

Abstract
The contribution of gut microbiota to human health and diseases has expanded our insights into how microbial composition and function impacts the human host. Heart failure has long been associated with splanchnic circulation congestion, leading to bowel wall edema and impaired intestinal barrier function. This is thought to heighten the overall inflammatory state via enhanced bacterial translocation and the presence of bacterial products in the systemic circulation. Recently, several metabolites produced by gut microbes from dietary metabolism have also been linked to pathologies such as atherosclerosis, hypertension, heart failure, chronic kidney disease, obesity and type 2 diabetes mellitus. These findings suggested that the gut microbiome functions like an endocrine organ by generating bioactive metabolites that can directly or indirectly impact host physiology. We will discuss several newly discovered gut microbial metabolic pathways, including the production of trimethylamine (TMA)/ trimethylamine N-oxide (TMAO), short-chain fatty acids, and secondary bile acids, that appear to participate in the development and progression of cardiovascular diseases, including heart failure. We will also discuss the gut microbiome as a novel therapeutic target for the treatment of cardiovascular disease, and potential strategies for targeting intestinal microbial processes.
Keywords: Gut microbiota, diet, heart failure
Introduction
Heart failure is the final common process that results from a variety of initial cardiac insults and subsequent imbalances in compensatory mechanisms and pathogenic processes1,2. It has long been recognized that heart failure is associated with altered intestinal function3,4. While this “gut hypothesis” in heart failure has prevailed over the years, the emphasis has focused on enhanced gut bacterial translocation and resultant heightened inflammatory responses as well as oxidative stress as a consequence of heart failure-induced ischemia and congestion within the intestines. However, research studies supporting the “gut hypothesis” thus far are associative in nature and have not demonstrated a causal link between gut microbial processes and heart failure pathogenesis. Also, the role of nutrition and diet in the causation, prevention, and management of heart failure remains poorly understood.
At birth, the human intestinal tract is relatively sterile, but as we are abruptly and continuously exposed to our environment, the gut becomes rapidly colonized with trillions of typically non-pathogenic bacteria (1013–1015)5–8. The stomach, due to its low pH, contains relatively few organisms, while the small intestines contain aero-tolerant microbes, mainly streptococci, and lactobacillus5,9. The colon, with a microbial density of 1011 to 1012 organisms per milliliter, is one of the most densely populated bacterial habitats on Earth5,7. In fact, gut microbiota contributes between 0.2 and 2 kg of the weight of a typical adult and constitutes nearly 50% of the dry weight of feces5,8. Gut microbiota remain in a symbiotic relationship within us, their human hosts, and provide crucial contributions that directly shape our health. For example, gut microbiota synthesize vitamin K and biotin, facilitate absorption of key nutrients, promote and shape the development of the mucosal immune system, and influence oral tolerance to ingested antigenic foods10–13. Gut microbiota also promote an essential barrier function by repressing the overgrowth or colonization of potentially harmful bacteria through competitive exclusion and production of diverse chemical mediators, such as quorum-sensing compounds and bacteriocins, which collectively keep the microbiota environment in check14–16.
A typical healthy human gut is colonized by over 500 different microbial species, and an even larger number of microbial strains17,18. Humans thus inherently possess an extended genome of microbial genes and functions that are collectively known as the microbiome. The number of genes within the gut microbiome is estimated to exceed the number in the human genome by two orders of magnitude. Also, while many gut community structural features are “inherited” early in life, especially from one’s mother during vaginal delivery19–21, the composition of gut microbiota varies considerably among individuals and host genetic variations may predispose individuals towards altered microbiota composition8,22. Due to this diversity in composition, the microbiome, in addition to host genetics, plays a significant role in individualized responses to major environmental exposures including our diet, xenobiotics and drug treatments23–25. The gut microbiome thus plays a role in person to person variations in disease susceptibility, and in responses to our diet25. Gut microbiota also impact how one metabolizes drugs, and can contribute to the production of pharmacologically active secondary metabolites that can foster adverse side effects or chemotherapeutic drug toxicities26,27.
In this Review, we discuss the long-standing hypothesis of how the modulation of inflammation by the gut barrier participates in the development of heart failure. We examine the emerging evidence for an altered gut microbiome environment in heart failure. Moreover, we discuss several notable gut microbiota mediated metabolic pathways and products that have recently been revealed to be involved in heart failure. These pathways and products include the trimethylamine (TMA)/ trimethylamine Noxide (TMAO) pathway, short-chain fatty acids pathway, and bile acid pathways. Finally, we consider potential therapeutic strategies targeting the intestinal microbiota to influence the susceptibility and progression of cardiovascular disease.
Impaired gut barrier function and inflammation in heart failure
Typically, a “healthy” gut microbial community maintains gut barrier function, including intact, tight junctions in the mucosa and normal mucosal immunity. Mice lacking adherens junction proteins will spontaneously develop colitis, highlighting the importance of cell-to-cell adhesion in maintaining a healthy mucosal barrier28. The current “leaky gut” hypothesis of heart failure3,4 posits that bowel wall edema and impairment in barrier function during heart failure leads to translocation of gut microbiota components into the host circulation, endotoxemia, and consequently, a heightened systemic inflammatory state29 (FIG. 1). Evidence exists wherein heightened bacterial translocation occurs during heart failure as a consequence of one or more mechanisms including structural and functional alterations of the gastrointestinal tract that occur with splanchnic congestion, as well as with abnormalities of host immunological defense.
Figure 1. Microbial-host meta-organismal pathway linking dietary metabolism, gut microbiota, and cardio-renal disease progression.
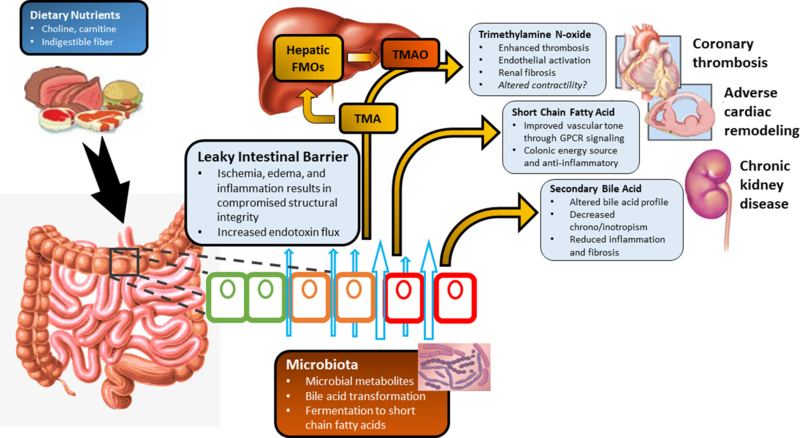
Legend: Poor cardiac output in heart failure results in intestinal ischemia, edema, and inflammation which leads to a “leaky” intestinal barrier. This allows for increased passage of inflammatory bacterial products to enter the bloodstream causing chronic low-grade inflammation. Furthermore, this alters the intestinal environment and impacts both the normal microbial community which resides in the gut and subsequently, the metabolic products from these bacteria. The metabolic pathways include fermentation of indigestible fiber to short chain fatty acids which have protective properties reducing inflammation and improving vascular tone. Dietary sources including choline, phosphatidylcholine, l-carnitine and other methylamine-containing nutrients provide substrates for microbiota mediated production of trimethylamine (TMA). TMA then enters the portal circulation and is converted by the hepatic host flavin-containing monooxygenase (FMO) family of enzymes to trimethylamine n-oxide (TMAO). TMAO can promote the development of atherogenesis, thrombosis, kidney disease, and heart failure. Additionally, the bacterial transformation of bile acids can result in altered bile acid profiles which then can affect systemic inflammatory and fibrotic processes. Collectively, these processes can influence the individual susceptibility, severity of heart failure.
An altered intestinal barrier function can arise from multiple factors, including as a consequence of a hypoperfused gut that allows gut microbiota components to be translocated into the bloodstream30. Decreased cardiac output and the resulting sympathetic vasoconstriction in heart failure stimulates adaptive re-distribution of the systemic circulation, leading to congestion in multiple end organs, including the bowel wall31. A decrease in perfusion particularly affects the structure of villi in the intestinal mucosa, which is susceptible to ischemia due to the countercurrent circulation present in the villus: the artery and vein run in parallel, thereby creating a descending tissue gradient in oxygen pressure and/or tension from the base to the tip of the villus32. As a result, intramucosal acidosis can be observed in approximately half of patients with decompensated heart failure33. Moreover, even light activity is observed to increase intragastric pCO2 in those with chronic heart failure34. Structurally, intestinal wall thickening with edema can be observed in patients with heart failure29. The collagen content in their mucosal walls is also increased, in proportion to the severity of heart failure35. Of interest, bowel wall thickening has been directly correlated with circulating levels of C-reactive protein, blood leukocytes, and increased markers of intestinal permeability29. Thus, a combination of both structural and functional intestinal alterations directly contribute to worsened enterocyte health, and as a result, the intestinal barrier integrity. Therefore, increased paracellular transport can be observed in heart failure29.
Compromise of the intestinal barrier can allow endotoxin, also known as lipopolysaccharide (LPS), to readily enter the bloodstream. LPS is found on the outer membrane of Gram-negative bacteria. It is a classic pathogen-associated molecular pattern molecule and can induce the expression of a wide array of inflammatory downstream products primarily through the Toll-like receptor 4 (TLR4) pattern recognition receptor36. Moreover, this effect occurs through multiple tissues and cell types, such as macrophages, dendritic cells, cardiomyocytes and cardiac fibroblasts37. Patients with decompensated heart failure have higher blood levels of endotoxin38. Moreover, targeted sampling studies have shown that LPS levels are higher in blood samples recovered from the hepatic vein as opposed to the systemic circulation (direct sampling from the ventricles), providing direct evidence that LPS can be translocated from the gut39. Additionally, LPS itself can further promote deterioration of mucosal barrier function40.
There is evidence that the intestinal absorption of endotoxin is a significant stimulus for systemic increases in inflammatory cytokines during heart failure41,42. In heart failure subjects, heightened circulating levels of multiple cytokines (e.g., TNF, IL-1, and IL-6) are associated with more severe clinical symptoms and can predict poorer patient survival43–45. Interestingly, while endotoxin levels in patients with heart failure have been shown to be reduced with diuretic or antibiotic therapy, plasma cytokine levels have not always been observed to change, suggesting a sustained effect as the disease process is triggered38,46. Strong preclinical evidence exists for these inflammatory mediators to participate in cardiac apoptosis, hypertrophy, and fibrosis47. For example, in mice, administration of TNF at pathologic concentrations is reported to result in a dilated cardiomyopathy phenotype48. Furthermore, transgenic overexpression of TNF in another mouse study resulted in cardiac hypertrophy, ventricular dilation, and fibrosis49.
Increased serum concentrations of TNF, IL-1β, and IL-6 have also been shown to be potent inducers of intestinal permeability50–53. This increased permeability complements the effects of endotoxin in the circulation and promotes a vicious feedforward cycle of increased endotoxin translocation and inflammatory cytokine accumulation. However, the results of clinical studies that tested this hypothesis (including numerous anti-TNF trials) reveal additional complexity (e.g., The Randomized Etanercept North American Strategy to Study Antagonism of Cytokines (RENAISSANCE), the Research into Etanercept Cytokine Antagonism in Ventricular Dysfunction (RECOVER), and Anti-TNF in Congestive Heart Failure (ATTACH))54,55. In brief, these studies revealed negative results and perhaps even increased cardiovascular events in the treatment group. By contrast, the Advance Chronic Heart Failure Clinical Assessment of Immune Modulation (ACCLAIM) trial utilized nonspecific immunomodulation therapy56. In this study, patient blood was stressed (via a combination of heat, ozonation and ultraviolet irradiation) to induce cell death, and the resulting material is re-injected into the patient to produce an anti-inflammatory stimulus. Although the primary endpoint of reduction in all-cause mortality or cardiovascular admission in this RCT was not met, the subgroup of patients with NYHA functional class II chronic heart failure (n=919) appeared to have significant reductions in all-cause mortality and hospital admissions for cardiovascular causes.
Although not in a heart failure cohort, groundbreaking findings from the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial, which involved the use of canakinumab, anIL-1β receptor antagonist, demonstrated for the first time in a large randomized clinical trial that targeting inhibition of the inflammatory pathway (through at least the IL-1β pathway) can reduce the risk of incident adverse cardiovascular events independently of lipid lowering57. In addition to an anti-inflammatory effect, it is conceivable that inhibiting the IL-1β receptor can also reduce intestinal epithelial tight junction permeability, leading to cardiovascular benefits due to its modulation of the microbiota. Additional studies (initiated after positive proof of concept findings58–61) involving both canakinumab in a subgroup of CANTOS patients, as well as anakinra (which blocks both IL-1α and IL-1β receptors), are underway to explore alternative anti-inflammatory targets that may benefit heart failure patients62,63.
In the gut hypothesis of heart failure, much focus has been placed on understanding how inflammation triggers heart failure and other related comorbidities. However, as we described, investigations of inflammatory cytokines have resulted in mixed evidence from clinical studies. The range of clinical findings demonstrate that targeting the inflammatory process through cytokine-driven pathways yields varied results; however, these undoubtedly complex inflammatory pathways are not discussed further as they have been thoroughly reviewed elsewhere64,65. These studies raise further interest in investigating the role of endotoxin-induced inflammation and the microbiota in the pathogenesis of heart failure. Indeed, we hope that the knowledge gained about the gut microbiome and its links to heart failure will lead to better risk stratification of heart failure subjects, as well as helping to identify novel pharmacologic strategies that target the microbiome and improve human health.
