

Abstract
The clinical utility of many peptide and protein drugs is limited by their short in-vivo half-life. To address this limitation, we report a new class of polypeptide-based materials that have a long plasma circulation time. The design of these polypeptides is motivated by the hypothesis that incorporating a zwitterionic sequence, within an intrinsically disordered polypeptide motif, would impart “stealth” behavior to the polypeptide and increase its plasma residence time, a behavior akin to that of synthetic stealth polymers. We designed these zwitterionic polypeptides (ZIPPs) with a repetitive (VPX1X2G)n motif, where X1 and X2 are cationic and anionic amino acids, respectively, and n is the number of repeats. To test this hypothesis, we synthesized a set of ZIPPs with different pairs of cationic and anionic residues with varied chain length. We show that a combination of lysine and glutamic acid in the ZIPP confer superior pharmacokinetics, for both intravenous and subcutaneous administration, compared to uncharged control polypeptides. Finally, to demonstrate their clinical utility, we fused the best performing ZIPP sequence to glucagon-like peptide-1 (GLP1), a peptide drug used for treatment of type-2 diabetes and show that the ZIPP-GLP1 fusion outperforms an uncharged polypeptide of the same molecular weight in a mouse model of type-2 diabetes.
Keywords: recombinant proteins, unstructured polypeptides, bioinspired materials, zwitterionic polypeptides
1. Introduction
The delivery of most peptide and protein drugs is limited by their short in vivo circulation, which can limit their therapeutic efficacy [1,2]. One way to address this limitation is to attach macromolecular carriers to these drugs, which can improve their plasma half-life and hence therapeutic efficacy [1–3]. The chemical conjugation of polyethylene glycol (PEG) to peptide and protein drugs —termed PEGylation— is an example of this strategy [1,4]. PEG is a “stealth” polymer that forms a ‘water cage’ around the drug, which then provides steric repulsion from blood components and prevents opsonization and premature clearance of the conjugate. This stealth property of PEG has made PEGylation one of the most commonly used approaches in the biopharmaceutical industry to improve the pharmacokinetics (PK) and pharmacodynamics (PD) of biologies [5]. In recent years, a new class of zwitterionic synthetic polymers have been discovered with similar stealth properties as PEG [6–8].
There are, however, four major shortcomings of all synthetic polymers as drug delivery vehicles. First, one of the limitations of PEG —the gold standard of synthetic “stealth” polymers —is that a substantial proportion of the human population has pre-existing anti-PEG antibodies from prior exposure to PEG that can trigger an adverse immune response [3,9,10]. Second, synthetic polymers are typically not biodegradable, and the long-term effect of their in vivo accumulation is still not well understood; for example, PEG is known to accumulate in vacuoles in the kidney [7,11]. Third, although there have been significant advances in the synthesis of synthetic polymers with low polydispersity, [3] they are intrinsically polydisperse in their molecular weights (MW), which limits the ability to precisely tune their in vivo behavior [12–14]. Fourth, the poorly controlled stoichiometry and site specificity of most chemical conjugation approaches, as well as limited yield, also limit the utility of synthetic polymer conjugates for protein and peptide delivery [13,15].
In contrast, recombinant peptide polymers, or polypeptides such as XTEN that do not have a discernible repeat unit, [16] can be attached to a peptide or protein drug with molecular precision, in terms of their sequence, stoichiometry, and chain length, as they are genetically encoded with the peptide or protein drug of interest [13,17–19]. Their PK and tissue biodistribution are less variable due to their precise sequence and monodispersity, which gives this class of macromolecules a significant advantage over most synthetic alternatives that are polydisperse. The polydispersity of synthetic polymers is undesirable because it often leads to a population of drug conjugates with different biological properties, especially with respect to their circulation half-life and immunogenicity [12].
The field of recombinant polypeptides as drug conjugates, however, is still in its infancy, with only a few candidates that have reached clinical trials, such as XTEN [16] and elastin-like polypeptides (ELPs) [18]. While previous studies with ELPs and XTEN have shown that an unstructured protein sequence can extend the half-life of the payload, they lack the “stealth” properties of polymers, such as PEG, that can impart favorable PK to peptide polymers. As with natural proteins, the PK of peptide polymers is governed by their physiochemical properties that is encoded by their amino acid sequence [20]. A systematic investigation to understand the impact of the amino acid sequence on the PK and PD of peptide polymers is a prerequisite to develop the next generation of recombinant polypeptides with optimized PK and PD.
To address this limitation, herein we systematically investigate the impact of amino acid sequence on plasma circulation and tissue biodistribution. Because the entire sequence space of peptide polymers is too large to experimentally interrogate, we focused our attention on zwitterionic peptide polymers that contain a cationic-anionic pair residue pair in the repeat unit. The choice of this narrow class of polypeptides was motivated by the hypothesis that incorporating a zwitterionic sequence within an intrinsically disordered polypeptide motif would impart “stealth” behavior to the polypeptide and increase its plasma residence time, a behavior akin to that of synthetic stealth polymers. This hypothesis stems from a previous study by Whitesides and coworkers, who showed that self-assembled monolayers on gold with kosmotropic head groups can prevent the adsorption of proteins, [21] and later studies that showed synthetic polymers with such zwitterionic monomers on surfaces can recapitulate this effect, [22,23] and when conjugated to proteins can prolong their plasma half-life [6,7,24]. Our work also builds upon the previous work by Jiang and co-workers, who showed that zwitterionic brushes made from natural amino acids have non-fouling behavior on surfaces in vitro [25,26] and when a poly-electrolyte peptide polymer made up of lysine (K) and glutamic acid (E) —poly(KE) — is recombinantly expressed with a protein, it can stabilize and protect the protein from extreme environments (heat and salt) [27]. However, longer sequences of poly(KE) are difficult to recombinantly synthesize due to the highly repetitive nature of such alternating dipeptide sequences [28]. The expression level of such minimal sequences is also typically low [29].
Motivated by their findings and the limitations, we explore herein the design of zwitterionic polypeptides (ZIPPs) with various combinations of oppositely charged residues that might exhibit long circulation in vivo. To ensure high expression levels and easy purification, we designed the ZIPP wherein the cationic-anionic dipeptide is embedded within a structurally disordered ELP scaffold for two reasons. First, because ELPs are known to express at high levels in E. coli as soluble proteins [30,31]. we hypothesized that the same would be true of ZIPPs. Second, we hypothesized that the ELP scaffold would impart thermally responsive phase behavior to the ZIPPs, which would enable easy non-chromatographic purification of the ZIPPs and their fusion proteins.
We tested the ZIPPs in vivo in mice upon i.v. and s.c. administration, and showed that the amino acid composition of polypeptides has a significant impact on their PK. The best performing ZIPP has a total plasma exposure and half-life that is two-fold greater than its uncharged control. We also showed the therapeutic utility of the optimal ZIPP in delivery of glucagon like peptide-1 (GLP1), a drug that is clinically used to treat type-2 diabetes. The GLP1-ZIPP conjugate reduced blood glucose levels for up to 3 days in a diet induced obesity model of type-2 diabetes in mice after a single s.c. injection, which is 70 times longer than an injection of the unmodified drug [32] and one and a half times longer than an uncharged ELP control.
2. Materials and Methods
2.1. Materials:
The pET-24+ cloning vector was purchased from Novagen Inc. Custom oligonucleotides were synthesized by Integrated DNA Technologies Inc. Restriction endonucleases, T4 DNA ligase, Quick ligase and calf intestinal phosphate (CIP) were purchased from New England Biolabs. DNA mini-prep and gel purification kits were purchased from Qiagen Inc. Escherichia coli EB5alpha and BL21 (DE3) cells were purchased from Edge BioSystems. All E. coli cultures were grown in 2XYT media, which comprises of sodium chloride (5gL−1; Alfa Aesar), tryptone (16 gL−1, Becton, Dickinson and Co.), and yeast extract (10 gL−1, Becton, Dickinson and Co.). Tris(2-carboxyethyl)phosphine (TCEP) hydrochloride was from Pierce Biotechnology, Inc. Kanamycin sulfate was purchased from EMD Millipore. Protein expression was induced with isopropyl β-d-l-thiogalactopyranoside (IPTG) from Gold Biotechnology. All the salts used for protein purification was purchased from Alfa Aesar. Alexa-488-maleimide (Cat # A10254) was purchased from Life Technologies.
2.2. Gene synthesis:
Genes encoding ELPs and ZIPPs were assembled by recursive directional ligation by plasmid reconstruction (Pre-RDL), [33] from single stranded oligonucleotides supplied by IDT Inc. The amino acid sequence of the ZIPPs and ELPs are shown in Table 1.
Table 1.
