

Abstract
Numerous variant alleles are associated with human acute myeloid leukemia (AML). However, the same variants are also found in individuals with no hematological disease, making their functional relevance obscure. Through NOD.Cg-Prkdcscid//2rgtmlWjl/Sz (NSG) xenotransplantation, we functionally identified pre-leukemic and leukemic stem cell populations present in FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD)+ AML patient samples. By single-cell DNA sequencing, we identified clonal structures and linked mutations with in vivo fates, distinguishing mutations permissive of non-malignant multilineage hematopoiesis from leukemogenic mutations. Although multiple somatic mutations co-existed at the single-cell level, inhibition of the mutation strongly associated with pre-leukemic to leukemic stem cell transition eliminated AML in vivo. Moreover, concurrent inhibition of BCL-2 uncovered a critical dependence of resistant AML cells on anti-apoptotic pathways. Co-inhibition of pathways critical for oncogenesis and survival may be an effective strategy that overcomes genetic diversity in human malignancies. This approach incorporating single-cell genomics with the NSG patient-derived xenograft (PDX) model may serve as a broadly applicable resource for precision target identification and drug discovery.
One-sentence summary:
Targeting leukemogenic mutations identified by functional single cell genomics eliminated human AML in vivo.
Introduction
Acute myeloid leukemia (AML) is a biologically and clinically heterogeneous entity. Recent studies using deep DNA and RNA sequencing have demonstrated intra-patient heterogeneity 1-4). Mutations in genes such as NPM1, TET2, WT1, IDH1/2, and DNMT3A are commonly found in AML(5-8), and next-generation DNA sequencing (NGS) suggests that certain mutations occur earlier than others based on variant allele frequencies (VAFs) (9-11). Pre-leukemic stem cells carrying these early somatic mutations may contribute to leukemogenesis and disease relapse(9, 10). On the other hand, large-scale population-based sequencing studies have revealed that hematopoietic cells in 5-18.4% of elderly subjects with non-malignant conditions such as diabetes mellitus and cardiovascular diseases harbored somatic mutations in genes including ASXL1, DNMT3A, and TET2(12-14). In these subjects, the mutations were associated with a 0.5-1% annual rate of progression to hematological malignancies. This raised some fundamental questions regarding leukemogenesis and treatment strategies. First, which among AML-associated recurrent mutations contribute to leukemogenesis? Second, which of the many mutations and pathways must be targeted for greatest clinical efficacy? To address these questions, we examined mutational profiles of phenotypically and functionally defined human AML cell populations to link mutations with in vivo fates. We then used a functional single-cell genomic approach to identify critical targets allowing in vivo elimination of human AML cells with multiple co-existing mutations.
Results
In vivo fates of human AML cells are linked with distinct mutational profiles through NSG xenotransplantation
We obtained bone marrow (BM) or peripheral blood (PB) samples from 27 patients with FLT3-ITD+AML (table S1). The majority of patients had poor prognostic factors such as complex chromosomal abnormalities in addition to FLT3-ITD mutation and/or had known aggressive disease (for example, primary resistance or relapse after multiple stem cell transplantations). Because AML-associated hematopoiesis consists of both normal and malignant cells, we profiled patterns of recurrent mutations in patient-derived cell populations purified according to cell surface phenotype that defines hematopoietic stem cells (HSCs), multipotent progenitor cells (MPPs), multi-lymphoid progenitors (MLPs), as well as mature lymphoid and myeloid cells (15). To link these mutations with in vivo fates, we transplanted the cell populations in newborn NSG mouse recipients (Fig. 1). If human lymphoid as well as myeloid subsets engrafted in NSG recipients (multilineage human hematopoietic repopulation), the transplanted subpopulation contained normal HSCs and/or pre-leukemic stem cells. If NSG recipients developed leukemia with uncontrolled proliferation of myeloid blasts and without lymphoid differentiation, the transplanted subpopulation contained leukemia-initiating cells (LICs). As expected, frequencies of hematopoietic subpopulations and their population-level mutational profiles varied among patients, and frequencies of mutated alleles varied among subpopulations within individual patients (representative data from six patients in Fig. 2 and fig. S1; sequence information in table S2). Upon transplantation, subpopulations with similar surface phenotypes isolated from different patients showed distinct behaviors in vivo. For instance, the Patient 21-derived CD34+CD38-CD90-CD45RA-cell population initiated AML in NSG mice and therefore contained LICs (Fig. 2A). In contrast, in Patients 20, 23, and 24, the CD34+CD38-CD90-CD45RA- cell population reconstituted multilineage human hematopoiesis in NSG mice and therefore contained multilineage-engrafting HSCs or pre-leukemic stem cells, whereas LICs were present in the CD34+CD38-CD90-CD45RA+ cell population (Fig. 2B, fig. S1, A and B). In Patient 13, the CD34+CD38- population reconstituted multilineage human hematopoiesis, and the CD34-CD33+ population contained LICs, whereas Patient 1-derived CD34+CD38- cells initiated AML in vivo (Fig. 2C, fig. S1C). These observations are consistent with recent reports showing variable cell surface phenotype of LICs (16). We next examined mutational profiles in these subpopulations with defined in vivo fates. In Patient 20, the same set of mutations (FLT3-ITD, DNMT3A, WT1) was present in CD34+CD38-CD90-CD45RA- and CD34+CD38-CD90-CD45RA+ subpopulations, but these subpopulations showed distinct in vivo fates (Fig. 2B). This functional difference may be due to uneven distribution of mutations identified in bulk AML cell populations in single-cell clones, resulting in disparate combinations of mutations and divergent in vivo fates. Therefore, we performed single-cell DNA sequencing of functionally-defined pre-leukemic stem cell- and LIC-containing subpopulations along with human multilineage hematopoietic cells and leukemia cells they generate in vivo, to define clonal structures and to identify mutation(s) associated with leukemia-initiating versus multilineage-engrafting function.
Figure 1.

Mutational profile defines in vivo fates of AML patient-derived cells
1. Somatic mutation profiles were identified in patient cell subpopulations defined by surface phenotype based on developmental hierarchy of human hematopoiesis. 2. In vivo fate of each subpopulation was determined through transplantation into newborn NSG mice. If repopulation by multilineage hematopoiesis occurred, the transplanted subpopulation contained hematopoietic stem cells or pre-leukemic stem cells; if AML engraftment occurred, the transplanted subpopulation contained LICs.
