

Abstract
Key points
Sickle cell disease (SCD) results in cardiopulmonary dysfunction, which may be exacerbated by prolonged exposure to environmental hypoxia.
It is currently unknown whether exposure to mild and moderate altitude exacerbates SCD associated cardiopulmonary and systemic complications.
Three months of exposure to mild (1609 m) and moderate (2438 m) altitude increased rates of haemolysis and right ventricular systolic pressures in mice with SCD compared to healthy wild‐type cohorts and SCD mice at sea level.
The haemodynamic changes in SCD mice that had lived at mild and moderate altitude were accompanied by changes in the balance between pulmonary vascular endothelial nitric oxide synthase and endothelin receptor expression and impaired exercise tolerance.
These data demonstrate that chronic altitude exposure exacerbates the complications associated with SCD and provides pertinent information for the clinical counselling of SCD patients.
Abstract
Exposure to high altitude worsens symptoms and crises in patients with sickle cell disease (SCD). However, it remains unclear whether prolonged exposure to low barometric pressures exacerbates SCD aetiologies or impairs quality of life. We tested the hypothesis that, relative to wild‐type (WT) mice, Berkley sickle cell mice (BERK‐SS) residing at sea level, mild (1609 m) and moderate (2438 m) altitude would have a higher rate of haemolysis, impaired cardiac function and reduced exercise tolerance, and that the level of altitude would worsen these decrements. Following 3 months of altitude exposure, right ventricular systolic pressure was measured (solid‐state transducer). In addition, the adaptive balance between pulmonary vascular endothelial nitric oxide synthase and endothelin was assessed in lung tissue to determine differences in pulmonary vascular adaptation and the speed/duration relationship (critical speed) was used to evaluate treadmill exercise tolerance. At all altitudes, BERK‐SS mice had a significantly lower percentage haemocrit and higher total bilirubin and free haemoglobin concentration (P < 0.05 for all). right ventricular systolic pressures in BERK‐SS were higher than WT at moderate altitude and also compared to BERK‐SS at sea level (P < 0.05, for both). Critical speed was significantly lower in BERK‐SS at mild and moderate altitude (P < 0.05). BERK‐SS demonstrated exacerbated SCD complications and reduced exercise capacity associated with an increase in altitude. These results suggest that exposure to mild and moderate altitude enhances the progression of SCD in BERK‐SS mice compared to healthy WT cohorts and BERK‐SS mice at sea level and provides crucial information for the clinical counselling of SCD patients.
Keywords: BERK mouse, hemolysis, critical speed, hypoxia
Key points
Sickle cell disease (SCD) results in cardiopulmonary dysfunction, which may be exacerbated by prolonged exposure to environmental hypoxia.
It is currently unknown whether exposure to mild and moderate altitude exacerbates SCD associated cardiopulmonary and systemic complications.
Three months of exposure to mild (1609 m) and moderate (2438 m) altitude increased rates of haemolysis and right ventricular systolic pressures in mice with SCD compared to healthy wild‐type cohorts and SCD mice at sea level.
The haemodynamic changes in SCD mice that had lived at mild and moderate altitude were accompanied by changes in the balance between pulmonary vascular endothelial nitric oxide synthase and endothelin receptor expression and impaired exercise tolerance.
These data demonstrate that chronic altitude exposure exacerbates the complications associated with SCD and provides pertinent information for the clinical counselling of SCD patients.
Introduction
A defining feature of clinical care for patients with sickle cell disease (SCD) is thorough counselling on the risks associated with time spent in regions of high elevation. Nonetheless, there are SCD patients who reside permanently and are treated at moderate altitude. Given these discrepancies, there is considerable debate amongst clinicians regarding whether SCD patients living at moderate elevations experience exacerbated haemolysis and worsening of the pulmonary and systemic vascular maladies emblematic of their disease.
The haemolysis, cardiopulmonary, vascular and microvascular abnormalities observed in SCD are the result of a genetic mutation in both β1‐ and β2‐haemoglobin genes that result in the expression of the haemoglobin S gene (Hb S) (Gladwin, 2016). Hb S polymerizes within the erythrocyte during deoxygenation to cause the classic sickle‐shaped morphology and the associated reduced red blood cell (RBC) deformability that leads to the impediment of microvascular blood flow and promotion of haemolysis (Gladwin, 2016; Khan et al. 2016; Mehari & Klings, 2016). The resulting extravascular haemolysis leads to increased spleen and hepatic iron accumulation, as well as increased plasma levels of lactate dehydrogenase and bilirubin. In addition, intravascular haemolysis elevates cell‐free plasma haemoglobin (Hb), leading to Hb extravasation into extravascular sites, where it can scavenge nitric oxide (NO), causing vasoconstriction, platelet activation and endothelial dysfunction (Dimmeler et al. 1999b; Iyamu et al. 2003b).
From a clinical perspective, acute altitude‐induced hypoxaemia (as might occur during mountain visits or rarely during aircraft travel) has long been associated with painful crises and sudden death in those with SCD (Mahony & Githens, 1979; Claster et al. 1981). This is not surprising when considering that (i) over 140 million people worldwide reside in areas with an elevation between ∼2000 and 2500 m (6000 and 8000 feet) (Cohen & Small, 1998) and that (ii) of the US population, the central rocky mountain region represents the most abundant concentration of SCD patients living at a mean altitude of ∼1800 m (∼5900 feet) (Hassell, 2010). Furthermore, when SCD patients travel acutely to areas of mild elevation of ∼2000 m (∼6500 feet), which is still below the average elevation for the US state of Colorado [∼2100 m (6800 feet)], approximately 25% of them develop crises (Mahony & Githens, 1979), which suggests that altitude exposure poses a serious threat to this patient population. Despite these facets, much less is known about the progression of the SCD associated pulmonary and systemic complications during chronic exposure to low vs. moderate altitudes. Thus, pertinent questions regarding the severity and progression of SCD, as well as its impact on the quality of life of individuals of this geographical population, remain unanswered.
The Berkeley mouse model of SCD (BERK‐SS) has emerged as a powerful preclinical tool by which to study the complex aetiologies associated with clinical SCD (Manci et al. 2006). As such, this model has been well characterized and exhibits multi‐organ pathologies that manifest spontaneously and include increased rates of haemolysis and resulting anaemia, as well as spleen, liver, lung and cardiac abnormalities (Manci et al. 2006; Hsu et al. 2007). With the BERK‐SS mouse model of SCD, it is possible to determine the impact of chronic exposure to moderate altitude on the cellular and molecular mechanisms of this disease. Furthermore, to aid in the clinical translation of these findings, determination of the running speed/duration relationship, known as the critical speed (CS), can also be achieved. The CS is a fundamental cross‐species exercise parameter first described in 1925 by the British physiologist, A. V. Hill (Hill, 1925) and defines the tolerable duration of severe‐intensity exercise, thus providing the functional significance of changes in cellular and molecular pathophysiology.