ZIPP and ELP libraries. ZIPP and ELP libraries along with their guest residues, number of pentapeptide repeats of (VPX1X2G), the theoretical molecular weight and hydrodynamic radius (Rh).
| X1 | X2 | Pentamers | MW (Da) | Rh (nm) | |
|---|---|---|---|---|---|
| 80 | 30532 | 4.7 ± 0.2 | |||
| VPGAG | G | A | 120 | 45789 | 5.6 ± 0.1 |
| 160 | 61047 | 6.5 ± 0.2 | |||
| VPKEG | K | E | 80 | 40865 | 7.6 ± 0.3 |
| 120 | 61288 | 9.2 ± 0.3 | |||
| VPREG | R | E | 80 | 43106 | 5.4 ± 0.2 |
| 120 | 64650 | 7.8 ± 0.3 | |||
| VPKDG | K | D | 80 | 39743 | 5.9 ± 0.3 |
| 120 | 59605 | 7.2 ± 0.2 | |||
| VPRDG | R | D | 80 | 41984 | 5.2 ± 0.3 |
| 120 | 62967 | 6.2 ± 0.2 |
2.3. Protein expression and purification:
ZIPPs and ELPs were expressed from a modified pET-24+ expression vector [33] that was transformed into E. coli strain BL21 (DE3). Liquid cultures of 50 mL E. coli harboring plasmids encoding ELP or ZIPP were inoculated from frozen DMSO stocks. After 16 h of culture, each 50 mL was used to inoculate six 1 L flasks of 2XYT media supplemented with 45 μg.mL−1 kanamycin. Each 1 L flask was then grown at 37 °C in a shaker incubator at 200 rpm for 8 h, at which time the cultures were induced with 1 mM IPTG and grown for an additional 16 h. Cells were harvested by centrifugation at 3000 × g for 10 min at 4 °C and resuspended in 20 mL of cold PBS. Cells were then lysed via sonication on ice for 3 min (10 s on, 40 s off) (Misonix) and DNA and cellular debris were precipitated by adding polyethylenimine (10%; MP Biomedical) and centrifugation at 20,000 × g at 4 °C. After removal of DNA and cellular debris, ZIPPs and ELPs were purified from the soluble fraction of the cell lysate using four rounds of inverse transition cycling with minor modifications [34,35]. The supernatant was heated at 60 °C for 10 min to trigger phase separation and precipitation of contaminating proteins as well as ELPs or ZIPPs. The solution was then cooled on ice for 10 mins to re-dissolve the ELPs or ZIPPs while the contaminating proteins that remain insoluble were precipitated by centrifuging at 20,000 × g at 4 °C. This process was followed by salt triggered phase separation at 37 °C by the addition of 3 M sodium chloride for ELPs or a combination of 3M sodium chloride and 2M ammonium sulfate for ZIPPs, followed by centrifugation at 20,000 × g for 15 min at 37 °C to obtain the polypeptide pellet. The pellet was then resuspended in cold PBS (or PBS supplemented with 30 mM TCEP if the sequence had cysteines), and any insoluble matter was removed by a centrifugation step at 20,000 × g 4 °C for 10 min. These steps, starting with incubation at 60 °C, were repeated three more times, until a single band corresponding to MW of the construct was observed in SDS-PAGE gel. For animal experiments, endotoxin was removed using a Detoxi-Gel resin (Thermo Scientific), following the protocol supplied.
2.4. Mass spectrometry:
The MW of ZIPPs and ELPs was measured by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS) on a Voyager DE-Pro MALDI-TOF instrument (Applied Biosystems) with 20 μM of the ZIPP or ELP and saturated sinapinic acid as the matrix.
2.5. Circular dichroism:
The secondary structure of ZIPPs was measured by circular dichroism (CD) spectroscopy in PBS on an Aviv Model 202 instrument by scanning a 0.2 mg/ml solution of the ZIPP in PBS from 260 nm to 180 nm in 1 nm steps with a 3 s averaging time in 1 mm quartz cells (Hellma). Data were used for analysis only if the Dynode voltage was below 500 V. Secondary structure percentages were quantified from the CD data (range 200–250nm) using BestSel [36,37].
2.6. Light scattering:
Dynamic light scattering (DLS) was carried out to determine the hydrodynamic radius (Rh) of the ZIPPs and ELPs using a DynaPro temperature controlled microsampler (Wyatt Technology). Solutions of each polypeptide at 25 μM in PBS and water were filtered through 0.02 μm Whatman Anotop sterile syringe filters (GE Healthcare Life Sciences) into a quartz crystal cuvette (12 μl, Wyatt Technology, Santa Barbara, CA). Five acquisitions were taken at 37°C, and the results are presented as the mean Rh of the sample. The error represents the polydispersity from the five data points.
2.7. Native-PAGE gel:
Native-PAGE was used to visualize any interaction between ZIPPs and mouse serum albumin (MSA) or human serum albumin (HSA). ZIPPs or ELPs were incubated with MSA or HSA (1:1 molar ratio) for 30 min at 25 °C. After 30 min, samples were diluted 1:2 with Native Sample Buffer (Bio-Rad) and run on Mini-PROTEAN TGX Stain-Free Gels (Bio-Rad) at 180 V for 40 min. The gels were visualized using ChemiDoc MP imager (Bio-Rad). An ELP with an albumin binding domain [38] was used as a positive control.
2.8. Zeta potential measurements:
Zeta potential measurements were carried out on a Zetasizer Nano ZS (Malvern) at 50 μM in PBS at 37° C using disposable folded capillary cells (Malvern). Each sample was measured ten times and the data represent the mean and standard error of the mean (SE) of the ten acquisitions. An uncharged ELP of the same MW as the ZIPP as well and ELP with the same number of repeats was used as a control.
2.9. Cloning, expression and purification of GLP-1 peptide fusions:
The gene encoding GLP-1 was purchased from Integrated DNA Technologies and was fused to the gene encoding (VPKEG)120 and (VPGAG)160 using plasmid reconstruction by recursive directional ligation (pre-RDL). The first amino acid in GLP-1 (His) is numbered 7, and the GLP-1 used in this study has point mutations to improve its stability in vivo [32,39]. An Ala to Gly mutation at the 8th residue confers dipeptidyl peptidase IV (DPPIV) resistance [40] and a Gly to Glu mutation at residue 22 stabilizes the helical structure of GLP-1 [41]. We also mutated Arg to Ala at the 26th position to prevent premature cleavage of GLP1 in the s.c. space by ubiquitous arginine targeting proteases [42]. The amino acid sequence of the GLP-1 variant used in the study can be found in Supplementary Information (Figure S2). GLP1-VPGAG160 and GLP-VPKEG120 fusions were purified by ITC.
2.10. In vitro cAMP ELISA:
The activity of native GLP-1 and conjugates were assessed in vitro by quantifying the increase in cyclic adenosine monophosphate (cAMP) levels in baby hamster kidney (BHK) cells that were stably transfected to constitutively express the rat GLP1 receptor (GLP1R) [43]. The transfected cells were a generous gift from Daniel Drucker (University of Toronto). GLP1R is a member of the glucagon receptor family of G protein-coupled receptor which binds specifically to GLP-1. Binding of GLP-1 stimulates the production of cAMP which then results in insulin synthesis and release of insulin, and a cAMP ELISA is a widely used assay to validate receptor activation as a response to stimulation by GLP-1 [43]. Cells were cultured in high glucose DMEM (Thermo Fisher) supplemented with 10% fetal bovine serum, 100 U.ml−1 penicillin, 100 μg.ml−1 streptomycin and 50 μg.ml−1 G418 (Thermo Fisher). Cells were passaged at least once before running the assay. In a 24 well plate, 30,000 cells were seeded without G418 and incubated for 48 h, until they were 80% confluent. Prior to running the assay, 50 μM of GLP1 or GLP1-ELP or GLP1-ZIPP fusions incubated with 0.5 μg of DPPIV (ProSpec) at 37°C overnight to remove the di-peptide leader. On the day of the assay, cells were first treated with 250 μM of 3-isobutyl-l-methylxanthine (IBMX) for 1 h to prevent cAMP degradation, followed by incubation with varying concentrations (0.001–100 nM) of GLP1 or conjugates for 10 min at 37 °C to trigger receptor activation. Intracellular amount of cAMP was then assayed using a competitive binding cAMP ELISA according to the manufacturer’s protocol (Enzo Life Sciences). Each treatment and concentration was assayed in triplicates and the data were analyzed using a four-parameter logistic, nonlinear regression model (GraphPad Prism 6). The calculated EC50 values represent the half-maximal effective concentration.
2.11. Animal studies:
All animal studies were done in accordance with the NIH Guide for the Care and Use of Laboratory Animals under protocols approved by the Duke Institutional Animal Care and Use Committee (IACUC). Mice were housed in a room maintained on a 12 h light and dark cycle with ad libitum access to food and water. Unless indicated otherwise, mice were fed a standard rodent diet (LabDiet 5001).
2.12. Pharmacokinetics:
Pharmacokinetics of ZIPPs and ELP were measured by fluorescently labeling a single cysteine residue at the N-terminus of the polypeptides with Alexa 488. A leader sequence (GCGYPG) with a cysteine residue (C) was added to the N-terminus of the ZIPPs and ELPs to site specifically conjugate the maleimide derivative of Alexa488 according to the manufacturer’s protocol. Unreacted free fluorophore was removed using Amicon Ultra-15 centrifugal filter units (MWCO: lOkDa, Millipore) at 1800 × g at 4 °C in 25% acetonitrile in PBS (v/v). Alexa labeled polypeptides were then diluted with unlabeled polypeptides to yield a fluorophore concentration of 100 μM (300 μM polypeptide concentration), a concentration previously determined to be optimal for the PK study of soluble polypeptides. Balb/c mice (Charles River) were then administered with the labeled polypeptides via either an i.v. tail vein injection or s.c. injection. 10 μl blood samples were collected into tubes with 100 μl of heparin at 40 s, 15 min, 0.5, 2, 4, 8, 24, 48 and 72 h after injection into the tail vein. The concentration of fluorescently labeled polymer in the blood was calculated using a standard curve of Alexa 488 (Figure S3 and S4). Blood concentration time-course data was analyzed with a standard two compartment PK model for i.v. PK data and non-compartmental model for s.c. PK data to ascertain the pharmacokinetic parameters.
2.13. Pharmacokinetic analysis:
The elimination half-life (t1/2β) was calculated from the slope of the linear regression fit to the elimination portion of the log concentration vs. time curve. For s.c. data, elimination half-life was calculated by calculating the slope of the time points after Cmax (maximum serum concentration). tmax for s.c. PK data was determined as the time required to reach Cmax. The area under the curve (AUC) was calculated by GraphPad Prism 6 software (La Jolla, CA) using the trapezoidal rule. The serum concentration at t=0 (Co), for the i.v. data was extrapolated by fitting a linear line through the early time points (45 s to 45 min range). The volume of distribution (VD) which reflects the degree of extravascular tissues and organs distribution of polypeptide, was calculated as VD= (Dose injected i.v.)/(Co). The clearance of polypeptide from systemic circulation (CL), was calculated as (dose × F)/AUC where F (bioavailability)= 1 for i.v. bolus injection. All pharmacokinetic parameters are reported as mean ± SE.