Figure 2.

Mutational profiles and in vivo engraftment of patient-derived subpopulations
Three additional patients are shown in fig. S1. In each patient sample, human CD45+CD3+CD19- T and human CD45+CD3-CD19+ B cells were identified. Within CD3-CD19- non-T non-B cells, subpopulations were identified based on CD34, CD38, CD90, and CD45RA surface expression. These populations underwent PCR for FLT3-ITD mutation and DNA sequencing for the other genes indicated. Variant allele frequencies are shown as heat maps. In Patient 21 (A) and Patient 20 (B), the CD34+CD38-CD90-CD45RA- and CD34+CD38-CD90-CD45RA+ subpopulations were identified. The in vivo fates of CD34+CD38-CD90-CD45RA- subpopulations differed between patients 20 and 21, showing engraftment with multilineage human hematopoiesis in Patient 20 (indicated by green rectangles) but initiation of AML in Patient 21 (indicated by red rectangles). In Patient 13 (C), the CD34+CD38- subpopulation showed multilineage repopulation, whereas CD34-CD33+ subpopulation with additional FLT3-ITD and NPM1 mutations initiated AML. AML engrafted recipients showed no B cell engraftment (indicated by gray dashed outlines on flow cytometry plots). Detailed information on variants found in each patient is shown in table S2.
Functional genomic approach combining PDX model and single cell DNA sequencing distinguishes leukemogenic from permissive mutations
We examined DNMT3A, TET2, NPM1, and WT1 mutations and FLT3-ITD among single cells isolated from multilineage-engrafting cell- and LIC-containing patient-derived populations and their in vivo progeny (Fig. 3A). Patient-derived multilineage-engrafting pre-leukemic stem cells showed mutational heterogeneity at the single-cell level, carrying combinations of multiple mutations (Fig. 3B,C). DNMT3A mutation was identified both in multilineage-engrafting CD34+CD38-CD90-CD45RA-pre-leukemic cells (Patient 20 and Patient 24) and AML-initiating CD34+CD38-CD90-CD45RA- cells (Patient 21) at single-cell level (Fig. 3B). In vivo generated single B cells from Patient 20- and Patient 24-derived pre-LSCs harbored DNMT3A mutation (and WT1 mutation in the case of Patient 20), whereas there were no FLT3-mutated B cell clones. There were no FLT3-ITD mutations in Patient 24-derived pre-leukemic stem cells and in vivo generated single B cells. Although there were FLT3-ITD-mutated single cells in the Patient 20-derived pre-leukemic stem cell population, those FLT3-ITD-mutated subclones did not contribute to normal lymphopoiesis. These findings indicate that DNMT3A and WT1 mutations are permissive and can co-exist in a single cell without hindering human multilineage differentiation. In contrast, FLT3-ITD was identified in substantial proportions of Patient 21-, Patient 20-, and Patient 24-derived LICs and engrafted AML cells at single-cell level. Likewise, CD34+CD38- cells derived from Patient 13 and Patient 1 exhibited distinct in vivo fates: At the single-cell level, the former carried WT FLT3, and the latter harbored FLT3-ITD (Fig. 3C). It is notable that, though FLT3-ITD+ single cells were a minority among the LIC population in Patient 13, every engrafted AML cell harbored FLT3-ITD mutation. In addition, FLT3-ITD+ single cells were enriched among LIC-containing CD34-CD33+ population at the time of relapse in Patient 13. These findings suggest that acquisition of the FLT3-ITD mutation acts as a critical trigger for leukemia initiation, working in cooperation with accumulated mutations in DNMT3A, TET2, NPM1, and/or WT1.
Figure 3.

Mutations contributing to distinct in vivo cell fates identified by single-cell functional genomics
(A) Patient-derived subpopulations with defined in vivo fates and their in vivo progeny underwent single-cell mutation profiling. Through this strategy, mutations present in patient-derived pre-leukemic stem cells and LIC clones were tracked and linked to in vivo fates. (B,C) Using samples from five patients, patient-derived multilineage-engrafting and LIC-containing population and engrafted B cells and AML cells were subjected to single-cell DNA sequencing for variants detected in each indicated gene in each patient. FLT3-ITD sequences with highly variable repeated sequence patterns were detected by single-cell PCR. In (B) and (C), each column of rectangles represents an individual cell. The presence or absence of mutations in each gene is shown by colors of rectangles as indicated.
Kinase inhibition effectively targets human AML with mutational diversity
Therapeutic efficacy of targeting such a mutation among multiple co-existing mutations is an important question for clinical translation. We addressed this by in vivo inhibition of the FLT3 kinase pathway in a NSG PDX model. To serve as a realistic platform for in vivo therapeutic testing, a PDX model must reflect the mutational diversity of human AML cells. At the single-cell level, we detected AML cells with various patterns of mutations in engrafted recipients. In addition, engrafted AML cells retained the mutations present in patient-derived LIC-containing cells from 12 patients examined, indicating that engrafted AML cells reflect mutational diversity of patient-derived AML cells (Fig. 4). Therefore, we went on to examine the effect of FLT3 pathway inhibition in human AML cells with multiple co-existing mutations by using RK-20449, a pyrrolo-pyrimidine-derivative inhibitor of Src family kinase HCK and FLT3 that we had previously identified (17). By treating 56 NSG mice engrafted with FLT3-ITD+ AML from 19 patients, we found significant responses to single agent RK-20449 in vivo in all 19 cases (data and associated p values in table S3). For five patients, RK-20449 completely eliminated AML cells in the BM, spleen, and PB of all recipient mice treated, despite the presence of mutations not directly targeted by RK-20449 (Patient 1: DNMT3A, NPM1, TET2; Patient 2: IDH1; Patient 16: FLT3 D835H point mutation, NRAS) (Fig. 5A, table S2). For 11 additional patients, RK-20449 treatment alone resulted in complete responses in the spleen in a majority of recipient mice tested (Fig. 5B). Although statistically significant (P<0.05) treatment effects were observed in group-wise comparisons, residual RK-20449-resistant AML cells were present in the BM of at least one treated mouse for 14 of 19 patients (Fig. 5, B and C).