Despite the likelihood that prolonged exposure to altitude exacerbates the progression of SCD‐induced pulmonary and systemic pathologies, investigations into this topic are lacking. Therefore, the present study aimed to test the hypothesis that BERK‐SS mice exposed to sea level, mild [1609 m (5280 feet), Denver altitude], or moderate [2438 m (8000 feet)] altitude for 2.5 months would demonstrate significantly higher rates of SCD related morbidity compared to age‐matched wild‐type (WT) controls. Additionally, we hypothesized that the rate of morbidity of BERK‐SS mice would be associated with the magnitude of altitude exposure and linked to more severe indices of haemolysis, right heart insufficiency, pulmonary hypertension (PH) and exercise intolerance. Understanding how altitude exposure impacts SCD progression is essential for resolving the current discrepancies in clinical recommendations and, thus, the results obtained in the present study may provide crucial links between exposure to moderate altitude and the increased pathophysiological progression of SCD.
Methods
Ethical approval and animal care
Young‐adult male and female C57Bl/6 WT and Berk‐SS mice (8 weeks old) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed and bred in an AAALAC accredited animal facility at the University of Colorado, Denver, Anschutz Medical campus and were maintained under a 12:12 h light/dark cycle with food and water available ad libitum. Female heterozygous Berk‐SS mice were bred with male homozygous Berk‐SS mice to generate homozygous offspring. Specifically, Berk mice with genotype Tg(Hu‐miniLCR α1 Gγ Aγ δ βs) Hba0/0 Hbb0/0 and the hemizygous with genotype Tg(Hu‐miniLCR α1 Gγ Aγ δ βs) Hba0/0 Hbb0 Hbb+ were littermates. Genotyping of mice used for breeding and experiments was performed by TransnetYX (Cordova, TN, USA). A total of 64 mice (WT n = 30, BERK‐SS: n = 34) were used in the present study and levels of discomfort and distress were monitored daily by the in‐house animal care staff, with a veterinarian available as needed. Mice presented no pain or discomfort associated with hypoxia or recovery measurements (e.g. treadmill exercise and ECG) and were alert and conducted normal eating, drinking, and grooming activities when housed. All experimental procedures were conducted in accordance with the guidelines recommended by The Journal of Physiology (Grundy, 2015) and were approved by the Institutional Animal Care and Use Committee at the University of Colorado, Denver, Anschutz Medical Campus (approval number 00218).
Materials
Moderate atmospheric hypoxia exposure
Mice were randomly divided into three groups and exposed continuously to simulated altitudes of sea level, as well as mild [1609 m (5280 feet)] or moderate [2438 m (8000 feet)] levels, in custom‐built rodent hypobaric/hyperbaric chambers. These chambers were pressurized to mimic either sea level or 2438 m (8000 feet) elevation (barometric pressures of 760 mmHg or 564 mmHg, respectively) as described previously (Buehler et al. 2012). Mice selected to reside at mild altitude were housed in normobaric conditions [located in Denver, Colorado, USA; 1609 m (5280 feet)]. All mice were exposed to individual altitudes for 3 months.
ECG
ECG analysis were performed at 2‐week intervals using a high‐resolution in vivo micro‐imaging Vevo770 system (Visual‐Sonics, Toronto, ON, Canada). Care was taken to follow the guidelines for measuring cardiac physiology in mice established by the American Physiological Society (Lindsey et al. 2018). For each analysis, mice were anaesthetized via an isoflurane/O2 mixture with induction at 4% and maintenance between 1.5% and 3% and the body temperature was maintained at 37°C via a heated surgical pad. As described by Cavasin et al. (2012), pulse‐wave Doppler imaging of the pulmonary outflow was recorded in the parasternal short‐axis view at the level of the aortic valve. Animals were then monitored during the recovery from anaesthesia and, upon full recovery, were returned to their respective housing.
Treadmill exercise and constant speed tests
Prior to the determination of CS, mice completed a treadmill familiarization phase, which consisted of seven ∼5 min runs on a motor‐driven rodent treadmill (Exer 3/6; Columbus Instruments, Columbus, OH, USA). For the first run out of several runs, the treadmill speed was maintained at 20 m min–1 (up a 5° grade, which was maintained throughout all of the treadmill tests). For the last run out of several runs, the speed of the treadmill was increased progressively over the last minute to ∼40–50 m min–1 to familiarize the mice with high speed running. Animals were encouraged to run with intermittent bursts of compressed room air aimed at the hind limbs from directly above the animal (so as not to push the mouse up the treadmill). All treadmill testing protocols were designed and conducted by experienced staff and strictly followed the guidelines set by the American Physiological Society's resource book for the design of animal exercise protocols (Kregel et al. 2006).
The CS was determined using a modified version of the methodology used by Copp et al. (2010) for rats. After completion of the treadmill familiarization period, each mouse performed three to five runs to exhaustion, in random order, at a constant speed that resulted in fatigue between at 1.5 and 15 min (speeds ranging from 30–50 m min–1). Each test was performed on separate days with a minimum of 24 h between tests. For each constant‐speed trial, mice were given a 2 min warm‐up period where they ran at 15–20 m min–1 followed by a 1 min period of quiet resting. To start the test, the treadmill speed was increased rapidly over a 10 s period to the desired speed at which point a stopwatch was started. Testing was terminated and time to exhaustion was measured to the nearest tenth of a second whenever the mouse could no longer maintain pace with the treadmill, despite obvious exertion of effort. A successful constant‐speed test was determined if (i) the mouse could quickly adapt to the treadmill speed at the beginning of the test (e.g., did not waste energy); (ii) a noticeable change in gait occurred preceding exhaustion (i.e. lowering of the hindlimbs and rising of the snout) and (iii) the animal's righting reflex was markedly attenuated when placed on their back in a supine position (an unexhausted quadruped will typically attempt to right themselves within ∼1 s).
Data modelling for the determination of CS
Following successful completion of the constant speed treadmill tests, the CS and finite distance capacity (D′) were calculated for each mouse using the linear 1/time model () as described previously (Poole et al. 1988; Gaesser et al. 1995; Copp et al. 2010). In this model, the treadmill speed used for the constant speed test is plotted as a function of the inverse of time to exhaustion and the y‐intercept of the regression line yields the CS and the slope is the D′.
Right ventricular systolic pressure (RVSP)
On the final day of data collection, mice were anaesthetized via isoflurane (as described above) and RVSP were measured via a direct right ventricle (RV) puncture with a 25‐gauge needle attached to a pressure transducer. Correct transducer placement was determined via live tracing by observing a significant rise in diastolic pressure as the catheter moved out of the ventricle. Pressures were acquired using the Cardiomax III Cardiac Output Computer (Columbus Instruments). At the end of the experimental protocol, mice were euthanized by cervical dislocation and exsanguination.