2.14. Biodistribution:
Organ distribution of optimal ZIPP and ELP were determined by radiolabeling a tyrosine residue at the N-terminus of the polypeptides with 125I, as previously described [31,44]. Briefly, 100 μl of ELP or ZIPP at 300 μM was added to a pre-coated IODO-Gen tube with 2 mCi [125I]Na on ice and incubated for 15 min. The reaction mixture was purified by a ZebaSpin desalting column (Thermo Fisher Scientific). Mice Balb/c (Charles river) were put on 0.4 wt% potassium iodide water to block radionuclide accumulation in the thyroid. After 7 days, they were randomly divided into 12 groups (n=3–4). Each mouse was weighed before injection. Each group received intravenous bolus injections of 10 μCi 125I equivalent of 200 μl of either (VPGAG)160 (MW control) or (VPKEG)120. The injected concentration of each polypeptide was 300 μM. 125I labeled samples were aliquoted at different concentrations at the time of injection to create a standard curve. Whole organs were harvested at 2, 6, 24, and 72 h and the amount of polypeptide in each organ was determined by measuring 125I activity with an automated gamma counter (LKB-Wallac) in counts per minute (CPM). The percent injected dose (%ID) was computed using the standard curve to relate the CPM to μCi and then normalized against the weight of the organ to calculate %ID/g of tissue.
2.15. GLP1 efficacy:
For GLP1 efficacy study, 6-week old C57BI/6J male mice (Jackson Laboratories) were used. Mice were put on 60 kcal% high calorie diet (Research Diets D12492) for 1 week to induce a diabetic phenotype, which was confirmed by measuring blood glucose before initiating the experiment. Previous studies have demonstrated this diet induced obesity (DIO) in C57BI/6J as an acceptable model for type 2 diabetes mellitus [45,46]. The mice were injected s.c. with a single dose of GLPl-VPKEG120 or GLP1-VPGAG160 fusions (150 nmol.kg−1 at 40 μM) or an equivalent volume of PBS. Blood glucose and body weight was measured periodically throughout the course of the experiments. Prior to measuring blood glucose, the tail was wiped with alcohol wipes and a small nick was created on the tail vein of the mice with a lancet. The first drop of blood was wiped off and the second drop was used to measure the blood glucose using an AlphaTrak2 handheld glucometer (Abbott). Glucose AUC was calculated using the trapezoidal method.
2.16. Data analysis:
Data are presented as mean and standard error of mean (SE). PK parameters were compared using one-way ANOVA followed by post-hoc Tukey’s multiple comparison test. Biodistribution data between the two treatment groups was analyzed using Student’s t-test correcting for multiple comparisons. Treatment effect on blood glucose level and percentage change in body weight were analyzed using two-way repeated measures analysis of variance (ANOVA), followed by post-hoc Dunnett’s multiple comparison test. Glucose AUC was compared using one-way ANOVA followed by post-hoc Dunnett’s test.
3. Results and Discussion
3.1. Molecular design, synthesis, and purification of zwitterionic polypeptides (ZIPPs)
We recombinantly synthesized a family of genetically encoded ZIPPs with various combinations of oppositely charged residues that are engineered into a V-P-X1X2-G motif (Table 1). We based this design on two principles: first, the underlying P-Xn-G motif imparts structural disorder [47] that increases the coil size of the polypeptide, which in turn leads to decreased renal clearance in vivo. Second, we added a flanking Val (V) residue in the repeat unit to mimic the VPGXG motif of ELPs that we chose as an uncharged polypeptide control, because ELPs are structurally disordered. Furthermore, eliminating the V residue would create a GP dipeptide between two repeat units (PX1X2GPX1X2G), which could lead to fibrillization. [47,48]
For the two promiscuous “guest residues” —X1 and X2— we chose pairs of a positively charged — lysine (K) or arginine (R)— residue and a negatively charged —glutamic acid (E) or aspartic acid (D)— residue to create zwitterionic dipeptides. This leads to the following sets of repeat units: (VPKEG). (VPREG), (VPKDG) and (VPRDG) (Table 1). These polypeptides are also reminiscent of the intrinsically disordered proteins that populate the disordered regions of the proteome, which are enriched in charged and polar residues that can confer high aqueous solubility. [49,50] This feature makes the disordered polypeptides with these sequence attributes attractive fusion partners for protein and peptide drugs.
For our uncharged polypeptide control, we chose glycine (G) and alanine (A) as the two guest residues, as we hypothesized that these residues would have minimal impact on the structural disorder we sought to impart to these polypeptides; the repeat unit of the control polymer is hence (VPGAG). As the charged amino acids in the ZIPPs have a higher molecular weight (MW) than G or A, we selected ELPs with two different MWs to control for both the MW and the number of repeats. The amino acid sequence of the repeat unit, number of repeats, and MW of each peptide polymer are shown in Table 1.
Synthetic genes encoding ZIPPs and ELPs were assembled from shorter oligonucleotides using recursive directional ligation by plasmid reconstruction (pre-RDL) method, a technique that was developed by our group to assemble synthetic genes that encode for long repeats of highly repetitive polypeptides [33]. All constructs were expressed in E. Coli from a plasmid-borne gene, and were purified using inverse transition cycling (ITC), a non-chromatographic method to purify ELPs, and their derivatives, that takes advantage of their aqueous phase behavior [35]. The purity of the ZIPPs and ELP controls were assessed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 1B), which showed that the ZIPPs and ELPs were >95% pure. The yield of purified ZIPPs ranged from 80 to 100 mg per liter of culture.
Figure 1. Design and synthesis of zwitterionic polypeptides.
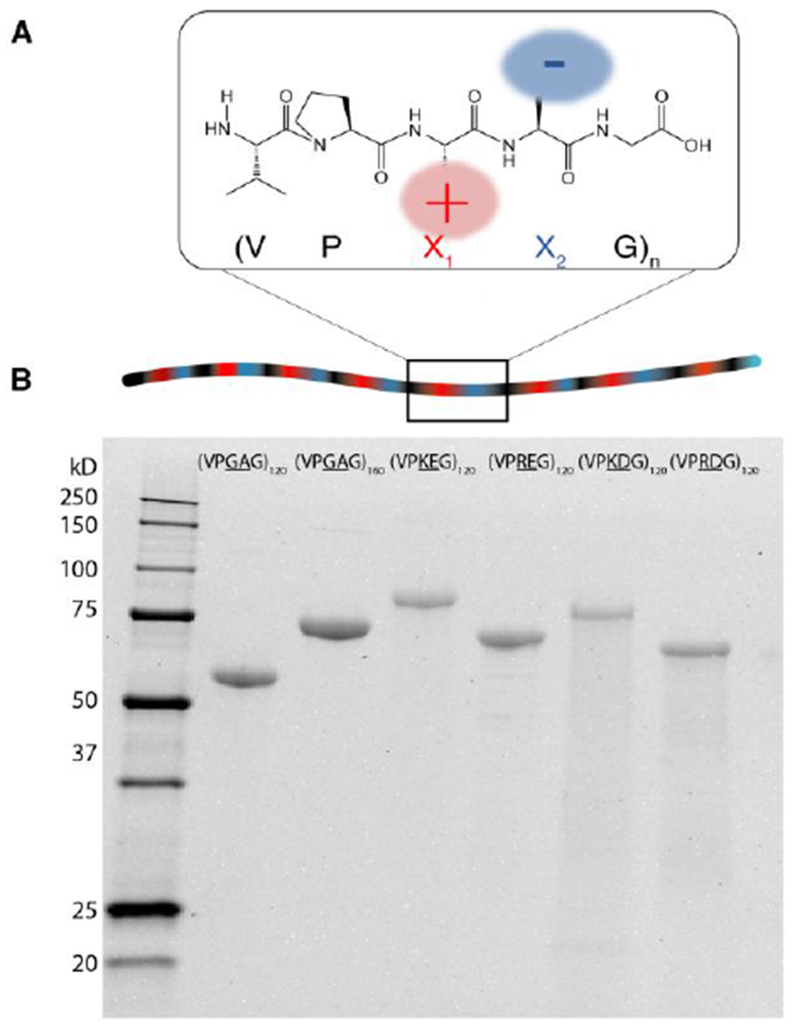
A. Schematic of the zwitterionic polypeptides (ZIPPs). ZIPPs are homopolymers of a VPX1X2G repeat unit, wherein X1 and X2 are positively and negatively charged amino acids respectively. B. SDS-PAGE of various ZIPPs sequences and the two ELP controls, (VPGAG)120 (control for equal number of repeat units) and (VPGAG)160 (MW controls). The polypeptide bands were compared to a standard MW ladder as a reference.
The molecular weight (MW) of unstructured polypeptides like ZIPPs and ELPs cannot be assessed from SDS-PAGE as they migrate anomalously compared to the globular proteins used in the standard ladder. Instead, the MW of the ZIPPs and ELPs were measured by matrix-assisted laser desorption/ionization time-of-fight mass spectrometry (MALDI-TOF-MS) (Figure S1). MALDI-TOF-MS showed that the measured mass of all ZIPPs and ELPs were within 0.3% of their calculated theoretical masses.