Figure 4.

Profiles of AML-associated mutations in nine genes in patient- and recipient-derived AML cells from twelve AML cases
Variant allele frequencies of indicated genes are represented as heat maps. P, LIC-containing population from the patient; 1’ and 2’, human AML cells from primary and secondary recipients, respectively. All patient-derived and recipient-derived leukemia populations were positive for FLT3-ITD by PCR. Non-ITD FLT3 mutations were identified by sequencing. Information on variants is shown in table S2.
Figure 5.

Induction of apoptosis via enhanced BCL-2 dependence in FLT3-ITD+ AML cells with diverse co-existing somatic mutations by in vivo kinase inhibition
Overall, treatment with RK-20449 resulted in significant reduction of AML cells in BM, spleen, and PB of recipients (p < 1E-19 for each; data tabulated in table S3). To document patient-to-patient variability, RK-20449 responses were classified as follows: Complete response (A), if all recipients treated showed residual BM human CD45+ chimerism < 5%; good response (B), if the case did not meet the criterion for complete responder but all recipients showed < 50% residual BM human CD45+; partial response (C), if at least one of the recipients showed greater than 50% residual BM human CD45+. For each response group, PB time-course of human AML chimerism (leftmost panels) for RK-20449 treated recipients and final BM (middle panels) and spleen (rightmost panels) human AML chimerism of RK-20449 treated and untreated recipients are shown. Pre-treatment PB human AML cell chimerism is shown at week 0. The numbers of recipients for each patient/each treatment group and pre-/post-treatment AML chimerism are shown in table S3. In all response groups, BM, spleen, and PB chimerism was significantly reduced with RK-20449 treatment. For BM and spleen, final chimerism for recipients in each treatment group was compared. For PB, pre- and post-treatment chimerism for RK-20449-treated recipients was compared. p < 5.8E-5 by two-tailed t-test for all comparisons. In each scatter graph, dotted lines are drawn at 50%, 5%, and 0%.
(D) Dynamics of apoptotic response to BIM peptide in the presence of RK-20449 was measured for six AML cases. Bars represent the BIM IC50 of cytochrome C loss in RK-20449-treated human CD45+ cells as percentages of IC50 in cells treated with DMSO alone. Enhancement of apoptotic response to BIM by RK-20449 showed substantial patient-to-patient variability.
(E) Dynamics of apoptotic response to BH3-only peptides BAD, HRK, and NOXA in the presence of RK-20449 or DMSO alone was measured for seven AML cases. Bars represent the percentage reduction of IC50 in the presence of RK-20449 compared with DMSO alone. RK-20449 enhanced apoptotic response to BAD and HRK peptides with substantial patient-to-patient variability, whereas apoptotic response to NOXA peptide was not substantially enhanced by RK-20449.
(F) Apoptotic response to a BCL-2 inhibitor ABT-199 was enhanced by RK-20449 in seven AML cases. Bars show increased cytochrome C loss in human AML cells treated with ABT-199 and RK-20449 compared with DMSO alone-treated human AML cells.
BCL-2 inhibition enhances mitochondrial priming to apoptosis induced by kinase inhibition
Resistance to RK-20449 may be mediated through pre-existing or newly acquired somatic mutations (18). However, enrichment or new acquisition of AML-associated somatic mutations was not identified in five AML cases after in vivo RK-20449 treatment (fig. S2). In addition, WT FLT3 gene was not identified in resistant cells, indicating that emergence of FLT3 WT cells is not a substantial mechanism for resistance. This is consistent with mutational profiles of patient samples serially obtained at primary presentation and at relapse (fig. S3). In six cases examined, neither emergence of FLT3 WT cells nor substantial increase in frequencies of somatic mutations were detected, with the exception of FLT3 D835H mutation+ cells in Patient 16 emerging at the time of relapse. In vivo RK-20449 treatment resulted in transcriptional up-regulation of S100A8 (associated with drug resistance in leukemia), HSPA5 (promotes cell survival under endoplasmic reticulum stress and suppresses ferroptosis), and IFI6 (negatively regulates apoptosis) (fig. S4) (19-23). Therefore, we functionally assessed dependence on anti-apoptotic mechanisms in RK-20449-resistant human AML cells by dynamic BH3 profiling (24). Some human malignancies are dependent on specific anti-apoptotic proteins for survival and therefore are sensitive to small molecule antagonists of those proteins (25-29). Dynamic BH3 profiling determines how “primed” cells are to apoptotic cell death and how changing conditions (such as exposure to drugs) affect baseline priming by quantifying mitochondrial cytochrome C release in response to BH3-only peptides that activate pro-apoptotic effectors BAX and BAK. Despite patient-to-patient variability, RK-20449 treatment lowered the IC50 of BIM peptide for mitochondrial cytochrome C release, indicating enhanced pro-apoptotic signaling in FLT3-ITD+ human AML cells (Fig. 5D). This was consistent with our finding that RK-20449 alone completely eliminated AML cells with DNMT3A, TET2, IDH1, and/or WT1 mutations. In addition, exposure to RK-20449 facilitated mitochondrial cytochrome C release in response to BAD and HRK peptides, indicating enhanced mitochondrial sensitivity to BCL-2 and BCL-XL inhibition (Fig. 5E). In addition, dynamic BH3 profiling showed that a selective BCL-2 inhibitor, ABT-199, enhanced RK-20449-induced apoptosis in FLT3-ITD+ human AML cells (Fig. 5F). This suggested that co-inhibition of anti-apoptotic signal, specifically BCL-2, might result in more robust eradication of human AML with diverse co-existing mutations.