Blood and organ collection
At the end of the experimental protocol, 0.8 mL of blood was collected and placed in an EDTA‐K+ vacutainer and a haematocrit (Hct) tube for analysis of plasma and Hct, respectively. The plasma was collected, snap frozen in liquid nitrogen and immediately stored at ‐80°C until analysis. Tissues were collected after heparinized saline perfusion as reported previously (Buehler et al. 2012). The hearts were removed and the RV and left ventricle with septum (LV+S) were weighed for the assessment of the Fulton index (RV/LV+S).
Western blot analysis
Protein was isolated from whole lung tissue and western blots were performed using 30 μg of sample protein run under denaturing and reducing conditions on Criterion Tris‐HCl 12.5% gels (Bio‐Rad, Hercules, CA, USA) with a SDS‐PAGE blot system (Bio‐Rad). For comparison, samples from each group at each altitude (e.g. both WT and BERK‐SS) were run on a single gel (18‐well gel). Endothelial nitric oxide synthase (eNOS) uncoupling analysis was performed under native and low‐temperature SDS‐PAGE conditions. All proteins except eNOS monomer:dimer were normalized to β‐actin.
The antibodies used were: mouse anit‐eNOS/NOS type III (catalogue number: 610297; dilution 1:500; Becton Dickinson Biosciences, Franklin Lakes, NJ, USA), mouse monoclonal eNOS pT495 (catalogue number: 612706; dilution 1:1000; Becton Dickinson Biosciences), rabbit polyclonal eNOS pS1177 (catalogue number: ab195944; dilution 1:500; Abcam, Cambridge, MA, USA) and rabbit polyclonal endothelin‐1 (ET‐1) (catalogue number: 117757; dilution 1:250; Abcam).
Spectrophotometric determination of bilirubin
Bilirubin in plasma was measured in semi‐microcuvettes using a Cary 60 UV‐Vis Spectrophotometer (Agilent Technologies Inc., Santa Clara, CA, USA). Absorbance was measured at 460 nm, and the concentration was determined using an extinction coefficient of 53,846 cm−1 m −1.
Spectrophotometric determination of plasma haemoglobin
Haemoglobin in plasma was measured in semi‐microcuvettes using a Cary 60 UV‐Vis Spectrophotometer (Agilent Technologies). Absorbance was measured at between 350 and 650 nm. Spectra were deconvoluted against standard extinction curves of pure substances using a non‐negative least square algorithm.
Histology and immunohistochemistry
Tissue sections were deparaffinized and rehydrated and processed as described previously (Buehler et al. 2012). Lung sections (5 μm) were stained with haematoxylin and eosin using standard procedures to assess the accumulation of perivascular cells, as well as vessel wall thickness, as described previously (Stacher et al. 2012).
For Masson's trichrome staining, tissue was re‐fixed in Bouin's solution, washed in deionized water, stained in Weigert's iron haematoxylin and Biebrich scarlet‐acid fuchsin solution and, finally, differentiated in phosphomolybdic‐phosphotungstic acid.
For Perls’ iron staining, non‐haeme ferric iron deposition was detected using the Perls’ method with diaminobenzidine (DAB) intensification. All sections were then rinsed in 0.1 m phosphate buffer (pH 7.4) incubated with DAB for 3 min, washed in deionized water and lightly counterstained with Gill's II haematoxylin.
Immunohistochemistry analysis of pulmonary resistant vessels from two animals per group with an outside diameter of <50 μm was performed using standard procedures for paraffin‐embedded tissues. In brief, antigen retrieval was performed on serial lung sections followed by incubation with anti‐eNOS/NOS type III (catalogue number: 6120297; dilution 1:500; Becton Dickinson Biosciences), anti‐smooth muscle actin (catalogue number: ab5694; dilution 1:100; Abcam) or anti‐ET‐1 (catalogue number: ab117757; dilution 1:250; Abcam). Lung sections were incubated with a fluorescent secondary rabbit or mouse antibody, Alexa Fluor 488 (catalogue numbers: 1726530 and 175350088; dilution 1:500, Life Technologies, Grand Island, NY, USA) or rabbit or mouse Alexa Fluor 555 (catalogue numbers: 1252795 and 1729804; dilution 1:500; Life Technologies). Afterwards, sections were scanned on an Aperio microscope and analyzed using the Aperio eSlide manager software, Imagescope (Leica, Buffalo Grove, IL, USA).
Statistical analysis
Data are presented as the mean ± SEM. Statistical comparisons for data measurements were completed using multifactorial analysis of variance (Kwon et al. 2015) and included a determination for strain effects. Main effects between mouse strains were determined by pooling all WT and BERK‐SS mice into respective groups. Post hoc analyses were completed with the Tukey–Kramer multiple comparison tests. Statistical analysis was completed using JMP, version 5 (SAS Institute, Cary, NC, USA) and Prism, version 6.0 (GraphPad Software Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
BERK‐SS mice
We observed an approximate 20% attrition rate of sickle cell mice before 6 weeks of age and a 100% attrition rate between 20 and 40 months as reported previously (Bakeer et al. 2016). During exposure to altitude, two BERK‐SS mice at both Denver and 2438 m (8000 feet) altitudes died (total attrition during study, n = 4) bringing the total number of BERK‐SS mice included in the analysis to 30. As anticipated, there were no deaths of WT mice in any group (analysis, n = 30).
Haemolysis
Analysis for markers of haemolysis showed significantly lower Hct, higher plasma concentrations of total bilirubin and free Hb (shown as μm haeme), and splenomegaly (Fig. 1 A). We observed moderately increased Hct in both WT and BERK‐SS mice housed at mild and moderate altitudes compared to sea level cohorts. Relative to sea level values, exposure to 2438 m (8000 feet) induced a rise in total bilirubin, extracellular Hb and spleen weight in BERK‐SS mice (Fig. 1 A).
Figure 1. Hematologic responses.