3.2. In vitro characterization of ZIPPs
To confirm the structural disorder of ZIPPs, circular dichroism (CD) spectroscopy was carried out to determine the secondary structure of ZIPPs, and to compare their secondary structure with that of ELP controls, which are known to be structurally disordered [32,47]. Despite differences in amino acid composition, ZIPPs —like ELPs— exhibited a negative peak at −197 nm, which is characteristic of intrinsically disordered polypeptides (Figure 2A) and a “shelf’ around 220 nm related to the presence of beta turns [51–53]. CD data was also used to calculate the secondary structure percentages using BestSel algorithm (Table S3), which indicated that there were no major differences in ZIPP secondary structures compared to ELPs.
Figure 2. Physiochemical characterization of ZIPPs.
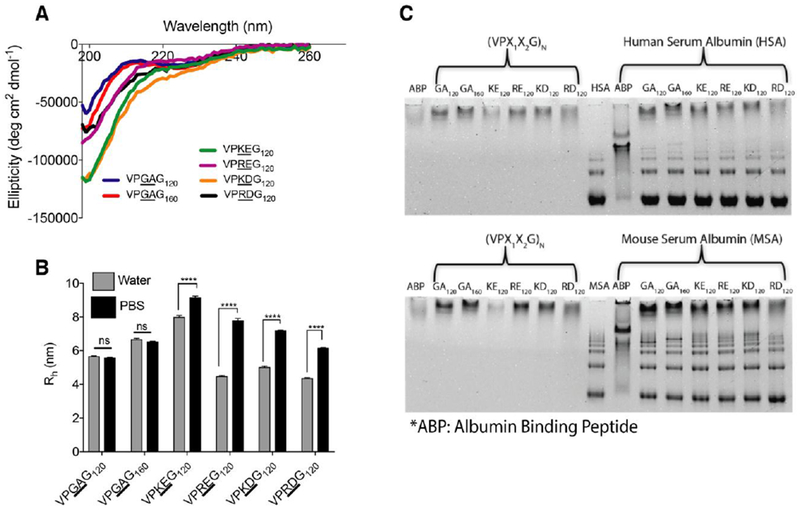
A. ZIPPs and ELPs show a high degree of disorder as seen from their CD spectra. The presence of a negative ellipticity peak ~197nm is characteristic of an intrinsically disordered polypeptide. B. Hydrodynamic radius (Rh) measured by Dynamic Light Scattering (DLS) shows that ZIPPs have anti-poly-electrolyte behavior, as they assume an extended conformation in a high ionic strength buffer such as PBS but have a collapsed structure in water. Uncharged ELPs, used as a control, do not display this behavior, as seen by the negligible change in Rh between water and PBS. (ns-not significant, ****p<0.0001 2-way repeated measures ANOVA, Sidak post-hoc test). C. Native PAGE shows that ZIPPs do not have any non-specific interaction with mouse serum albumin as well as human serum albumin, as seen by the similar mobility of ZIPPs in the presence or absence of albumin. An Albumin binding peptide (ABP) fused to an ELP was used as a positive control.
Next, the anti-polyelectrolyte property of ZIPPs was assessed by measuring their hydrodynamic radius (Rh) using dynamic light scattering (DLS) in PBS and water. The anti-polyelectrolyte property of polyzwitterions is manifested by an extended conformation in the presence of salt, due to the screening of their charges and a collapsed conformation in pure water due to dipole-dipole interactions between oppositely oriented dipoles [54–56].
As seen in Figure 2B, there is negligible change in the Rh of ELPs in PBS compared to water, whereas ZIPPs have a 15 – 60% larger Rh in PBS as compared to water (****p< 0.0001, 2-way repeated measures ANOVA and Sidak’s post-hoc test). This indicates that ZIPPs assume an expanded conformation in PBS, due to the screening of their charges in the presence of salt, and a more collapsed conformation in pure water. Notably, ZIPP sequences with lysine (K) have the largest Rh compared to ELPs in PBS, after controlling for both MW and number of repeats, suggesting that they have the most extended conformation in PBS. We also confirmed the overall neutral charge of ZIPPs in physiological conditions (PBS, pH 7.4) using zeta-potential measurements (Table S2).
Finally, to test whether densely charged polypeptides such as ZIPPs interact with serum proteins, we ran a native PAGE gel of ZIPPs incubated with the most abundant serum protein —albumin— from both mice and humans. Native PAGE gel shows that ZIPPs do not interact with human or mouse serum albumin (Figure 2C). As albumin is negatively charged, it migrates towards the positive electrode while ELP and ZIPPs have an overall neutral charge so that they do not enter the gel. We used an albumin binding peptide (ABP) fused to an ELP as a positive control [38]. When incubated with albumin, only ABP hinders the migration pattern of albumin which confirms their interaction, while ELPs and ZIPPs have no effect on the migration of albumin. The native PAGE results show that ZIPPs do not exhibit any non-specific interaction with the most abundant serum protein—albumin.
3.3. In vivo pharmacokinetics of ZIPPs
We investigated the effect of various zwitterionic dipeptide motifs (KE, KD, RD, RE) in the ZIPPs on the half-life and cumulative plasma exposure after i.v. and s.c. administration in Balb/c mice (Figure 3). We chose to investigate both i.v. and s.c. routes of administration, because the route of administration will depend on the type of drug the ZIPP is chemically conjugated to, or recombinantly fused to. To minimize the effect of renal clearance on the PK, we designed ZIPPs such that their MW was slightly above the renal threshold of 60 kDa, so that any differences in their PK that are controlled by their sequence would become readily apparent [57,58]. We performed a single i.v. injection of fluorescently labeled ZIPPs and ELP120 (control for the number of repeats) and ELP160 (MW control) and collected blood at various time points up to 72 h (Figure 3A). The concentration of ZIPPs and ELPs in plasma at various time points was then calculated using a standard curve that relates fluorescence intensity to ZIPP or ELP concentration (Figure S3). Data from the i.v. pharmacokinetic study were then fit to a two-compartment model [59] to calculate various PK parameters, shown in Table 2.
Figure 3. Pharmacokinetics and biodistribution of ZIPPs.
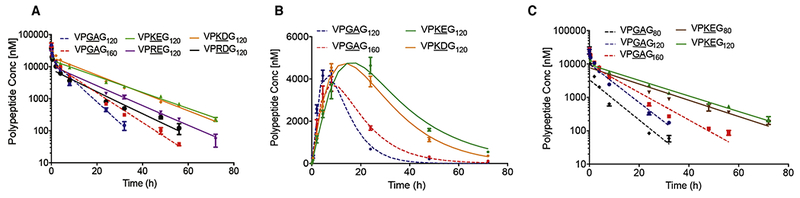
A. Circulating ZIPPs and ELPs in plasma as a function of time following a single i.v. injection. The data represent mean ± SE (n=5). The plasma concentration was fit to a two-compartment model, which yielded the pharmacokinetic parameters in Table 2. B. Plasma pharmacokinetics upon s.c. administration, shown as mean ± SE (n=3-4). Data from the s.c. administration study was fit to a non-compartmental model to determine the parameters in Table 3. C. Plasma pharmacokinetics upon i.v. administration for a shorter ZIPP, (VPKEG)80. Data represent mean ± SE (n=4).
Table 2.
Intravenous pharmacokinetic parameters
| AUC [nM × hr] | t1/2 (h) | CL (ml/h) | VD (ml) | |
|---|---|---|---|---|
| VPGAG120 | 113,579 ± 8,967 | 4.5 ± 0.2 | 0.58 ± 0.04 | 1.48 ± 0.14 |
| VPGAG160 | 146,100 ± 9,125 | 6.6 ± 0.3 | 0.45 ± 0.03 | 1.63 ± 0.19 |
| VPKEG120 | 282,555 ± 8,176 | 12.0 ± 0.4 | 0.23 ± 0.01 | 1.32 ± 0.10 |
| VPREG120 | 162,578 ± 4,248 | 9.6 ± 0.5 | 0.40 ± 0.01 | 1.91 ± 0.26 |
| VPKDG120 | 323,380 ± 7,939 | 10.8 ± 0.2 | 0.20 ± 0.01 | 1.65 ± 0.08 |
| VPRDG120 | 113,066 ± 3,758 | 8.2 ± 0.3 | 0.57 ± 0.02 | 1.85 ± 0.11 |
The 12 h elimination half-life (t1/2β) of (VPKEG)120 is ~three-fold greater than the 4.5 h t1/2β of (VPGAG)120, a repeat-unit matched control (p<0.001, one-way ANOVA and Tukey’s post-hoc test) (Table 2). Even when compared to the longer ELP control —(VPGAG)160— a MW-matched control that has a t1/2β of 6.6 h, the elimination half-life of (VPKEG)120 is almost two-fold greater (p<0.001, one-way ANOVA and Tukey’s test). The total cumulative blood exposure of the polypeptide, measured by the area under the plasma concentration versus time curve (AUC), is perhaps the parameter of greatest interest. It parallels the plasma elimination half-life data, as it is 2–3 times greater for VPKEG120 (282,555 nM.h) and VPKDG120 (323,380 nM.h) compared to the ELP controls —113,579 nM.h for (VPGAG)120 and 146,100 nM.h for (VPGAG)160 (p<0.001, one-way ANOVA and Tukey’s post-hoc test). Interestingly, ZIPPs with arginine (R) as the cationic residue —(VPREG)120 and (VPRDG)120 had a similar PK profile to that of the ELP controls (p=0.61, one-way ANOVA and Tukey’s post-hoc test).
These results show that some ZIPPs have significantly improved pharmacokinetics compared to the uncharged ELP controls. The clearance (CL), which is a measure of the plasma volume cleared of drug per unit time, was two-fold higher for ELP controls compared to ZIPPs with lysine (K) as the cationic residue —(VPKEG)120 and (VPKDG)120 (0.45 ml/h) versus (0.22 ml/h) (Table 2) (p<0.001, one-way ANOVA and Tukey’s post-hoc test).