Dysbiosis in heart failure
The intestine is unique in its constant exposure to trillions of microbial antigens; this creates an environment constantly at risk of barrier dysfunction. The microbiota might be able to directly modify the intestinal barrier66,67. Therefore, a better understanding of the gut microbiota (a primary contributor to underlying inflammation) will help to tease out beneficial net effects for heart failure in this complex multi-level network.
Changes in the proportions of specific taxa of gut microbiota have been observed in various disease states, including obesity68,69, inflammatory bowel disease70, type 1 and type 2 diabetes mellitus71,72, biliary disease73, and intestinal cancers74,75 among many others. Animal studies have shown that the intestinal microbial environment can affect the efficiency of harvesting energy from the diet and affect obesity69. It has been demonstrated that the gut microbiota can play a role in the development of metabolic abnormalities such as insulin resistance and nonalcoholic fatty liver disease76,77. Furthermore, psychiatric disorders, including autism and depression, have also been associated with alterations in gut microbiota in what is known as the “gut-brain hypothesis.”78–80
Research has now gone further by demonstrating a mechanistic link via direct microbial transplantation, and evidence of transmission of disease susceptibility. Turnbaugh et al. demonstrated in a seminal study that adiposity could be transferred by microbiota transplantation69. In the setting of cardiovascular disease, both atherosclerosis and increased platelet activation were shown to be transmittable with microbial transplantation and linked to elevated trimethylamine N-oxide (TMAO) production, a novel microbiota-obligate metabolite that we will review in detail in later sections81,82. Moreover, several studies have demonstrated that microbes can transmit alterations in vascular tone and blood pressure. Pluznick et al. demonstrated that microbiota could produce SCFA to modulate blood pressure through short-chain fatty acid interaction with the host G protein-coupled receptors (GPCRs) Olfr78 and Gpr41; antibiotic treatment can then alter blood pressure in Olfr78 knockout mice83. A salt-sensitive rat model with cecal microbial transplant demonstrated altered blood pressure and longevity in the recipient84. Similarly, an obstructive sleep apnea (OSA) model also demonstrated that cecal microbial transplantation from the OSA mice transmitted elevated blood pressure to the recipient85. Finally, a probiotic therapy (Goodbelly) study reported that delivery of Lactobacillus plantarum 299v reduced circulating leptin and improved ventricular function and remodeling after left anterior descending artery ligation86. However, there have only been a limited number of studies examining the role of gut microbiota in heart failure.
Alterations in gut microbial composition
Several small cohort studies have demonstrated that alterations in microbial communities are present in heart failure patients (Table 2). In addition to correlations with inflammation and intestinal permeability, Sandek et al. also observed bacterial overgrowth consisting of mucosal biofilm and increased bacterial adhesion in heart failure patients29,87. Newer studies showed that more pathogenic microbes such as Candida, Campylobacter, Shigella, and Yersinia could be detected in the stools of heart failure patients and correlated with heart failure severity88. Supporting these findings, using a national inpatient data registry, Mamic et al. observed significant increases in Clostridium difficile infections amongst patients with a heart failure diagnosis at discharge after controlling for relevant patient and hospital characteristics89. Moreover, across these hospitalizations, patients admitted with urinary tract infections, pneumonia, or sepsis with concomitant C. difficile infection had higher in-hospital mortality compared to those without89.
Table 2.
Summary of Contemporary Studies Investigating Heart Failure and Alterations in Microbiota
| Author | Sample Groups and Measurements | Results Summary |
|---|---|---|
| Sandek et al.29 | FISH of mucosal biofilm samples (20 CHF, 22 Ctrl) collected by sigmoidoscopy | Elevated bacterial concentration and increased strain diversity of mucosal biofilm in CHF patients; Increased detection frequency (from 38 FISH probes) of Bacteroides/Prevotella, Eubacterium Rectale, Faecalibacterium Prausnitzii in CHF; Increased gut permeability as measured by lactulose-mannitol test. |
| Sandek et al.87 | FISH of mucosal bacterial film (22 CHF; 20 Ctrl) collected by sigmoidoscopy; Stool samples (21 CHF; 17 Ctrl) | Concentrations and proportions of both anaerobic and aerobic bacteria in the stool were not significantly different; Trend of positive correlation between increased anaerobic juxtamucosal bacteria and decreased intestinal blood flow. |
| Pasini et al.88 | Stable CHF patients with NYHA I-II (n=30), NYHA III-IV, (n=30), and matched healthy control (n=20) had stool samples collected to measure bacteria and Candida species using traditional culture techniques. | The CHF population (NYHA III-IV in particular) had large increases in detected pathogenic bacteria including Campylobacter, Shigella, Salmonella, Yersinia enterolytica, and Candida species; Increased gut permeability as measured by cellobiose sugar test |
| Mamic et al.89 | 2012 Healthcare cost and utilization project National Inpatient Sample data | Clostridium difficile infection rates were higher in hospitalizations with discharge diagnosis of HF compared with those without HF after controlling for patient demographics and comorbidities and hospital characteristics. |
| Luedde et al.90 | Bacterial 16S rRNA gene sequencing of fecal samples from 20 patients with heart failure with reduced ejection fraction due to ischemic or dilated cardiomyopathy and matched controls selected from the PopGen study. | Tendency for decreased bacterial diversity in HF with significant differentiation between HF and control patients; Those with HF showed significant decreases in Coriobacteriaceae, Erysipelotrichaceae and Ruminococcaceae on the family level and decreases in Blautia, Collinsella, uncl. Erysipelotrichaceae and uncl. Ruminococcaceae on the genus level. |
| Cui et al.91 | Bacterial 16S rRNA gene sequencing of fecal samples from consecutive recruitment of 53 ischemic and dilated cardiomyopathy CHF patients (94% of NYHA III-IV) and 41 controls. | Significant differentiation between CHF and controls but similar between ischemic and dilated cardiomyopathy patients; Faecalibacterium prausnitzii decrease and Ruminococcus gnavus increase were the essential characteristics identified in this CHF patient cohort. |
| Kamo et al.92 | Bacterial 16S rRNA gene sequencing of fecal samples from 12 HF patients and 12 age-matched controls; 12 HF patients younger than 60 and 10 HF patients 60 or older. | Significant differentiation between HF and controls; Those with HF showed significant decreases in Clostridium and Dorea at the genus level and decreases in Eubacterium Rectale and Dorea longicatena at the species level; Older HF patients had depletion of Faecalibacterium and enriched Lactobacillus genus compared to younger HF patients. |
| Kummen et al.93 | 2 independent cohorts of stable HFrEF cohorts (discovery, n = 40; and validation, n = 44; NYHA II–IV) and population-based control subjects (n = 266, randomly allocated to HF cohorts for comparison). | Decreased bacterial diversity in HF even after risk factor adjustments; Increase in genus Prevotella, Hungatella, Succinclasticum; Decrease in multiple genera belonging to Lachnospiraceae family - Anaerostipes, Blautia, Coprococcus (3), Fusicatenibacter, Lachnospiraceae FCS020, NCS2004, ND3007, Pseudobutyrivibrio, Eubacterium Hallii group; Decrease in Rumminococcaceae Faecalibacterium and Bifidobactericeae Bifidobacterium |
CHF-Congestive Heart Failure; FISH – fluorescent in situ hybridization; HF – Heart Failure; NYHA – New York Heart Association; Uncl. - unclassified
The use of 16s rRNA gene sequencing is now a standard methodology to identify proportions of bacterial taxa within the gut. However, such analyses rarely delve deep enough to identify individual species, and certainly cannot provide strain-specific data. Metagenomics and deep sequencing provide greater depth of classification of the microbial community members present and has provided new insight into our study of the microbiota as we can now better detect species that were previously not able to be cultured. These methods have only recently been utilized in heart failure studies. Using 16s rRNA sequencing, Luedde et al. showed in a small cohort of age and sex-matched patients that heart failure patients tended towards decreased intra-individual taxonomic diversity (alpha-diversity) whereas the inter-individual diversity was able to distinguish between heart failure and control patients90. Extending Luedde et al’s findings, Cui et al. analyzed the metagenomic data of 53 heart failure patients and was also able to distinguish patients with congestive heart failure from controls91. However, in this small study, additional differentiation between ischemic and dilated cardiomyopathy based on gut microbiota composition was not found. In the heart failure patients, the authors observed enrichments in gut microbial genes involved in LPS synthesis as well as genes necessary for the generation of the pro-atherogenic gut metabolite, TMAO (discussed in detail below). Furthermore, they observed decreases in Faecalibacterium prausnitzii that have previously been characterized in elderly congestive heart failure patients92. This bacterium is an abundant butyrate-producing species, and these findings could be demonstrative of this bacterium’s importance in anti-inflammatory functions. Finally, a more comprehensive analysis of microbiota from Kummen et al. used discovery and validation cohorts (n=44 and n=40 respectively) along with 266 population-based controls and adjustment for multiple risk factors to create a more robust heart failure microbiota signature93. They reaffirmed the decreased microbial diversity in disease, identified changes in fifteen core taxa, and emphasized a depletion of the Lachnospiraceae family (of which several members are butyrate producers) that was inversely associated with increased levels of soluble CD25, a marker for T-cell and macrophage activation. This once again highlights the potential importance of the microbial modulation of inflammation through SCFA production in disease states. Lastly, in their small cohort, the authors found that depletion of Eubacterium hallii94, another important commensal SCFA producer, and increased soluble CD25, was associated with death or heart transplantation.
These studies are important first steps into categorizing the microbiota profile in heart failure. However, it is important to note that studies examining the microbial diversity in heart failure is still a budding field. Multiple discrepancies can be noted when comparing major bacterial groups identified in studies summarized in Table 2. For example, Eubacterium Rectale, a gut symbiont and butyrate producer was found to be increased by Sandek et al. using fluorescent in situ hybridization in gut mucosal biofilms but decreased by Kamo et al29,92. It can be noted that this bacteria has also been associated w/ decreases in atherosclerosis95. Similarly, Luedde et al. observed that Collinsella was decreased in the HF cohort90 while this genus has been found to be enriched in atherosclerosis95. Lastly, Kummen et al. observed multiple genus belonging to the Lachnospiraceae family to be decreased in HF93, but interestingly, this family has also consistently demonstrated positive associations with plasma TMAO levels82,96. As methodologies become more uniform, reproducibility must be affirmed in future studies with adequate power and more importantly, clinically homogenous heart failure diagnoses.
Environmental and treatment-related factors
One limitation of the current studies seeking to link specific gut microbial taxa to disease presence, susceptibility, or severity is the confounding that is always present from both environmental and intrinsic factors related to the presence of the disease and its treatment. Regarding environmental factors – diet, medications, and the surrounding milieu play substantial roles in shaping our gut microbiome103. Similarly, the intrinsic physiology of heart failure, in addition to the multi-factorial etiologies which lead to this state, also require scrutiny. However, these potential confounders can also lead us to novel questions in the investigation of microbiome differences. For example, determining how medication-microbiome interactions alter or perhaps drive specific changes in microbial composition is of relevance104. In addition to the focus on diet and the microbiota, numerous new studies have already demonstrated dramatic effects driven by widely used medications such as metformin and statins on the bile acid pool, as well as on microbial signatures associated with disease105,106. The reverse can also occur wherein alterations in gut microbiota influence drug metabolism, with potential impact on the host. For example, digoxin may be inactivated by the actinobacterium Eggerthella lenta107. Similarly, in a recent study of the human gut microbiome in 1,135 Dutch individuals, the metabolism of heart failure management drugs (such as angiotensin inhibitors, β-blockers, and angiotensin II receptor antagonists) was associated with inter-individual variations in gut microbiota composition108. Gut microbiota can even alter levels of host hepatic drug metabolism enzymes, thereby modulating an individual’s circulating drug levels and responses to therapeutics109–111. In an intriguing recent study examining nearly 1,000 clinically used medications, an estimated 24% of the drugs were found to suppress or impact the growth rate of at least one human commensal in culture104. These studies raise significant new questions about the enormous potential interplay between our gut microbiome and inter-individual differences in drug metabolism, as well as the potential development of antibiotic resistance104,112. It remains to be seen whether further understanding of how the gut microbiome may influence the inter-individual variability of drug responses, impact personalized medicine and the management of heart failure patients.
Gut microbiota-generated metabolites known to impact cardiometabolic processes in the host
Short-chain fatty acids
Gut microbiota within the distal intestinal tract foster fermentation processes that often produce short-chain fatty acids (SCFAs), a major nutrient source for the ceco-colonic epithelium113. These organic fatty acids, defined by being from one to six carbons in length, are important end-products of colonic fermentation and are derived from macronutrients such as dietary fiber, resistant starches, as well as sugars or proteins that escape digestion in the upper intestinal tract. While many branched chain and hydroxylated SCFA of various structures can be made, the majority have yet to be fully characterized. Notably, the vast majority of SCFAs produced in the colon appears to be the simple aliphatic two, three and four carbon SCFAs acetate, propionate and butyrate, with an assumed approximate 60%, 20%, and 20% distribution, respectively69,113. A significant proportion of the SCFAs produced in the distal bowel is reabsorbed from the gut lumen by facilitated apical solute transport via multiple different transporters114–117 as well as via passive (concentration-dependent) diffusion118. Because of their abundance within the distal intestinal tract, SCFAs also play a role in colonic acid-base status119,120. Furthermore, myriad different SCFA products have been readily measured in blood121. In recent studies, SCFAs have been mechanistically linked via specific host receptor recognition (including Gpr41, Gpr43, Olfr78) to development of hypertension through alterations in blood pressure in the host (FIG. 2)83,122,123. Moreover, acetate-producing bacteria have also recently been identified in preclinical studies as a potential protective intervention for the treatment of hypertension, adverse cardiac hypertrophy, and fibrosis development124. It was demonstrated that both high fiber and acetate feeding in a deoxycorticosterone acetate-salt hypertension mouse model resulted in decreased blood pressure, decreased cardiac and renal fibrosis, and improved cardiovascular function. This study also concluded that SCFA receptors, including Gpr43 and Olfr78, were not readily detectable in the cardiac transcriptome. Therefore, additional independent mechanisms by which SCFA modulate cardiac function likely exist and require further investigation. In addition, it is important to note that SCFA pharmacokinetics (such as production and tissue utilization) vary significantly between humans and smaller animals which limits the direct extrapolation of SCFA peripheral effects from animal models to human disease125.