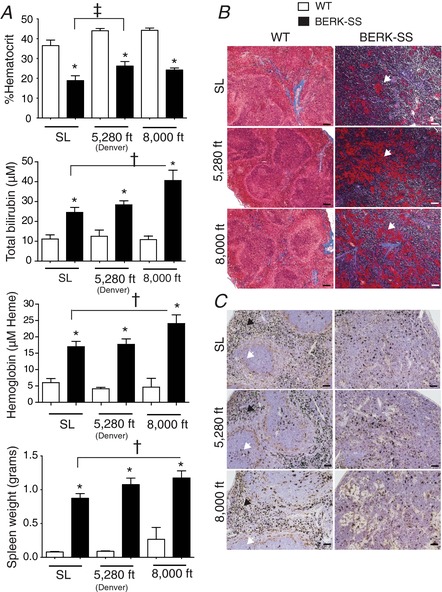
A, top: total %Hct for WT (open bars) and BERK‐SS mice (solid bars). * P < 0.0001 vs. WT. ‡P = 0.011 vs. BERK‐SS + sea level (n = 6–8). A, upper‐middle: total bilirubin for WT (open bars) and BERK‐SS mice (solid bars). * P < 0.001 vs. WT. †P = 0.026 vs. BERK‐SS + sea level (n = 6–8). A, lower‐middle: plasma haemoglobin concentrations for WT (open bars) vs. BERK‐SS mice (solid bars). * P < 0.0001 vs. WT. †P = 0.044 vs. BERK‐SS + sea level (n = 6–8). A, lower‐panel: spleen weights for WT (open bars) and BERK‐SS mice (solid bars). * P < 0.0001 vs. WT. †P = 0.031 vs. BERK‐SS + sea level (n = 6–8). B, Masson's Trichrome and fibrinogen staining of spleens from WT and BERK‐SS mice. White arrows show deposits of fibrinogen in collagen rich regions of the spleen red/white pulp. Magnification 10× (scale bar = 100 μm). C, Perls’ DAB iron staining of spleens from WT and BERK‐SS mice. White arrows show intact structures of the white pulp, whereas dark arrows show intact structure of the red pulp. Magnification 20× (scale bar = 50 μm). [Color figure can be viewed at wileyonlinelibrary.com]
Histopathology showed diffuse fibrosis in Berk‐SS but not WT mouse spleens based on Masson's Trichrome staining for collagen (blue/purple) and the precursor to fibrin, fibrinogen (Fig. 1 B, patchy red areas). Perls’ iron staining showed defined red pulp (black arrows) and white pulp (white arrows) in WT mouse spleens, whereas BERK‐SS mouse spleens showed a progressive loss of these anatomical regions (Fig. 1 C).
Haemodynamics and cardiac responses
ECG was performed on all mice at 2‐week intervals to assess the progression of cardiopulmonary disease. No altitude based within‐group differences were observed. However, after combining WT (n = 25) and BERK‐SS mice (n = 20), comparisons between strains revealed that BERK‐SS mice had a higher cardiac output, stroke volume and RV thickness (systole and diastole) compared to WT mice (Fig. 2 A–D).
Figure 2. Haemodynamic and ventricular characterization of pulmonary hypertension.
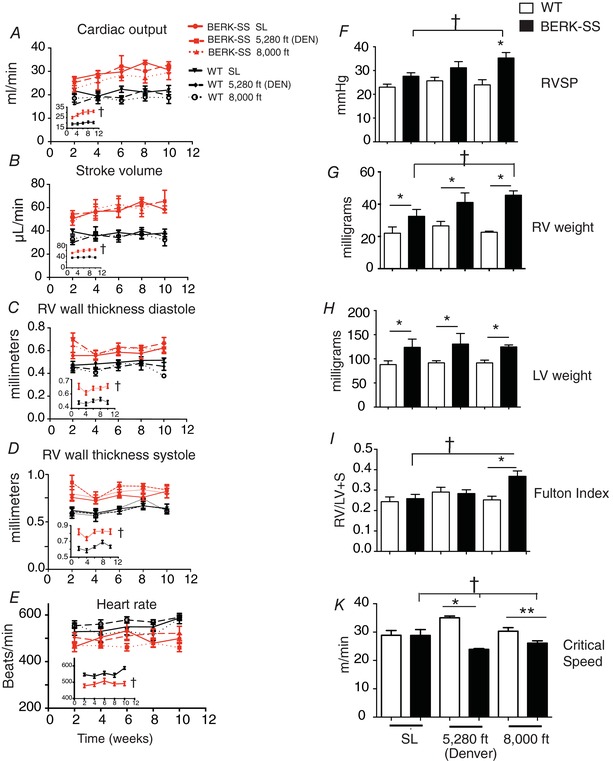
A–E, serial ECG of differences between WT (black lines) vs. BERK‐SS mice (red lines). Insets all WT groups combined vs. all BERK‐SS groups combined. †P < 0.0001 (n = 6–8). F, RVSP. * P < 0.01 BERK‐SS 8000 feet (solid bars) vs. WT mice (open bars) for all elevations (n = 6–8). †P < 0.01, BERK‐SS 8000 feet vs. Berk‐SS (sea level) (n = 6–8). G, RV weight. * P < 0.04 for BERK‐SS (solid bars) vs. WT mice (open bars) at each corresponding altitude. †P < 0.01 (n = 6–8) BERK‐SS 8000 feet vs. Berk‐SS (sea level). H, left ventricular weight. * P < 0.03 (n = 6–8) for BERK‐SS (solid bars) vs. WT mice (open bars) at each corresponding altitude. I, Fulton index. * P = 0.01 (n = 6–8) BERK‐SS vs. WT mice housed at 8000 feet. †P < 0.01 (n = 6–8) BERK‐SS mice housed at 8000 feet vs. BERK‐SS (solid bars) and WT mice (open bars) housed at sea level. J, CS. * P = 0.001 (n = 5) BERK‐SS (solid bars) vs. WT (open bars) housed at 5280 feet. ** P = 0.021 (n = 5) BERK SS (solid bars) vs. WT (open bars) housed at 8000 feet. †P ≤ 0.037 (n = 5) BERK‐SS housed at sea level vs. BERK‐SS housed at 5280 feet and 8000 feet. [Color figure can be viewed at wileyonlinelibrary.com]
As a direct determinant of pulmonary vascular disease, we evaluated RVSP and RV/LV+S at 3 months. Compared to WT cohorts, BERK SS mice had elevated RVSP at all altitudes, with the greatest difference occurring in BERK‐SS mice exposed to 2438 m (8000 feet) (Fig. 2 E). Heart mass was greater in the BERK‐SS compared to WT mice (Fig. 2 F and G). The Fulton index revealed that RV/LV+S ratios were greater in the BERK‐SS exposed to 2438 m (8000 feet) compared to WT cohorts and BERK‐SS mice exposed to sea level and Denver altitudes (Fig. 2 H). BERK‐SS mice housed at 2438 m (8000 feet) demonstrated a significantly increased RVSP, RV weight and Fulton index compared to BERK‐SS mice at sea level.
Exercise Capacity
Figure 2 K depicts the CS for WT and BERK mice at all altitudes. There were no significant between‐group differences in CS for mice housed at sea level. However, BERK‐SS mice housed at 1609 m (5280 feet) (Denver) and 2438 m (8000 feet) altitudes had a significantly lower CS compared to their respective WT controls (P < 0.05 for all) and a lower CS compared to both WT and BERK‐SS mice housed at sea level (P < 0.05).
Pulmonary histology
Lung slides from each animal were stained with haematoxylin and eosin to assess inflammation and pulmonary vascular remodelling. Assessment of inflammation revealed that more monocytes/neutrophils were surrounding pulmonary arteries in BERK‐SS mice compared to WT cohorts (Fig. 3 A). We also noted fewer monocytes/neutrophils in BERK‐SS mice exposed to sea level conditions compared to the mild and moderate altitude cohorts (Fig. 3 A). We observed only a modest degree of pulmonary vascular wall thickening in medium size vessels (50–100 μm) in BERK‐SS mice exposed to mild and moderate altitude compared to WT cohorts (Fig. 3 B), although we did not observe any differences in small (25–50 μm) or large (100–250 μm) vessels between BERK‐SS or WT mice.