These results are especially notable, because they also show that not all polyzwitterions behave identically, as their PK are highly dependent upon the amino acid sequence of the zwitterionic dipeptide, something that has hitherto not been reported in the literature. ZIPPs with lysine (K), as the cationic residue in the guest dipeptide of the repeat unit, have enhanced PK compared to arginine (R).
The lower CL values for ZIPPs with lysines —(VPKEG)120 and (VPKDG)120— when comparered to those with arginines, or the ELP controls also highlight their pharmacological benefits as drug carriers, reduced clearance and longer persistance in circulation.In contrast, the choice of the anionic residue in the guest dipepetide of the repeat unit had little impact, as the pharmacokinetics of ZIPPs with aspartic acid (D) or glutamic acid (E) —(VPKDG)120 and (VPKEG)120— were almost indistinguishable.
Although we do not have an explanation as to why ZIPPs with arginine (R) residues performed worse than lysine (K), there are several plausible hypotheses. First, the guanidium head group of arginine has a delocalized charge, which makes arginine a relatively hydrophobic residue compared to lysine [60– 62]. Second, the guanidium moiety in arginine can disrupt hydrogen bonding between water molecules and hence binds to water less tightly than lysine [63]. This may directly impact the ability of water to create a hydration layer around the polypeptide, which is essential for its stealth property. Third, sequences with lysines take up an extended conformation as indicated by a larger Rh in PBS (Table 1), and higher percentages of random coil structures (Table S3), which results in slower excretion and longer circulation time. Finally, arginine residues are commonly observed in cell penetrating peptides (CPP), [64,65] as they strongly interact with cell membranes, which could negatively impact the PK by causing premature loss of the ZIPPs with arginine from circulation.
We next investigated the blood circulation of the two best performing sequences, (VPKDG)120 and (VPKEG)120 after a single s.c. injection (Figure 3B). The data were fit to a non-compartmental model to determine the PK parameters shown in Table 3. The AUC of both ZIPPs are significantly greater than those of the ELP controls (p<0.001, one-way ANOVA and Tukey’s test). The 180,387 nM.h AUC of (VPKEG)120 is almost twice that of the MW-matched ELP control, (VPGAG)160, with an AUC of 92,914 nM.h. Similar to the i.v. PK data, the 15.6 h elimination half-life (t1/2β) of (VPKEG)120 was also significantly longer than that of the ELPs [10.6 h and 12.4 h for (VPGAG)120 and (VPGAG)160 respectively] (p<0.01, one-way ANOVA and Tukey’s test). However, the elimination half-life of (VPKDG)120 was similar to (VPGAG)160 (p=0.75, one-way ANOVA and Tukey’s post-hoc test).
Table 3.
Sub-cutaneous pharmacokinetic parameters
| AUC [nM × hr] | t1/2 (h) | tmax (h) | |
|---|---|---|---|
| VPGAG120 | 77,636 ± 3,367 | 10.6 ± 1.6 | 5.6 ± 2.2 |
| VPGAG160 | 92,914 ± 6,599 | 12.4 ± 0.8 | 8.0 ± 0.0 |
| VPKEG120 | 180,387 ± 16,415 | 15.6 ± 0.6 | 18 ± 0.0 |
| VPKDG120 | 164,419 ± 8,298 | 11.8 ± 1 | 14 ± 0.0 |
Another interesting observation is the difference in tmax —defined as the time it takes for a drug to reach its maximum plasma concentration after administration— between ZIPPs and ELPs. As seen from Figure 3B and Table 3, the tmax of ZIPPs is 2–3-fold longer than that of the ELP controls (p<0.001, one-way ANOVA and Tukey’s post-hoc test). This suggests that ZIPPs are absorbed slowly into systemic circulation. This is beneficial for s.c. drug delivery because the slow release profile of ZIPPs prevents wasted drug, eliminates a spike in plasma concentration of the drug and can minimize side effects that are caused by a spike in blood concentration. From these PK studies, we conclude that a ZIPP with K and E as the two residues in the guest dipeptide of the ZIPP repeat unit have significantly longer-elimination half-life, higher bioavailability, and slower adsorption profile than the other ZIPPs and the ELP controls. This suggests that (VPKEG)120 as the optimal sequence for drug delivery via both i.v. and s.c. administration routes.
In a separate cohort of mice, we also looked at the length dependence of our best performing ZIPP — (VPKEG)120. To do so, we synthesized a shorter variant —(VPKEG)80 with a MW of 40 kDa— so that its size is nominally below the renal cutoff of ~60 kDa. We compared its pharmacokinetics with (VPKEG)120 that has a MW of 61 kDa, which is closer to the renal threshold, as well as with ELPs of similar repeat units and MW (Figure 3C). As expected, (VPKEG)80 was superior to (VPGAG)80 (no. repeat unit control) as well as (VPGAG)120 (MW control), with a larger AUC and longer elimination half-life (p<0.001, one-way ANOVA and Tukey’s post-hoc test). The trend observed previously, where the AUC and elimination half-life of (VPKEG)120 were almost double to that of the ELP controls, holds true even with the shorter (VPKEG)80 (Figure 3C, Table S1). Interestingly, the difference in the elimination half-life of (VPKEG)80 vs. (VPKEG)120 was not significant, but the difference in AUC, a measure of the cumulative polypeptide exposure in the body, was significantly greater for (VPKEG)120 than (VPKEG)80 (p<0.001, one-way ANOVA and Tukey’s post-hoc test).
The i.v. pharmacokinetic study of (VPKEG)80 is also interesting because we can compare the half-life of the(VPKEG)80–40kDa to PEG-48kDa, which has been reported in the literature [1,66]. PEG-48kDa has an elimination half-life of 14 h, which is close to the half-life of (VPKEG)80 [1,66]. Their similar half-lives is encouraging as it demonstrates the feasibility of matching the pharmacokinetics of PEG — the pharmaceutical gold standard of “stealth” polymers— with an optimized ZIPP of the same MW. It is also interesting to compare our results with XTEN, which has been touted as a recombinant alternative to PEG [67]. XTEN432 (~40kDa), which has a similar MW weight as (VPKEG)80, has an elimination half-life of 9 h, which is less than the 12.5 h t1/2β of (VPKEG)80, suggesting that ZIPPs can match and perhaps even outperform XTEN [67].
3.4. Biodistribution of ZIPPs
We carried out a biodistribution study of our best performing ZIPP —(VPKEG)120— after i.v. injection and compared it with a MW-matched ELP control —(VPGAG)160. There were no major differences in the organ distribution of (VPKEG)120 and (VPGAG)160 (Figure 4A). However, the radioactivity detected in the blood was significantly higher for (VPKEG)120, 6 h post-administration, which is consistent with the PK data (p<0.0001, unpaired t-test). The ZIPP also showed a trend towards significant lower non-specific accumulation in the heart and lungs at 6 h and 24 h respectively. ZIPP also had lower accumulation in the organs of the reticuloendothelial system (RES) such as liver and kidney, although this accumulation was not statistically significant compared to ELP. In order to examine if the accumulation in the RES organs is dependent on the polypeptide concentration in circulation, we calculated the organ-to-blood ratio to normalize the amount present in the RES organs to the amount in blood (Figure 4B). We found that (VPKEG)120 had significantly lower retention in the liver compared to (VPGAG)160 at 6, 24 and 72 h (p<0.001, unpaired t-test), which hints at the possibility of lower uptake of this ZIPP by phagocytic cells residing in these organs. Moreover, less than 0.5 % ID/g organ of conjugates accumulated in the heart, lung, liver, and kidney at 72 h post-administration (Figure 4A). This result is in sharp contrast with those of synthetic polymers, like PEG and polycarboxybetaine, that are non-biodegradable and localize in organs for long durations [9,68]. The relative lack of accumulation of the ZIPP in healthy tissue, together with its enhanced pharmacokinetics, suggest that (VPKEG)120 can be an attractive alternative to synthetic polymers for half-life extension of pharmaceuticals.
Figure 4. Biodistribution of ZIPPs.
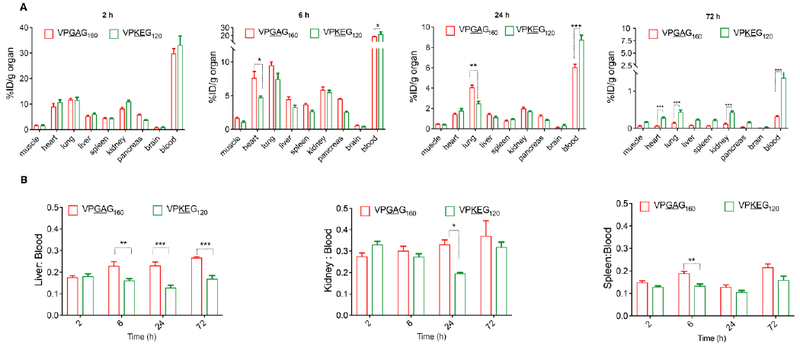
A. Biodistribution of ELP and ZIPP after i.v. injection at 2 h, 6 h, 24 h and 72 h revealed that there is not a large difference in organ distribution between ZIPP and ELP, with a few exceptions at several time points that are marked in the figure. Data shown as mean ± SE (n=3-4). (*p<0.05, ** p<0.01, ***p<0.0001, unpaired t-test). B. Retention of ELP and ZIPP in the organs of the reticuloendothelial system (RES) expressed as organ to blood ratio, calculated by normalizing the amount present in each organ to that in the blood. Each bar is the mean ± SE (n=3-4). (*p<0.05, **p<0.01, ***p<0.0001, unpaired t-test).