Figure 2. Downstream Effects of Gut Microbiota-generated Short-chain Fatty Acid (SCFA) in Cardiovascular System.
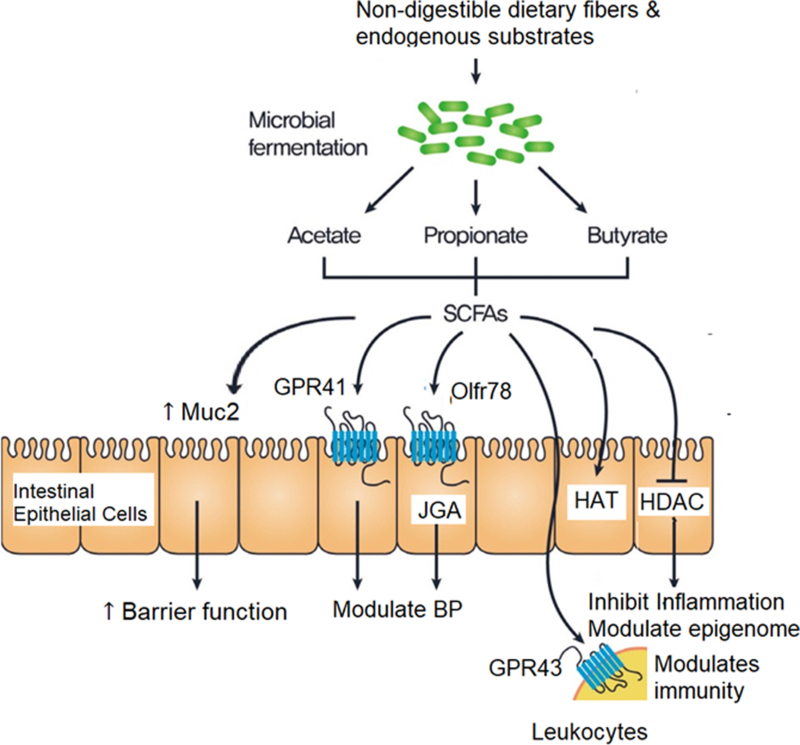
Legend: SCFAs produced by gut microbiota exert their cardiovascular effects by 1) indirectly improving intestinal barrier function by promoting mucous production; 2) activate Olfr78 in mouse (or hOR51E2 in humans) in the renal juxtaglomerular apparatus (JGA) and peripheral vasculature, that leads to increased renin release, and raised blood pressure thereby counteracting hypotensive responses mediated by GPR41; and 3) activate histone acetyltransferases (HAT) and inhibit histone deacetylases (HDACs) thereby inhibiting inflammation, balancing gene regulation (epigenome) and modulates immune cell activation.
Bile acids
Bile acids are traditionally viewed as emulsifiers of fats and fat-soluble vitamins to aid intestinal absorption. The primary bile acids, such as cholic and chenodeoxycholic acid (CDCA), are synthesized through the oxidation of cholesterol in the liver126. These primary bile acids are typically reabsorbed (>95%)127. However, microbiota in the colon can further metabolize any unrecycled bile acids to produce secondary bile acids such as deoxycholate (DCA), lithocholate (LCA), ursodeoxycholate (UDCA), and numerous (at least 50) others128,129. Nearly all of the bile acid pool in the large intestine comprises secondary bile acids with concentrations up to 1,000µM. These secondary bile acids also enter the enterohepatic circulation with any remaining excretion occurring through the feces130.
Studies suggest that both the composition and pool size of bile acids are altered in subjects with heart failure. For example, in a recent small cohort study of heart failure patients and sex-matched controls (n=142 and n=20, respectively), it was reported that the ratio of primary to secondary bile acids was decreased in heart failure subjects131. The difference in bile acid ratios was driven mainly by decreased levels of primary bile acids. Moreover, there was a shift in the secondary bile acid profile, though the total secondary bile acid level remained similar. There is no doubt that bile acids and microbiota are inextricably linked as primary bile acids are biochemically modified by the microbiota while the resulting composition of the bile acid pool can conversely affect the microbiota profile. This raises questions of how these shifts in profile may affect host physiology in general, as well as how they affect processes relevant to cardiac physiology and disease susceptibility. The interactions between bile acids and the microbiota can be both direct and indirect. As an example, in disease states such as cholestasis, bile acids will reach supraphysiologic levels. Early studies of primary bile acids have been shown to exert direct negative myocardial chronotropic effects in a dose-dependent fashion132,133. Though at lower concentrations relative to primary bile acids even in cholestatic states, secondary bile acids can also exert similar effects. It has been shown that at increased concentrations of bile acids (10−3M), significant reductions in both receptor density and affinity of beta-adrenoreceptors were observed134. Although it was not specifically compared in this adrenoreceptor study, the reductions in receptor density and affinity appeared greater for DCA, a secondary bile acid. Moreover, there are many mechanistic connections between bile acids, lipid signaling and biochemical pathways as outlined below.
Our understanding of bile acid physiology was greatly expanded by the discovery of bile acid responsive receptors, such as Farnesoid nuclear receptor (FXR)135,136 and various G-protein coupled receptors (GPCRs) including muscarinic receptors, TGR5, and sphingosine phosphate receptor137–139. Among these receptors, FXR is the most well-studied example of a bile acid signaling receptor that modulates metabolism and inflammation140. FXR responds to CDCA as the most potent endogenous agonist (amongst major bile acids) followed by DCA and LCA. Furthermore, FXR mediates a negative feedback control on the synthesis of bile acids through ileal increases of FGF15141. The majority of literature on FXR is focused on its metabolic effects in obesity and liver pathophysiology140. Nonetheless, the role of FXR has been explored in atherosclerotic disease, and its activating ligands have been shown to antagonize inflammatory responses through repression of nuclear factor-kappaB (NF-kB)142,143. Given the potential for NF-kB to cause cardiac hypertrophy independent of processes such as hypertension, this presents indirect evidence for the potential of FXR-targeted therapies to improve heart failure144,145. However, the converse has also been observed. Antagonism of FXR has also been suggested to improve cardiac function. For example, in vitro studies on cardiomyocytes pinpoint FXR as a mediator of apoptosis through mitochondrial signaling146. Moreover, FXR knockout mice demonstrated better recovery following myocardial infarction through reduced apoptosis and fibrosis146.
Because of the many mechanistic links between bile acids and processes relevant to cardiovascular disease, various bile acids have been explored for their potential clinical applications. However, like the physiological role of bile acids in cardiovascular disease, the therapeutic studies have thus far experienced mixed results. A prominent example is obeticholic acid (OCA), a synthetic bile acid derived from CDCA and the first FXR agonist utilized in human intervention studies. OCA is currently FDA approved for the treatment of primary biliary cholangitis147–149. Moreover, its ability to reverse fibrosis in patients with nonalcoholic steatohepatitis (NASH) was demonstrated in a proof-of-concept study followed by a randomized control trial (RCT)150,151. These findings give confidence for speculation that targeting FXR for optimization of metabolic parameters might then also reduce cardiovascular complications. However, in the OCA trial for NASH, additional metabolic derangements such as increased blood lipid levels were also observed, in contrast to the findings of other proof of concept studies152,153. In a divergent study, the effect of supplementation with another secondary bile acid, UDCA, on endothelial function and inflammatory markers in chronic heart failure patients was investigated in a double-blind RCT154. As UDCA is capable of forming mixed micelles around LPS, it was hypothesized that decreased endotoxin levels and a corresponding reduction in inflammation would result, improving peripheral blood flow in subjects with chronic heart failure155. The results showed limited but positive improvements in peripheral blood flow. However, these effects appeared to occur independently from inflammation, as markers such as tumor necrosis factor (TNF) and IL-6 were unchanged. Together, these findings not only show a need for additional validation of currently studied pathways to advance bile acid therapies, but they also highlight the numerous gaps in our understanding of bile acid physiology.
Trimethylamine and trimethylamine N-oxide
Recent work from our group has demonstrated, for the first time, that the trimethylamine (TMA)/trimethylamine N-oxide (TMAO) pathway represents a novel union between diet, gut microbiota, and both atherosclerosis and thrombosis risks82,96,156–159. TMAO is generated from dietary precursors that contain the TMA moiety such as phosphatidylcholine, choline, and L-carnitine. These nutrients, found in high-fat foods, yield TMA through the action of multiple microbial enzyme complexes (described in a later section) and enters the portal circulation where further metabolism by host enzymes in the liver, called flavin-containing monooxygenases (FMO and primarily the FMO3 isoform), produce TMAO (FIG. 3). Initial studies revealed a strong association between gut-microbiota-dependent phospholipid metabolism and atherosclerosis risks mediated by generation of the pro-atherosclerotic metabolite TMAO.
Figure 3: Microbial Generation of Dietary-Induced Trimethylamine (TMA) and Trimethylamine N-oxide (TMAO).
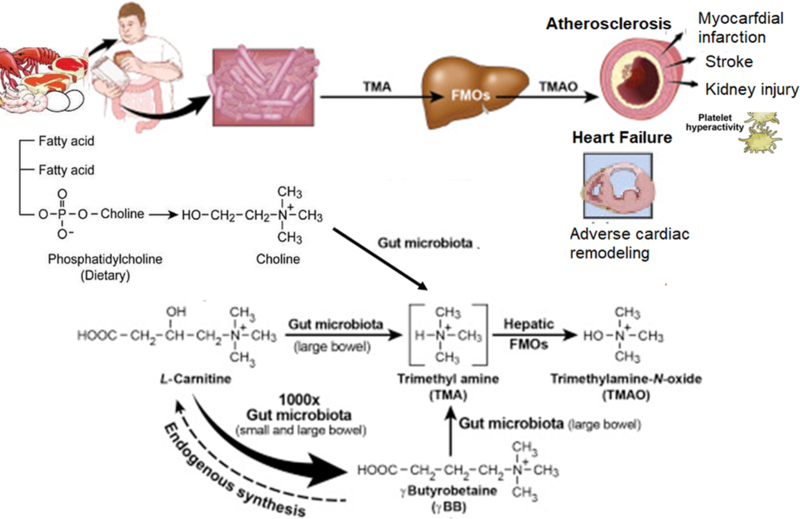
Legend: Diets rich in choline/phosphatidylcholine and carnitine can be metabolized by certain species of gut microbiota to TMA that is then converted to TMAO by hepatic flavin mono-oxygenase (FMO, especially FMO3) and excreted via the kidneys from the circulation. Accumulation of TMAO levels has been associated with atherogenesis, platelet hyperactivation, and future development of adverse cardiac events such as myocardial infarction, stroke, and death. Adverse cardiac remodeling is also associated with elevated TMAO levels. Gamma butyrobetaine, a precursor for endogenous synthesis of carnitine, can also be converted to TMA (Modified from Wang et al, Nature 2011; Koeth et al, Nat Med 2013; Koeth et al, Nat Med 2014)156,158,227
Using untargeted metabolomics as a discovery platform, increased plasma levels of small-molecule metabolites, including TMAO, were shown to predict an increased risk of CVD in stable patients undergoing elective cardiac evaluation who subsequently experienced a myocardial infarction, stroke or death158. The top candidate metabolites were subjected to confirmation studies in an independent validation cohort of 1,876 patients. Subsequent animal model studies showed that this pathway was mechanistically linked to atherosclerosis156,158, and heart failure susceptibility160. For example, it was demonstrated that direct supplementation of TMAO or its dietary precursors, choline158 or carnitine156,159, both resulted in enhanced macrophage foam cell formation and atherosclerotic plaque development in aortic roots of atherosclerosis-prone apolipoprotein E−/− mice. These findings were accompanied by observations of increases in both the scavenger receptor CD36 and scavenger receptor A1 (SRA1) protein, correlating with the findings of increased atherosclerosis burden156,158. A relationship between heightened TMAO and increased risk for myocardial infarction, stroke or death was then validated in an independent cohort of 4,007 patients, which showed a 2.5-fold increased risk for major adverse cardiovascular events in patients in the highest quartile of TMAO157. To prove causality, antibiotic abrogation of gut microbiota was shown to result in improved phenotypes, and microbiota transplantation studies with low versus high TMA generating community confirmed that one could successfully transmit TMA production potential and correspondingly, transmit enhanced diet dependent pro-atherogenic phenotype, thereby, satisfying a fundamental criteria of Koch’s postulates81. Similarly, microbial transplantation studies have confirmed that heightened TMAO generation, platelet responsiveness, and thrombosis potential may be transmitted from donor to recipient82.
Commonly discussed confounders to the TMAO story concern dietary intakes of fish (which can contain TMAO) and L-carnitine. We note that much work needs to be done in this field as we pick apart these dietary links to TMAO and determine what macronutrients may be beneficial or harmful. Despite commentary on this factor, few studies have directly investigated the relationship between fish, TMAO, and cardiovascular disease. First, TMAO content correlates with high salinity and deep, cold environments161,162. In fact, most fish do not contain TMAO, just as most fish do not contain significant amounts of omega-3 fatty acids163,164. Thus, broad sweeping generalizations about the health benefits or otherwise of eating fish need to be more nuanced. Nonetheless, high dietary fish intakes, especially of deep sea (and cold water) varieties of fish, which are more likely to contain TMAO, is associated with increases in urinary TMAO excretion165. However, it is also clear that in people with adequate renal function, this rise in TMAO is transient. Of note, a recent study supplementing fish protein to a western diet in Apolipoprotein E−/− mice showed increases in atherosclerotic plaque area and calcification when compared to the matched soy or casein protein fed counterparts166. The fish protein in this study was derived from the turbot, a highly consumed fish in Europe; its fatty acid profile contained high levels of monounsaturated fats, and a low omega-6 to omega-3 ratio, while the TMA level was 20-fold higher than that in the comparison diets. It was observed that although blood lipid levels in the mice remained similar between all diet groups, TMAO was significantly elevated in the fish protein group.
