

Abstract
Solute carrier family 7 member 2 (SLC7A2, also known as CAT2) is an inducible transporter of the semi-essential amino acid L-arginine (L-Arg), which has been implicated in wound repair. We have reported that both SLC7A2 expression and L-Arg availability are decreased in colonic tissues from inflammatory bowel disease patients and that mice lacking Slc7a2 exhibit a more severe disease course when exposed to dextran sulfate sodium (DSS) compared to wild-type (WT) mice. Here, we present evidence that SLC7A2 plays a role in modulating colon tumorigenesis in the azoxymethane(AOM)-DSS model of colitis-associated carcinogenesis (CAC). SLC7A2 was localized predominantly to colonic epithelial cells in WT mice. Utilizing the AOM-DSS model, Slc7a2–/– mice had significantly increased tumor number, burden, and risk of high-grade dysplasia versus WT mice. Tumors from Slc7a2–/– mice exhibited significantly increased levels of the proinflammatory cytokines/chemokines IL-1β, CXCL1, CXCL5, IL-3, CXCL2, CCL3, and CCL4, but decreased levels of IL-4, CXCL9, and CXCL10 compared to tumors from WT mice. This was accompanied by a shift toward pro-tumorigenic M2 macrophage activation in Slc7a2-deficient mice, as marked by increased colonic CD11b+F4/80+ARG1+ cells with no alteration in CD11b+F4/80+NOS2+ cells by flow cytometry and immunofluorescence microscopy. The shift toward M2 macrophage activation was confirmed in bone marrow-derived macrophages from Slc7a2–/– mice. In bone marrow chimeras between Slc7a2–/– and WT mice, the recipient genotype drove the CAC phenotype, suggesting the importance of epithelial SLC7A2 in abrogating neoplastic risk. These data reveal that SLC7A2 has a significant role in the protection from CAC in the setting of chronic colitis, and suggest that the decreased SLC7A2 in inflammatory bowel disease (IBD) may contribute to CAC risk. Strategies to enhance L-Arg availability by supplementing L-Arg and/or increasing L-Arg uptake could represent a therapeutic approach in IBD to reduce the substantial long-term risk of colorectal carcinoma.
Keywords: SLC7A2, colon cancer, inflammation, tumorigenesis, M2 macrophages
INTRODUCTION
With more than 1.6 million Americans affected1, inflammatory bowel disease (IBD) causes significant morbidity and leads to increased risk of colon cancer. As many as 20% of patients with IBD will develop colitis-associated carcinogenesis (CAC) over the course of their disease2–4, and those that develop CAC have an approximately a 2-fold increase in mortality compared with the general population of patients with colon cancer5. The pathophysiology of IBD is not completely defined, but is postulated to arise from the interplay between luminal bacteria and the colonic mucosa6, 7 leading to an abnormal immune response that is linked to numerous potential genetic associations8. Studies to define the mechanisms underlying the abnormal immune system response are ongoing with the hope that new therapeutics may arise, as the severity of inflammation when assessed on colonoscopic and histologic grading is strongly correlated with cancer risk9. Unfortunately, a cohort analysis reported that from 1998 to 2010, patients with IBD continued to have a significantly increased incidence of colorectal cancer (CRC) compared to the general public, and this risk remained stable despite advances in disease management5. Therefore, there is an ongoing need to develop therapies that lead to improved quality of life and decreased risk for progression to cancer.
We have previously shown that L-arginine (L-Arg) metabolism is altered in mouse models of experimental colitis10, 11 as well as in patients with ulcerative colitis12. L-Arg can be synthesized by the body13, thus dietary intake of arginine is not essential for maintaining heath12. However, under conditions of stress including injury and inflammation, L-Arg may become depleted12, 14, 15. The active transport of L-Arg across the cell membrane and into the cytosol is mediated by y+ transporters, which includes the cationic amino acid transporter (CAT) protein family12, 16. The two main CAT proteins are the constitutively expressed transporter, CAT1/SLC7A1, and the inducible transported SLC7A212, 17. Though the UniProt designation for the inducible protein, which is encoded by the Slc7a2 gene is CAT2, for ease of understanding we will denote the protein as SLC7A2. In animal models, we have shown SLC7A2 to be upregulated in the setting of experimental colitis10, 18, 19. However, we have recently reported that SLC7A2 expression is decreased in colon tissues from patients with active ulcerative colitis (UC) or Crohn’s disease (CD)12. Further, tissue L-Arg levels in UC were also found to be decreased; both SLC7A2 expression and L-Arg tissue levels were inversely correlated with the UC disease activity index12. L-Arg metabolism is modulated by four enzymes: arginase, nitric oxide (NO) synthase (NOS), arginine:glycine amidinotransferase, and arginine decarboxylase12, 20. There are two isoforms of arginase; arginase I (ARG1) and arginase II (ARG2), which metabolize L-Arg into L-ornithine and urea21. The arginase enzymes directly compete for L-Arg with inducible NOS (NOS2), which metabolizes L-Arg to NO and L-citrulline12.
We have previously shown that L-Arg supplementation improved survival and body weight loss, and led to reduced colonic permeability and the number of colonic myeloperoxidase-positive neutrophils after exposure to dextran sulfate sodium (DSS)10. In addition, there was a marked reduction in proinflammatory cytokine/chemokine expression with L-Arg treatment10. Furthermore, SLC7A2-deficient (Slc7a2−/−) mice exposed to DSS exhibited worsening of survival, body weight loss, colon weight, and histological injury with loss of the clinical benefits of L-Arg supplementation that were observed in wild-type (WT) mice18. DSS-induced colitis led to increased infiltration of colonic macrophages, dendritic cells, granulocytes, and T cells with associated increases in proinflammatory cytokines pointing to a shift from an IFN-γ- to an IL-17-predominant T cell response in Slc7a2–/– versus WT mice18. While SLC7A2 is predominantly known as an inducible L-Arg transporter in macrophages17, 22, we have reported that SLC7A2 can also be upregulated in colonic epithelial cells, and that SLC7A2-mediated L-Arg uptake is essential for colonic epithelial wound repair23.
We now report that in the azoxymethane (AOM)-DSS murine model of colitis-associated tumorigenesis, SLC7A2 expression localizes mainly to the colonic epithelium, and loss of SLC7A2 has a profound effect on the development of CAC. Slc7a2–/– mice exhibit increased tumor number and burden, alterations in the tumor microenvironment with differences in cytokine levels, and a shift toward more pro-tumorigenic M2 colonic macrophages. Importantly, studies in bone marrow chimeric mice pointed to loss of epithelial-derived SLC7A2 as an important driver of the increased tumorigenesis. Thus, SLC7A2 has a protective role in prevention of CAC, and the loss of SLC7A2 that occurs in IBD may contribute to the risk for development of CAC.
Results
SLC7A2 expression is increased in colonocytes in response to AOM-DSS
SLC7A2 contributes to the epithelial response to injury23 and is induced in the setting of acute DSS-induced colitis10; there is increased histologic injury and decreased survival in Slc7a2–/– mice18. Given this phenotype in the acute DSS model, we assessed whether SLC7A2 levels were altered in a chronic injury model associated with tumorigenesis via the AOM-DSS model. Immunohistochemistry demonstrated that SLC7A2 protein was mainly increased in epithelial cells in WT mice exposed to AOM-DSS with modest lamina propria cell expression (Figure 1) that was not present in Slc7a2–/– mice (Figure 1). We then examined mRNA levels of Slc7a2 by in situ hybridization (RNAscope), and found increased Slc7a2 mRNA levels in WT mice exposed to AOM-DSS, predominantly in epithelial cells (Supplementary Figure 1), though there was some lamina propria cell detection (Supplementary Figure 1). Slc7a2 mRNA levels were not detectable in control or AOM-DSS treated Slc7a2−/− mice (Supplementary Figure 1).