3.5. In vitro activity of GLP1 fusions
Having observed a significant improvement in the PK by incorporating zwitterionic motifs into the repeat unit of an intrinsically disordered peptide polymer, we next investigated the ability of a ZIPP to deliver a peptide drug and compared it with a MW-matched uncharged ELP control. We chose an incretin hormone glucagon-like peptide-1 (GLP-1) as our model drug. GLP1 is used to treat type 2 diabetes due to its glucose-dependent mode of action, which results in secretion of insulin only when blood glucose is high [69]. This prevents hypoglycemia that is a common issue with other type 2 diabetes-based therapies, which makes it attractive as a drug. GLP1 is also an attractive therapy for type 2 diabetes because it has been shown to prevent apoptosis and enhance proliferation of pancreatic β cells, which can help to slow disease progression [70]. However, like many peptide drugs, the clinical utility of GLP-1 is limited because of its rapid clearance, as it has a very short half-life of only 2 min in humans [69]. We hence decided to investigate if fusion of GLP-1 to (VPKEG)120 could enhance its temporal duration of efficacy.
To that end, we recombinantly expressed a GLP1 -(VPKEG)120 fusion as well as GLP1 -(VPGAG)160 as a control (Figure S2 and S6). We tested the in vitro potency of these two fusions by a cAMP ELISA, [43] as described in the methods section. The half-maximal effective concentration (EC50) of native GLP-1 was 0.3 nM, which is close to that reported elsewhere, [39,71] while GLP1-(VPKEG)120 and GLP1-(VPGAG)160 had EC50’s of 17 nM and 10 nM, respectively (Figure 5A, Table 4). The larger EC50 of the GLP-1 fusions compared to GLP1 is to be expected because macromolecular conjugates of GLP-1, such as an ELP or ZIPP, can sterically hinder the interaction between GLP-1 and its receptor and reduce their potency. This finding is also consistent with the 30–500 lower potency of PEGylated GLP1 relative to the unmodified peptide [72].
Figure 5. GLP1-ZIPP fusions are active in vitro and in vivo.
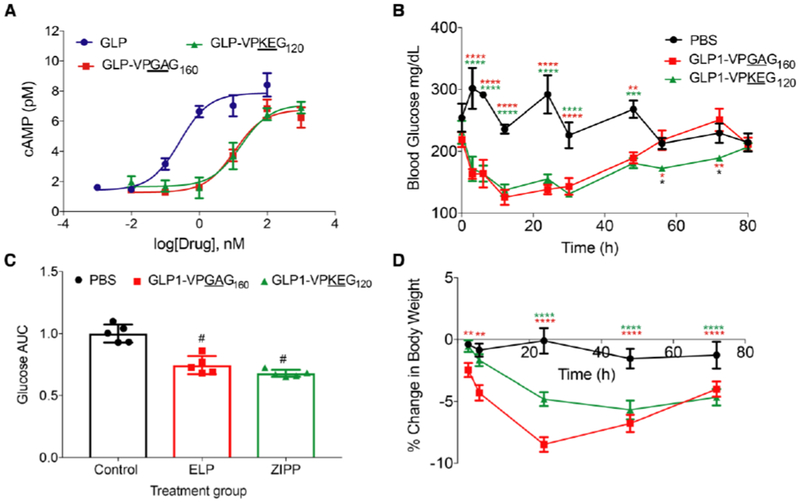
A. cAMP response of native GLP1 and GLPl-(VPKEG)120, GLPl-(VPGAG)160 was tested in BHK cells expressing the GLP-1R (n=3). B. Blood glucose was monitored for 3 days after treating 7-week-old DIO mice (n=5) with a single s.c. injection of GLP1-(VPKEG)120. GLP1-(VPGAG)160 or PBS. C. Glucose AUC was calculated for each mouse up to 72 h and normalized to the mean of the PBS control group. (#p<0.0001, one-way ANOVA and Dunnett’s test). D. Percent body weight change relative to weight at time of injection, t=0 h. For B and D (*p<0.05, **p<0.01, ***p<0.001, ****p< 0.0001 2-way repeated measures ANOVA, Tukey’s post-hoc test).
Table 4.
In vitro activity of GLP1 and GLP1 conjugates
| GLP1 | GLP1-VPGAG160 | GLP1-VPKEG120 | |
|---|---|---|---|
| EC50 (nM)* | 0.07 (0.12 to 0.62) | 10 (5.19 to 22.71) | 17 (6.57 to 46.67) |
Values reported are the mean with 95% CI range in parentheses
3.6. In vivo therapeutic efficacy of GLP1 fusions.
We next examined whether a single s.c. injection of a GLP1-(VPKEG)120 fusion could provide extended temporal reduction of blood glucose compared to an ELP fusion in mice. Seven-week-old diet-induced obese (DIO) mice were treated s.c. with 150 nmol kg−1 of each GLP-1 fusion at 40 μM or were administered an equivalent volume of PBS as a sham control. Blood glucose was monitored throughout the first day and then twice a day for the next 3 days. The most common side effects from GLP-1 agonist therapy are decreased appetite, nausea, vomiting and diarrhea, which result in weight loss. It is believed that this anorexic side-effect is due to peptide’s activity in the hypothalamus [58,59]. The body weight of the treated mice was hence measured daily to monitor these side effects of GLP-1 fusions [73,74].
The ZIPP fusion —GLP1-(VPKEG)120— lowered the blood glucose for up to 72 h. In contrast, the GLP1-(VPGAG)160, uncharged control, only exhibited glucose control for 48 h (Figure 5B). The glucose AUC, which is a measure of total glucose exposure, was calculated for the duration of efficacy (72 h) by integrating the blood glucose concentration over time and was then normalized to the PBS control. The AUC values were significantly lower for both GLP1 fusion groups when compared to the PBS control group (p<0.001, one-way ANOVA and Dunnett’s Test) (Figure 5C). Another relevant finding is that the GLP1-(VPKEG)120 only showed a 4.8% weight loss, whereas GLP1-(VPGAG)160 showed a greater weight loss of about 8.5% within 24 h after injection, presumably because of its bolus release that leads to a sharp spike in drug concentration (Figure 5D) (p<0.0001, 2-way repeated measured ANOVA). This data is consistent with the s.c. pharmacokinetics of the ZIPP and ELP (Table 3, Figure 3B), as the tmax of (VPKEG)120 was ~2-fold longer than the MW-matched ELP control — (VPGAG)160. We believe that the slower absorption profile of (VPKEG)120 reduced the peak concentration of GLP-1 in circulation for GLP1-(VPKEG)120 relative to (VPGAG)160. which in turn reduced nausea, the most common side effect of GLP-1 administration, that leads to reduced food uptake and causes sudden weight loss in mice. Overall, these data show that a zwitterionic polypeptide elongated the temporal window of the efficacy of GLP-1 by one and half fold after a single s.c. injection and significantly outperformed GLP1-VPGAG160 constructs in controlling blood glucose for 3 days with reduced side-effects.
4. Conclusion
In conclusion, this paper tested the hypothesis that incorporation of a zwitterionic motif into the repeat unit of an intrinsically disordered peptide polymer can increase the plasma circulation of the polypeptide. We identified lysine and glutamic acid as the optimal pair of cationic-anionic residues for incorporation into a ZIPP. The optimal ZIPP that contains these two residues as a dipeptide has a plasma exposure and half-life that is two-fold greater than its uncharged control. We also showed the therapeutic utility of the optimal ZIPP in delivery of glucagon-like peptide-1 (GLP1-), a drug that is clinically used to treat type-2 diabetes. The GLP1-ZIPP conjugate reduces blood glucose levels for up to 3 days in a diet induced obesity model of type-2 diabetes in mice after a single s.c. injection, which is 70 times longer than the unmodified drug [32] and one and a half times longer than an uncharged ELP control.
The identification of a ZIPP with optimal PK will allow the development of long-circulating fusion peptides and protein therapeutics by genetically encoded fusion and recombinant expression. The design of the ZIPP wherein the cationic-anionic dipeptide is embedded within a structurally disordered ELP scaffold is important for their clinical translation, as it enables overexpression of ZIPPs —and their fusions with peptide or protein drugs— in E. coli, and allows easy and scalable non-chromatographic purification by ITC, thereby eliminating the need for chromatographic purification that is time-consuming and expensive. The recombinant expression of ZIPP fusions —like all recombinant fusions— also eliminates the need for chemical conjugation of the carrier that is required for synthetic “stealth” polymers such as PEG. Chemical conjugation provides limited control over the site of conjugation and stoichiometry, has a low yield, and requires extensive purification of product from reactants. Finally, the ability to precisely control the sequence and chain length, at the gene level with ZIPP, allows us to fine-tune pharmacokinetics with relative ease, with a precision that is impossible to achieve with the synthetic polymers.
There are also some drawbacks of ZIPPs. Like all recombinant fusions, they are restricted to the N-or C-terminus of peptides and proteins, and they have the potential to be immunogenic. A deeper understanding of the long-term, efficacy, toxicity, and immunogenicity of ZIPPs is hence necessary for their clinical translation as drug carriers and will be addressed in future studies.
Supplementary Material
Acknowledgements
The authors would like to thank the Duke Proteomics Facility and the Chemistry Instrumentation Facility for help with mass spectrometry, Dr. Michael R. Zalutsky, Duke University, for providing laboratory space and equipment for radiolabeling, and Dr. Terrence G. Oas, Duke University, for providing equipment for the circular dichroism studies. S.B. would like to thank Dr. Felipe Garcia Quiroz for insightful discussions on purifying ZIPP constructs, Dr. Kelli M. Luginbuhl for help with the diabetes in vivo study design and Dr. Jayanta Bhattacharya for helpful discussions with in vivo experimental design and assistance with i.v. PK study. S.B. would also like to acknowledge the Pratt School of Engineering at Duke University for support from the Pratt-Gardner Fellowship. This work was supported by the NIH through grant R01-DK091789 to A.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Supporting Information is attached. The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
Conflict of interest:
A.C. is a scientific advisor and serves on the board of directors for PhaseBio Pharmaceuticals, Inc., which has licensed the ELP technology for drug delivery applications from Duke University. The remaining authors declare no competing financial interests.