Combined inhibition of kinase and anti-apoptosis pathways eliminates AML with genetic complexity and clonal diversity
To examine whether combining RK-20449 and ABT-199 eliminates resistant AML cells, we chose cases that were not completely eradicated in vivo by RK-20449 alone. In the majority of cases, BCL-2 inhibitor ABT-199 alone resulted in transient and limited responses. In contrast, in all 12 cases, combination treatment significantly reduced human AML chimerism in PB, BM, and spleen (Fig. 6, A and B, tables S4-S6; p<0.05 in all comparisons). Combination treatment completely eliminated human AML cells in vivo without targeting co-existing mutations in 9 of 12 cases (Fig. 6B; Cases 3, 5, 6, 7, 8, 9, 10, 13, and 14). We compared residual leukemia-initiating capacity by transplanting a pre-determined fraction of viable human CD45+ AML cells from treated mice into secondary NSG recipients (Fig. 6C). We found that BM treated with RK-20449 alone or ABT-199 alone contained enough viable LICs to initiate AML in every secondary recipient transplanted, whereas AML cells remaining after combination treatment engrafted in only one of 23 mice, indicating that combination treatment more effectively reduced the frequency of LICs in vivo (Fig. 6C). On the other hand, combination treatment did not have significant effects on human leucocytes, T cells, B cells, and myeloid cells in human CB HSC-engrafted NSG recipients (table S7).
Figure 6.

Eradication of FLT3-ITD+ AML cells in vivo through combined inhibition of kinase and anti-apoptotic pathways
(A,B) Mice engrafted with AML derived from 12 patients associated with good or partial responses to RK-20449 alone received four different treatments (no treatment, ABT-199 alone, RK-20449 alone, combination). The numbers of recipients for each patient/each treatment group and pre-/post-treatment AML chimerism are shown in table S4. (A) PB human CD45+ chimerism is shown over time. Recipients were phlebotomized weekly, and pre-treatment PB human CD45+ AML chimerism is shown at time 0. Mean PB human CD45+ chimerism for each patient/each treatment group and statistics comparing treatment groups are shown in table S5.
(B) Final BM and spleen human AML chimerism is shown for mice engrafted with AML derived from 12 patients. 9 cases showed complete responses and 3 cases showed good responses to combination treatment. Each circle represents an AML-engrafted recipient. Mean BM/spleen human CD45+ chimerism for each patient/each treatment group and statistics comparing treatment groups are shown in table S6. In each scatter graph, dotted lines are drawn at 50%, 5%, and 0%.
(C) Residual human AML initiation capacity of human CD45+ cells after in vivo treatment was assessed by serial transplantation for four treatment groups. To compare the amount of residual LICs in recipients after in vivo treatment, each secondary recipient was transplanted with human CD45+ cells sorted from 2.5% (by cell number) of viable BM cells remaining in treated recipients. Mean and SEM for human CD45+ AML cell chimerism in the BM of secondary recipients are shown. Each circle represents a secondary recipient.
Discussion
Multilineage-engrafting pre-leukemic HSCs carrying somatic mutations are thought to be poised for leukemogenesis and may act as a reservoir for leukemic progression and relapse (9, 10). To prevent such events, early-occurring somatic mutations or founder mutations with high variant allele frequencies have been considered as critical therapeutic targets (30). However, some of these somatic mutations were found in individuals with advanced age with no apparent hematological disease, associated with a relatively low rate of progression to hematological malignancies (Fig. S5 upper left) (31). In this study, we demonstrated that contribution of these somatic mutations to normal hematopoietic differentiation or to leukemogenesis could inform therapeutic target selection in AML. By integrating population- and single-cell-level genomics and in vivo functional assessment in PDXs, we found that relatively late acquisition of FLT3-ITD on the background of permissive earlier-occurring mutations in DNMT3A, TET2, NPM1, and WT1, alone or in combination, triggered in vivo leukemogenesis (Fig. S5, upper right). Moreover, acquisition of FLT3-ITD triggered leukemogenesis along a broad spectrum of hematopoietic differentiation. Through single-cell sequencing, we found substantial mutational heterogeneity in patient-derived pre-leukemic cells and NSG-engrafted human lymphoid cells, whereas patient-derived LICs were less mutationally heterogeneous at the single-cell level, with substantial enrichment of FLT3-ITD+ single cells. This is consistent with the hypothesis that late acquisition of FLT3-ITD mutation results in selective clonal expansion. Single-cell RNA sequencing may help fully characterize clonal structures of pre-leukemic and leukemic stem cells.
In a recent clinical study, midostaurin, a small molecule inhibitor of FLT3, improved both 5-year event-free and overall survivals in FLT3-ITD+ AML patients when combined with standard chemotherapy, making it the first FDA-approved targeted agent in AML (32-34). Our findings are consistent with this clinical finding: Targeting FLT3-ITD alone resulted in effective elimination of AML in vivo despite co-existing earlier-occurring mutations (Fig. S5, lower left and right). Furthermore, dynamic BH3 profiling identified an additional target, BCL-2, in kinase inhibitor-resistant AML cells. Co-inhibition of BCL-2 resulted in complete eradication of human AML cells resistant to kinase inhibition. It is particularly notable that this approach was effective in cases with poor prognoses and highly aggressive clinical courses, multiple co-existing mutations, and/or complex chromosomal abnormalities. With the advent of high-throughput sequencing technologies and the genomic information gained from each patient, precision medicine is becoming more and more feasible. By functionally connecting genomic information with in vivo fate and behavior of patient-derived cells at the level of single cells through a PDX model, we could identify therapeutic targets with improved precision to promote more effective drug discovery in genetically complex human malignancies.
Materials and Methods
Study design
The overall objective of this study was to explore strategies for eliminating FLT3-ITD+ human AML by 1) tracing profiles of known somatic mutations associated with AML and other malignancies in single-cell clones of patient-derived hematopoietic cells to human hematopoietic cells engrafting in NSG xenotransplantation recipients to identify mutations associated with leukemia initiation, 2) examining whether the anti-apoptosis pathway can be a therapeutic target independent of leukemogenic somatic mutations, and 3) testing whether targeting these pathways by small molecule inhibitors would result in efficient elimination of human AML in vivo. To do so, we isolated various subpopulations of patient-derived hematopoietic cells by FACS and performed next-generation sequencing (NGS) of bulk and single-cell genomic DNA for known malignancy-associated somatic mutations in parallel with NSG xenotransplantation. By performing NGS of bulk and single-cell DNA in human hematopoietic cells that engrafted in NSG recipients, we traced and identified somatic mutations affiliated with in vivo leukemia-initiating AML cell clones and pre-leukemic cell clones that reconstituted non-malignant human hematopoiesis in vivo. To determine the degree to which human AML cells resistant to kinase inhibition were dependent on the anti-apoptotic pathway, we performed dynamic BH3-profiling. To test the efficacy of kinase inhibition and inhibition of the anti-apoptotic pathway, we treated human FLT3-ITD+ AML-engrafted NSG recipients with a dual kinase inhibitor RK-20449 and BCL-2 inhibitor ABT- 199, and assessed human AML cell chimerism by flow cytometry in the PB weekly during treatment and BM, spleen, and PB at the time the animal was sacrificed.