Figure 1. SLC7A2 expression is increased in inflammation-associated tumorigenesis.
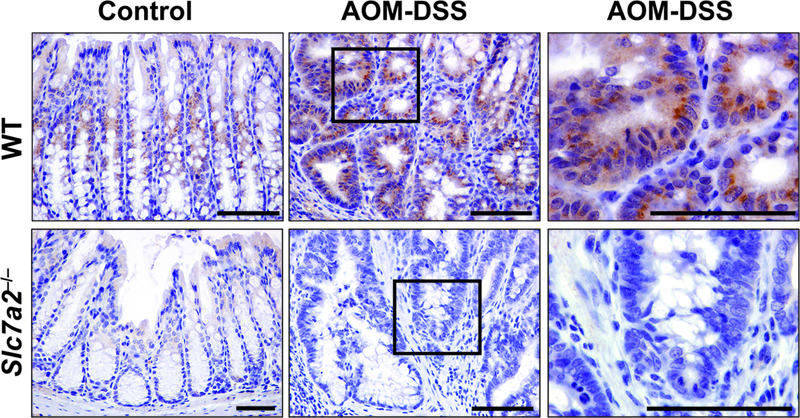
C57BL/6 WT and Slc7a2–/– mice were exposed to the AOM-DSS protocol (AOM-DSS) or not (Ctrl) and colonic tissues were harvested at day 77. Colon sections were stained for SLC7A2 protein by immunohistochemistry. Scale bar = 100 μm. Representative images of at least 3 mice per group.
Loss of SLC7A2 leads to increased tumorigenesis
When WT or Slc7a2–/– mice were subjected to the AOM-DSS protocol (Supplementary Figure 2) with repeated cycles of 4% DSS, Slc7a2–/– mice exhibited significantly more tumors with increased tumor burden compared to WT mice (Supplementary Figure 3a, b). Interestingly, in the mice exposed to 3 cycles of DSS alone, the Slc7a2–/– mice had increased histologic injury compared to WT mice (Supplementary Figure 3c) similar to the pattern we previously described in the setting of acute DSS-induced colitis18. However, there was no significant difference in the histologic injury scores in the non-tumor areas in mice exposed to AOM-DSS (Supplementary Figure 3c). In addition, all of the Slc7a2–/– mice had evidence of dysplasia, with the majority (7/8) exhibiting high grade dysplasia, whereas, 2/18 of the WT mice had no adenomas and were more likely to exhibit low grade dysplasia (10/18) with a lower frequency of high grade dysplasia (6/18) as shown in Supplementary Figure 3e, f (P < 0.05 by Chi square test). The lack of significant difference between the histologic injury scores between the two groups of mice was likely affected by the significant mortality that occurred in the Slc7a2–/– mice which reached 60% by the end of the 77 day protocol and therefore limited the number of colons that could be assessed (Supplementary Figure 3d).
As the Slc7a2–/– mice were very sensitive to the combination of AOM followed by 3 cycles of 4% DSS, we decreased the dosage of DSS to 2.5% for 3 cycles and used this dosage for the remainder of the data presented in this study. When subjected to the AOM-DSS protocol with repeated cycles of 2.5% DSS, Slc7a2–/– mice continued to exhibit significantly more tumors and increased tumor burden compared to WT mice (Figure 2a, b), with improved survival (Figure 2d). However, the Slc7a2–/– mice exposed to 3 cycles of 2.5% DSS alone did not develop increased histologic injury compared to WT mice (Figure 2c). Interestingly, there was a significant decrease in the histologic injury scores in the non-tumor areas of Slc7a2–/– mice exposed to AOM-DSS (Figure 2c, f). In addition, with the lower dosage of DSS, tumors in WT mice had no histology greater than low grade dysplasia, and more than half of the mice (12/22) had no adenomas (Figure 2e). However, the Slc7a2–/– mice were significantly more likely to have adenomas with low grade dysplasia (13/23) or high grade dysplasia (5/23) as shown in Figure 2e, f. In addition, assessment via Ki67 staining revealed increased epithelial proliferation in Slc7a2–/– tumors compared to WT tumors (Supplementary Figure 4).
Figure 2. Slc7a2–/– mice exhibit increased tumorigenesis after exposure to the AOM-DSS model of CAC using 2.5% DSS.
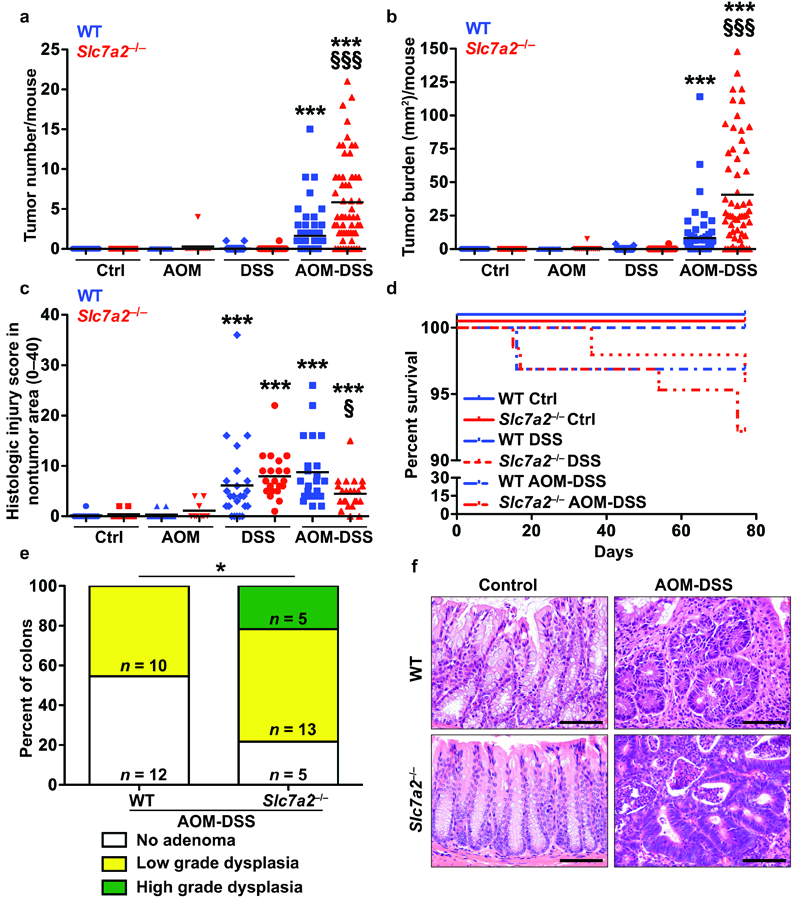
(a) Tumor number was assessed by gross visual inspection, utilizing a dissecting microscope. (b) Tumor burden was determined by the addition of the calculated area of each identified tumor, as assessed with an electronic caliper for both length and width. (c) Histologic colitis score in nontumor area. In (a), (b), and (c), ***P < 0.001 versus WT Ctrl; §P < 0.05 and §§§P < 0.001 versus WT AOM-DSS by one-way ANOVA with Student-Newman-Keuls posthoc multiple comparisons test. (d) Survival curve assessed by log-rank (Mantel-Cox) test. In (a), (b), (c), and (d), n = 25–29 control, 48–55 DSS-treated, and 61–62 AOM-DSS-treated mice per genotype. (e) Percentage of cases with either no adenoma, low grade dysplasia, and high grade dysplasia determined by a gastrointestinal pathologist (M.K.W.) in a blinded manner. By Chi Square test, *P = 0.0161. The number of mice with each diagnosis is shown on the graph. (f) Representative H&E-stained images from AOM-DSS-treated mice. Scale bar = 100 μm.