Contributor Information
Samagya Banskota, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA.
Dr. Parisa Yousefpour, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA
Nadia Kirmani, Department of Biology, Trinity College of Arts and Sciences, Duke University, Durham, NC 27708, USA.
Dr. Xinghai Li, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA
Prof. Ashutosh Chilkoti, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA
References
- [1].Caliceti P, Veronese FM, Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates., Advanced Drug Delivery Reviews. 55 (2003) 1261–77. doi: 10.1016/S0169-409X(03)00108-X. [DOI] [PubMed] [Google Scholar]
- [2].Leader B, Baca QJ, Golan DE, Protein therapeutics: a summary and pharmacological classification., Nature Reviews. Drug Discovery. 7 (2008) 21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- [3].Qi Y, Simakova A, Ganson NJ, Li X, Luginbuhl KM, Ozer I, Liu W, Hershfield MS, Matyjaszewski K, Chilkoti A, A brush-polymer/exendin-4 conjugate reduces blood glucose levels for up to five days and eliminates polyethylene glycol) antigenicity, Nature Biomedical Engineering. 1 (2017)25–29. doi: 10.1038/s41551-016-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alconcel SNS, Baas AS, Maynard HD, FDA-approved poly(ethylene glycol)-protein conjugate drugs, Polymer Chemistry. 2 (2011) 1442. doi: 10.1039/clpy00034a. [DOI] [Google Scholar]
- [5].Harris JM, Martin NE, Modi M, Pegylation: a novel process for modifying pharmacokinetics., Clinical Pharmacokinetics. 40 (2001) 539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- [6].Yang W, Liu S, Bai T, Keefe AJ, Zhang L, Ella-Menye JR, Li Y, Jiang S, Poly(carboxybetaine) nanomaterials enable long circulation and prevent polymer-specific antibody production, Nano Today. 9 (2014) 10–16. doi: 10.1016/j.nantod.2014.02.004. [DOI] [Google Scholar]
- [7].Zhang P, Jain P, Tsao C, Yuan Z, Li W, Li B, Wu K, Hung H, Lin X, Jiang S, Polypeptide with High Zwitterion density for Safe and Effective Therapeutics., Angewandte Chemie (International Ed. in English). (2018). doi: 10.1002/anie.201802452. [DOI] [PubMed] [Google Scholar]
- [8].Keefe AJ, Jiang S, Poly(zwitterionic)protein conjugates offer increased stability without sacrificing binding affinity or bioactivity, Nature Chemistry. 4 (2012) 59–63. doi: 10.1038/nchem.l213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Knop K, Hoogenboom R, Fischer D, Schubert US, Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives., Angewandte Chemie (International Ed. in English). 49 (2010) 6288–308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- [10].Ishida T, Kiwada H, Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes, International Journal of Pharmaceutics. 354 (2008) 56–62. doi: 10.1016/j.ijpharm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- [11].Rudmann DG, Alston JT, Hanson JC, Heidel S, High molecular weight polyethylene glycol cellular distribution and PEG-associated cytoplasmic vacuolation is molecular weight dependent and does not require conjugation to proteins., Toxicologic Pathology. 41 (2013) 970–83. doi: 10.1177/0192623312474726. [DOI] [PubMed] [Google Scholar]
- [12].Veronese FM, Pasut G, PEGylation, successful approach to drug delivery., Drug Discovery Today. 10(2005) 1451–8. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- [13].Uli B, Arne S, Half-Life Extension of Therapeutic Proteins via Genetic Fusion to Recombinant PEG Mimetics, in: Therapeutic Proteins, Wiley-Blackwell, 2012: pp. 63–80. doi: 10.1002/9783527644827.ch4. [DOI] [Google Scholar]
- [14].Kontos S, Hubbell JA, Improving protein pharmacokinetics by engineering erythrocyte affinity, Molecular Pharmaceutics. 7 (2010) 2141–2147. doi: 10.1021/mpl001697. [DOI] [PubMed] [Google Scholar]
- [15].Roberts MJ, Bentley MD, Harris JM, Chemistry for peptide and protein PEGylation., Advanced Drug Delivery Reviews. 54 (2002) 459–76. doi:10.1016/S0169-409X(02)00022-4. [DOI] [PubMed] [Google Scholar]
- [16].Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, Cleland JL, Silverman J, Stemmer WPC, A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner, Nature Biotechnology. 27 (2009) 1186–1190. doi: 10.1038/nbt,1588. [DOI] [PubMed] [Google Scholar]
- [17].Hassouneh W, MacEwan SR, Chilkoti A, Fusions of elastin-like polypeptides to pharmaceutical proteins, 1st ed., Elsevier Inc., 2012. doi: 10.1016/B978-0-12-416039-2.00024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].MacEwan SR, Chilkoti A, Applications of elastin-like polypeptides in drug delivery, Journal of Controlled Release. 190 (2014) 314–330. doi: 10.1016/j.jconrel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nagarsekar A, Ghandehari H, Genetically engineered polymers for drug delivery., Journal of Drug Targeting. 7 (1999) 11–32. doi: 10.3109/10611869909085489. [DOI] [PubMed] [Google Scholar]
- [20].Monfardini C, Veronese FM, Stabilization of substances in circulation., Bioconjugate Chemistry. 9 (1998) 418–50. doi: 10.1021/bc970184f. [DOI] [PubMed] [Google Scholar]
- [21].Kane RS, Deschatelets P, Whitesides GM, Kosmotropes form the basis of protein-resistant surfaces, Langmuir. 19 (2003) 2388–2391. doi: 10.1021/la020737x. [DOI] [Google Scholar]
- [22].Cao Z, Jiang S, Super-hydrophilic zwitterionic poly ( carboxybetaine ) and amphiphilic non-ionic poly ( ethylene glycol ) for stealth nanoparticles, (2012) 404–413. [Google Scholar]
- [23].Ladd J, Zhang Z, Chen S, Hower JC, Jiang S, Nonspecific Protein Adsorption from Human Serum and Plasma, Interface. 9 (2008). doi: 10.1021/bm701301s. [DOI] [PubMed] [Google Scholar]
- [24].Bhattacharjee S, Liu W, Wang WH, Weitzhandler I, Li X, Qi Y, Liu J, Pang Y, Hunt DF, Chilkoti A, Site-Specific Zwitterionic Polymer Conjugates of a Protein Have Long Plasma Circulation, ChemBioChem. 16 (2015)2451–2455. doi: 10.1002/cbic.201500439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen S, Cao Z, Jiang S, Ultra-low fouling peptide surfaces derived from natural amino acids., Biomaterials. 30 (2009) 5892–6. doi: 10.1016/j.biomaterials.2009.07.001. [DOI] [PubMed] [Google Scholar]
- [26].Keefe AJ, Caldwell KB, Nowinski AK, White AD, Thakkar A, Jiang S, Screening nonspecific interactions of peptides without background interference., Biomaterials. 34 (2013) 1871–7. doi: 10.1016/j.biomaterials.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu EJ, Sinclair A, Keefe AJ, Nannenga BL, Coyle BL, Baneyx F, Jiang S, EKylation: Addition of an Alternating-Charge Peptide Stabilizes Proteins., Biomacromolecules. 16 (2015) 3357–61. doi: 10.1021/acs.biomac.5b01031. [DOI] [PubMed] [Google Scholar]
- [28].Tang NC, Chilkoti A, Combinatorial codon scrambling enables scalable gene synthesis and amplification of repetitive proteins, Nature Materials. 15 (2016) 419–424. doi: 10.1038/nmat4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rudorf S, Lipowsky R, Protein Synthesis in E. coli: Dependence of Codon-Specific Elongation on tRNA Concentration and Codon Usage., PloS One. 10 (2015) e0134994. doi: 10.1371/journal.pone.0134994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hassouneh W, Christensen T, Chilkoti A, Elastin-like polypeptides as a purification tag for recombinant proteins, Current Protocols in Protein Science. (2010) 1–20. doi: 10.1002/0471140864.ps0611s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Asai D, Xu D, Liu W, Quiroz F. Garcia, Callahan DJ, Zalutsky MR, Craig SL, Chilkoti A, Protein polymer hydrogels by in situ, rapid and reversible self-gelation., Biomaterials. 33 (2012) 5451–8. doi: 10.1016/j.biomaterials.2012.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Amiram M, Luginbuhl KM, Li X, Feinglos MN, Chilkoti A, A depot-forming glucagon-like peptide-1 fusion protein reduces blood glucose for five days with a single injection, Journal of Controlled Release. 172 (2013) 144–151. doi: 10.1016/j.jconrel.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McDaniel JR, MacKay JA, Quiroz FG, Chilkoti A, Recursive Directional Ligation by Plasmid Reconstruction allows Rapid and Seamless Cloning of Oligomeric Genes, Biomacromolecules. 11 (2010) 944–952. doi: 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacEwan SR, Hassouneh W, Chilkoti A, Non-chromatographic Purification of Recombinant Elastin-like Polypeptides and their Fusions with Peptides and Proteins from Escherichia coli, Journal of Visualized Experiments. (2014) e51583. doi: 10.3791/51583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meyer DE, Chilkoti A, Purification of recombinant proteins by fusion with thermally-responsive polypeptides., Nature Biotechnology. 17 (1999) 1112–5. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- [36].Micsonai A, Wien F, Bulyaki E, Kun J, Moussong E, Lee Y-H, Goto Y, Refregiers M, Kardos J, BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra., Nucleic Acids Research. 46 (2018) W315–W322. doi: 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Micsonai A, Wien F, Kernya L, Lee Y, Goto Y, Refregiers M, Kardos J, Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy., Proceedings of the National Academy of Sciences of the United States of America. 112 (2015) E3095–103. doi: 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jonsson A, Dogan J, Herne N, Abrahmsen L, Nygren P-A, Engineering of a femtomolar affinity binding protein to human serum albumin., Protein Engineering, Design & Selection : PEDS. 21 (2008) 515–27. doi: 10.1093/protein/gzn028. [DOI] [PubMed] [Google Scholar]