We did not predetermine the numbers of mice in each treatment group. To ensure each comparison was sufficiently powered, we performed power calculation for each comparison (either two-tailed t-test or paired two-tailed t-test) using StatMate (GraphPad). We found that the comparisons deemed statistically significant (p<0.05) were powered at 85-99% in detecting the differences observed using the statistical test used. PB sampling was done before treatment and every week after start of treatment. Recipients were checked daily and sacrificed when they showed signs of progressive disease such as ruffled fur and weakness.
No randomization and blinding were performed and there were no exclusions. For in vivo treatment experiments, recipients with similar extent of PB human AML engraftment chimerism were chosen as a set for indicated treatments. Pre-treatment human CD45 chimerism and statistical data are shown in tables S3-S6. There were no significant differences found in pre-treatment human AML engraftment between treatment groups.
Because it is logistically difficult and undesirable from the standpoint of animal health and comfort to obtain replicate samples from highly immunosuppressed NSG recipients, PB was sampled only once every week for all recipients in all experiments. Human AML chimerism obtained at the time of sacrifice was also evaluated once in BM, spleen, and PB of the recipients. Overall experimental replication for in vivo treatment studies was ensured by the numbers of patient samples tested as well as the numbers of recipients treated for each group. For each patient sample, treatment experiments were performed as a set of untreated control, treatment A, treatment B, and/or treatment C, with A, B, and C being ABT-199 alone, RK-20449 alone, or combination. Therefore, there were at least three experimental replicates for each patient sample.
Ethical Statements
Written informed consent was obtained from all patients. The study was performed with authorization from the Institutional Review Board for Human Research at RIKEN and Toranomon Hospital, in accordance with the ethical standards of responsible committees on human experimentation at each institution. CB samples were purchased from Lonza.
All experiments using NSG mice (35, 36) were performed with authorization from and according to guidelines established by the Institutional Animal Committees at RIKEN and The Jackson Laboratory.
Mice
Mice were bred and maintained under defined flora at the animal facility of RIKEN and at The Jackson Laboratory. Both female and male NSG mice received transplants at two days of age. Treatment studies were conducted when sufficient engraftment was observed, at approximately 6 weeks of age.
Flow cytometry
The following monoclonal antibodies (mAbs) were used for flow cytometry: Mouse anti-human CD19 (Catalog No. 562653, 555412, 341093), CD3 (Catalog No. 563800, 562426, 555341), CD33 (Catalog No. 562854, 555450), CD34 (Catalog No. 348791), CD38 (Catalog No. 340439, 555459), CD4 (Catalog No. 555348), and CD45 (Catalog No. 563204, 641399, 563204); Rat anti-mouse Ter119 (Catalog No. 557915) and CD45 (Catalog No. 563891, 563410) (BD Biosciences). Analyses were performed with FACSAriaIII and FACSCantoII (BD). To obtain cells for xenogeneic transplantation, BV786-conjugated anti-CD3 mAb, BV605-conjugated anti-CD19 mAb, BV421-conjugated anti-CD33 mAb, PE-Cy7-conjugated anti-CD34 mAb, APC-conjugated anti-CD38 mAb, FITC-conjugated anti-CD90 mAb, and PE-conjugated anti-CD45RA mAb were used. For single-cell sorting, a 100 μm nozzle was used.
Transplantation
NSG newborn mice received 1.5 Gy total body irradiation followed by intravenous injection of purified human cells. For primary transplantation shown in Figures 2, 3, and 4, cells of the indicated phenotype were sorted from BM or PB cells obtained from the patients, and 5×102 – 2×105 cells were transplanted per recipient, depending on the frequency of the cell population. For in vivo treatment experiments, the known LIC population from each patient was transplanted to create human AML-engrafted recipients. Donor cells were purified according to their cell surface phenotype using monoclonal antibodies against human CD34, CD38, CD90, CD45RA, CD3, CD19, and CD33. The extent of engraftment of human cells in the NSG recipients was assessed by retro-orbital phlebotomy and flow cytometry.
Genome analysis
DNA was extracted from human cells purified from patient samples or recipient organs using DNeasy Blood & Tissue Kit (QIAGEN). PCR detection of FLT3-ITD was performed using TaKaRa PCR FLT3/ITD Mutation Detection Set (Takara).
The bulk DNA sequences were determined by next-generation DNA sequencing (NGS). After shearing with a Covaris S220 (Covaris), the fragmented genomic DNA (10 ng) was converted to NGS sequencing library with a KAPA Hyper Prep Kit (KAPA Biosystems) according to the protocol provided by the supplier. Targeted sequencing of AML-related genes was carried out by a hybridization-capture method with xGen AML Cancer Panel v1.0 (Integrated DNA Technologies) according to the protocol provided by the supplier. The hybridization-captured DNA library was subjected to NGS in a paired-end read mode (200 cycles) with an Illumina HiSeq 1500 (Illumina). The obtained DNA sequences were mapped onto human genome sequence (hg19) using BWA (http://bio-bwa.sourceforge.net/; v0.7.12), then realigned with a Realigner Target Creator in Genome Analysis Toolkit (https://www.broadinstitute.org/gatk/index.php; v. 1.6-13). After treatment with Fix Mate Information in Picard (http://broadinstitute.github.io/picard/; v.1.119) and Quality Score Covariate and Table Recalibration in Genome Analysis Toolkit (v.1.6-13), variants were detected with VarScan (http://varscan.sourceforge.net/somatic-calling.html; v2.3.6, read depth>10).