Also, fish oil itself may counteract the harmful effects of TMAO. It appears that in co-feeding experiments with fish oil and TMAO, the inflammatory effect of elevated TMAO feeding was ameliorated with the addition of fish oil167. The debate regarding the relationship between TMAO levels and eating fish have mostly been encapsulated to the macro scale. However, it is more challenging to ascertain micronutrient contents in such capacities. Moreover, individuals at significant risk for plasma TMAO retention, namely the CKD cohort, are advocated to reduce their protein intake168,169.
L-carnitine is a popular supplement that has been studied for its potential to improve lipid profiles and metabolism170. In a study conducted by investigators associated with manufacturers of L-carnitine, their findings suggested that L-carnitine feeding resulted in increased TMAO but a lower atherosclerotic burden. This finding has often been cited as opposing evidence to the numerous studies which have associated TMAO with poor clinical outcomes. In their study, male mice were supplemented with Lcarnitine or vehicle controls. A surprising inverse correlation between TMAO level and lesion area in the thoracic aorta was observed. It is interesting that the model of choice was male mice since significant sexual dimorphism exists for trimethylamine monooxygenase (encoded by Fmo3) expression in mice: male mice display 100-fold decreased liver Fmo3 mRNA compared to females, resulting in far lower natural levels of plasma TMAO171. Given this understanding, it is unlikely that L-carnitine feeding in male mice will induce significant changes in plasma TMAO levels. If one converted the parts per million units from this study to the more conventionally utilized micromolar to report plasma TMAO, we observe very similar values of TMAO in this study to that of a typical C57BL6 male mouse171. In fact, the highest subgroup cutoff of 0.2ppm converts to 2.6µM. These low levels of TMAO have never been implicated in disease. Although critical commentaries are essential for discussion, criticism must be met with an objective evaluation of the data at hand.
Overall, in the discussion of microbial metabolites, TMAO is likely only the tip of the iceberg. In the untargeted metabolomics study by Wang et al., there were a total of 18 unidentified peaks that were found to be associated with major adverse cardiovascular events (refer to the supplement in ref. 153)158. Moreover, beyond the three metabolites found to be in the TMAO biochemical pathway, the remaining unknown metabolites also demonstrated strong hazard ratios in unadjusted analysis. Similar discovery approaches can conceivably be undertaken for heart failure patients as well, though careful patient selection is a must as heart failure has a broad definition and significant technical hurdles exist in the chemical derivatization of unknown metabolites. Nonetheless, there likely exists additional microbial generated metabolites with potential links to heart failure and cardiometabolic disease.
The meta-organismal TMA/TMAO pathway
Since the first studies demonstrating that the TMA/TMAO microbial-human metabolic pathway can generate disease-relevant metabolites that contribute to atherogenesis156–158, a multitude of pathophysiologic associations have now been linked with TMAO (FIG. 1). Here, we summarize basic and clinical findings, namely revolving around thrombosis, inflammation, and chronic kidney disease, which are entwined in the upstream etiologies that contribute to heart failure of ischemic or non-ischemic origin. Furthermore, new data also suggest that even in acute settings, cardiovascular function, specifically hemodynamics, may be altered by TMAO, as well.
Atherogenesis and altered platelet function.
Extending the original findings of the relationship between TMAO and atherogenesis, we have also demonstrated that plasma TMAO levels are associated with incident thrombotic event risk82,172. Mechanistic studies show that TMAO acts to potentiate platelet reactivity through alterations in stimulus-dependent calcium signaling. Increased calcium release from platelets and enhanced activation was observed following stimulation from sub-maximal levels of multiple different agonists such as ADP, thrombin, and collagen82. This finding suggests that TMAO-dependent enhancement in stimulus-dependent platelet activation occurs via a target down-stream from these platelet agonists but proximal to stimulus-dependent release of intracellular calcium stores. Finally, fecal transplantation further demonstrated that choline diet-mediated heightened thrombosis potential in vivo is a transmittable trait82.
The mouse findings were replicated in both a large scale (n=4,007) clinical observational study showing circulating TMAO levels are strongly associated with incident thrombotic event risks157, as well as within a small prospective human cohort study involving the taking of choline supplementation172. In both omnivores and vegans alike, choline supplementation resulted in heightened TMAO levels and increased ADPdependent aggregation responses. Moreover, a dose-dependent association was observed between plasma TMAO levels and heightened platelet responsiveness172. Surprisingly, TMAO dependent enhancement in platelet responsiveness was shown to attenuate low-dose aspirin-induced reduction in platelet aggregation172. Thus, in subjects taking low-dose aspirin, elevation in TMAO levels by choline supplementation overcame the aspirin-induced reduction in platelet responsiveness, suggesting that socalled aspirin resistance may in part be attributed to heightened generation of prothrombotic factors by the gut microbiota. These findings also are consistent with clinical studies showing that elevated TMAO levels can help to explain the residual elevation in cardiovascular disease risk in subjects on traditional preventive cardiovascular disease medications, including antiplatelet agents. The findings of platelet hyper-reactivity with elevated TMAO levels are also complemented by studies discovering that TMAO is capable of inciting a robust inflammatory response in aortic tissue in vivo173. This inflammatory response is believed to be mediated in part by endothelial cell NF-kB activation, with small-molecule inhibitor studies suggesting the involvement of a GPCR. A pro-inflammatory signaling effect of TMAO has been independently replicated in studies utilizing human umbilical vein endothelial cells, as well as in studies exploring additional mechanisms such as the NLRP3 inflammasome174,175.
Despite overwhelming clinical and animal model data supporting a link between TMAO and CVD, many questions remain. Most importantly, the nature of the “TMAO receptor,” if one indeed exists, remains unknown. TMAO is known to function as a protein conformation altering small molecule (i.e., a protein chaperone mimetic)176. It may, therefore, impact signaling pathways not only via a classic receptor-ligand interaction but as an allosteric modifier. Thus, detailed molecular understanding of the exact mechanism of action by which TMAO exerts its pro-inflammatory and prothrombotic effects remains undiscovered. It is known that the TMAO precursor, TMA, is detected through the GPCR TAAR5 chemosensory receptor, to which TMA binds with high affinity, though TAAR5 is known not to recognize TMAO177. Therefore, there is optimism for the discovery of a potential receptor-mediated effect of TMAO, as well. It is of interest that TMAO has been reported to potentiate alternative signal transduction processes such as intracellular calcium signaling and platelet reactivity, or sustained increases in blood pressure after angiotensin infusion82,178.
Direct participation in heart failure susceptibility.
In the heart failure setting, animal experiments from Organ et al. found that either choline supplementation (which raises circulating TMAO), or direct TMAO feeding in mice, resulted in higher systemic TMAO levels, enhanced myocardial fibrosis and worsened hemodynamic and anatomic parameters after trans-aortic constriction (TAC)160. Furthermore, in separate studies, it is reported that removal of the TMAO diet after TAC resulted in a greater myocardial recovery in the mice179. These findings were corroborated in a different system to induce cardiovascular dysfunction by Chen et al180. Mice fed a western-diet were given 3,3-dimethyl-1-butanol, a TMA-lyase inhibitor, to reduce the production of TMA and hence, conversion to TMAO. In this study, it was found that inhibition of TMA and TMAO production resulted in significantly improved ventricular remodeling and hemodynamic parameters of mice on a western diet180.
Beyond animal model studies showing heightened TMAO is associated with worse adverse ventricular remodeling and function in heart failure models, our group has reported findings further exploring clinical links between TMAO and heart failure by examining sequential subjects with a history of heart failure who underwent cardiac evaluations181. We observed that subjects with heart failure have significantly higher levels of TMAO compared to age- and sex-matched non-heart failure subjects. We further showed that elevated plasma TMAO in subjects with heart failure was associated with worse long-term survival, even after adjusting for cardio-renal parameters (FIG. 4), and regardless of underlying etiology181. These findings suggest that understanding why TMAO levels are elevated in the setting of heart failure may provide valuable insights into how gut microbiota may contribute to disease progression in heart failure. In a separate study, we observed similar findings in chronic systolic heart failure patients182. The most intriguing aspect of this association was that heightened TMAO levels correlate with adverse diastolic indices such as increased left atrial volume index, suggesting that perhaps an accumulation of this metabolite may affect tissue mechanics. Similar findings have subsequently been reported in an independent Norwegian cohort where plasma TMAO levels were elevated in the heart failure cohort and predictive of transplant-free survival183. The authors observed associations between TMAO and increased pulmonary artery pressure, as well as wedge pressure, which represent indirect estimates of stress on the left atrium. Most recently, Schuett et al. independently validated the association of TMAO with poor heart failure outcomes, specifically in heart failure with reduced ejection fraction patients184. Moreover, they found that TMAO was able to better predict outcomes compared to BNP in this population, after adjusting for traditional risk factors and renal function. One can speculate that in the setting of volume overload, TMAO, as an organic osmolyte, could be drawing fluid into spaces where they usually do not reside, or through a yet unelucidated receptor-mediated mechanism.
Figure 4. The relationship between plasma levels of TMAO and 5yr mortality risk in Patients with Chronic Heart Failure.
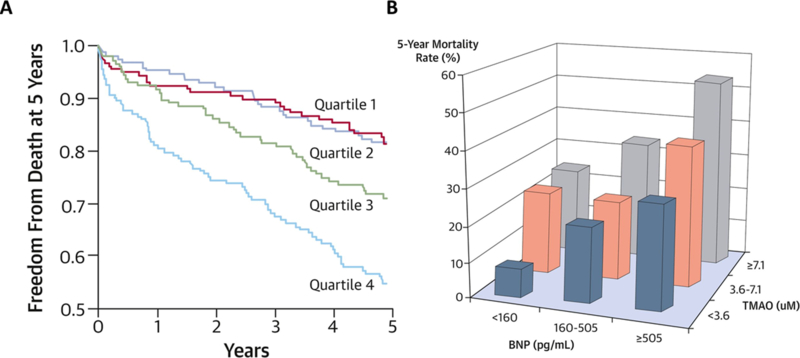
Legend: Patients with a history of heart failure stratified according to quartiles of fasting TMAO levels and circulating B-type natriuretic peptide (BNP) levels showing an increased incidence of 5-year mortality with rising TMAO levels, independent of BNP levels (from Tang WH et al., J Am Coll Cardiol 2014)181
Multiple studies in the chronic setting thus suggest that TMAO impacts heart failure susceptibility and incident risks, in part through increased myocardial fibrosis, and accompanying diastolic dysfunction. One obvious question then is whether there is a similar relationship between TMAO and adverse outcomes in the acute decompensated heart failure (ADHF) setting. In support of this, recent clinical data in a study of ADHF patients from Suzuki et al. show that inclusion of TMAO into risk models provided improved risk stratification for in-hospital mortality in combination with clinical scores185. Additionally, the authors also found that utilizing TMAO levels concurrently with elevated NT-proBNP improved outcome prediction. These clinical studies are summarized in Table 1. Novel evidence from Savi et al. observed worsened cardiomyocyte contractility in the presence of TMAO in vitro186. However, different findings were observed in a CD1 mouse model from Oakley et al. which showed that high levels TMAO has direct effects on increases in myocardial contractility and calcium flux in a dose-dependent manner187. These findings suggest that TMAO could potentially impact myocardial contractility in the acute setting. However, it is difficult to reconcile the two studies given the substantial differences in TMAO dose used (20100uM vs. 300–3000uM) as well as the experimental settings (in vitro vs. ex vivo). Therefore, additional studies are necessary to elucidate these mechanisms further.
Table 1.
Summary of Clinical Investigations Between TMAO and Cardiovascular Related Outcomes in Heart Failure Cohorts
| Group | Subjects | Study Population Criteria | Endpoint of Interest | Results |
|---|---|---|---|---|
| Tang et al.181 | 720 | Stable cardiac patients with a history of heart failure undergoing elective coronary angiographic evaluation | All-cause mortality over 5-year of highest quartile TMAO | Adjusted HR 1.75, CI 1.07–2.86, P < 0.001 |
| Tang et al.182 | 112 | Stable but symptomatic, chronic systolic heart failure (LVEF ≤ 35%) with comprehensive echocardiographic evaluation | Composite endpoint all-cause mortality and cardiac transplantation over 5 years per SD ln TMAO | Adjusted HR 1.46, CI 1.03 2.14, P=0.03 |
| Suzuki et al.185 | 972 | Patients with AHF based on progressive worsening or new onset of shortness of breath along with clinical signs of pulmonary edema, peripheral edema, or elevated jugular venous pressure | All-cause mortality; Composite of death or rehospitalization due to HF at 1 year of highest TMAO tertile | Adjusted HR 1.16, CI 1.01–1.33, P=0.037; non-significant after inclusion of renal function |
| Trøseid et al.183 | 155 | Patients with stable HF for >6 months in NYHA functional class II–IV | Transplant free survival over median 5.2 years of highest TMAO tertile | Unadjusted HR 2.25, CI 1.28–3.92, P=0.005; non-significant after adjustment HR 1.79, CI 0.90–1.79, P = 0.097 |
| Schuett et al.184 | 823 | Patients referred for coronary angiography with heart failure diagnosis in accordance with European guidelines | Death and CV death over mean follow-up of 9.7 years of highest TMAO tertile | Adjusted death HR 1.84, CI 1.44–2.36; CV death HR 1.98, CI 1.47–2.68; remained significant after additional adjustment |
AHF – Acute Heart Failure; CAC – Coronary Artery Calcium; cIMT – carotid intima-media thickness; CI – 95% Confidence Interval; COXPH – Cox Proportional Hazard; HR - Hazard Ratio; LVEF – Left Ventricular Ejection Fraction; OR- Odds Ratio; RR – Rate Ratio
Renal disease and heart failure susceptibility.