Figure 3. Pulmonary artery remodelling.
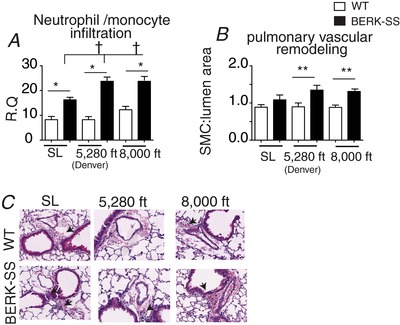
A, quantification of neutrophil / monocyte cells surrounding pulmonary arteries. * P < 0.001, BERK‐SS (solid bars) vs. WT mice (open bars). †P < 0.01 (n = 6–8) BERK‐SS (solid bars) housed at Denver and 8000 feet vs. BERK‐SS mice housed at sea level. B, quantification of medial hypertrophy in medium sized vessels (50–100 μm). ** P = 0.03 (n = 6–8) BERK‐SS (solid bars) housed at Denver and 8000 feet vs. WT cohorts (open bars). C, representative microphotographs of medium sized vessel (50 to 100 μm) stained for haemtoxylin and eosin (H&E). [Color figure can be viewed at wileyonlinelibrary.com]
Lung eNOS and ET‐1
Based on the results shown in Fig. 3 and the known opposing vasodilatory and vasoconstrictive effects of NO and ET‐1, respectively, we aimed to determine whether eNOS and ET‐1 were differentially expressed in the lungs of WT and BERK‐SS mice adapting to either mild or moderate altitude compared to sea level cohorts.
eNOS expression profile
Relative to WT, total eNOS expression was lower in BERK‐SS at all altitudes (Fig. 4 A). When compared with sea level or mild cohorts, both WT and BERK mice exposed to moderate altitude had a lower total eNOS expression. Evaluation of post‐translational modifications of eNOS showed that BERK‐SS mice demonstrated a different amino acid phosphorylation profile compared to WT mice (Fig. 4 B and C). All groups of BERK‐SS mice had higher ratios of active or phosphorylated serine (pS1177) to total eNOS compared to WT mice (Fig. 4 D). However, BERK‐SS mice exposed to either mild or moderate altitude had greater ratios of phosphorylated threonine 495 (pT495) to total eNOS (Fig. 4 E). By contrast to BERK‐SS mice, where both active and deactivated eNOS states existed at mild and moderate altitude, WT mice had a higher pS1177 to total eNOS ratio at moderate altitude in the absence of any significant levels of deactivated eNOS (pT495) (Fig. 4 D and E). Immunohistochemistry staining for eNOS demonstrated greater expression in the pulmonary resistance vessels ≤50 μm in WT compared to BERK‐SS mice across all altitudes (Fig. 5 A).
Figure 4. eNOS and ET‐1 lung expression.
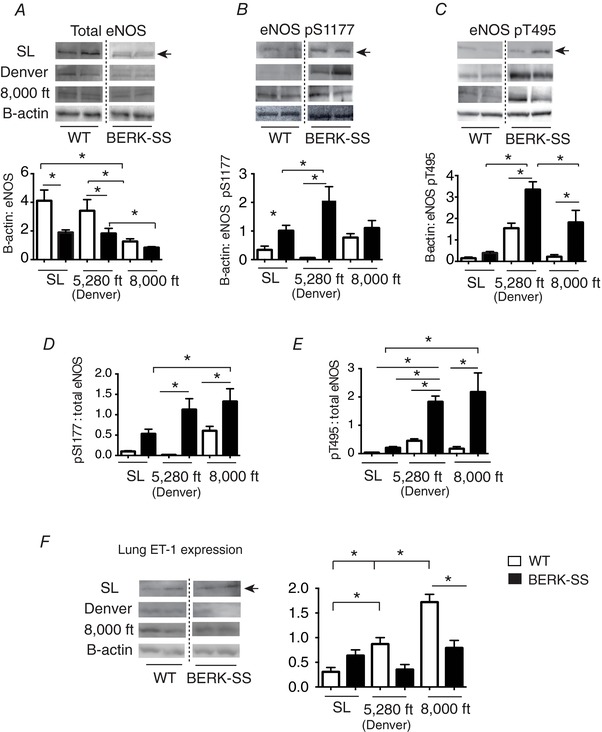
A, total eNOS expression in BERK‐SS (solid bars) vs. WT mice (open bars). * P ≤ 0.03 (n = 6–8). B, phosphorylated serine 1177 (pS1177) eNOS in BERK‐SS (solid bars) vs. WT mice (open bars). * P ≤ 0.01, (n = 6–8). C, phosphorylated threonine 495 (pT495) eNOS in BERK‐SS (solid bars) vs. WT (open bars) P ≤ 0.012 (n = 6–8). D, ratio of pS1177 eNOS to total eNOS. * P ≤ 0.04 (n = 6–8). E, ratio of pT495 eNOS to total eNOS. * P ≤ 0.002 (n = 6–8). F, lung ET‐1 expression. * P ≤ 0.04 (n = 6–8).
Figure 5. Localization of lung eNOS and ET‐1 expression from a representative animal.
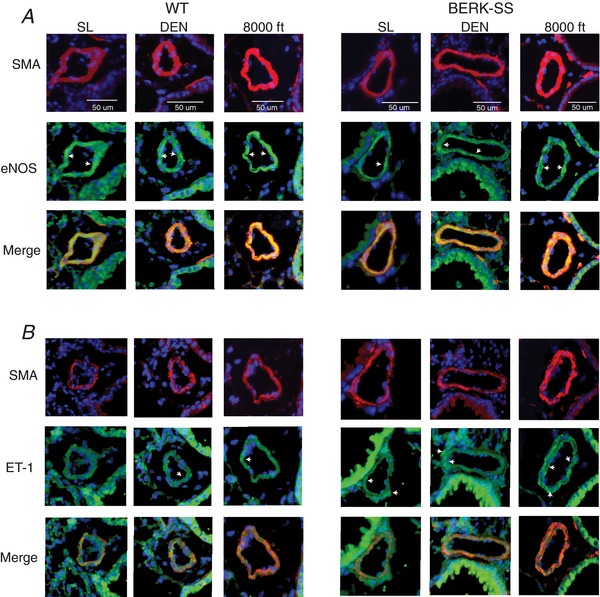
Microphotographs of serial lung sections are from distal pulmonary arteries ∼50 μm in diameter showing (A) eNOS expression and (B) ET‐1 expression in resistant arteries. Images were originally captured at 20×. White arrows show areas of eNOS or ET‐1 expression. Red: smooth muscle actin (SMA). Green: eNOS or ET‐1. Blue: 4′,6‐diamidino‐2‐phenylindole. [Color figure can be viewed at wileyonlinelibrary.com]
ET‐1 expression profile
The ET‐1 expression increased with altitude exposure in WT mice, with the highest expression occurring in the moderate altitude cohort. Interestingly, ET‐1 levels trended higher (P = 0.06) in the BERK‐SS mice housed at sea level compared to WT cohorts. In addition, although there were no significant differences between WT and BERK‐SS mice living at mild attitude, ET‐1 expression was significantly higher in WT than BERK‐SS in the moderate altitude cohort. There were no significant across‐altitude differences in ET‐1 expression in BERK‐SS mice (Fig. 4 F). To determine whether ET‐1 protein expression also occurred in the same pulmonary resistant vessels as eNOS, we stained and visualized serial lung sections. The altitude exposure‐based distribution of eNOS and ET‐1 in pulmonary resistant vessels is shown in relation to smooth muscle actin in Fig. 5 A and B.