Single-cell variation analysis was carried out for single cells sorted on a BD FACS Aria into 96-well plates. Single-cell whole-genome amplification by Multiple Annealing and Looping-Based Amplification Cycles (MALBAC) (37), associated with a substantially low allelic dropout rate at ~1% and a false-positive rate of ~4×10−5, was used (37-39). After single-cell genome amplification with a MALBAC Single Cell WGA Kit (Yikon Genomics), target regions of genes of interest were PCR-amplified with primers including well indexes by PCR. The first-round gene-specific PCR was conducted using gene-specific primers by 25-cycle PCR with Gflex DNA Polymerase (Takara Bio) in a single-plex mode. The second-round PCR was performed to link Illumina anchor sequences at both sides of the first-round PCR products. PCR conditions were as in the first-round PCR, except that PCR primers were replaced with those for attaching Illumina anchoring sequences. Because of low PCR efficiency, the first-round PCRs for WT1 and CEBPA were carried out as follows: For WT1, PCR was performed over 40 cycles using Taq DNA polymerase (Qiagen) under a 3-step thermal cycling of 94°C 30 sec-55°C 30 sec-72°C 30 sec; for CEPBA, PCR was performed over 36 cycles using Taq DNA polymerase and Q-solution (Qiagen) under a 2-step thermal cycling of 95°C 1 min-68°C 6 min. Primer sequences are described in table S8. The PCR products were sequenced in a paired-end read mode (300 cycles) on an Illumina MiSeq. The obtained DNA sequences were mapped onto human genome sequence (hg19) with BWA (v0.7.12) and paired-end reads were merged with SAMtools (http://samtools.sourceforge.net/; v1.0, read depth>100). Variant detection and frequency calculation were done with mpileup in SAMtools. In this study, variation genotype was assigned on assumption that sequencing error rate is lower than 2% and apparent variation with allele frequency lower than 2% was regarded as wild-type. Three independent amplification and sequencing rounds were performed before calling a locus wild-type. No germline tissue was available for evaluation of somatic status of mutations. CEBPA, DNMT3A, FLT3, IDH1, TET2, and WT1 variants were included or excluded according to gene-specific characteristics as described by Lindsley et al. (40).
For RNA-seq analysis, total RNA was extracted from FACS-purified cells treated with TRIzol (ThermoFisher Scientific). RNA-seq libraries were prepared using the SureSelect Strand-Specific RNA library Preparation Kit (Agilent Technologies) according to the manufacturer’s protocol and were sequenced by HiSeq 1500 (Illumina). The sequence reads were mapped to the human genome (hg19) using TopHat2 software (version 2.0.8). Cufflinks (version 2.1.1) was run with the same reference annotation with TopHat2 to generate FPKM values. Statistical evaluation of gene expression change was performed using edgeR algorithm with read counts on exons determined using R program.
In vivo treatment
In vivo treatment experiments were performed with AML-engrafted NSG recipients using RK-20449 (17) and ABT-199 (41, 42). The recipients were treated with RK-20449 (30 mg/kg) intraperitoneally twice a day, ABT-199 (70 mg/kg) orally once a day, or both RK-20449 and ABT-199. The mice were euthanized when they became moribund or after 4-6 weeks of treatment, and human AML chimerism in BM, spleen, and PB was determined using flow cytometry. In secondary transplantation experiments, each mouse received 7-AAD(−) viable human CD45+ cells from 2.5% of total BM that remained in AML-engrafted recipients at the time of sacrifice, to simulate relapse occurring from residual viable AML cells. All treated recipients and their pre- and post-treatment engraftment data are tabulated in tables S3 and S4. No sample size pre-estimation was performed. To ensure that each comparison was sufficiently powered, we performed power calculation for each comparison (either two-tailed t-test or paired two-tailed t-test) using StatMate (GraphPad). Each comparison deemed significant (p<0.05) was powered at 85-99% for detecting the differences observed.
Dynamic BH3 profiling
Cells were harvested from BM of recipients engrafted with AML derived from patients with FLT3-ITD+ AML, and BH3 profiling was performed using the plate-based assay previously described (43, 44). For dynamic BH3 profiling, 2×106 harvested cells were exposed to 500 nM RK-20449 in 2 ml HPGM supplemented with 50 ng/mL SCF, 50 ng/mL FLT3 ligand, and 50 ng/mL TPO for four hours at 37 C in humidified atmosphere containing 5% CO2. After surface labeling with anti-human CD45 and dead cell exclusion with Zombie NIR (BioLegend), the cells were permeabilized, exposed to BH3 peptides (0.39 μM BIM, 80 μM BAD, 80 μM HRK, or 80 μM NOXA) or 1 μM ABT-199, and retained intracellular cytochrome C was measured by flow cytometry using anti-cytochrome C antibody.
Statistical analysis
For in vivo treatment experiments, numerical data are presented as means +/− SEM. The differences were examined with two-tailed t test (GraphPad Prism, GraphPad). Statistical methods for genomic analyses are included in Genome analysis section.
For in vivo treatment experiments, pre- and post-treatment data were obtained from PB of each mouse. To detect differences between pre- and post-treatment data obtained from the same mouse, paired two-tailed t-test (pairing pre- and post-treatment values of each mouse) was used. Because pre-treatment data from BM/spleen of the mice are not available, differences in BM and spleen of the mice were evaluated by unpaired two-tailed t-test. We designed the experiment such that there were 3 to 6 engrafted recipients per treatment group from a given patient. Therefore, in comparisons restricted to mice engrafted with AML from a particular patient, n was insufficient (3 to 6 in each group) for meaningful tests of normality or variation. In all such comparisons, performing non-parametric tests did not yield contradicting results on the significance of the differences detected, and the comparisons were sufficiently powered even though n was small, because the sizes of the differences detected were sufficiently large and the values observed within each group were sufficiently tightly distributed (standard deviation was sufficiently low). For comparisons of treatment groups including all mice (across patient samples), we chose to use the t-test because parametric tests on non-Gaussian data are robust as long as sample sizes are sufficient. In these cases, using non-parametric tests also did not yield contradicting results.