Beyond a myocardium centered focus on heart failure pathogenesis, a contributory and synergistic cardio-renal connection has long been recognized188. Importantly, a substantial proportion (40–50%) of individuals with heart failure also demonstrate some extent of kidney dysfunction189,190. Many pathophysiologic processes contribute to this connection, including altered fluid status, neuro-hormonal responses, and metabolic dysregulation. Therefore, appropriate management of heart failure also requires paying close attention to any underlying renal dysfunction.
In our initial studies of TMAO, we hypothesized that an efficient potential mechanism for conferring protection from cardiovascular disease and heart failure development could be via prevention in the accumulation of TMAO157. Subsequent studies showed that TMAO is rapidly cleared from the body after ingestion157,165,191. The strong relationship between kidney function and TMAO has been recognized in multiple early studies on rat models showing primarily renal excretion greater than 95% after radiolabel ingestion192. More detailed renal physiology evidence so far only exists in models of deep-sea fish or chicken from decades ago, and have demonstrated tubular secretion, reabsorption, or passive transport as potential mechanisms of renal excretion193–195. However, it is difficult to extrapolate these findings to a mammalian system. TMAO was incidentally found but not further characterized in several studies investigating broad-scale metabolites associated with CKD. This association was first seen in small cohorts of dialysis, transplant or late-stage CKD patients196. Then in a larger cohort from the Framingham Heart Study, elevated circulating levels of both choline and TMAO were observed to predict the incident development of CKD in subjects with normal renal function at the time of enrollment and sample collection197.
To investigate the relationship between TMAO and CKD development, our group observed that high levels of TMAO in patients with CKD (defined as eGFR <60mL/min) portended a significantly increased mortality risk with HR 1.93 (95%CI 1.13–3.29)198. Translating findings to mice models, we observed that both chronic TMAO and choline feeding resulted in striking development of renal fibrosis, along with increased levels of tubular injury markers such as cystatin C, pSMAD, and KIM-1/tubulin ratio198. These observations have since been replicated in multiple independent observational cohorts across a spectrum of CKD patients199–203. Furthermore, in the past two years, multiple large observational studies have been published that recapitulate our initial observations which we have reviewed in detail.
In a small comparative GWAS study involving n=1,892 subjects with relatively preserved renal function, it was suggested that genetic variant play a relatively weak role in determining TMAO levels204. Interestingly, in a clinical cohort from the Seattle Kidney Study involving n=339 subjects, it was reported that specific polymorphisms of FMO3 were associated with elevated circulating TMAO. In this latter study, the recruited patients also had impaired kidney function and thus clearance, which may have heightened the sensitivity for the detection of genetic variants associated with variations in circulating levels of TMAO, despite its smaller sample size. Furthermore, another recent study also observed increased FMO3 expression associated with decreased kidney function in mice, suggesting genetic impact on plasma TMAO variability in specific settings such as decreased renal function205.
In addition, questions exist whether genetic differences in ethnicity may also play a role in the relationship between TMAO levels and observed genetic variants associated with plasma levels – Shafi et al. reported that black CKD populations tended to have better survival than their white CKD counterparts within the same quintile of TMAO levels201.
Overall, beyond CKD and heart failure patients, the observational clinical data relating elevated TMAO to poorer prognosis has been robust. Most recently, several independent meta-analyses arrived at similar conclusions of elevated TMAO being prognostic for cardiovascular events or mortality, independent of renal function and cardiovascular comorbidities206–208.
Gut microbiota-relevant interventions for heart failure
Prebiotic and probiotic therapy
Prebiotics are non-digestible food products (non-culturable) that can alter microbial composition and function, or induce the growth of select micr209oorganisms. Probiotics, on the other hand, represent live microorganisms. These supplements are thought to be able to improve the intestinal microbial ‘balance’ by altering the microbial composition and community structure. Some preliminary evidence suggests that administration of select probiotics can be cardioprotective. For example, treatment of rats with a beverage containing L. plantarum 299v 24 hours before coronary ligation reportedly produced reduced infarct sizes and improved left ventricular function86. In a separate study, administration of Lactobacillus rhamnosus GR-1 also demonstrated similar results in a rat myocardial ischemia model210. A decrease in the adipokine leptin was attributed as a potential mechanism of the probiotic benefits. However, it is notable that the 16s sequencing in the GR-1 study showed that the treatment strain did not appear to colonize or alter the gut community structure/composition. Of interest, a randomized control pilot study with Saccharomyces boulardii was reported to show benefits in heart failure patients, with left ventricular ejection fraction being improved in the short term, along with decreases in serum creatinine and inflammatory markers211. These findings are very preliminary and must be balanced with the concerns of probiotic administration in a population that tends to become more critically ill. Though probiotics have an excellent safety record, the lack of regulation across supplements could potentially raise the risk of probiotic translocation and associated sepsis212,213.
Dietary intervention
Dietary interventions to alter the progression of heart failure have primarily focused on the reduction of salt, to prevent hypertension and maintain volume homeostasis1. A formally adopted strong recommendation from the American College of Cardiology/American Heart Association guidelines is the Dietary Approaches to Stop Hypertension (DASH) eating plan consisting of a diet that is rich in fruits, vegetables, whole grains, and low-fat dairy foods; includes meat, fish, poultry, nuts, and beans; and is limited in sugar-sweetened foods and beverages, red meat, and added fats. Multiple observational studies suggest that a DASH diet reduces heart failure incidence214–217. A small randomized, controlled clinical trial of 48 patients assigned to either a DASH diet (n=24) or general guidelines for HF management (n=24), those in the DASH group demonstrated better 6 minute walk performance, quality of life, and trended towards increased arterial elasticity after a three month intervention. Interestingly, there appeared to be little difference in the salt intake between groups, which suggests that other dietary components require further investigation218. Moreover, biomarker and cardiac morphometry data from another small pilot study named DASH-Diastolic Heart Failure (DASH-HF), showed that the 13 females patients on the DASH diet had significant weight loss, improved cardiac function along with improvements in multiple surrogate markers such as blood pressure, BNP, urinary isoprostanes and acylcarnitines219–221. Beyond the DASH diet, there is still a gap in the literature for additional dietary interventions as a definitive therapeutic route for heart failure. Proper nutrition such as the Mediterranean diet, can certainly aid the prevention of cardiovascular events and the subsequent development of heart failure. Despite the recent retraction and revision, the benefits of the Mediterranean diet for primary prevention of cardiovascular diseases are still supported by the Prevención con Dieta Mediterránea (PREDIMED) study, and there are also data showing that adherence to a Mediterranean diet is more likely to result in lower levels of TMAO222. This may be because of the presence of 3,3-dimethyl-1-butanol (DMB) (which we will discuss later), which inhibits TMAO production and is found in extra virgin olive oil, a major component of the Mediterranean diet. Two recent studies explored the potential of the Mediterranean diet to improve the prognosis of acute heart failure. A pre-specified secondary outcome for heart failure incidence was investigated in PREDIMED, but there was no association between diet and heart failure incidence223. However, this population was not powered to provide validated conclusions. In the MEDIT-AHF study, which prospectively followed emergency department acute heart failure admissions, the authors did not see long-term benefits in mortality224. However, the patients did have decreased risk for rehospitalization.
Microbial TMA-lyase inhibitors
With strong associations in heart failure, we look ahead and discuss strategies for targeted inhibition of TMAO production as a framework for other potential microbiota targets. Much attention has been focused on the reduction of TMAO levels, with an emphasis focusing on the meta-organismal production pathway225. Thus, both microbial and host liver enzymes represent potential targets for reducing TMAO production, either as a therapeutic target or as scientific proof of principal in understanding the importance of the TMAO meta-organismal pathway to host pathophysiology. Following ingestion of a TMA containing nutrient (the choline moiety being the most abundant in a typical diet), distinct microbial enzyme systems participate in the generation of TMA, the precursor of TMAO158,171 (FIG. 5). The choline utilization gene cluster contains catalytic and regulatory polypeptides, CutC/D, which appear to serve as the primary microbial catabolic enzyme system for choline conversion into TMA (i.e., choline TMA lyase) within the human gut microbiome226. Additional microbial enzyme systems that contribute to TMA generation are now known to include CntA/B and yeaW/X for conversion of carnitine into TMA226–230. In a study by Wang et al., DMB was developed as a competitive inhibitor against CutC catalyzed choline TMA-lyase activity to reduce the production of TMAO96. This inhibitor was found to be non-lethal to human commensals possessing CutC and fostered suppression of both TMA production in cultured human commensals and TMAO lowering in mice on a choline supplemented diet. Most importantly, exposure to DMB was shown to also effectively reduce atherosclerosis progression in the apolipoprotein E−/− mouse model without significant adverse toxicity96. Interestingly, DMB was also observed to be present in some cold-pressed extra-virgin olive oils96, a major component of the Mediterranean diet. Hence, there may be some mechanistic link between microbial inhibition of TMAO production and the benefits of Mediterranean diet. However, it is unclear if the reduction in TMA as a result of TMA-lyase inhibition may contribute to as of yet unrecognized or unintended effects. Other less validated examples of potential TMA generating inhibitors include meldonium, an analog of γ-butyrobetaine and carnitine, that is reported to be able to reduce carnitine to TMA conversion, or resveratrol, which while not an inhibitor, is suggested to reduce TMAO levels by reorganization of microbial community structure231,232.
Figure 5. Microbial Metabolic Pathways as Potential Druggable Targets.
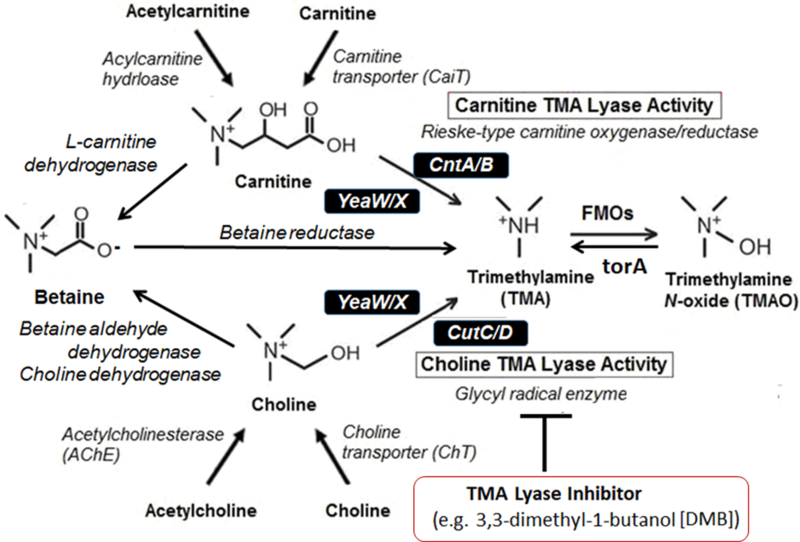
Caption: Carnitine (and gamma butyrobetaine) can be converted by gut microbiota to trimethylamine (TMA) via carnitine TMA lyases such as CntA/B or YeaW/X, whereas choline can be converted to TMA via choline TMA lyases such as CutC/D or YeaW/X. Betaine conversion to TMA is in much lower quantities. Many of these enzymes are unique in some microbial species, making them attractive druggable targets for inhibition to reduce TMA production (such as 3,3-dimethyl-1-butanol [DMB] as choline TMA lyase inhibitors (Wang et al., Cell 2015).
Future directions
The realization that gut microbiota can contribute to both normal health and disease susceptibility provides a novel perspective and permits expansion in our approach to disease research and therapeutic opportunities. The next steps in future investigations include teasing out, at a mechanistic level, how the gut microbiota contribute to heart failure and cardiovascular disease. This includes defining chemical mediators, microbemicrobe as well as microbe-host interactions within the complex microbiota ecosystem. Much work remains beyond the swath of association studies regarding understanding host receptors which perpetuate signaling processes that lead to altered host physiology. Moreover, are there specific genetic factors which may predispose individuals towards disease susceptibility in the combination of both specific nutrient depletion/excess and microbiota functional capability? Finally, translation of “microbiome-centred” findings will be necessary through well-designed prospective, longitudinal studies.
The most relevant phenotypes must be considered to bring microbial information to clinical practice. Although sequencing of the microbial genome is becoming more common in research practice, the utility of this methodology for clinical practice is still unclear. Because sequencing information, unless the more expensive meta-genomic option, is performed, cannot estimate at all the phenotype pattern of the microbiome but rather only the “potential” phenotype based on the genes present. Rather, measurement of metabolic profiles in the blood or urine to guide appropriate dietary recommendations and to provide targeted interventions will result in more translational findings. These interventions in the future can take numerous forms. Beyond dietary recommendations, one can imagine sequestrants or binding resins as one form, intercepting the microbial metabolites of interest before absorption into the host. Bile acid sequestrants are an early lipid-lowering medication, and thus this approach, albeit for an alternative class of intestinal luminal metabolites, has precedence. Yet another potential approach will be non-lethal microbial inhibitors, such as DMB for TMAO. Other more potent TMA-lyase inhibitors are under investigation, and the use of a non-lethal microbial enzyme inhibitor has the potential theoretical benefit of being less likely to foster the development of microbial resistance because the selective pressure for developing resistance is reduced compared to an antibiotic. Finally, the use of pre or probiotics – to result in “terraforming” of the gut microbial community and function to achieve changes in microbial metabolites is an alternative approach and an area of active investigation.