Discussion
The principal novel finding of the present study was that BERK‐SS mice exposed to sea level, mild [1609 m (5280 feet)] and moderate [2438 m (8000 feet)] altitude for 3 months demonstrated significantly greater cardiopulmonary morbidity and associated SCD complications compared to WT controls. Specifically, and in accordance with our original hypothesis, the magnitude of morbidity of BERK‐SS mice was associated with the degree of altitude exposure and was accompanied by more severe indices of haemolysis, right heart insufficiency and PH. Crucially, these altitude induced exacerbations of SCD also translated to a lower exercise tolerance, as measured by CS, further suggesting that exposure to high altitude may significantly impair functionality and quality of life in patients with SCD.
Ecological validity and relationship with the existing literature
It is well established that acute exposure to high elevation can affect the hypoxic manifestations of sickle cell crisis and other SCD sequela. In the present study, the responses to moderate altitude exposure over an extended period (3 months) were examined in a murine model that approximates the pathophysiology of SCD in humans (Manci et al. 2006). It is essential to take into consideration that hypoxia exposure in mouse models of SCD is often fulfilled using 5–10% (de Franceschi et al. 2003; Iyamu et al. 2003a) and these severely low levels of O2, which are consistent with elevations of 5486–9144 m (18000–30,000 feet), represent the extreme altitude domain and are typically used to induce crisis. Although these types of experiments provide essential mechanistic insight into acute crises scenarios, they are not necessarily reflective of the environmental exposure that patients with SCD experience during their lifetime. Therefore, the results of the present study are especially novel when considering that many patients with SCD will never venture into the very high or extreme altitude domains, although they do routinely travel to and live in regions of moderate elevations and fly in a commercial aircraft pressurized to a barometric pressure approximating 2000 m (Mahony & Githens, 1979). Consequently, the simulated altitudes utilized in the present study [1609 m (5280 feet) and 2438 m (8000 feet) represent an ecologically relevant dose of hypoxia exposure, thus improving the clinical utility of this investigation.
Haematological and vascular effects of altitude exposure
As expected, BERK‐SS mice had a significantly lower Hct (e.g. ∼45% lower than WT at all altitudes), higher total bilirubin and plasma haemoglobin concentrations, as well as higher spleen weights, compared to WT. BERK‐SS mice exposed to 8000 feet (2438 m) of altitude for 2.5 months also demonstrated significantly greater indices of accelerated extra‐ and intravascular haemolysis compared to BERK‐SS mice residing at sea level. The extent of splenic fibrosis and the loss of red and white pulp structure were also visually different over increasing altitudes. This observation suggests that increasing intra‐ and extravascular haemolysis with splenic changes occurs over prolonged exposure to moderate altitude and differs from the responses to acute altitude exposure in humans. These results further indicate that Hb SC and Hb S‐β‐thalassaemia patients exposed to moderate altitude have a higher probability of experiencing splenic complications compared to Hb SS‐SCD patients (Claster et al. 1981).
In SCD, the depletion of NO, as well as reduced eNOS function, is assumed to be a critical mechanism of PH and the development of right heart failure (Hsu et al. 2007). Phosphorylation of eNOS at serine 1177 by serine/threonine protein kinase has been demonstrated to increase the activity of the enzyme, independent of calcium concentration (Dimmeler et al. 1999a). Conversely, AMP‐activated kinase phosphorylates eNOS at threonine 495, leading to diminished activity of the enzyme (Fleming et al. 2001). Therefore, it is the concomitant phosphorylation of both threonine 495 and serine 1177 that ultimately determines the activity of eNOS. However, the general belief that threonine 495 and serine 1177 exist in duality, where the activation of one is associated with the dephosphorylation of another, has been challenged by the results of several studies (Li et al. 2007; Hsu et al. 2010; Rafikov et al. 2011). Consequently, although an overall reduction in eNOS function may help tip the balance of vascular tone to that of vasoconstriction by ET‐1 expression and enhance the development of PH (Yoon et al. 2012), the relative changes in threonine 495 and serine 1177 observed in the present study do not provide conclusive evidence that eNOS function is indeed attenuated. Thus, the structural and functional changes in the pulmonary vasculature observed in the present study probably reflect the combined changes in NOS function and increased rates of Hb induced NO scavenging. This notion is further supported by the results of a recent investigation by Ferguson et al. (2018), which shows a robust effect of cell‐free Hb on vascular control in the skeletal muscle.
Crucially, the hypoxic pulmonary vasoconstrictive response associated with ventilation/perfusion matching during hypoxia exposure is regulated in part by ET‐1; therefore, as NO bioavailability is reduced (e.g. via decreased eNOS or increased NO scavenging by Hb), ET‐1 expression increases (Swenson & Bartsch, 2012; Sylvester et al. 2012). These effects of ET‐1 are well documented in SCD, support the use of ET‐1 receptor antagonists with respect to lessening the vascular consequences associated with SCD (Bourque et al. 2012), and have been studied extensively in preclinical SCD and PH models, as well as both patient populations (Minniti et al. 2009; Barst et al. 2010; Kasztan et al. 2017).
Differences in WT and BERK‐SS eNOS and ET‐1 suggest discordant adaptation amongst BERK‐SS and WT mice. This is supported by differences in ECG and the Fulton index between WT and BERK‐SS mice, as well as BERK‐SS mice housed at 2438 m (8000 feet) relative to BERK‐SS mice living at sea level. These data are consistent with decreased NO bioavailability and increased ET‐1 expression in WT mice with increasing altitude exposure. Moreover, significant pulmonary vascular remodelling was observed in BERK‐SS mice residing at mild and moderate altitude, further indicating that PH in BERK‐SS mice is exacerbated, at least in part, as a result of reduced NO bioavailability and sustained vasoconstriction and smooth muscle cell proliferation (Hsu et al. 2007). Importantly, the development of PH sequelae was also accompanied by a reduced functional capacity observed in both BERK‐SS mice residing at altitude.