Because n was relatively small (< 20) for groups restricted to a particular patient sample, independent data points were plotted in all relevant figures, and all data are tabulated in supplementary tables. In comparisons of whole treatment groups (containing experiments using samples from multiple patients), normality testing using two different tests (D'Agostino-Pearson omnibus test and Shapiro-Wilk test) yielded variable results. However, because n is relatively large, detected differences are relatively large, and deviations among data points within each group are relatively small, parametric test (two-tailed t-test) should be robust. Performing non-parametric tests on these datasets did not contradict the results of the parametric two-tailed t-test.
In comparisons between groups restricted to individual patients, n was insufficient to obtain a meaningful estimate on the variance. For aggregate data of mice engrafted with AML from multiple patients, n was sufficiently large; however, in such comparisons, we did not expect variances to be similar because patients have highly heterogeneous disease characteristics and biology. As expected, F test to compare variances showed similar variances between some groups but not others. In comparisons between groups restricted to individual patients, n was insufficient to obtain a meaningful estimate on the variance. For aggregate data across multiple patient samples and comparisons made on mice engrafted with AML from multiple patients with highly heterogeneous disease characteristics and biology, we did not expect variances to be similar.
Because n is relatively small (< 20) for groups restricted to a particular patient sample, independent data points are plotted in all relevant figures and all data are tabulated in Supplementary tables. In comparisons of whole treatment groups (containing experiments using samples from multiple patient), normality testing using two different tests (D'Agostino-Pearson omnibus test and Shapiro-Wilk test) yielded variable results. However, since n is relatively large, detected differences are relatively large and deviations among data points within each group are relatively small; therefore, parametric test (two-tailed t-test) should be robust. Performing non-parametric tests on these datasets did not contradict the results of the parametric two-tailed t-test.
In comparisons between groups restricted to individual patients, n is insufficient to obtain a meaningful estimate on the variance. As aggregate data of mice engrafted with AML from multiple patients, n becomes sufficiently large; however, in such comparisons, we do not expect variances to be similar because patients have highly heterogeneous disease characteristics and biology. As expected, F test to compare variances showed similar variances between some groups but not in others. In comparisons between groups restricted to individual patients, n is insufficient to obtain a meaningful estimate on the variance. For aggregate data across multiple patient samples, comparisons are made on mice engrafted with AML from multiple patients with highly heterogeneous disease characteristics and biology, we do not expect variances to be similar.
Supplementary Material
Figure S1. Identification of hematopoietic subpopulations in patient samples by surface phenotype and in vivo function in three additional patients
Figure S2.Mutational profiles of AML cells with and without in vivo RK-20449 treatment
Figure S3.Mutational profiles of AML patient samples obtained serially during clinical course
Figure S4.Altered transcription profiles of human AML cells with in vivo RK-20449 treatment
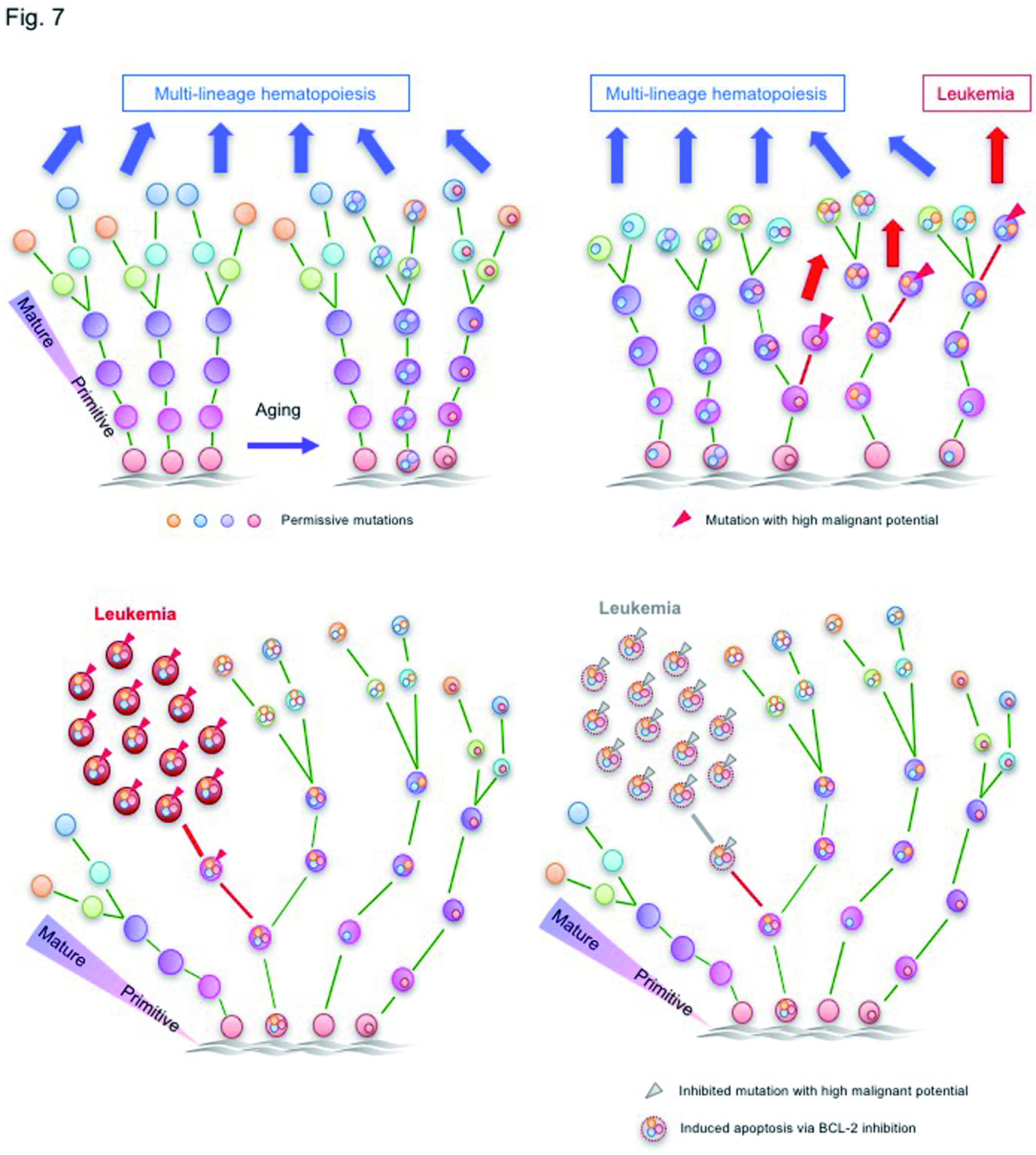
Figure S5.Overcoming genetically-complex AML by targeting leukemia-initiating mutation and anti-apoptotic BCL-2 pathway
{kind=link}
Table S1. Patient characteristics
Table S2. Information of identified variants
Table S3. Human AML chimerism in PB, BM and spleen of AML-engrafted recipients treated with RK-20449 alone
Table S4. Human AML chimerism in PB, BM and spleen of AML-engrafted recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S5. Statistics comparing pre- and post-treatment PB human AML chimerism in recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S6. Statistics comparing BM and spleen human AML chimerism in recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S7. Effect of in vivo exposure to combined RK-20449 and ABT-199 on human multilineage hematopoiesis
Table S8. PCR primers for targeted sequencing of MALBAC products
Acknowledgments
We would like to acknowledge and thank the patients and the clinical care team at Toranomon Hospital whose cooperation and effort made this study possible. We thank Dr. R. Coleman Lindsley and Dr. Jacqueline S. Garcia for critical review of the manuscript.