Conclusions
Undoubtedly, there is a complex association between gut microbiota and the cardiovascular system that can influence and even determine cardiovascular diseases. Understanding the role of the gut microbiota is of paramount importance--- chronic inflammation, underlying comorbidities, antibiotics, diet, and probiotics can drastically alter the long-term composition of the gut microbiota and thereby impact our health. Similarly, increasing clinical use of pre- and probiotics may have unappreciated effects. Future research must elucidate the mechanisms behind these complex associations to determine whether a causal link exists between this “forgotten organ” and heart failure. Because the gut microbiota represents a relatively uncharted ground for novel pharmacologic targets, we need to develop a solid understanding of these underlying mechanisms to pave the way for targeted therapeutic strategies in heart failure.
Text Box 1. Categorization of the Gut Microbiota -.
Several approaches that attempted to classify bacteria as “beneficial” or “harmful” have been highly cited despite being over-simplified, such as the three enterotypes paradigm (designated as enrichment in Bacteroidetes phylum, or Prevotella, or Ruminococcus genus based on multidimensional cluster and principal component analysis of 61 fecal samples) or the Firmicutes to Bacteroidetes ratios97,98. Although these approaches provided an initial paradigm for the study of microbiota effects on health, such broad classification schemes were difficult to replicate, as gradients rather than distinct groups arose as a more accurate descriptor99,100. Furthermore, such characterizations belie an appreciation of the nuances that an individual microbial species or strain can under certain circumstances be considered healthful and essential in supporting host health, yet in other situations (e.g., an extreme increase or decrease in level) be considered a potential contributor to disease.
Endeavors such as the Human Microbiome Project and MetaHIT aim to create a catalog of bacteria in the healthy human gut microbiome6,101. Through deep sequencing, it is possible to predict functional groups of the bacteria within our gut. This knowledge is essential as it has been shown that functional genes may not be present in bacteria of the same species from different individuals (though they may be shared amongst communities)102. Further, such analyses only relay relative functional capacity and thus may not reliably predict global functional capacity. Therefore, progression to direct functional interrogation (e.g. metabolomics, peptidomics, and direct microbial transplantation studies) beyond any sequence-dependent measurement (metagenomics or metatranscriptomics) is needed to move beyond quantification of microbe copy number, or predicted gene presence, to better demonstrate function, disease associations, and participation.
Key points.
The “gut hypothesis” of heart failure postulates that bowel wall edema and impaired intestinal barrier function in heart failure can promote bacterial translocation and heightened inflammation
Changes in the composition and diversity of gut microbiota have been observed in patients with heart failure
Microbial metabolites, especially those derived from dietary nutrients, can generate paracrine and endocrine effects that can also lead to an increased susceptibility to heart failure
Novel therapeutic strategies targeting microbial metabolic pathways and/or metabolites, as well as altering microbial composition, have the potential to modulate cardiovascular disease susceptibility and prevent progression to heart failure
Acknowledgments
The authors’ research work is supported by grants from the National Institutes of Health (NIH) and the Office of Dietary Supplements including R01HL103866 (S.L.H.), P20HL113452 (W.H.T. & S.L.H.), R01DK106000 (W.H.T. & S.L.H.), R01HL126827 (W.H.T. & S.L.H.).
Competing interests
Drs. Tang and Li have no relationships relevant to the contents of this paper to disclose. Dr. Hazen is named as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr. Hazen is a paid consultant for Proctor & Gamble. Dr. Hazen has received research funds from Proctor & Gamble and Roche Diagnostics. Dr. Hazen is eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland Heart Lab/Quest Diagnostics or Proctor & Gamble. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Glossary
- Meta-organism
Polygenomic organism consisting of a community of living beings of different species (e.g. microbiota and host) associated by interspecies symbiosis.
- Metagenomics
The study of the collective genome of microorganisms from an environmental sample
- Metatranscriptomics
The study of the collective transcripts of microorganisms from an environmental sample
References
- 1.Yancy CW et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128, 1810–1852, doi: 10.1161/CIR.0b013e31829e8807 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Yancy CW et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 70, 776–803, doi: 10.1016/j.jacc.2017.04.025 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Anker SD et al. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol 79, 1426–1430 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Krack A, Sharma R, Figulla HR & Anker SD The importance of the gastrointestinal system in the pathogenesis of heart failure. Eur Heart J 26, 2368–2374, doi: 10.1093/eurheartj/ehi389 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Sender R, Fuchs S & Milo R Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol 14, e1002533, doi: 10.1371/journal.pbio.1002533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turnbaugh PJ et al. The human microbiome project. Nature 449, 804–810, doi: 10.1038/nature06244 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dethlefsen L, Eckburg PB, Bik EM & Relman DA Assembly of the human intestinal microbiota. Trends Ecol Evol 21, 517–523, doi: 10.1016/j.tree.2006.06.013 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, doi: 10.1038/nature11234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booijink CC, Zoetendal EG, Kleerebezem M & de Vos WM Microbial communities in the human small intestine: coupling diversity to metagenomics. Future Microbiol 2, 285–295, doi: 10.2217/17460913.2.3.285 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Gorbach SL in Medical Microbiology (eds th & Baron S) (1996). [PubMed]
- 11.Hooper LV et al. Molecular analysis of commensal host-microbial relationships in the intestine. Science 291, 881–884, doi: 10.1126/science.291.5505.881 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Marcobal A et al. Consumption of human milk oligosaccharides by gut-related microbes. J Agric Food Chem 58, 5334–5340, doi: 10.1021/jf9044205 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belkaid Y & Hand TW Role of the microbiota in immunity and inflammation. Cell 157, 121–141, doi: 10.1016/j.cell.2014.03.011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujiya M et al. The Bacillus subtilis quorum-sensing molecule CSF contributes to intestinal homeostasis via OCTN2, a host cell membrane transporter. Cell Host Microbe 1, 299–308, doi: 10.1016/j.chom.2007.05.004 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Buffie CG & Pamer EG Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 13, 790–801, doi: 10.1038/nri3535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corr SC et al. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci U S A 104, 7617–7621, doi: 10.1073/pnas.0700440104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajilic-Stojanovic M, Smidt H & de Vos WM Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol 9, 2125–2136, doi: 10.1111/j.1462-2920.2007.01369.x (2007). [DOI] [PubMed] [Google Scholar]
- 18.Yang X, Xie L, Li Y & Wei C More than 9,000,000 unique genes in human gut bacterial community: estimating gene numbers inside a human body. PLoS One 4, e6074, doi: 10.1371/journal.pone.0006074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer C, Bik EM, DiGiulio DB, Relman DA & Brown PO Development of the human infant intestinal microbiota. PLoS Biol 5, e177, doi: 10.1371/journal.pbio.0050177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominguez-Bello MG et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107, 11971–11975, doi: 10.1073/pnas.1002601107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu DM et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med 23, 314–326, doi: 10.1038/nm.4272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall AB, Tolonen AC & Xavier RJ Human genetic variation and the gut microbiome in disease. Nat Rev Genet 18, 690–699, doi: 10.1038/nrg.2017.63 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Koppel N, Maini Rekdal V & Balskus EP Chemical transformation of xenobiotics by the human gut microbiota. Science 356, doi: 10.1126/science.aag2770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnenburg JL & Backhed F Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64, doi: 10.1038/nature18846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown JM & Hazen SL Targeting of microbe-derived metabolites to improve human health: The next frontier for drug discovery. J Biol Chem 292, 8560–8568, doi: 10.1074/jbc.R116.765388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saitta KS et al. Bacterial beta-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44, 28–35, doi: 10.3109/00498254.2013.811314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace BD et al. Structure and Inhibition of Microbiome beta-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem Biol 22, 1238–1249, doi: 10.1016/j.chembiol.2015.08.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermiston ML & Gordon JI Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 270, 1203–1207 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Sandek A et al. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol 50, 1561–1569, doi: 10.1016/j.jacc.2007.07.016 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Fink MP, Kaups KL, Wang HL & Rothschild HR Maintenance of superior mesenteric arterial perfusion prevents increased intestinal mucosal permeability in endotoxic pigs. Surgery 110, 154–160; discussion 160–151 (1991). [PubMed] [Google Scholar]
- 31.Triposkiadis F et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 54, 1747–1762, doi: 10.1016/j.jacc.2009.05.015 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Jodal M & Lundgren O Countercurrent mechanisms in the mammalian gastrointestinal tract. Gastroenterology 91, 225–241 (1986). [DOI] [PubMed] [Google Scholar]
- 33.Maynard N et al. Assessment of splanchnic oxygenation by gastric tonometry in patients with acute circulatory failure. JAMA 270, 1203–1210 (1993). [PubMed] [Google Scholar]
- 34.Krack A et al. Studies on intragastric PCO2 at rest and during exercise as a marker of intestinal perfusion in patients with chronic heart failure. Eur J Heart Fail 6, 403–407, doi: 10.1016/j.ejheart.2004.03.002 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Arutyunov GP, Kostyukevich OI, Serov RA, Rylova NV & Bylova NA Collagen accumulation and dysfunctional mucosal barrier of the small intestine in patients with chronic heart failure. Int J Cardiol 125, 240–245, doi: 10.1016/j.ijcard.2007.11.103 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Lu YC, Yeh WC & Ohashi PS LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151, doi: 10.1016/j.cyto.2008.01.006 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Frangogiannis NG The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11, 255–265, doi: 10.1038/nrcardio.2014.28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niebauer J et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet 353, 1838–1842, doi: 10.1016/S0140-6736(98)09286-1 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Peschel T et al. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail 5, 609–614 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Hietbrink F et al. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock 32, 374–378, doi: 10.1097/SHK.0b013e3181a2bcd6 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Munger MA, Johnson B, Amber IJ, Callahan KS & Gilbert EM Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 77, 723–727 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Paulus WJ How are cytokines activated in heart failure? Eur J Heart Fail 1, 309–312 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Deswal A et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103, 2055–2059 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Rauchhaus M et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 102, 3060–3067 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Ferrari R et al. Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation 92, 1479–1486 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Conraads VM et al. Selective intestinal decontamination in advanced chronic heart failure: a pilot trial. Eur J Heart Fail 6, 483–491, doi: 10.1016/j.ejheart.2003.12.004 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Mann DL Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116, 1254–1268, doi: 10.1161/CIRCRESAHA.116.302317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bozkurt B et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 97, 1382–1391 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Kubota T et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res 81, 627–635 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Al-Sadi R et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One 9, e85345, doi: 10.1371/journal.pone.0085345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Sadi RM & Ma TY IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol 178, 4641–4649 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma TY, Boivin MA, Ye D, Pedram A & Said HM Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol 288, G422–430, doi: 10.1152/ajpgi.00412.2004 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Al-Sadi R, Guo S, Ye D & Ma TY TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol 183, 1871–1884, doi: 10.1016/j.ajpath.2013.09.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung ES et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107, 3133–3140, doi: 10.1161/01.CIR.0000077913.60364.D2 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Mann DL et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 109, 1594–1602, doi: 10.1161/01.CIR.0000124490.27666.B2 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Torre-Amione G et al. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): a placebo-controlled randomised trial. Lancet 371, 228–236, doi: 10.1016/S0140-6736(08)60134-8 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Ridker PM et al. Anti-inflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131, doi: 10.1056/NEJMoa1707914 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Abbate A et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail 12, 319–322, doi: 10.1093/eurjhf/hfq017 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Abbate A et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117, 2670–2683, doi: 10.1161/CIRCULATIONAHA.107.740233 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Van Tassell BW et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol 55, 117–122, doi: 10.1097/FJC.0b013e3181c87e53 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Van Tassell BW et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 10, doi: 10.1161/CIRCHEARTFAILURE.117.004373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Tassell BW et al. Interleukin-1 blockade in heart failure with preserved ejection fraction: rationale and design of the Diastolic Heart Failure Anakinra Response Trial 2 (D-HART2). Clin Cardiol 40, 626–632, doi: 10.1002/clc.22719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abbate A Interleukin-1 blockade with canakinumab to improve exercise capacity in patients with chronic systolic heart failure and elevated high sensitivity creactive protein (Hs-CRP), <https://clinicaltrials.gov/ct2/show/NCT01900600> (2013).
