

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting upper and lower motor neurons (MNs), resulting in paralysis and precocious death from respiratory failure. Although the causes of ALS are incompletely understood, the role of alterations in RNA metabolism seems central. MicroRNAs (miRNAs) are noncoding RNAs implicated in the regulation of gene expression of many relevant physiological processes, including cell death. The recent model of programmed cell death (PCD) encompasses different mechanisms, from apoptosis to regulated necrosis (RN), in particular necroptosis. Both apoptosis and necroptosis play a significant role in the progressive death of MNs in ALS. In this review, we present key research related to miRNAs that modulate apoptosis and RN pathways in ALS. We also discuss whether these miRNAs represent potential targets for therapeutic development in patients.
Keywords: amyotrophic lateral sclerosis, apoptosis, microRNAs, motor neurons, necroptosis, therapy
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disease characterized by the progressive degeneration of both upper and lower motor neurons (MNs), which results in muscle paralysis and ultimately death from respiratory failure within a median of 3 years.1 At present, no effective therapy is available for ALS. Riluzole and Edaravone are the sole two drugs approved for ALS but only modestly slow disease progression.1, 2
Patients affected by idiopathic ALS and without a family history are classified as sporadic (sALS). A fraction of cases (approximately 10%) are familial (fALS), because of mutations in genes involved in a wide range of cellular functions, encompassing roles in motor neuronal and non‐neuronal cells.3, 4
Since the first identification, in 1993, of superoxide dismutase 1 (SOD1) gene mutations as being responsible for some forms of fALS,5 many other genes linked to ALS have been identified.4 The most frequent altered gene in both sALS and fALS is C9ORF72.6 Other relevant genes are TAR DNA‐binding protein (TARDBP), encoding for TDP‐43, and fused in sarcoma (FUS), which encode for proteins implicated in the regulation of RNA processing and expression.4 Intracellular inclusions of TDP‐43 are the main pathological substrate of sporadic and familial forms of ALS, except for those associated with SOD1 mutations.7, 8, 9
Understanding the etiology of ALS and the factors that influence its progression is crucial for the implementation of effective therapeutic strategies that are urgently needed. Although specific causes leading to ALS are unknown, different cellular mechanisms were proposed to mediate MN degeneration, such as glutamate excitotoxicity, mitochondrial dysfunction, protein aggregation, proteasomal and autophagic dysfunction, neuroinflammation, altered axonal transport, and impaired RNA metabolism.1 In this context, the role of alterations of RNA metabolism seems particularly central, especially considering that TDP43 and FUS are key components of coding and noncoding RNA processing.
MicroRNAs (miRNAs) are short noncoding RNAs that exert a pivotal role in the regulation of gene expression of many relevant physiological processes and, also, in MNs.10, 11 miRNAs bind to Argonaute (AGO) proteins to form a ribonucleoprotein (RNP) complex and recognize complementary sequences of messenger RNA (mRNA) via base‐pairing, inducing the down‐regulation of RNA targets.10, 12 However, because physiological cellular processes need a complex regulatory mechanism, dysregulation of these molecular pathways, typically occurring in chronic diseases, lead to a similarly complex dysregulation of the miRNA expression profile. Indeed, the alterations of each specific miRNA and, consequently, of pathways in which they are involved are variable in different tissues and different disorders.