Slc7a2–/– mice exhibit altered tumor chemokine and cytokine levels
In the Slc7a2–/– mice, the tumors were not only more advanced in dysplasia level (Figure 2e, f), but the tumors themselves exhibited significant intra-tumoral inflammation with crypt abscesses (Figure 2f). To provide further insight into the immunologic effects of loss of SLC7A2 in the tumor microenvironment, we utilized paired pieces of tumor and adjacent non-tumor colonic tissue in mice exposed to AOM-DSS. Of the 32 cytokines/chemokines that were assessed via a Luminex Multiplex Array, 27 were significantly increased in the isolated tumor areas of either WT or Slc7a2–/– mice compared to adjacent non-tumor areas (Figure 3, Supplementary Table 1). There were no differences detected between control and DSS alone, or between DSS alone and non-tumor areas from AOM-DSS-treated mice (Figure 3, Supplementary Table 1). Consistent with the lack of difference in histologic injury in the chronic DSS groups, there were no significant differences in the cytokine levels between non-tumor areas between the WT and Slc7a2–/– mice (Figure 3, Supplementary Table 1).
Figure 3. Slc7a2–/– mice have significantly altered cytokine and chemokine production within colon tumors.
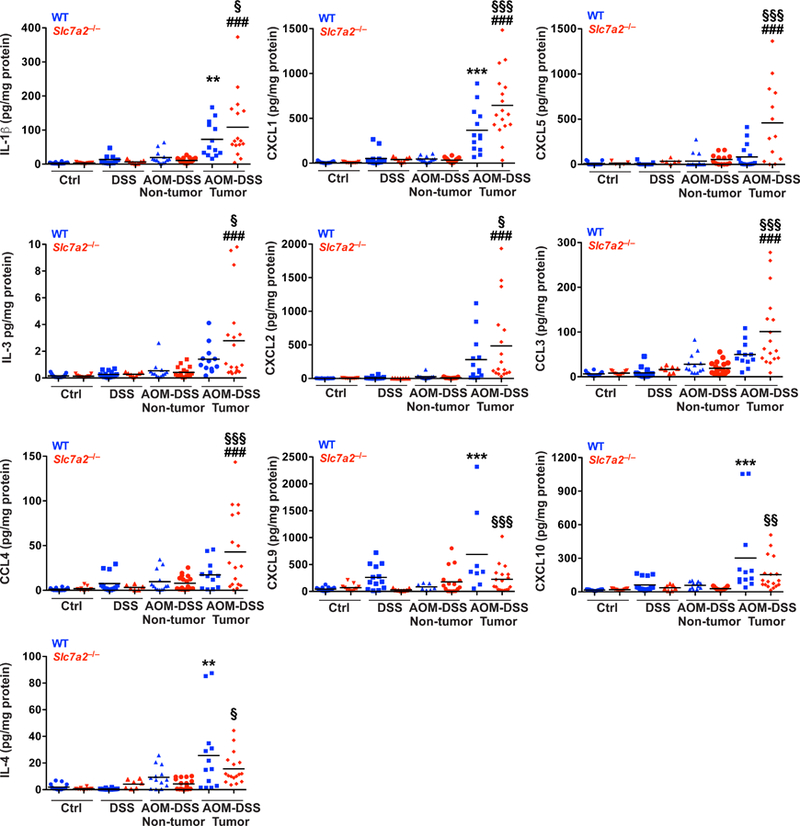
Protein levels were assessed by Luminex Multiplex Array from colonic tissues 77 days post-AOM injection. In all panels, **P < 0.01, ***P < 0.001 versus WT AOM DSS non-tumor; ###P < 0.001 versus Slc7a2–/– AOM-DSS non-tumor; and §P < 0.05, §§P < 0.01, and §§§P < 0.001 versus WT AOM-DSS tumor by one-way ANOVA with Student-Newman-Keuls posthoc multiple comparisons test. n = 11–15 control, 7–15 DSS-treated, and 13–18 AOM-DSS-tumors with paired non-tumor area per genotype.
Importantly, when tumors from WT or Slc7a2–/– mice were compared, there were significant alterations. In the tumors from Slc7a2–/– mice, there were significant increases in the innate cytokine IL-1β, and the chemokines CXCL2 (MIP2), CCL3 (MIP-1α), CCL4 (MIP-1β), and CXCL1 (KC, GRO-α), the murine equivalent of CXCL8 (IL-8)24, which are chemoattractants for innate immune cells, and are produced by macrophages25 and epithelial cells26, 27(Figure 3). However, CXCL9 (MIG) and CXCL10 (IP-10), which are also primarily produced by macrophages28 and induce T cell infiltration29, were decreased in Slc7a2–/– tumors (Figure 3). Multiple cytokines can contribute to angiogenesis, which is essential for tumor growth30, including vascular endothelial growth factor (VEGF) A, CXCL5, and CXCL1 (KC, GRO-α). CXCL1 was increased in WT tumors, and was significantly further increased in Slc7a2–/– tumors, and CXCL5 was increased only in Slc7a2–/– tumors (Figure 3), while VEGF was increased to a similar degree in both WT and Slc7a2–/– tumors (Supplementary Table 1). In addition, CXCL10 can function to inhibit angiogenesis31; thus, the significant decrease in CXCL10 in Slc7a2–/– tumors (Figure 3) could lead to further promotion of angiogenesis. Taken together, there was an overall increase in the inflammatory response within tumor areas in Slc7a2–/– mice compared to WT mice, indicating that increased inflammation is a driver of the increased tumorigenesis seen in the Slc7a2–/– mice.