- [39].Luginbuhl KM, Schaal JL, Umstead B, Mastria EM, Li X, Banskota S, Arnold S, Feinglos M, D’Alessio D, Chilkoti A, One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer., Nature Biomedical Engineering. 1 (2017). doi: 10.1038/s41551-017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Burcelin R, Dolci W, Thorens B, Long-lasting antidiabetic effect of a dipeptidyl peptidase IV-resistant analog of glucagon-like peptide-1., Metabolism: Clinical and Experimental. 48 (1999) 252–8. doi: 10.1016/S0026-0495(99)90043-4. [DOI] [PubMed] [Google Scholar]
- [41].Miranda LP, Winters KA, Gegg CV, Patel A, Aral J, Long J, Zhang J, Diamond S, Guido M, Stanislaus S, Ma M, Li H, Rose MJ, Poppe L, Veniant MM, Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity., Journal of Medicinal Chemistry. 51 (2008) 2758–65. doi: 10.1021/jm701522b. [DOI] [PubMed] [Google Scholar]
- [42].Amiram M, Luginbuhl KM, Li X, Feinglos MN, Chilkoti A, Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control., Proceedings of the National Academy of Sciences of the United States of America. 110 (2013) 2792–7. doi: 10.1073/pnas.1214518110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baggio LL, Huang Q, Brown TJ, Drucker DJ, A recombinant human glucagon-like peptide (GLP)-l-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis., Diabetes. 53 (2004) 2492–500. http://www.ncbi.nlm.nih.gov/pubmed/15331566. [DOI] [PubMed] [Google Scholar]
- [44].Schaal JL, Li X, Mastria E, Bhattacharyya J, Zalutsky MR, Chilkoti A, Liu W, Injectable polypeptide micelles that form radiation crosslinked hydrogels in situ for intratumoral radiotherapy., Journal of Controlled Release : Official Journal of the Controlled Release Society. 228 (2016) 58–66. doi: 10.1016/j.jconrel.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Surwit RS, Kuhn CM, Cochrane C, McCubbin J. a., Feinglos MN, Diet-induced type II diabetes in C57BL/6J mice., Diabetes. 37 (1988) 1163–7. doi: 10.2337/diabetes.37.9.1163. [DOI] [PubMed] [Google Scholar]
- [46].Winzell MS, Ahren B, The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes., Diabetes. 53 Suppl 3 (2004) S215–9. doi: 10.2337/diabetes.53.suppl_3.S215. [DOI] [PubMed] [Google Scholar]
- [47].Quiroz FG, Chilkoti A, Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers, Nature Materials. 14 (2015) 1164–1171. doi: 10.1038/nmat4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rauscher S, Baud S, Miao M, Keeley FW, Pomes R, Proline and glycine control protein selforganization into elastomeric or amyloid fibrils., Structure (London, England : 1993). 14 (2006) 1667–76. doi: 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- [49].van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM, Classification of intrinsically disordered regions and proteins., Chemical Reviews. 114 (2014) 6589–631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Galea CA, High AA, Obenauer JC, Mishra A, Park C-G, Punta M, Schlessinger A, Ma J, Rost B, Slaughter CA, Kriwacki RW, Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome., Journal of Proteome Research. 8 (2009)211–26. doi: 10.1021/pr800308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Muiznieks LD, Keeley FW, Proline periodicity modulates the self-assembly properties of elastin-like polypeptides, Journal of Biological Chemistry. 285 (2010) 39779–39789. doi: 10.1074/jbc.M110.164467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fandrich M, Dobson CM, The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation, EMBO Journal. 21 (2002) 5682–5690. doi: 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gilroy CA, Roberts S, Chilkoti A, Fusion of fibroblast growth factor 21 to a thermally responsive biopolymer forms an injectable depot with sustained anti-diabetic action., Journal of Controlled Release : Official Journal of the Controlled Release Society. 277 (2018) 154–164. doi: 10.1016/j.jconrel.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Georgiev GS, Kamenska EB, Vassileva ED, Kamenova IP, Georgieva VT, Iliev SB, Ivanov IA, Self-assembly, antipolyelectrolyte effect, nonbiofouling properties of polyzwitterions, Biomacromolecules. 7 (2006) 1329–1334. doi: 10.1021/bm050938q. [DOI] [PubMed] [Google Scholar]
- [55].Delgado JD, Schlenoff JB, Static and Dynamic Solution Behavior of a Polyzwitterion Using a Hofmeister Salt Series, Macromolecules. 50 (2017) 4454–4464. doi: 10.1021/acs.macromol.7b00525. [DOI] [Google Scholar]
- [56].Lowe AB, McCormick CL, Synthesis and solution properties of zwitterionic polymers, Chemical Reviews. 102 (2002) 4177–4189. doi: 10.1021/cr020371t. [DOI] [PubMed] [Google Scholar]
- [57].Kontermann RE, Half-Life Modulating Strategies-An Introduction, in: Therapeutic Proteins, Wiley-VCH Verlag GmbH & Co. KGaA, 2012: pp. 1–21. doi: 10.1002/9783527644827.chl. [DOI] [Google Scholar]
- [58].Ruggiero A, Villa CH, Bander E, Rey DA, Bergkvist M, Batt CA, Manova-Todorova K, Deen WM, Scheinberg DA, McDevitt MR, Paradoxical glomerular filtration of carbon nanotubes, Proceedings of the National Academy of Sciences. 107 (2010) 12369–12374. doi: 10.1073/pnas.0913667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jambhekar S, Breen P, Basic Pharmacokinetics, 2009.
- [60].Baud F, Karlin S, Measures of residue density in protein structures., Proceedings of the National Academy of Sciences of the United States of America. 96 (1999) 12494–9. doi: 10.1073/pnas.96.22.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moon CP, Fleming KG, Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers, Proceedings of the National Academy of Sciences. 108 (2011) 10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Acids A, Dougherty D. a, Cation-p Interactions Involving Aromatic, The Journal of Nutrition. 137(2007) 1504–1508. [DOI] [PubMed] [Google Scholar]
- [63].White AD, Keefe AJ, Ella-Menye J-R, Nowinski AK, Shao Q, Pfaendtner J, Jiang S, Free energy of solvated salt bridges: a simulation and experimental study., The Journal of Physical Chemistry. B. 117 (2013) 7254–7259. doi: 10.1021/jp4024469. [DOI] [PubMed] [Google Scholar]
- [64].MacEwan SR, Chilkoti A, Digital switching of local arginine density in a genetically encoded self-assembled polypeptide nanoparticle controls cellular uptake., Nano Letters. 12 (2012) 3322–8. doi: 10.1021/nl301529p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].MacEwan SR, Chilkoti A, Controlled apoptosis by a thermally toggled nanoscale amplifier of cellular uptake., Nano Letters. 14 (2014) 2058–64. doi: 10.1021/nl5002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yamaoka T, Tabata Y, Ikada Y, Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice, Journal of Pharmaceutical Sciences. 83 (1994) 601–606. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- [67].Podust VN, Balan S, Sim BC, Coyle MP, Ernst U, Peters RT, Schellenberger V, Extension of in vivo half-life of biologically active molecules by XTEN protein polymers, Journal of Controlled Release. 240 (2016) 52–66. doi: 10.1016/j.jconrel.2015.10.038. [DOI] [PubMed] [Google Scholar]
- [68].Longley CB, Zhao H, Lozanguiez YL, Conover CD, Biodistribution and excretion of radiolabeled 40 kDa polyethylene glycol following intravenous administration in mice., Journal of Pharmaceutical Sciences. 102 (2013)2362–70. doi: 10.1002/jps.23506. [DOI] [PubMed] [Google Scholar]
- [69].Meloni AR, DeYoung MB, Lowe C, Parkes DG, GLP-1 receptor activated insulin secretion from pancreatic β-cells: mechanism and glucose dependence., Diabetes, Obesity & Metabolism. 15 (2013) 15–27. doi: 10.111l/j,1463-1326.2012.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Drucker DJ, Glucagon-like peptide-1 and the islet beta-cell: augmentation of cell proliferation and inhibition of apoptosis., Endocrinology. 144 (2003) 5145–8. doi: 10.1210/en.2003-1147. [DOI] [PubMed] [Google Scholar]
- [71].Runge S, Schimmer S, Oschmann J, Schiodt CB, Knudsen SM, Jeppesen CB, Madsen K, Lau J, Thogersen H, Rudolph R, Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain., Biochemistry. 46 (2007) 5830–40. doi: 10.1021/bi062309m. [DOI] [PubMed] [Google Scholar]
- [72].Chae SY, Chun GY, Lee S, Jin CH, Lee ES, Lee KC, Youn YS, Pharmacokinetic and pharmacodynamic evaluation of site-specific pegylated glucagon-like peptide-1 analogs as flexible postprandial-glucose controllers, Journal of Pharmaceutical Sciences. 98 (2009) 1556–1567. doi: 10.1002/jps.21532. [DOI] [PubMed] [Google Scholar]
- [73].Prasad-Reddy L, Isaacs D, A clinical review of GLP-1 receptor agonists: Efficacy and safety in diabetes and beyond, Drugs in Context. 4 (2015) 1–19. doi: 10.7573/dic.212283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Thiele TE, Van Dijk G, Campfield LA, Smith FJ, Bum P, Woods SC, Bernstein IL, Seeley RJ, Central infusion of GLP-1, but not leptin, produces conditioned taste aversions in rats., The American Journal of Physiology. 272 (1997) R726–R730. doi: 10.1152/ajpregu.l997.272.2.R726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.