- 64.Gullestad L et al. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 122, 23–35, doi: 10.1159/000338166 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Dick SA & Epelman S Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res 119, 159–176, doi: 10.1161/CIRCRESAHA.116.308030 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Jakobsson HE et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep 16, 164–177, doi: 10.15252/embr.201439263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tlaskalova-Hogenova H et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 8, 110–120, doi: 10.1038/cmi.2010.67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ley RE, Turnbaugh PJ, Klein S & Gordon JI Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023, doi: 10.1038/4441022a (2006). [DOI] [PubMed] [Google Scholar]
- 69.Turnbaugh PJ et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031, doi: 10.1038/nature05414 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79, doi: 10.1186/gb-2012-13-9r79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wen L et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113, doi: 10.1038/nature07336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin J et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60, doi: 10.1038/nature11450 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Kummen M et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 66, 611–619, doi: 10.1136/gutjnl-2015-310500 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Sears CL & Garrett WS Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328, doi: 10.1016/j.chom.2014.02.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arthur JC et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123, doi: 10.1126/science.1224820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Roy T et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794, doi: 10.1136/gutjnl-2012-303816 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Dumas ME et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 103, 12511–12516, doi: 10.1073/pnas.0601056103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hsiao EY et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463, doi: 10.1016/j.cell.2013.11.024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foster JA & McVey Neufeld KA Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci 36, 305–312, doi: 10.1016/j.tins.2013.01.005 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Naseribafrouei A et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil 26, 1155–1162, doi: 10.1111/nmo.12378 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Gregory JC et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 290, 5647–5660, doi: 10.1074/jbc.M114.618249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu W et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 165, 111–124, doi: 10.1016/j.cell.2016.02.011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pluznick JL et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A 110, 4410–4415, doi: 10.1073/pnas.1215927110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mell B et al. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics 47, 187–197, doi: 10.1152/physiolgenomics.00136.2014 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durgan DJ et al. Role of the Gut Microbiome in Obstructive Sleep Apnea-Induced Hypertension. Hypertension 67, 469–474, doi: 10.1161/HYPERTENSIONAHA.115.06672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lam V et al. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J 26, 1727–1735, doi: 10.1096/fj.11-197921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sandek A et al. Intestinal blood flow in patients with chronic heart failure: a link with bacterial growth, gastrointestinal symptoms, and cachexia. J Am Coll Cardiol 64, 1092–1102, doi: 10.1016/j.jacc.2014.06.1179 (2014). [DOI] [PubMed] [Google Scholar]
- 88.Pasini E et al. Pathogenic Gut Flora in Patients With Chronic Heart Failure. JACC Heart Fail 4, 220–227, doi: 10.1016/j.jchf.2015.10.009 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Mamic P, Heidenreich PA, Hedlin H, Tennakoon L & Staudenmayer KL Hospitalized Patients with Heart Failure and Common Bacterial Infections: A Nationwide Analysis of Concomitant Clostridium Difficile Infection Rates and In-Hospital Mortality. J Card Fail 22, 891–900, doi: 10.1016/j.cardfail.2016.06.005 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Luedde M et al. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail 4, 282–290, doi: 10.1002/ehf2.12155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cui X et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep 8, 635, doi: 10.1038/s41598-017-18756-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamo T et al. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS One 12, e0174099, doi: 10.1371/journal.pone.0174099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kummen M et al. Gut Microbiota Signature in Heart Failure Defined From Profiling of 2 Independent Cohorts. J Am Coll Cardiol 71, 1184–1186, doi: 10.1016/j.jacc.2017.12.057 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Engels C, Ruscheweyh HJ, Beerenwinkel N, Lacroix C & Schwab C The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Front Microbiol 7, 713, doi: 10.3389/fmicb.2016.00713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karlsson FH et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 3, 1245, doi: 10.1038/ncomms2266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Z et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 163, 1585–1595, doi: 10.1016/j.cell.2015.11.055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Johnson EL, Heaver SL, Walters WA & Ley RE Microbiome and metabolic disease: revisiting the bacterial phylum Bacteroidetes. J Mol Med (Berl) 95, 1–8, doi: 10.1007/s00109-016-1492-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arumugam M et al. Enterotypes of the human gut microbiome. Nature 473, 174–180, doi: 10.1038/nature09944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jeffery IB, Claesson MJ, O’Toole PW & Shanahan F Categorization of the gut microbiota: enterotypes or gradients? Nat Rev Microbiol 10, 591–592 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Knights D et al. Rethinking “enterotypes”. Cell Host Microbe 16, 433–437, doi: 10.1016/j.chom.2014.09.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32, 834–841, doi: 10.1038/nbt.2942 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Martinez-del Campo A et al. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio 6, doi: 10.1128/mBio.00042-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rothschild D et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215, doi: 10.1038/nature25973 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Maier L et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature, doi: 10.1038/nature25979 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caparros-Martin JA et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome 5, 95, doi: 10.1186/s40168-017-0312-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu H et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23, 850–858, doi: 10.1038/nm.4345 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Haiser HJ et al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341, 295–298, doi: 10.1126/science.1235872 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569, doi: 10.1126/science.aad3369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Selwyn FP, Cui JY & Klaassen CD RNA-Seq Quantification of Hepatic Drug Processing Genes in Germ-Free Mice. Drug Metab Dispos 43, 1572–1580, doi: 10.1124/dmd.115.063545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bjorkholm B et al. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One 4, e6958, doi: 10.1371/journal.pone.0006958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Claus SP et al. Colonization-induced host-gut microbial metabolic interaction. MBio 2, e00271–00210, doi: 10.1128/mBio.00271-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Spanogiannopoulos P, Bess EN, Carmody RN & Turnbaugh PJ The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14, 273–287, doi: 10.1038/nrmicro.2016.17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Topping DL & Clifton PM Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 81, 1031–1064, doi: 10.1152/physrev.2001.81.3.1031 (2001). [DOI] [PubMed] [Google Scholar]
- 114.Hadjiagapiou C, Schmidt L, Dudeja PK, Layden TJ & Ramaswamy K Mechanism(s) of butyrate transport in Caco-2 cells: role of monocarboxylate transporter 1. Am J Physiol Gastrointest Liver Physiol 279, G775–780, doi: 10.1152/ajpgi.2000.279.4.G775 (2000). [DOI] [PubMed] [Google Scholar]
- 115.Mascolo N, Rajendran VM & Binder HJ Mechanism of short-chain fatty acid uptake by apical membrane vesicles of rat distal colon. Gastroenterology 101, 331–338 (1991). [DOI] [PubMed] [Google Scholar]
- 116.Vidyasagar S, Barmeyer C, Geibel J, Binder HJ & Rajendran VM Role of short-chain fatty acids in colonic HCO(3) secretion. Am J Physiol Gastrointest Liver Physiol 288, G1217–1226, doi: 10.1152/ajpgi.00415.2004 (2005). [DOI] [PubMed] [Google Scholar]
- 117.den Besten G et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54, 2325–2340, doi: 10.1194/jlr.R036012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sellin JH SCFAs: The Enigma of Weak Electrolyte Transport in the Colon. News Physiol Sci 14, 58–64 (1999). [DOI] [PubMed] [Google Scholar]
- 119.Duncan SH, Louis P, Thomson JM & Flint HJ The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 11, 2112–2122, doi: 10.1111/j.1462-2920.2009.01931.x (2009). [DOI] [PubMed] [Google Scholar]
- 120.Cherrington CA, Hinton M, Pearson GR & Chopra I Short-chain organic acids at ph 5.0 kill Escherichia coli and Salmonella spp. without causing membrane perturbation. J Appl Bacteriol 70, 161–165 (1991). [DOI] [PubMed] [Google Scholar]
- 121.Pouteau E, Meirim I, Metairon S & Fay LB Acetate, propionate and butyrate in plasma: determination of the concentration and isotopic enrichment by gas chromatography/mass spectrometry with positive chemical ionization. J Mass Spectrom 36, 798–805, doi: 10.1002/jms.181 (2001). [DOI] [PubMed] [Google Scholar]
- 122.Yang T et al. Gut dysbiosis is linked to hypertension. Hypertension 65, 1331–1340, doi: 10.1161/HYPERTENSIONAHA.115.05315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pluznick J A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 5, 202–207, doi: 10.4161/gmic.27492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marques FZ et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 135, 964–977, doi: 10.1161/CIRCULATIONAHA.116.024545 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Pouteau E, Nguyen P, Ballevre O & Krempf M Production rates and metabolism of short-chain fatty acids in the colon and whole body using stable isotopes. Proc Nutr Soc 62, 87–93, doi: 10.1079/PNS2003208 (2003). [DOI] [PubMed] [Google Scholar]
- 126.Lefebvre P, Cariou B, Lien F, Kuipers F & Staels B Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89, 147–191, doi: 10.1152/physrev.00010.2008 (2009). [DOI] [PubMed] [Google Scholar]
- 127.Dawson PA, Lan T & Rao A Bile acid transporters. J Lipid Res 50, 2340–2357, doi: 10.1194/jlr.R900012-JLR200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Devlin AS & Fischbach MA A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol 11, 685–690, doi: 10.1038/nchembio.1864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ridlon JM, Kang DJ & Hylemon PB Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259, doi: 10.1194/jlr.R500013-JLR200 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Hamilton JP et al. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol 293, G256–263, doi: 10.1152/ajpgi.00027.2007 (2007). [DOI] [PubMed] [Google Scholar]
- 131.Mayerhofer CCK et al. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J Card Fail 23, 666–671, doi: 10.1016/j.cardfail.2017.06.007 (2017). [DOI] [PubMed] [Google Scholar]
- 132.Binah O, Rubinstein I, Bomzon A & Better OS Effects of bile acids on ventricular muscle contraction and electrophysiological properties: studies in rat papillary muscle and isolated ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol 335, 160–165 (1987). [DOI] [PubMed] [Google Scholar]
- 133.Joubert P An in vivo investigation of the negative chronotropic effect of cholic acid in the rat. Clin Exp Pharmacol Physiol 5, 1–8 (1978). [DOI] [PubMed] [Google Scholar]
- 134.Gazawi H et al. The effects of bile acids on beta-adrenoceptors, fluidity, and the extent of lipid peroxidation in rat cardiac membranes. Biochem Pharmacol 59, 1623–1628 (2000). [DOI] [PubMed] [Google Scholar]
- 135.Wang H, Chen J, Hollister K, Sowers LC & Forman BM Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3, 543–553 (1999). [DOI] [PubMed] [Google Scholar]
- 136.Forman BM et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81, 687–693 (1995). [DOI] [PubMed] [Google Scholar]
- 137.Kawamata Y et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem 278, 9435–9440, doi: 10.1074/jbc.M209706200 (2003). [DOI] [PubMed] [Google Scholar]
- 138.Rosen H, Gonzalez-Cabrera PJ, Sanna MG & Brown S Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem 78, 743–768, doi: 10.1146/annurev.biochem.78.072407.103733 (2009). [DOI] [PubMed] [Google Scholar]
- 139.Raufman JP, Chen Y, Zimniak P & Cheng K Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology 65, 215–221, doi: 10.1159/000064347 (2002). [DOI] [PubMed] [Google Scholar]
- 140.Calkin AC & Tontonoz P Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13, 213–224, doi: 10.1038/nrm3312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sayin SI et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235, doi: 10.1016/j.cmet.2013.01.003 (2013). [DOI] [PubMed] [Google Scholar]
- 142.Wang YD et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–1643, doi: 10.1002/hep.22519 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li YT, Swales KE, Thomas GJ, Warner TD & Bishop-Bailey D Farnesoid x receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler Thromb Vasc Biol 27, 2606–2611, doi: 10.1161/ATVBAHA.107.152694 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Purcell NH et al. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A 98, 6668–6673, doi: 10.1073/pnas.111155798 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gordon JW, Shaw JA & Kirshenbaum LA Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res 108, 1122–1132, doi: 10.1161/CIRCRESAHA.110.226928 (2011). [DOI] [PubMed] [Google Scholar]
- 146.Pu J et al. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J 34, 1834–1845, doi: 10.1093/eurheartj/ehs011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ali AH, Carey EJ & Lindor KD Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 3, 5, doi: 10.3978/j.issn.2305-5839.2014.12.06 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pellicciari R et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 45, 3569–3572 (2002). [DOI] [PubMed] [Google Scholar]
- 149.Nevens F et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med 375, 631–643, doi: 10.1056/NEJMoa1509840 (2016). [DOI] [PubMed] [Google Scholar]
- 150.Neuschwander-Tetri BA et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965, doi: 10.1016/S0140-6736(14)61933-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mudaliar S et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 e571, doi: 10.1053/j.gastro.2013.05.042 (2013). [DOI] [PubMed] [Google Scholar]
- 152.Xu Y et al. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology 64, 1072–1085, doi: 10.1002/hep.28712 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cipriani S, Mencarelli A, Palladino G & Fiorucci S FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res 51, 771–784, doi: 10.1194/jlr.M001602 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.von Haehling S et al. Ursodeoxycholic acid in patients with chronic heart failure: a double-blind, randomized, placebo-controlled, crossover trial. J Am Coll Cardiol 59, 585–592, doi: 10.1016/j.jacc.2011.10.880 (2012). [DOI] [PubMed] [Google Scholar]
- 155.Aouad K et al. Immunosuppressive effects of endotoxins and bile acids in vivo in the rat. Eur J Clin Invest 26, 45–48 (1996). [DOI] [PubMed] [Google Scholar]