Slc7a2–/– colons exposed to AOM-DSS exhibit increased M2 macrophages
Immune cells, including macrophages, play key roles in homeostasis, pro-inflammatory and anti-inflammatory responses, and wound repair32–34. Thus, macrophages are functionally important in the initiation and promotion of tumorigenesis35, 36. We have previously shown in the acute DSS model that Slc7a2–/– mice exhibit decreased tissue L-Arg uptake in the setting of increased colonic macrophages18. Here, our goal was to assess the balance of macrophage phenotypes present, as macrophage function can be altered based on the method of activation32–34, 37–39. M1 macrophages are considered to exert anti-tumoral effects due to tissue and tumor destructive abilities, while M2 macrophages are generally considered to exert pro-tumorigenic effects by increasing angiogenesis, promoting wound healing, and suppressing immune responses34, 35. There were no significant differences between the two genotypes in the number of CD11b+F4/80+ARG1+ macrophages or CD11b+F4/80+NOS2+ macrophages in colon tissues from control mice or mice treated with DSS alone (Figure 4a, b), as determined by flow cytometry. However, consistent with the increased tumorigenesis related to loss of SLC7A2, we found significantly increased numbers of CD11b+F4/80+ARG1+ M2 macrophages (Figure 4a, d) with no significant change in CD11b+F4/80+NOS2+ M1 macrophages (Figure 4b, d) in the colon tissues of Slc7a2–/– versus WT mice treated with AOM-DSS. Thus, there was a significant shift in the M2/M1 ratio in the Slc7a2–/– colons exposed to AOM-DSS (Figure 4c, d). To assess if this shift in macrophage phenotype was associated with the tumor tissue in situ, we performed immunofluorescence staining for co-localization of the macrophage marker, CD68, with the M2 marker, ARG1 (Figure 5a), or the M1 marker, NOS2 (Figure 5b). This showed that CD68+ARG1+ macrophages are markedly increased, but the abundance of CD68+NOS2+ macrophages are not changed in the tumor areas in Slc7a2–/– versus WT mice.
Figure 4. AOM-DSS exposure leads to increased M2 colonic macrophages in Slc7a2–/– mice.
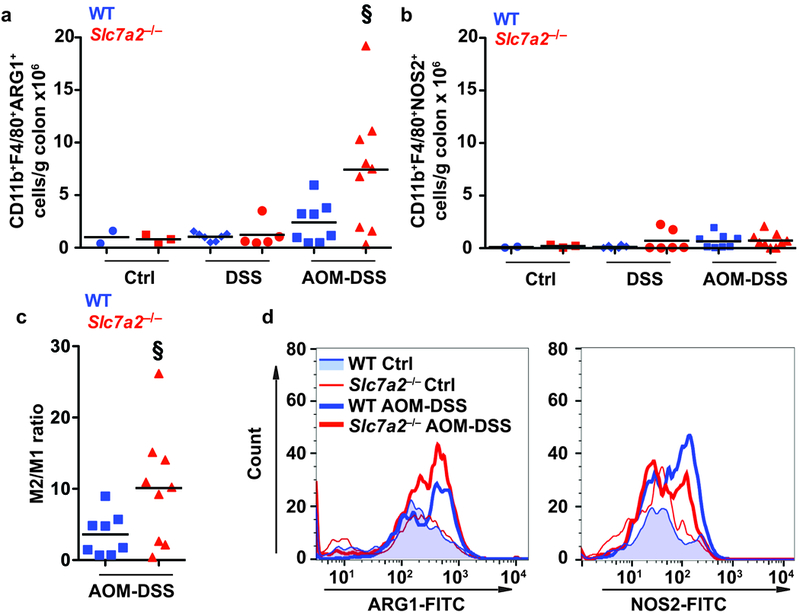
(a-b) Quantification of CD11b+F4/80+ARG1+ macrophages and CD11b+F4/80+NOS2+ macrophages per gram of colon tissue. (c) Ratio of CD11b+F4/80+ARG1+/CD11b+F4/80+NOS2+ in the same colon. (d) Representative flow plots showing the increase in CD11b+F4/80+ARG1+ macrophages in Slc7a2–/– AOM-DSS colon compared to WT AOM-DSS colon. In a-c, §P < 0.05 versus WT AOM-DSS by one-way ANOVA with Student-Newman-Keuls posthoc multiple comparisons test. n = 2–3 control, 5–7 DSS-treated, and 7–9 AOM-DSS-treated per genotype.
Figure 5. ARG1+CD68+ macrophages are increased in Slc7a2–/– AOM-DSS tumors.
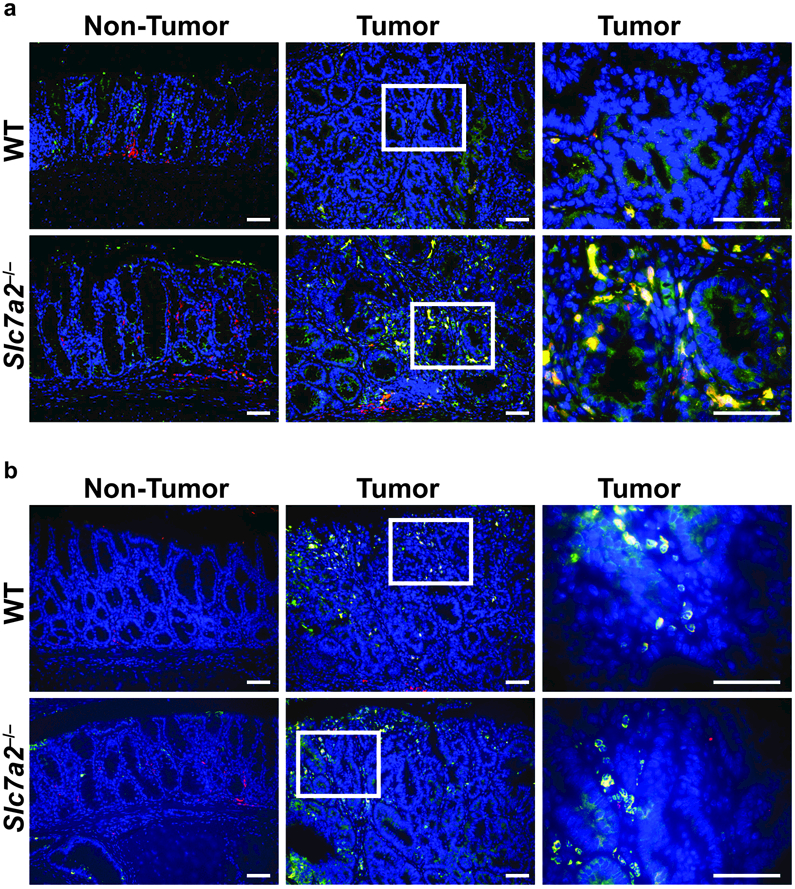
Representative images of immunofluorescence staining for (a) macrophage marker, CD68 (red); M2 marker, ARG1 (green); CD68+ARG1+ cells (yellow, merge). Cell nuclei, DAPI (blue). (b) Macrophage marker, CD68 (red); M1 marker, NOS2 (green); CD68+NOS2+ cells (yellow, merge). Cell nuclei, DAPI (blue). Representative merged images of at least 3 per group. Scale bars = 50 μm.
