xie XS, Nat. Methods, 2011, 8, 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le TT, Yue S and Cheng J-X, J. Lipid Res, 2010, 51, 3091–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu Y, Ramachandran PV and Wang MC, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids, 2014, 1841, 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nan X, Cheng J-X and Xie XS, J. Lipid Res, 2003, 44, 2202–2208. [DOI] [PubMed] [Google Scholar]
- 77.Le TT and Cheng JX, PLoS One, 2009, 4, e5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Manen H-J, Kraan YM, Roos D and Otto C, Proc. Natl. Acad. Sci. USA, 2005, 102, 10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie XS, Yu J and Yang WY, Science (80-. )., 2006, 312, 228–230. [DOI] [PubMed] [Google Scholar]
- 80.Yamakoshi H, Dodo K, Okada M, Ando J, Palonpon A, Fujita K, Kawata S and Sodeoka M, J. Am. Chem. Soc, 2011, 133, 6102–6105. [DOI] [PubMed] [Google Scholar]
- 81.Yamakoshi H, Dodo K, Palonpon A, Ando J, Fujita K, Kawata S and Sodeoka M, J. Am. Chem. Soc, 2012, 134, 20681–20689. [DOI] [PubMed] [Google Scholar]
- 82.Wei L, Hu F, Shen Y, Chen Z, Yu Y, Lin C-C, Wang MC and Min W, Nat. Methods, 2014, 11, 410–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li J and Cheng J-X, Sci. Rep, 2015, 4, 6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu F, Chen Z, Zhang L, Shen Y, Wei L and Min W, Angew. Chemie - Int. Ed, 2015, 54, 9821–9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andresen ER, Berto P and Rigneault H, Opt. Lett, 2011, 36, 2387. [DOI] [PubMed] [Google Scholar]
- 86.Beier HT, Noojin GD and Rockwell BA, Opt. Express, 2011, 19, 18885. [DOI] [PubMed] [Google Scholar]
- 87.Fu D, Holtom G, Freudiger C, Zhang X and Xie XS, J. Phys. Chem. B, 2013, 117, 4634–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang D, Wang P, Slipchenko MN, Ben-Amotz D, Weiner AM and Cheng J-X, Anal. Chem, 2013, 85, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, Matei D and Cheng JX, Cell Stem Cell, 2017, 20, 303–314.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alfonso-García A, Pfisterer SG, Riezman H, Ikonen E and Potma EO, J. Biomed. Opt, 2015, 21, 061003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kraemer FB, Mol. Cell. Endocrinol, 2007, 265–266, 42–45. [DOI] [PubMed] [Google Scholar]
- 92.Khor VK, Ahrends R, Lin Y, Shen WJ, Adams CM, Roseman AN, Cortez Y, Teruel MN, Azhar S and Kraemer FB, PLoS One, 2014, 9, e105047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu D, Yu Y, Folick A, Currie E, Farese RV, Tsai TH, Xie XS and Wang MC, J. Am. Chem. Soc, 2014, 136, 8820–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Otsu N, IEEE Trans. Syst. Man. Cybern, 1979, SMC-9, 62–66. [Google Scholar]
- 95.dos Anjos A and Shahbazkia H, in Proc. of ICBED, BIOSIGNALS, 2008, vol. 2, pp. 70–76. [Google Scholar]
- 96.Szeliski R, Computer Vision: Algorithms and Applications (Texts in Computer Science), Springer, London, 2011. [Google Scholar]
- 97.Canny J, IEEE Trans. Pattern Anal. Mach. Intell, 1986, PAMI-8, 679–698. [PubMed] [Google Scholar]
- 98.Hough P, US Pat., US3069654A, 1962. [Google Scholar]
- 99.Ballard DH, Pattern Recognit, 1981, 13, 111–122. [Google Scholar]
- 100.Beucher S and Lantuejoul C, Int. Work. Image Process. Real-time Edge Motion Detect, 1979, 12–21. [Google Scholar]