SCD and functional capacity
The inability to sustain muscular exercise represents a hallmark feature in a plethora of chronic diseases (Jones & Poole, 2013) and, although impaired exercise tolerance has been well established in patients with SCD (Machado et al. 2007; Rees et al. 2010; Sachdev et al. 2011), the fundamental mechanisms underlying this dysfunction remain poorly understood. This is the first investigation to measure exercise tolerance in a preclinical mouse model of SCD and, consistent with our original hypothesis, CS was significantly lower in BERK‐SS mice compared to WT at both altitudes above sea level and was significantly lower compared to both sea level groups. This impaired exercise tolerance in BERK‐SS is probably the consequence of many facets, including cardiopulmonary maladaptation and reduced Hct, the latter of which would be expected to impair skeletal muscle capillary haemodynamics (Poole et al. 2013). In addition, recent evidence also suggests that elevated plasma levels of cell‐free Hb disrupt peripheral skeletal muscle vascular and metabolic control during contractions (Ferguson et al. 2018) and therefore probably contribute further to the lower CS observed in BERK‐SS in the present study.
As noted in the Introduction, the CS represents a powerful cross‐species tool that affords the ability to explore and understand the physiological and pathophysiological mechanisms of fatigue in health and disease (Poole et al. 2016). This information is essential for resolving the severely compromised functional capacity of SCD patients whose maximal oxygen uptake () mimics patients with severe heart failure (∼15 mL min–1 kg–1) (Anthi et al. 2007). Accordingly, the CS values reported in the present study define the tolerable rate of exercise for this pre‐clinical model of SCD and therefore provide a platform by which to assess the efficacy of future therapeutic interventions. Indeed, the reductions in NO bioavailability caused by alterations in NOS function and Hb‐NO interactions are probably reflected by reductions in circulating nitrate and nitrite, which would suggest a potential therapeutic application of dietary nitrate supplementation to improve vascular and metabolic control in SCD (Ferguson et al. 2016; Jones et al. 2016).
What is striking about these results is that CS in BERK‐SS mice was not significantly different at sea level despite a robust attenuation being observed at both higher elevations. This observation further supports the notion that moderate altitude exposure exacerbates the complications associated with SCD and, considering that the CS remains highly conserved across species and forms of exercise, suggests that patients living at moderate elevation suffer from a reduced ability to sustain exercise and thus an impaired quality of life. However, it is worth noting that the analysis of CS performed in the present study was designed to play a supportive role and only included a total of 15 mice (e.g. five in each group). Thus, a full characterization of the CS and assessment of the associated mechanistic underpinnings of exercise intolerance in BERK‐SS mice, particularly at sea level, should be performed in a future studies.
Conclusions
This is the first investigation to assess the impact of chronic mild and moderate altitude exposure on SCD associated sequelae and functional capacity in a preclinical model of SCD. Consistent with our original hypothesis, these data suggest that altitude exposure impacts cardiopulmonary function, which may be associated with BERK‐SS mice uniquely adapting their active eNOS to ET‐1 balance in an altitude dose‐dependent manner. The structural and functional changes in cardiopulmonary physiology are similar to those observed in the clinical PH and SCD population and thus further strengthen the utility of this preclinical SCD model. Therefore, the results of the present study provide a crucial mechanistic linkage between the impaired cardiopulmonary function and reduced tolerance to exercise observed in SCD patients and establish a basis for future comprehensive investigations into the associated cellular and molecular mechanisms. In addition, these results provide important considerations for healthcare providers of SCD patients residing at moderate altitude and an evaluation of the clinical progression such patients is warranted.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SKF, AY, RN, KH, PWB and DCI were responsible for the conception and design of the experiments. SKF, AY, KR, JWH, JHB, DIP, ZL, DH, PE, EG, RN, KRS, PWB and DCI were responsible for the collection, analysis and interpretation of the data. SKF, JWH, RN, KH, KRS, ENG, PWB and DCI were responsible for the drafting the article and revising it critically for intellectual content. All authors have approved the final version of this manuscript submitted for publication.
Funding
The present study was supported by the National Heart, Lung and Blood Institute Grants 1R01HL125642‐01A1 (Irwin DC), 1R01HL086680‐07 (Nozik‐Grayck E), 5P01HL014985‐38 (Stenmark KR) and T32‐HL007171 (Stenmark KR), Colorado Sickle Cell Treatment and Research Center (Hassell K and Nuss R). The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Acknowledgements
We thank Dr Maria Cavasin for her expert ECG analysis.
Biography
Scott K. Ferguson is a Postdoctoral Fellow in the Department of Medicine at the University of Colorado, Anschutz Medical Center. His work focuses on oxygen transport in health and disease, with an emphasis on cardiopulmonary and skeletal muscle function during exercise. He completed his graduate training in the Departments of Kinesiology and Anatomy and Physiology at Kansas State University, which provided a holistic understanding of integrative and applied physiology. Scott's current research focuses on uncovering the mechanistic basis for exercise intolerance in patients with sickle cell disease, with the goal of identifying novel therapeutic modalities to improve microvascular function.

Edited by: Laura Bennet & Harold Schultz
This is an Editor's Choice article from the 15 February 2019 issue.
Linked articles: This article is highlighted in a Perspectives article by Kato. To read this article, visit https://doi.org/10.1113/JP276705.