Loss of Slc7a2 leads a shift toward a M2 macrophage activation pattern
To assess if SLC7A2 loss resulted in a baseline alteration in macrophage activation pattern, we isolated bone marrow-derived macrophages (BMmacs) from WT and Slc7a2–/– mice and stimulated them ex vivo with either classical M1 stimuli (IFN-γ + LPS) or M2 stimuli (IL-4 + IL-10). Markers of M1 and M2 macrophage activation status were then assessed. IFN-γ + LPS stimulation resulted in robust expression of the M1 markers Nos2 and Il1b (Figure 6a) in both WT and Slc7a2–/– BMmacs. However, there was no difference between BMmacs from the WT and Slc7a2–/– mice. As expected, there was decreased nitric oxide (NO) production (measured as NO2–) in the Slc7a2–/– cells, consistent with the loss of L-Arg availability due to deletion of the SLC7A2 transporter, as we have described in a macrophage cell line with knockdown of Slc7a240. However, there was no effect of Slc7a2 deletion on IL-1β expression by ELISA (Figure 6c). IL-4 + IL-10 stimulation led to a significant induction of mRNA expression of the M2 markers Arg1 and chitinase-like 3 (Chil3) in both WT and Slc7a2–/– BMmacs (Figure 6b). Importantly, Arg1 and Chil3 levels were significantly further increased in Slc7a2–/– BMmacs (Figure 6b). Increased CHIL3 expression was confirmed at the protein level by Western blot analysis (Figure 6d). Furthermore, increased L-Arg uptake was detected in WT BMmacs stimulated with either M1 or M2 stimuli, but not in Slc7a2–/– BMmacs (Supplementary Figure 5). Accordingly, when assessed in the acute DSS18 or the injury and recovery DSS models (Supplementary Figure 5), L-Arg uptake was reduced in colonic tissues from Slc7a2–/– versus WT mice. These data suggest that SLC7A2 contributes to macrophage polarization, but is not the only factor controlling polarization; it does not appear to be critical for M1 gene expression, under M1 polarizing conditions, but loss of SLC7A2 can specifically affect NO levels (Figure 6c).
Figure 6. Bone marrow-derived macrophages (BMmacs).
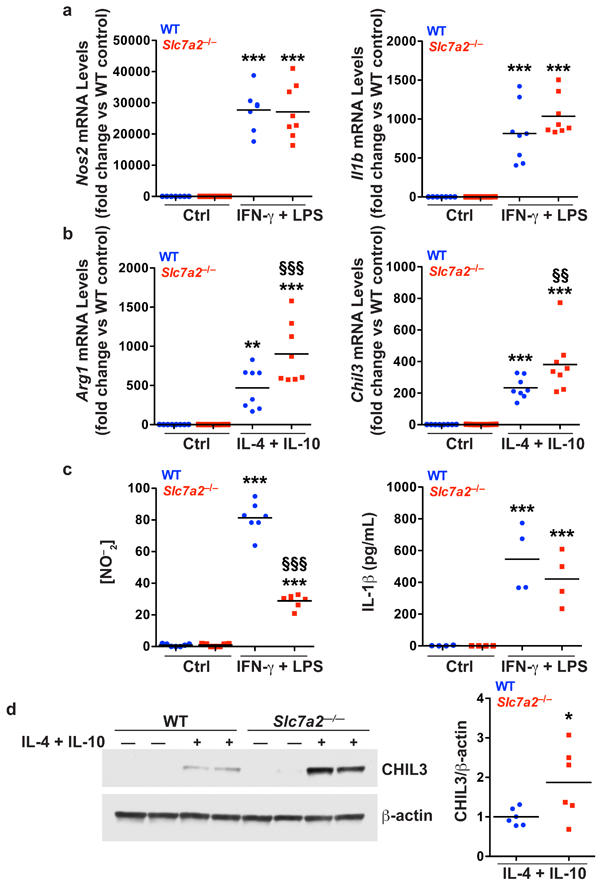
BMmacs were stimulated with classical M1 stimuli, IFN-γ (200 U/mL) and LPS (100 ng/mL), or M2 stimuli, IL-4 (10 ng/mL) and IL-10 (10 ng/mL), for 24 hours. Cells were collected for isolation of RNA and protein. The supernatants were collected. (a) mRNA levels of M1 markers, Nos2 and Il1b, were assessed by qRT-PCR in unstimulated (Ctrl) or stimulated cells. (b) mRNA levels of M2 markers, Arg1 and Chil3, were assessed by qRT-PCR in unstimulated (Ctrl) or stimulated cells. (c) Concentration of NO–2 in the supernatants assessed by Griess assay, and IL-1β in the supernatant assessed by ELISA, in unstimulated (Ctrl) or stimulated cells. n = 4–8 biological replicates per genotype. In (a-c), **P < 0.01 and ***P < 0.001 versus WT unstimulated; §§P < 0.01 and §§§P < 0.001 versus WT stimulated, by one-way ANOVA with Newman-Keuls post-test. (d) Representative Western blot of CHIL3 levels in BMmacs activated with M2 stimulus for 24 hours. n = 3 biological replicates. Combined densitometry shown. In (d), *P < 0.05 versus WT stimulated by Student’s t test.
Loss of hematopoietic SLC7A2 does not promote tumorigenesis
While SLC7A2 is known as a macrophage L-Arg transporter22, our data indicate that SLC7A2 expression is mainly localized to the epithelial cells in the setting of AOM-DSS induced CAC (Figure 1). In addition, we have previously shown that SLC7A2 expression enhanced epithelial wound repair23. Thus, effects of Slc7a2 deletion observed in intra-tumoral immune cell populations could be due to its absence in either the epithelial or immune cell compartments and drive the pro-tumorigenic microenvironment alterations observed in Slc7a2–/– mice. To test whether the increased tumorigenesis was due to an SLC7A2 hematopoietic cell-autonomous effect, we exposed WT and Slc7a2–/– mice to the AOM-DSS model 8 weeks following bone marrow transplants. We confirmed the efficiency of the transplantation by genotyping (Supplementary Figure 6). However, the transfer of WT bone marrow into Slc7a2–/– recipient mice did not rescue the Slc7a2–/– AOM-DSS phenotype of increased tumorigenesis (Figure 7). Similar in overall pattern to that observed with non-transplanted mice, Slc7a2–/– mice receiving Slc7a2–/– marrow exhibited increased tumor number and tumor burden (Figure 7a, b) versus WT mice receiving WT marrow, and had similar mortality (Figure 7d). Slc7a2–/– mice receiving WT marrow and WT mice receiving Slc7a2–/– marrow phenocopied the Slc7a2–/– mice receiving Slc7a2–/– marrow and WT mice receiving WT marrow, respectively, in terms of tumor number and burden (Figure 7a, b). While no differences were seen in the histologic injury scores in the non-tumor areas between the groups (Figure 7c), Slc7a2–/– mice receiving WT marrow had dysplasia in 11/12 mice (10 low grade, 1 high grade) while WT mice receiving Slc7a2–/– marrow had dysplasia in only 3/9 mice, and all were low grade (P < 0.05, Figure 7e). There was no inflammation or epithelial injury detected in any of the control groups in the bone marrow transplant experiments (data not shown). Taken together, these data demonstrate that the exacerbated CAC phenotype in the AOM-DSS model caused by Slc7a2 deletion is mediated by the recipient mouse genotype; this indicates that the protection from CAC due to SLC7A2 derives from the non-hematopoietic cells, consistent with the predominantly epithelial SLC7A2 localization that we have observed in the AOM-DSS CAC model.
Figure 7. Bone marrow transplant between WT and Slc7a2–/– mice.