- 101.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A, Nat. Methods, 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dejgaard SY and Presley JF, J. Histochem. Cytochem, 2014, 62, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Varinli H, Osmond-McLeod MJ, Molloy PL and Vallotton P, J. Lipid Res, 2015, 56, 2206–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fukushima K, Biol Cybern, 1980, 36, 193–202. [DOI] [PubMed] [Google Scholar]
- 105.LeCun Y, Bottou L, Bengio Y and Haffner P, Proc. IEEE, 1998, 86, 2278–2323. [Google Scholar]
- 106.Litjens G, Kooi T, Bejnordi BE, Setio AAA, Ciompi F, Ghafoorian M, van der Laak JAWM, van Ginneken B and Sánchez CI, Med. Image Anal, 2017, 42, 60–88. [DOI] [PubMed] [Google Scholar]
- 107.Eulenberg P, Köhler N, Blasi T, Filby A, Carpenter AE, Rees P, Theis FJ and Wolf FA, Nat. Commun, , DOI: 10.1038/s41467-017-00623-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen CL, Mahjoubfar A, Tai LC, Blaby IK, Huang A, Niazi KR and Jalali B, Sci. Rep, , DOI: 10.1038/srep21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fu D and Xie XS, Anal. Chem, 2014, 86, 4115–4119. [DOI] [PubMed] [Google Scholar]
- 110.Di Napoli C, Pope I, Masia F, Langbein W, Watson P and Borri P, Anal. Chem, 2016, 88, 3677–3685. [DOI] [PubMed] [Google Scholar]
- 111.Masia F, Glen A, Stephens P, Borri P and Langbein W, Anal. Chem, 2013, 85, 10820–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jüngst C, Klein M and Zumbusch A, J. Lipid Res, 2013, 54, 3419–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang C, Li J, Lan L and Cheng JX, Anal. Chem, 2017, 89, 4502–4507. [DOI] [PubMed] [Google Scholar]
- 114.Sbalzarini IF and Koumoutsakos P, J. Struct. Biol, 2005, 151, 182–195. [DOI] [PubMed] [Google Scholar]
- 115.Medyukhina A, Meyer T, Schmitt M, Romeike BFM, Dietzek B and Popp J, J. Biophotonics, 2012, 5, 878–888. [DOI] [PubMed] [Google Scholar]
- 116.Cao C, Zhou D, Chen T, Streets AM and Huang Y, Anal. Chem, 2016, 88, 4931–4939. [DOI] [PubMed] [Google Scholar]
- 117.Marcy Y, Ouverney C, Bik EM, Losekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA and Quake SR, Proc. Natl. Acad. Sci, 2007, 104, 11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Streets AM, Zhang X, Cao C, Pang Y, Wu X, Xiong L, Yang L, Fu Y, Zhao L, Tang F and Huang Y, Proc. Natl. Acad. Sci, 2014, 111, 7048–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A and McCarroll SA, Cell, 2015, 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA and Kirschner MW, Cell, 2015, 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fan HC, Fu GK and Fodor SPA, Science (80-. )., , DOI: 10.1126/science.1258367. [DOI] [Google Scholar]
- 122.Yuan J and Sims PA, Sci. Rep, , DOI: 10.1038/srep33883. [DOI] [Google Scholar]
- 123.Lane K, Van Valen D, DeFelice MM, Macklin DN, Kudo T, Jaimovich A, Carr A, Meyer T, Pe’er D, Boutet SC and Covert MW, Cell Syst, 2017, 4, 458–469.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gierahn TM, Wadsworth MH, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Christopher Love J and Shalek AK, Nat. Methods, 2017, 14, 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bose S, Wan Z, Carr A, Rizvi AH, Vieira G, Pe’er D and Sims PA, Genome Biol, 2015, 16, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang C, Huang K-C, Rajwa B, Li J, Yang S, Lin H, Liao C, Eakins G, Kuang S, Patsekin V, Robinson JP and Cheng J-X, Optica, 2017, 4, 103. [Google Scholar]