- 156.Koeth RA et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19, 576–585, doi: 10.1038/nm.3145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tang WH et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368, 1575–1584, doi: 10.1056/NEJMoa1109400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang Z et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63, doi: 10.1038/nature09922 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhao Y et al. The Effect of Different l-Carnitine Administration Routes on the Development of Atherosclerosis in ApoE Knockout Mice. Mol Nutr Food Res 62, doi: 10.1002/mnfr.201700299 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Organ CL et al. Choline Diet and Its Gut Microbe-Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload-Induced Heart Failure. Circ Heart Fail 9, e002314, doi: 10.1161/CIRCHEARTFAILURE.115.002314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Seibel BA & Walsh PJ Trimethylamine oxide accumulation in marine animals: relationship to acylglycerol storage. J Exp Biol 205, 297–306 (2002). [DOI] [PubMed] [Google Scholar]
- 162.Parab SN & Rao SB DISTRIBUTION OF TRIMETHYLAMINE OXIDE IN SOME MARINE AND FRESHWATER FISH. Current Science 53, 307–309 (1984). [Google Scholar]
- 163.Cladis DP, Kleiner AC, Freiser HH & Santerre CR Fatty acid profiles of commercially available finfish fillets in the United States. Lipids 49, 1005–1018, doi: 10.1007/s11745-014-3932-5 (2014). [DOI] [PubMed] [Google Scholar]
- 164.Weaver KL et al. The content of favorable and unfavorable polyunsaturated fatty acids found in commonly eaten fish. J Am Diet Assoc 108, 1178–1185, doi: 10.1016/j.jada.2008.04.023 (2008). [DOI] [PubMed] [Google Scholar]
- 165.Svensson BG, Akesson B, Nilsson A & Paulsson K Urinary excretion of methylamines in men with varying intake of fish from the Baltic Sea. J Toxicol Environ Health 41, 411–420, doi: 10.1080/15287399409531853 (1994). [DOI] [PubMed] [Google Scholar]
- 166.Yazdekhasti N et al. Fish protein increases circulating levels of trimethylamine-N-oxide and accelerates aortic lesion formation in apoE null mice. Mol Nutr Food Res 60, 358–368, doi: 10.1002/mnfr.201500537 (2016). [DOI] [PubMed] [Google Scholar]
- 167.Gao X et al. Fish oil ameliorates trimethylamine N-oxide-exacerbated glucose intolerance in high-fat diet-fed mice. Food Funct 6, 1117–1125, doi: 10.1039/c5fo00007f (2015). [DOI] [PubMed] [Google Scholar]
- 168.Kasiske BL, Lakatua JD, Ma JZ & Louis TA A meta-analysis of the effects of dietary protein restriction on the rate of decline in renal function. Am J Kidney Dis 31, 954–961 (1998). [DOI] [PubMed] [Google Scholar]
- 169.Pedrini MT, Levey AS, Lau J, Chalmers TC & Wang PH The effect of dietary protein restriction on the progression of diabetic and nondiabetic renal diseases: a meta-analysis. Ann Intern Med 124, 627–632 (1996). [DOI] [PubMed] [Google Scholar]
- 170.Collins HL et al. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis 244, 29–37, doi: 10.1016/j.atherosclerosis.2015.10.108 (2016). [DOI] [PubMed] [Google Scholar]
- 171.Bennett BJ et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab 17, 49–60, doi: 10.1016/j.cmet.2012.12.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhu W, Wang Z, Tang WHW & Hazen SL Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation 135, 1671–1673, doi: 10.1161/CIRCULATIONAHA.116.025338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Seldin MM et al. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J Am Heart Assoc 5, doi: 10.1161/JAHA.115.002767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sun X et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res Commun 481, 63–70, doi: 10.1016/j.bbrc.2016.11.017 (2016). [DOI] [PubMed] [Google Scholar]
- 175.Ma G et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci Rep 37, doi: 10.1042/BSR20160244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bennion BJ & Daggett V Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: a chemical chaperone at atomic resolution. Proc Natl Acad Sci U S A 101, 6433–6438, doi: 10.1073/pnas.0308633101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liberles SD & Buck LB A second class of chemosensory receptors in the olfactory epithelium. Nature 442, 645–650, doi: 10.1038/nature05066 (2006). [DOI] [PubMed] [Google Scholar]
- 178.Ufnal M et al. Trimethylamine-N-oxide: a carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can J Cardiol 30, 1700–1705, doi: 10.1016/j.cjca.2014.09.010 (2014). [DOI] [PubMed] [Google Scholar]
- 179.Organ CL et al. Abstract 19293: Removal of Dietary Trimethylamine N-Oxide (TMAO) Attenuates Cardiac Dysfunction in Pressure Overload Induced Heart Failure. Circulation 134, A19293–A19293 (2016). [Google Scholar]
- 180.Chen K, Zheng X, Feng M, Li D & Zhang H Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front Physiol 8, 139, doi: 10.3389/fphys.2017.00139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tang WH et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol 64, 1908–1914, doi: 10.1016/j.jacc.2014.02.617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tang WH et al. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail 21, 91–96, doi: 10.1016/j.cardfail.2014.11.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Troseid M et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med 277, 717–726, doi: 10.1111/joim.12328 (2015). [DOI] [PubMed] [Google Scholar]
- 184.Schuett K et al. Trimethylamine-N-oxide and Heart Failure With Reduced Versus Preserved Ejection Fraction. J Am Coll Cardiol 70, 3202–3204, doi: 10.1016/j.jacc.2017.10.064 (2017). [DOI] [PubMed] [Google Scholar]
- 185.Suzuki T, Heaney LM, Bhandari SS, Jones DJ & Ng LL Trimethylamine N-oxide and prognosis in acute heart failure. Heart 102, 841–848, doi: 10.1136/heartjnl-2015-308826 (2016). [DOI] [PubMed] [Google Scholar]
- 186.Savi M et al. Trimethylamine-N-Oxide (TMAO)-Induced Impairment of Cardiomyocyte Function and the Protective Role of Urolithin B-Glucuronide. Molecules 23, doi: 10.3390/molecules23030549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Oakley C et al. The Uremic Metabolite, Trimethylamine-N-Oxide (TMAO), Directly Alters Cardiac Contractile Function. The FASEB Journal 31, 688.683 (2017). [Google Scholar]
- 188.Schefold JC, Filippatos G, Hasenfuss G, Anker SD & von Haehling S Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nat Rev Nephrol 12, 610–623, doi: 10.1038/nrneph.2016.113 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Hillege HL et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 113, 671–678, doi: 10.1161/CIRCULATIONAHA.105.580506 (2006). [DOI] [PubMed] [Google Scholar]
- 190.van Deursen VM et al. Co-morbidities in patients with heart failure: an analysis of the European Heart Failure Pilot Survey. Eur J Heart Fail 16, 103–111, doi: 10.1002/ejhf.30 (2014). [DOI] [PubMed] [Google Scholar]
- 191.Cho CE et al. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol Nutr Food Res 61, doi: 10.1002/mnfr.201600324 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Smith JL, Wishnok JS & Deen WM Metabolism and excretion of methylamines in rats. Toxicol Appl Pharmacol 125, 296–308, doi: 10.1006/taap.1994.1076 (1994). [DOI] [PubMed] [Google Scholar]
- 193.Forster RP, Berglund F & Rennick BR Tubular secretion of creatine, trimethylamine oxide, and other organic bases by the aglomerular kidney of Lophius americanus. J Gen Physiol 42, 319–327 (1958). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cohen JJ, Krupp MA, Chidsey CA 3rd & Biltz CI Effect of trimethylamine and its homologues on renal conservation of trimethylamine oxide by the spiny dogfish, Squalus acanthias. Am J Physiol 196, 93–99 (1959). [DOI] [PubMed] [Google Scholar]
- 195.Acara M, Camiolo S & Rennick B Renal N-oxidation of trimethylamine in the chicken during tubular excretion. Drug Metab Dispos 5, 82–90 (1977). [PubMed] [Google Scholar]
- 196.Bell JD et al. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: identification of trimethylamine-N-oxide. Biochim Biophys Acta 1096, 101–107 (1991). [DOI] [PubMed] [Google Scholar]
- 197.Rhee EP et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol 24, 1330–1338, doi: 10.1681/ASN.2012101006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tang WH et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 116, 448–455, doi: 10.1161/CIRCRESAHA.116.305360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Stubbs JR et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J Am Soc Nephrol 27, 305–313, doi: 10.1681/ASN.2014111063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kim RB et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int 89, 1144–1152, doi: 10.1016/j.kint.2016.01.014 (2016). [DOI] [PubMed] [Google Scholar]
- 201.Shafi T et al. Trimethylamine N-Oxide and Cardiovascular Events in Hemodialysis Patients. J Am Soc Nephrol 28, 321–331, doi: 10.1681/ASN.2016030374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Missailidis C et al. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS One 11, e0141738, doi: 10.1371/journal.pone.0141738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gruppen EG et al. TMAO is Associated with Mortality: Impact of Modestly Impaired Renal Function. Sci Rep 7, 13781, doi: 10.1038/s41598-017-13739-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hartiala J et al. Comparative genome-wide association studies in mice and humans for trimethylamine N-oxide, a proatherogenic metabolite of choline and L-carnitine. Arterioscler Thromb Vasc Biol 34, 1307–1313, doi: 10.1161/ATVBAHA.114.303252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Johnson C, Prokopienko AJ, West RE 3rd, Nolin TD & Stubbs JR Decreased Kidney Function is Associated with Enhanced Hepatic Flavin Monooxygenase Activity and Increased Circulating Trimethylamine N-oxide Concentrations in Mice. Drug Metab Dispos, doi: 10.1124/dmd.118.081646 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Heianza Y, Ma W, Manson JE, Rexrode KM & Qi L Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J Am Heart Assoc 6, doi: 10.1161/JAHA.116.004947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Qi J et al. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: a systematic review and meta-analysis of 11 prospective cohort studies. J Cell Mol Med, doi: 10.1111/jcmm.13307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Schiattarella GG et al. Gut microbe-generated metabolite trimethylamine-Noxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J, doi: 10.1093/eurheartj/ehx342 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Robinson-Cohen C et al. Association of FMO3 Variants and Trimethylamine N-Oxide Concentration, Disease Progression, and Mortality in CKD Patients. PLoS One 11, e0161074, doi: 10.1371/journal.pone.0161074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gan XT et al. Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ Heart Fail 7, 491–499, doi: 10.1161/CIRCHEARTFAILURE.113.000978 (2014). [DOI] [PubMed] [Google Scholar]
- 211.Costanza AC, Moscavitch SD, Faria Neto HC & Mesquita ET Probiotic therapy with Saccharomyces boulardii for heart failure patients: a randomized, double-blind, placebo-controlled pilot trial. Int J Cardiol 179, 348–350, doi: 10.1016/j.ijcard.2014.11.034 (2015). [DOI] [PubMed] [Google Scholar]
- 212.Kochan P et al. Lactobacillus rhamnosus administration causes sepsis in a cardiosurgical patient--is the time right to revise probiotic safety guidelines? Clin Microbiol Infect 17, 1589–1592, doi: 10.1111/j.1469-0691.2011.03614.x (2011). [DOI] [PubMed] [Google Scholar]
- 213.Liong MT Safety of probiotics: translocation and infection. Nutr Rev 66, 192–202, doi: 10.1111/j.1753-4887.2008.00024.x (2008). [DOI] [PubMed] [Google Scholar]
- 214.Salehi-Abargouei A, Maghsoudi Z, Shirani F & Azadbakht L Effects of Dietary Approaches to Stop Hypertension (DASH)-style diet on fatal or nonfatal cardiovascular diseases--incidence: a systematic review and meta-analysis on observational prospective studies. Nutrition 29, 611–618, doi: 10.1016/j.nut.2012.12.018 (2013). [DOI] [PubMed] [Google Scholar]
- 215.Levitan EB, Wolk A & Mittleman MA Consistency with the DASH diet and incidence of heart failure. Arch Intern Med 169, 851–857, doi: 10.1001/archinternmed.2009.56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Levitan EB, Wolk A & Mittleman MA Relation of consistency with the dietary approaches to stop hypertension diet and incidence of heart failure in men aged 45 to 79 years. Am J Cardiol 104, 1416–1420, doi: 10.1016/j.amjcard.2009.06.061 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nguyen HT et al. DASH eating pattern is associated with favorable left ventricular function in the multi-ethnic study of atherosclerosis. J Am Coll Nutr 31, 401–407 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Rifai L, Pisano C, Hayden J, Sulo S & Silver MA Impact of the DASH diet on endothelial function, exercise capacity, and quality of life in patients with heart failure. Proc (Bayl Univ Med Cent) 28, 151–156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mathew AV, Seymour EM, Byun J, Pennathur S & Hummel SL Altered Metabolic Profile With Sodium-Restricted Dietary Approaches to Stop Hypertension Diet in Hypertensive Heart Failure With Preserved Ejection Fraction. J Card Fail 21, 963–967, doi: 10.1016/j.cardfail.2015.10.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hummel SL et al. Low-sodium DASH diet improves diastolic function and ventricular-arterial coupling in hypertensive heart failure with preserved ejection fraction. Circ Heart Fail 6, 1165–1171, doi: 10.1161/CIRCHEARTFAILURE.113.000481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hummel SL et al. Low-sodium dietary approaches to stop hypertension diet reduces blood pressure, arterial stiffness, and oxidative stress in hypertensive heart failure with preserved ejection fraction. Hypertension 60, 1200–1206, doi: 10.1161/HYPERTENSIONAHA.112.202705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Estruch R et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med 378, e34, doi: 10.1056/NEJMoa1800389 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Papadaki A et al. Mediterranean diet and risk of heart failure: results from the PREDIMED randomized controlled trial. Eur J Heart Fail 19, 1179–1185, doi: 10.1002/ejhf.750 (2017). [DOI] [PubMed] [Google Scholar]
- 224.Miro O et al. Adherence to Mediterranean Diet and All-Cause Mortality After an Episode of Acute Heart Failure: Results of the MEDIT-AHF Study. JACC Heart Fail 6, 52–62, doi: 10.1016/j.jchf.2017.09.020 (2018). [DOI] [PubMed] [Google Scholar]
- 225.Falony G, Vieira-Silva S & Raes J Microbiology Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine. Annu Rev Microbiol 69, 305–321, doi: 10.1146/annurev-micro-091014-104422 (2015). [DOI] [PubMed] [Google Scholar]
- 226.Craciun S & Balskus EP Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A 109, 21307–21312, doi: 10.1073/pnas.1215689109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Koeth RA et al. gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab 20, 799–812, doi: 10.1016/j.cmet.2014.10.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Meyer M, Granderath K & Andreesen JR Purification and characterization of protein PB of betaine reductase and its relationship to the corresponding proteins glycine reductase and sarcosine reductase from Eubacterium acidaminophilum. Eur J Biochem 234, 184–191 (1995). [DOI] [PubMed] [Google Scholar]
- 229.Wagner M et al. Substrate-specific selenoprotein B of glycine reductase from Eubacterium acidaminophilum. Biochemical and molecular analysis. Eur J Biochem 260, 38–49 (1999). [DOI] [PubMed] [Google Scholar]
- 230.Zhu Y et al. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc Natl Acad Sci U S A 111, 4268–4273, doi: 10.1073/pnas.1316569111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kuka J et al. Suppression of intestinal microbiota-dependent production of pro-atherogenic trimethylamine N-oxide by shifting L-carnitine microbial degradation. Life Sci 117, 84–92, doi: 10.1016/j.lfs.2014.09.028 (2014). [DOI] [PubMed] [Google Scholar]
- 232.Chen ML et al. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. MBio 7, e02210–02215, doi: 10.1128/mBio.02210-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]