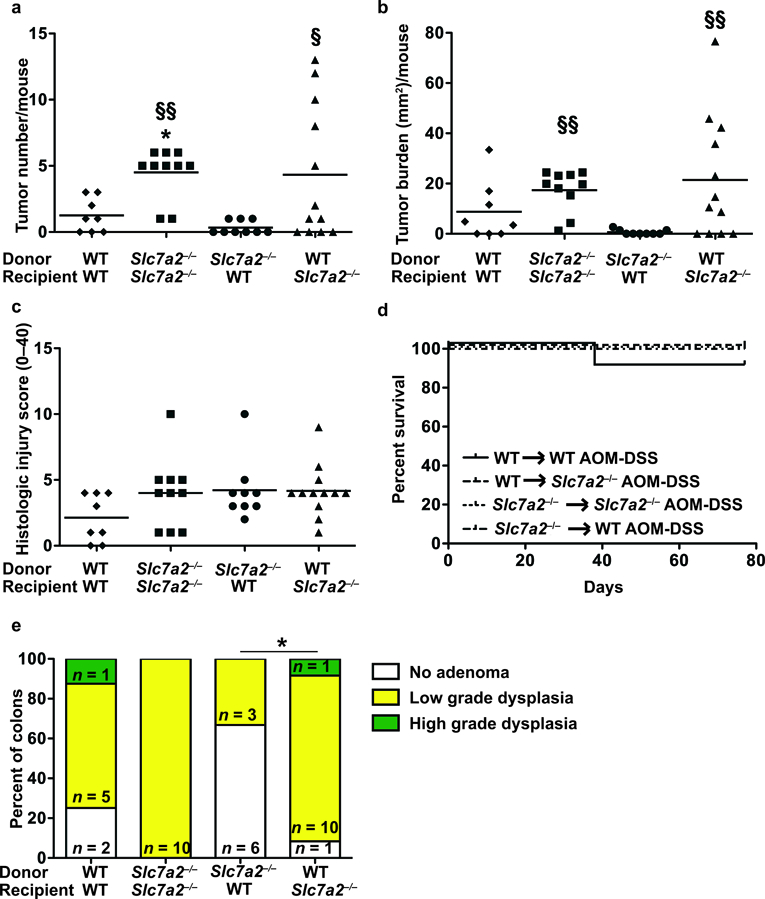
Irradiated animals were given bone marrow-derived hematopoietic cells and were treated with AOM-DSS. (a) Tumor number was assessed by gross visual inspection, utilizing a dissecting microscope. (b) Tumor burden was determined by the addition of the calculated area of each identified tumor, as assessed with an electronic caliper for both length and width. (c) Histologic colitis was determined by a gastrointestinal pathologist (M.K.W.) in a blinded manner. In (a), (b), and (c), *P < 0.05 versus WT to WT AOM-DSS; §P < 0.05 and §§P < 0.01 versus Slc7a2–/– to WT AOM-DSS, by one-way ANOVA with Student-Newman-Keuls posthoc multiple comparisons test. (d) Survival curve assessed by log-rank (Mantel-Cox) test. (e) Percentage of cases with either no adenoma, low grade dysplasia, and high grade dysplasia determined by a gastrointestinal pathologist (M.K.W.) in a blinded manner. By Chi Square test, *P < 0.05. n = 8–12 AOM-DSS-treated animals per transplant type.
Discussion
We have previously shown that SLC7A2 expression is decreased in colonic tissues from UC patients in a disease activity-specific manner as well as being decreased in patients with active CD12. Given this, we performed studies in mice lacking SLC7A2 as a model to mimic the changes identified in patients with IBD. We have reported that Slc7a2−/− mice exhibit profound exacerbation of DSS-induced colitis18, and that colonic epithelial restitution is dependent on the transport of L-Arg into cells by SLC7A223. Based on the exacerbation of acute colitis and alteration in epithelial restitution identified in the setting of loss of SLC7A2, we hypothesized that SLC7A2 may play a key role in modulating the risk for colon tumorigenesis as the inflammatory response is thought to drive the development of tumors in the AOM-DSS model37, 41, and disease duration of UC in humans is associated with increased risk for colonic dysplasia and carcinoma42.
In this study, we demonstrate that Slc7a2–/– mice have dramatically increased tumor number and burden, epithelial proliferation, and likelihood of high-grade dysplasia compared to WT mice in the AOM-DSS model. Furthermore, this phenotype was associated with significant increases in pro-inflammatory cytokines, the majority that were further increased in the Slc7a2–/– tumors are chemokines, which recruit immune cells to the site of injury or insult. It is possible that the infiltrating monocytes/macrophages are predisposed to M2 polarization, and we found that under polarizing conditions Slc7a2–/– BMmacs exhibited a propensity toward an M2 (tumor-associated) macrophage phenotype, not an M1 (proinflammatory, antitumor) macrophage phenotype. Since both M1 and M2 stimuli led to increased L-Arg uptake that was abrogated with Slc7a2 deletion, it may be that SLC7A2 modulates macrophage response via other methods than just L-Arg uptake. It should be noted that when we attempted to determine the effect of Slc7a2 loss on isolated colonic macrophages, there was not a sufficient yield of cells to conduct the L-Arg uptake assay. Notably, we did see a similar shift in the colonic tissues from Slc7a2–/– mice, as indicated by a shift in L-Arg metabolism from NOS2 to ARG1 by flow cytometry.
However, this phenotype is not an immune cell autonomous effect. Bone marrow transplant studies indicated that the increased tumorigenesis relies on loss of epithelial Slc7a2 expression, which is consistent with our prior findings that L-Arg enhances epithelial restitution relying on SLC7A218. Thus, it may be that the epithelial loss of SLC7A2 alters the signaling milieu present, leading to M2 polarization, which may result in less host surveillance and a tumor microenvironment that supports growth of aberrant epithelial cells. Our data do suggest that SLC7A2 contributes to regulation of macrophage polarization and that the driving force of the epithelial differences in the AOM-DSS model may be a key regulator of that process. Interestingly, in the acute DSS model, SLC7A2 was more robustly expressed in colonic macrophages18, whereas, we now show in the chronic AOM-DSS model that SLC7A2 expression is mainly in the epithelial cells with modest lamina propria cell expression. This may indicate a shift in the expression profile of SLC7A2 in the setting of acute versus chronic inflammation. We have previously shown that L-Arg supplementation is beneficial in acute DSS colitis10, but that this benefit was lost in the setting of total tissue knockout of Slc7a218.
In our IBD patients, we have shown variable expression of SLC7A2, not absolute deficiency12. Interestingly, when SLC7A2 polymorphisms were assessed in patients with colorectal adenomas and healthy controls, no polymorphisms were directly associated with risk for adenoma development43. However, the rs2720574 polymorphism was found to significantly interact with the ratio of calcium to magnesium intake leading to altered odds of adenoma development, particularly the development of multiple or advanced adenomas43. However, neither SLC7A2 expression nor function were assessed in this cohort, thus further studies are needed. It should be noted that we have tested all commercially available antibodies (10 antibodies from 6 different companies), and we have not been able to successfully obtain immunohistochemical detection of SLC7A2 in human colon tissues, thus it has not been possible to immunolocalize SLC7A2 in humans with IBD or CAC. However, our previous SLC7A2 mRNA and tissue L-Arg quantification in IBD tissues, showing diminished SLC7A2 function, clearly supports the relevance of our current findings. While studies to further assess the role that SLC7A2 plays in patients at risk for progression of IBD to colon cancer are needed, adjunctive approaches including L-Arg supplementation or other methods to enhance colonic L-Arg uptake may represent a potential therapeutic strategy in patients with IBD to reduce the risk for development of CAC.
Materials and Methods
Reagents
Cell culture reagents were obtained from Invitrogen (Carlsbad, CA, USA). RNA extraction reagents were purchased from Qiagen (Valencia, CA, USA). Reagents for cDNA synthesis and qRT-PCR were from Bio-Rad (Hercules, CA, USA). Murine M-CSF, IFN-γ, IL-4, and IL-10 were obtained from PeproTech (Rocky Hill, NJ, USA). Azoxymethane and LPS were obtained from Sigma-Aldrich (St. Louis, MO, USA). The DSS used was purchased from TdB Consultancy (Uppsala, Sweden). 14C L-Arg was obtained from Perkin-Elmer (Waltham, MA, USA).