- 127.Liao C-S, Huang K-C, Hong W, Chen AJ, Karanja C, Wang P, Eakins G and Cheng J-X, Optica, 2016, 3, 1377. [Google Scholar]
- 128.He R, Liu Z, Xu Y, Huang W, Ma H and Ji M, Opt. Lett, 2017, 42, 659. [DOI] [PubMed] [Google Scholar]
- 129.Liao C-S, Slipchenko MN, Wang P, Li J, Lee S-Y, Oglesbee RA and Cheng J-X, Light Sci. Appl, 2015, 4, e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Slipchenko MN, Oglesbee RA, Zhang D, Wu W and Cheng JX, J. Biophotonics, 2012, 5, 801–807. [DOI] [PubMed] [Google Scholar]
- 131.Alshaykh MS, Liao C-S, Sandoval OE, Gitzinger G, Forget N, Leaird DE, Cheng J-X and Weiner AM, Opt. Lett, 2017, 42, 1548. [DOI] [PubMed] [Google Scholar]
- 132.Hashimoto K, Takahashi M, Ideguchi T and Goda K, Sci. Rep, 2016, 6, 21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tamamitsu M, Sakaki Y, Nakamura T, Podagatlapalli GK, Ideguchi T and Goda K, Vib. Spectrosc, 2017, 91, 163–169. [Google Scholar]
- 134.Coluccelli N, Vicentini E, Gambetta A, Howle CR, Mcewan K, Laporta P and Galzerano G, Opt. Express, 2018, 26, 18855. [DOI] [PubMed] [Google Scholar]
- 135.Chen KH, Boettiger AN, Moffitt JR, Wang S and Zhuang X, Science (80-. )., , DOI: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, Turczyk BM, Yang JL, Lee HS, Aach J, Zhang K and Church GM, Nat. Protoc, 2015, 10, 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M and Cai L, Nat. Methods, 2014, 11, 360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cunningham S, Leslie P, Hopwood D, Illingworth P, Jung RT, Nicholls DG, Peden N, Rafael J and Rial E, Clin. Sci. (Lond), 1985, 69, 343–8. [DOI] [PubMed] [Google Scholar]
- 139.Lean ME, Proc. Nutr. Soc, 1989, 48, 243–56. [DOI] [PubMed] [Google Scholar]
- 140.Hany TF, Gharehpapagh E, Kamel EM, Buck A, Himms-Hagen J and von Schulthess GK, Eur. J. Nucl. Med. Mol. Imaging, 2002, 29, 1393–1398. [DOI] [PubMed] [Google Scholar]
- 141.Nedergaard J, Bengtsson T and Cannon B, Am. J. Physiol. Metab, 2007, 293, E444–E452. [DOI] [PubMed] [Google Scholar]
- 142.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto N-J, Enerbäck S and Nuutila P, N. Engl. J. Med, 2009, 360, 1518–1525. [DOI] [PubMed] [Google Scholar]
- 143.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, Schrauwen P and Teule GJJ, N. Engl. J. Med, 2009, 360, 1500–1508. [DOI] [PubMed] [Google Scholar]
- 144.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM and Ronald Kahn C, Obstet. Gynecol. Surv, 2009, 64, 519–520. [Google Scholar]
- 145.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR and Spiegelman BM, Nature, 2008, 454, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang A-H, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerbäck S, Schrauwen P and Spiegelman BM, Cell, 2012, 150, 366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, Huang TL, Roberts-Toler C, Weiner LS, Sze C, Chacko AT, Deschamps LN, Herder LM, Truchan N, Glasgow AL, Holman AR, Gavrila A, Hasselgren P-O, Mori MA, Molla M and Tseng Y-H, Nat. Med, 2013, 19, 635–9. [DOI] [PMC free article] [PubMed] [Google Scholar]