References
- Anthi A, Machado RF, Jison ML, Taveira‐DaSilva AM, Rubin LJ, Hunter L, Hunter CJ, Coles W, Nichols J & Avila NA (2007). Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Resp Crit Care Med 175, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa SK, Lorenz JN, Osinska H, Backer K, Huby AC, Shrestha A, Niss O, Fleck R, Quinn CT, Taylor MD, Purevjav E, Aronow BJ, Towbin JA & Malik P (2016). Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A 113, E5182‐5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barst RJ, Mubarak KK, Machado RF, Ataga KI, Benza RL, Castro O, Naeije R, Sood N, Swerdlow PS & Hildesheim M (2010). Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol 149, 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque sea level , Whittingham HA, Brien SE, Davidge ST & Adams MA (2012). Role of endothelin‐1 in the hyper‐responsiveness to nitrovasodilators following acute NOS inhibition. Br J Pharmacol 165, 1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler PW, Baek JH, Lisk C, Connor I, Sullivan S, Kominsky D, Majka S, Stenmark KR, Nozik‐Grayck E, Bonventura J & Irwin D (2012). Free hemolgobin induction of pulmonary vascular disease: evidence for and inflammatory mechanism. Am J Physiol Lung Cell Mol Physiol 303, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasin MA, Demos‐Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser‐Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR & McKinsey TA (2012). Selective class I histone deacetylase inhibition suppresses hypoxia‐induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 110, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claster S, Godwin MJ & Embury SH (1981). Risk of altitude exposure in sickle cell disease. West J Med 135, 364–367. [PMC free article] [PubMed] [Google Scholar]
- Cohen JE & Small C (1998). Hypsographic demography: the distribution of human population by altitude. Proc Natl Acad Sci U S A 95, 14009–14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp SW, Hirai DM, Musch TI & Poole DC (2010). Critical speed in the rat: implications for hindlimb muscle blood flow distribution and fibre recruitment. J Physiol 588, 5077–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Franceschi L, Baron A, Scarpa A, Adrie C, Janin A, Barbi S, Kister J, Rouyer‐Fessard P, Corrocher R, Leboulch P & Beuzard Y (2003). Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease‐specific lung injury induced by hypoxia/reoxygenation. Blood 102, 1087–1096. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R & Zeiher AM (1999a). Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399, 601–605. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R & Zeiher AM (1999b). Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399, 601. [DOI] [PubMed] [Google Scholar]
- Ferguson SK, Glean AA, Holdsworth CT, Wright JL, Fees AJ, Colburn TD, Stabler T, Allen JD, Jones AM & Musch TI (2016). Skeletal muscle vascular control during exercise: impact of nitrite infusion during nitric oxide synthase inhibition in healthy rats. J Cardiovasc Pharmacol Ther 21, 201–208. [DOI] [PubMed] [Google Scholar]
- Ferguson SK, Harral JW, Pak DI, Redinius KM, Stenmark KR, Schaer DJ, Buehler PW & Irwin DC (2018). Impact of cell‐free hemoglobin on contracting skeletal muscle microvascular oxygen pressure dynamics. Nitric Oxide 76, 29–36. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE & Busse R (2001). Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin‐dependent endothelial nitric oxide synthase activity. Circ Res 88, E68–E75. [DOI] [PubMed] [Google Scholar]
- Gaesser GA, Carnevale TJ, Garfinkel A, Walter DO & Womack CJ (1995). Estimation of critical power with nonlinear and linear models. Med Sci Sports Exerc 27, 1430–1438. [PubMed] [Google Scholar]
- Gladwin MT (2016). Cardiovascular complications and risk of death in sickle‐cell disease. Lancet 387, 2565–2574. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell KL (2010). Population estimates of sickle cell disease in the U.S. Am J Prev Med 38, S512‐521. [DOI] [PubMed] [Google Scholar]
- Hill AV (1925). The physiological basis of athletic records. Sci Mon 21, 409–428. [Google Scholar]
- Hsu J‐H, Oishi P, Wiseman DA, Hou Y, Chikovani O, Datar S, Sajti E, Johengen MJ, Harmon C & Black SM (2010). Nitric oxide alterations following acute ductal constriction in the fetal lamb: a role for superoxide. Am J Physiol Lung Cell Mol Physiol 298, L880‐L887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LL, Champion HC, Campbell‐Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, Schimel DM, Cochard AE, Wang X, Schechter AN, Noguchi CT & Gladwin MT (2007). Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 109, 3088–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyamu EW, Turner EA & Asakura T (2003a). Niprisan (Nix‐0699) improves the survival rates of transgenic sickle cell mice under acute severe hypoxic conditions. Br J Haematol 122, 1001–1008. [DOI] [PubMed] [Google Scholar]
- Iyamu EW, Turner EA & Asakura T (2003b). Niprisan (Nix‐0699) improves the survival rates of transgenic sickle cell mice under acute severe hypoxic conditions. Br J Haematol 122, 1001–1008. [DOI] [PubMed] [Google Scholar]
- Jones AM, Ferguson SK, Bailey SJ, Vanhatalo A & Poole DC (2016). Fiber type‐specific effects of dietary nitrate. Exerc Sport Sci Rev 44, 53–60. [DOI] [PubMed] [Google Scholar]
- Jones AM & Poole DC (2013). Oxygen Uptake Kinetics in Sport, Exercise and Medicine. Routledge, Abington U.K. [Google Scholar]
- Kasztan M, Fox BM, Speed JS, De Miguel C, Gohar EY, Townes TM, Kutlar A, Pollock JS & Pollock DM (2017). Long‐term endothelin‐a receptor antagonism provides robust renal protection in humanized sickle cell disease mice. J Am Soc Nephrol 28, 2443–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Damanhouri G, Ali A, Khan SA, Khan A, Bakillah A, Marouf S, Al Harbi G, Halawani SH & Makki A (2016). Precipitating factors and targeted therapies in combating the perils of sickle cell disease–a special nutritional consideration. Nutr Metab (Lond) 13, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kregel KC, Allen DL, Booth FW, Fleshner MR, Henriksen EJ, Musch T, O'Leary D, Parks C, Poole D & Ra'anan A (2006). Resource Book for the Design of Animal Exercise Protocols. American Physiological Society, Rockville, MD, USA. [Google Scholar]
- Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, Iqbal S, Ivanova S, Gerzanich V & Simard JM (2015). Methemoglobin is an endogenous toll‐like receptor 4 ligand‐relevance to subarachnoid hemorrhage. Int J Mol Sci 16, 5028–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ruan L, Sood SG, Papapetropoulos A, Fulton D & Venema RC (2007). Role of eNOS phosphorylation at Ser‐116 in regulation of eNOS activity in endothelial cells. Vasc Pharmacol 47, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Kassiri Z, Virag JA, de Castro Brás LE & Scherrer‐Crosbie M (2018). Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol 314, H733‐H752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RF, Kyle Mack A, Martyr S, Barnett C, MacArthur P, Sachdev V, Ernst I, Hunter LA, Coles WA & Nichols JP (2007). Severity of pulmonary hypertension during vaso‐occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol 136, 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony BS & Githens JH (1979). Sickling crises and altitude: Occurrence in the Colorado patient population. Clin Pediatr 18, 431–435. [DOI] [PubMed] [Google Scholar]
- Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA & Coller BS (2006). Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood 107, 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehari A & Klings ES (2016). Chronic pulmonary complications of sickle cell disease. Chest 149, 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minniti CP, Machado RF, Coles WA, Sachdev V, Gladwin MT & Kato GJ (2009). Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol 147, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC, Burnley M, Vanhatalo A, Rossiter HB & Jones AM (2016). Critical power: an important fatigue threshold in exercise physiology. Med Sci Sports Exerc 48, 2320–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC, Copp SW, Ferguson SK & Musch TI (2013). Skeletal muscle capillary function: contemporary observations and novel hypotheses. Exp Physiol 98, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC, Ward SA, Gardner GW & Whipp BJ (1988). Metabolic and respiratory profile of the upper limit for prolonged exercise in man. Ergonomics 31, 1265–1279. [DOI] [PubMed] [Google Scholar]
- Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, Fulton D & Black SM (2011). eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol 210, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Williams TN & Gladwin MT (2010). Sickle‐cell disease. The Lancet 376, 2018–2031. [DOI] [PubMed] [Google Scholar]
- Sachdev V, Kato GJ, Gibbs JSR, Barst RJ, Machado RF, Nouraie M, Hassell KL, Little JA, Schraufnagel DE & Krishnamurti L (2011). Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 124, 1452–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD & Tuder RM (2012). Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson ER & Bartsch P (2012). High‐altitude pulmonary edema. Compr Physiol 2, 2753–2773. [DOI] [PubMed] [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI & Ward JP (2012). Hypoxic pulmonary vasoconstriction. Physiol Rev 92, 367–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Zuccarello M & Rapoport RM (2012). pCO(2) and pH regulation of cerebral blood flow. Front Physiol 3, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]