Antibodies
Detailed information about the antibodies utilized is provided in Supplementary Table 2.
Cells and Culture Conditions
BMmacs were isolated and differentiated from murine femurs as previously described37, 38, 44. Differentiated BMmacs were stimulated with classical M1 stimuli, IFN-γ (200 U/mL) and LPS (100 ng/mL), or M2 stimuli, IL-4 (10 ng/mL) and IL-10 (10 ng/mL), for 24 hours as described37.
Animal Studies
WT and Slc7a2–/– C57BL/6 mice were utilized as described18, 19. WT and Slc7a2–/– bone marrow chimera mice were created, and this was confirmed using DNA isolated from spleens as described19. The primers for Slc7a2 and the bacterial neomycin resistance cassette (neo) genes are provided in Supplementary Table 3. Representative genotyping is shown in Supplementary Figure 6.
These studies were approved by the Institutional Animal Care and Use Committee at Vanderbilt University and the Research and Development Committee of the Veterans Affairs Tennessee Valley Healthcare System under IACUC protocol M/08/124.
Samples sizes were determined based on our prior experience with AOM-DSS studies37, 38, 44, 45. All studies were performed in male mice, aged 7–12 weeks. The mice remained in the cages they were weaned into with no other selection or randomization performed. WT, Slc7a2–/–, and bone marrow chimera mice were subjected to the AOM-DSS colon tumorigenesis model37, 45. Mice received one intraperitoneal AOM injection (12.5 mg/kg) on Day 0 followed by three cycles of 4% or 2.5% DSS in their drinking water on Days 5, 26, and 47 as described37, 38, 44, 45. In addition, a subset of WT and Slc7a2–/– mice were subjected to an injury and recovery model of DSS as described46.
At the time of sacrifice, numbered colons were assessed randomly by visual inspection using a dissecting microscope to determine tumor multiplicity and tumor burden as described37, 45, so the investigator was blinded to the genotype and the treatment. The histologic injury score and severity of dysplasia were determined by a gastrointestinal pathologist, M.K.W., in a blinded manner37, 45.
Immunohistochemistry Staining for SLC7A2
Staining for SLC7A2 in mouse colonic tissues was performed as described19.
Luminex Multiplex Array
Colon tissues were harvested at the time of sacrifice and assessed using a 32-analyte MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (MCYTMAG-70K-PX32, EMD Millipore, Billerica, MA) with a FLEXMAP 3D instrument (Luminex, Austin, TX) 10, 18, 19, 37, 45. Data were standardized to tissue protein concentrations measured by the bicinchoninic acid (BCA) Protein Assay Kit (Pierce, Rockford, IL) 10, 18, 19, 37, 45.
Isolation of colonic lamina propria cells
Colonic lamina propria immune cells were isolated by enzymatic digestion as described previously18, 19, 45.
Identification and Assessment of colonic macrophages by flow cytometry
Isolated lamina propria cells were analyzed for CD11b and F4/80 with NOS2 or ARG1 by flow cytometry as described18 using the antibodies listed in Supplementary Table 2.
Immunohistochemistry Staining for Ki67 and Image Analysis
Staining for Ki67 was performed as described47. Immunostained tissue slides were imaged, and positive cells were identified using standard Ariol® analysis scripts (Leica) as described48.
Immunofluorescence Staining for ARG1, NOS2, and CD68
Staining for ARG1, NOS2, and CD68 in murine tissues was performed as described18, 37 using the antibodies listed in Supplementary Table 2.
RNAscope
RNAscope assessment of Slc7a2 expression in mouse colon tissues was performed as described19.
L-Arg Uptake
14C L-Arg uptake studies in BMmacs and whole colon tissue were performed as described18.
Real-Time PCR
RNA was isolated from cells and tissues as described10, 37. cDNA was prepared and PCR was performed as described10, 37. All primers used in this study are listed in Supplementary Table 3.
Western Blot Analysis
Western blotting was performed as described37, 38. The primary and secondary antibodies utilized are listed in Supplementary Table 2. Densitometry was performed using ImageStudio, version 4.0.21 (LI-COR)38.
Measurement of NO2−
The concentration of nitrite (NO2−), the oxidized metabolite of NO, was assessed by the Griess reaction as previously described32–34, 37–40.
IL-1β ELISA
IL-1β was assessed using an R&D DuoSet ELISA kit (DY401) according to kit instructions.
Statistical Analysis
Data are displayed as mean ± S.E.M. Outlier testing was done using the Grubbs method, also known as the extreme studentized deviate method49. As previously described, when comparisons between multiple groups were made, analysis of variance with the Student-Newman-Keuls posthoc multiple comparisons test was performed10. When data were not normally distributed, log or square root transformation was performed45. When comparisons between only 2 groups were made, Student’s t test was performed10. All survival analyses were performed using the log-rank (Mantel-Cox) test10. The significance of the number of cases of mice displaying dysplasia grades was determined by using the χ2 test. All statistics were performed in Prism 5.0 (GraphPad Software, San Diego, CA, USA). A P value of < 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by National Institutes of Health (NIH) grants R01AT004821, 3R01AT004821–02S1, R01DK053620, R01CA190612, P01CA116087, and P01CA028842 (K.T.W), Veterans Affairs Merit Review grant I01BX001453 (K.T.W.), the Thomas F. Frist Sr. Endowment (K.T.W.), and the Vanderbilt Center for Mucosal Inflammation and Cancer (K.T.W.). L.A.C. was supported by NIH training grant 5T32DK007673, a Vanderbilt Physician Scientist Development Award, and a Veterans Affairs Career Development Award 1IK2BX002126. D.M.H. was supported by T32GM008554 and F31DK10715. Additional support was provided by NIH Grant P30DK058404 (Vanderbilt Digestive Disease Research Center). Whole slide imaging and quantification were performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center (http://www.mc.vanderbilt.edu/dhsr).