Funding
This study was supported by the Basic Science and Platform Technology Program for Innovative Biological Medicine (FI) and the Project for Development of Innovative Research on Cancer Therapeutics (FI) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology, The Japan Agency for Medical Research and Development (FI), and The Maine Cancer Foundation and National Institutes of Health grant CA034196 (LDS).
Footnotes
Competing interests
Yoriko Saito and Fumihiko Ishikawa are inventors on patent application (62/394871) submitted by RIKEN that covers combinatory use of RK-20449 and BCL-2 inhibitors for AML. All other authors declare that they have no competing interests.
Data and materials availability
Raw sequence data reported in this paper are available at NBDC Human database (Japan) (accession number: hum0116). NSG mice are available from The Jackson Laboratory under a material transfer agreement.
References and notes
- 1.N. Cancer Genome Atlas Research, Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kandoth C et al. , Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klco JM et al. , Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 25, 379–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller CA, Wilson RK, Ley TJ, Genomic landscapes and clonality of de novo AML. N Engl J Med 369, 1473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.N. Cancer Genome Atlas Research, Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delhommeau F et al. , Mutation in TET2 in myeloid cancers. N Engl J Med 360, 2289–2301 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Ley TJ et al. , DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363, 2424–2433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PS et al. , The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R, Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 111, 2548–2553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shlush LI et al. , Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welch JS et al. , The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G et al. , Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371,2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaiswal S et al. , Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371,2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie M et al. , Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Notta F et al. , Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quek L et al. , Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med 213, 1513–1535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito Y et al. , A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci Transl Med 5, 181ra152 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Shlush LI et al. , Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Hetz C, The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13, 89–102 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Spijkers-Hagelstein JA et al. , Elevated S100A8/S100A9 expression causes glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia 26, 1255–1265 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Yang M et al. , S100A8 contributes to drug resistance by promoting autophagy in leukemia cells. PLoS One 9, e97242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Yang XY et al. , High expression of S100A8 gene is associated with drug resistance to etoposide and poor prognosis in acute myeloid leukemia through influencing the apoptosis pathway. Onco Targets Ther 9, 4887–4899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S et al. , HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res 77, 2064–2077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montero J et al. , Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160, 977–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chonghaile TN et al. , Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov 4, 1074–1087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Del Gaizo Moore V et al. , Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 117, 112–121 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly GL et al. , Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 28, 58–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ, Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 6, 241–249 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Vo TT et al. , Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brumatti G, Lalaoui N, Wei AH, Silke J, 'Did He Who Made the Lamb Make Thee?' New Developments in Treating the 'Fearful Symmetry' of Acute Myeloid Leukemia. Trends Mol Med 23, 264–281 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Steensma DP et al. , Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia JS, Stone RM, The Development of FLT3 Inhibitors in Acute Myeloid Leukemia. Hematol Oncol Clin North Am 31, 663–680 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Sallman DA, Lancet JE, What are the most promising new agents in acute myeloid leukemia? Curr Opin Hematol 24, 99–107 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Stone RM et al. , Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikawa F et al. , Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 106, 1565–1573 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shultz LD et al. , Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Zong C, Lu S, Chapman AR, Xie XS, Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grun D, van Oudenaarden A, Design and Analysis of Single-Cell Sequencing Experiments. Cell 163, 799–810 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Leung ML, Wang Y, Waters J, Navin NE, SNES: single nucleus exome sequencing. Genome Biol 16, 55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsley RC et al. , Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med 376, 536–547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan R et al. , Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4, 362–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Souers AJ et al. , ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19, 202–208 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Ryan J, Letai A, BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryan JA, Brunelle JK, Letai A, Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A 107, 12895–12900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Li M, Hakonarson H, ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of hematopoietic subpopulations in patient samples by surface phenotype and in vivo function in three additional patients
Figure S2.Mutational profiles of AML cells with and without in vivo RK-20449 treatment
Figure S3.Mutational profiles of AML patient samples obtained serially during clinical course
Figure S4.Altered transcription profiles of human AML cells with in vivo RK-20449 treatment
Figure S5.Overcoming genetically-complex AML by targeting leukemia-initiating mutation and anti-apoptotic BCL-2 pathway
Table S1. Patient characteristics
Table S2. Information of identified variants
Table S3. Human AML chimerism in PB, BM and spleen of AML-engrafted recipients treated with RK-20449 alone
Table S4. Human AML chimerism in PB, BM and spleen of AML-engrafted recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S5. Statistics comparing pre- and post-treatment PB human AML chimerism in recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S6. Statistics comparing BM and spleen human AML chimerism in recipients treated with ABT-199 alone, RK-20449 alone or combination
Table S7. Effect of in vivo exposure to combined RK-20449 and ABT-199 on human multilineage hematopoiesis
Table S8. PCR primers for targeted sequencing of MALBAC products