Footnotes
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
References:
- 1.Shivashankar R, Tremaine WJ, Harmsen WS, Loftus EV Jr. Incidence and prevalence of Crohn’s disease and ulcerative colitis in Olmsted County, Minnesota from 1970 through 2010. Clin Gastroenterol Hepatol 2017; 15: 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brower V Feeding the flame: new research adds to role of inflammation in cancer development. J Natl Cancer Inst 2005; 97: 251–253. [DOI] [PubMed] [Google Scholar]
- 3.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol 2009; 6: 297–305. [DOI] [PubMed] [Google Scholar]
- 4.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology 2010; 138: 2101–2114 e2105. [DOI] [PubMed] [Google Scholar]
- 5.Herrinton LJ, Liu L, Levin TR, Allison JE, Lewis JD, Velayos F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 2012; 143: 382–389. [DOI] [PubMed] [Google Scholar]
- 6.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet 2007; 369: 1627–1640. [DOI] [PubMed] [Google Scholar]
- 7.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 2007; 369: 1641–1657. [DOI] [PubMed] [Google Scholar]
- 8.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011; 474: 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M et al. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004; 126: 451–459. [DOI] [PubMed] [Google Scholar]
- 10.Coburn LA, Gong X, Singh K, Asim M, Scull BP, Allaman MM et al. L-arginine supplementation improves responses to injury and inflammation in dextran sulfate sodium colitis. PLoS One 2012; 7: e33546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gobert AP, Cheng Y, Akhtar M, Mersey BD, Blumberg DR, Cross RK et al. Protective role of arginase in a mouse model of colitis. J Immunol 2004; 173: 2109–2117. [DOI] [PubMed] [Google Scholar]
- 12.Coburn LA, Horst SN, Allaman MM, Brown CT, Williams CS, Hodges ME et al. L-Arginine Availability and Metabolism Is Altered in Ulcerative Colitis. Inflamm Bowel Dis 2016; 22: 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose WC. Amino acid requirements of man. Fed Proc 1949; 8: 546–552. [PubMed] [Google Scholar]
- 14.Castillo L, Ajami A, Branch S, Chapman TE, Yu YM, Burke JF et al. Plasma arginine kinetics in adult man: response to an arginine-free diet. Metabolism 1994; 43: 114–122. [DOI] [PubMed] [Google Scholar]
- 15.Castillo L, Chapman TE, Sanchez M, Yu YM, Burke JF, Ajami AM et al. Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets. Proc Natl Acad Sci U S A 1993; 90: 7749–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakuda DK, Sweet MJ, Mac Leod CL, Hume DA, Markovich D. CAT2-mediated L-arginine transport and nitric oxide production in activated macrophages. Biochem J 1999; 340 ( Pt 2): 549–553. [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholson B, Manner CK, Kleeman J, MacLeod CL. Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J Biol Chem 2001; 276: 15881–15885. [DOI] [PubMed] [Google Scholar]
- 18.Singh K, Coburn LA, Barry DP, Asim M, Scull BP, Allaman MM et al. Deletion of cationic amino acid transporter 2 exacerbates dextran sulfate sodium colitis and leads to an IL-17-predominant T cell response. Am J Physiol Gastrointest Liver Physiol 2013; 305: G225–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh K, Al-Greene NT, Verriere TG, Coburn LA, Asim M, Barry DP et al. The L-Arginine transporter solute carrier family 7 member 2 mediates the immunopathogenesis of attaching and effacing bacteria. PLoS Pathog 2016; 12: e1005984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coman D, Yaplito-Lee J, Boneh A. New indications and controversies in arginine therapy. Clin Nutr 2008; 27: 489–496. [DOI] [PubMed] [Google Scholar]
- 21.Mori M Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr 2007; 137: 1616S–1620S. [DOI] [PubMed] [Google Scholar]
- 22.Martin L, Comalada M, Marti L, Closs EI, MacLeod CL, Martin del Rio R et al. Granulocyte-macrophage colony-stimulating factor increases L-arginine transport through the induction of CAT2 in bone marrow-derived macrophages. Am J Physiol Cell Physiol 2006; 290: C1364–1372. [DOI] [PubMed] [Google Scholar]
- 23.Singh K, Coburn LA, Barry DP, Boucher JL, Chaturvedi R, Wilson KT. L-arginine uptake by cationic amino acid transporter 2 is essential for colonic epithelial cell restitution. Am J Physiol Gastrointest Liver Physiol 2012; 302: G1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hol J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J Leukoc Biol 2010; 87: 501–508. [DOI] [PubMed] [Google Scholar]
- 25.Chandrasekar B, Deobagkar-Lele M, Victor ES, Nandi D. Regulation of chemokines, CCL3 and CCL4, by interferon gamma and nitric oxide synthase 2 in mouse macrophages and during Salmonella enterica serovar typhimurium infection. J Infect Dis 2013; 207: 1556–1568. [DOI] [PubMed] [Google Scholar]
- 26.Zimmerman NP, Vongsa RA, Wendt MK, Dwinell MB. Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflamm Bowel Dis 2008; 14: 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh K, Chaturvedi R, Barry DP, Coburn LA, Asim M, Lewis ND et al. The apolipoprotein E-mimetic peptide COG112 inhibits NF-kappaB signaling, proinflammatory cytokine expression, and disease activity in murine models of colitis. J Biol Chem 2011; 286: 3839–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A 2009; 106: 14978–14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valbuena G, Bradford W, Walker DH. Expression analysis of the T-cell-targeting chemokines CXCL9 and CXCL10 in mice and humans with endothelial infections caused by rickettsiae of the spotted fever group. Am J Pathol 2003; 163: 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rmali KA, Puntis MC, Jiang WG. Tumour-associated angiogenesis in human colorectal cancer. Colorectal Dis 2007; 9: 3–14. [DOI] [PubMed] [Google Scholar]
- 31.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 1995; 182: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosser DM. The many faces of macrophage activation. J Leukoc Biol 2003; 73: 209–212. [DOI] [PubMed] [Google Scholar]
- 34.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014; 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erreni M, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron 2011; 4: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Isidro RA, Appleyard CB. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol 2016; 311: G59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardbower DM, Coburn LA, Asim M, Singh K, Sierra JC, Barry DP et al. EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene 2017; 36: 3807–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagomez D, Barry DP et al. EGFR regulates macrophage activation and function in bacterial infection. J Clin Invest 2016; 126: 3296–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hardbower DM, Asim M, Luis PB, Singh K, Barry DP, Yang C et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc Natl Acad Sci U S A 2017; 114: E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP et al. Polyamines impair immunity to Helicobacter pylori by inhibiting L-arginine uptake required for nitric oxide production. Gastroenterology 2010; 139: 1686–1698, 1698 e1681–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett CW, Fingleton B, Williams A, Ning W, Fischer MA, Washington MK et al. MTGR1 is required for tumorigenesis in the murine AOM/DSS colitis-associated carcinoma model. Cancer Res 2011; 71: 1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med 2015; 372: 1441–1452. [DOI] [PubMed] [Google Scholar]
- 43.Sun P, Zhu X, Shrubsole MJ, Ness RM, Hibler EA, Cai Q et al. Genetic variation in SLC7A2 interacts with calcium and magnesium intakes in modulating the risk of colorectal polyps. J Nutr Biochem 2017; 47: 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardbower DM, Asim M, Murray-Stewart T, Casero RA Jr., Verriere T, Lewis ND et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to Helicobacter pylori infection. Amino Acids 2016; 48: 2375–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh K, Coburn LA, Asim M, Barry DP, Allaman MM, Shi C et al. Ornithine decarboxylase in macrophages exacerbates colitis and promotes colitis-associated colon carcinogenesis by impairing M1 immune responses. Cancer Res 2018; doi: 10.1158/0008-5472.CAN-18-0116. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gobert AP, Al-Greene NT, Singh K, Coburn LA, Sierra JC, Verriere TG et al. Distinct immunomodulatory effects of spermine oxidase in colitis induced by epithelial injury or infection. Front Immunol 2018; 9: 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagy TA, Wroblewski LE, Wang D, Piazuelo MB, Delgado A, Romero-Gallo J et al. beta-Catenin and p120 mediate PPARdelta-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011; 141: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sierra JC, Asim M, Verriere TG, Piazuelo MB, Suarez G, Romero-Gallo J et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut 2018; 67(7):1247–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grubbs FE. Procedures for detecting outlying observations in samples. Technometrics 1969; 11: 1–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.