The finding that miRNAs are essential to MN physiology and survival, supported by the observation that transgenic mice that do not process miRNAs and show hallmarks of MN degeneration,13 prompted the investigation of the role of miRNAs in MN diseases, in particular ALS and type 1 spinal muscular atrophy (SMA) (reviewed by us elsewhere).14, 15 In both these conditions, the miRNA expression profile was shown to be deregulated both in the central nervous system (CNS) and peripheral tissues, with ALS displaying a more profound global dysregulation.14 Deregulation in the miRNA expression pattern was reported in several tissues from ALS patients, in particular in the spinal cord, brain, blood, and cerebrospinal fluid (CSF), and it was also demonstrated in induced pluripotent stem cells (iPSCs) generated from affected patients.14 The human spinal cord miRNA expression profile showed a substantial global down‐regulation in motor neuron disease,16 and this alteration was then demonstrated to be particular to MNs.17 These findings have been related to the inhibition of DICER, an endoribonuclease involved in the processing of pre‐miRNA molecules.17 Remarkably, miRNA reduction was found also in fibroblasts and was confirmed in serum, plasma, CSF, and the motor cortex.18, 19, 20, 21, 22
The most constant finding in ALS mutant SOD1 rodents is an increase in miR‐9 and miR‐206.23, 24, 25, 26 In particular, the miR‐9 level is increased in ALS rodent spinal cords,24, 26 whereas the miR‐206 level is increased in skeletal muscles in both SOD1 and SMA murine models.27 Remarkably, the reduction in miR‐206 has a negative effect on the ALS disease course in mice, suggesting that it could exert a protective role.23 Additionally, a constant increase in the miR‐206 level in skeletal muscle28, 29 and in serum has been described in ALS patients.25, 29 This finding may be associated with clinical progression, suggesting that miR‐206 may represent a biomarker in motor neuron disorders, particularly ALS.29
Furthermore, miRNAs are key elements in the regulation of cell death, which is the crucial event in the pathophysiology of MN disorders. Programmed cell death (PCD), which has been considered an analogue to apoptosis until recently, was actually extended to include other mechanisms of regulated necrosis (RN), specifically necroptosis. Both apoptosis and necroptosis exert a significant role in ALS MN death.30, 31, 32 In fact, although ALS can represent a rather heterogeneous group of diseases, one of the final common pathogenic elements is represented by MN death, which can serve also as a common therapeutic target in addition to the etiology of ALS.
In this review, we present relevant information pertaining to the miRNA role as modulators of apoptosis and RN pathways in ALS (Figure 1, Table 1), considering both their possible pathogenic role and their use as therapeutic targets.
Figure 1.
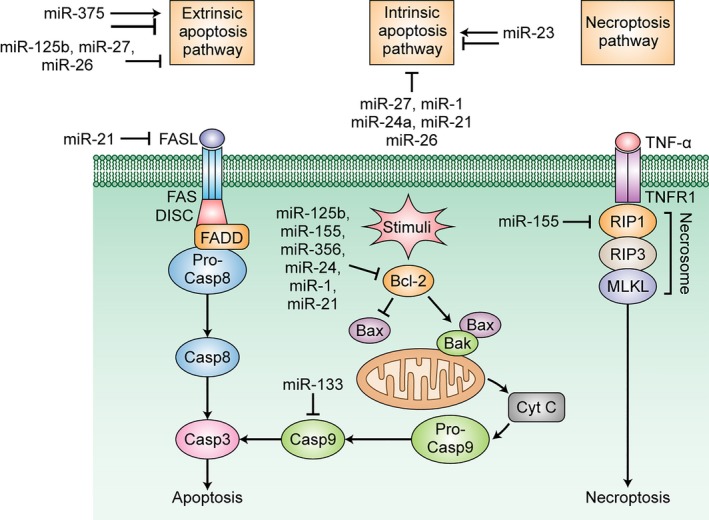
Cell death pathways currently implicated in motor neuron cell death in ALS and related miRNAs. (A) Extrinsic apoptosis. Fas ligand (FASL) activation of the cognate FAS death receptor induces intracellular formation of the death‐inducing signalling complex (DISC), which in turn interacts with Fas‐associated protein with death domain (FADD) through their death effector domains, inducing the recruitment and dissociation of pro‐caspase‐8 from the DISC. Activated caspase‐8 initiates the caspase cascade and leads to apoptosis through caspase‐3 activation. miR‐375 can promote the extrinsic pathway by various mechanisms. miR‐375, miR‐125b, and miR‐27a/b can inhibit the extrinsic pathway by silencing p53. miR‐21 can inhibit the extrinsic pathway by targeting FasL. (B) Intrinsic apoptosis. Following cytotoxic stimuli, pro‐apoptotic BH3‐only proteins silence the anti‐apoptotic protein BCL2, effectively allowing BAX and BAK to dimerize. This dimerization makes the outer mitochondrial membrane permeable and allows cytochrome_c (cyt_c) release into the cytoplasm. The cyt_c binding to apoptotic protease activating factor‐1 (Apaf‐1) prompts formation of the apoptosome, which activates caspase‐9 and the caspase cascade, in which apoptosis is ultimately driven by caspase‐3 activation. MiR‐125b, miR‐155, miR‐365, miR‐24, miR‐1, and miR‐21 promote apoptosis by silencing BCL2. MiR‐133a inhibits apoptosis by silencing caspase‐9. MiR‐23, miR‐27, miR‐1, miR‐21, and miR‐26 inhibit intrinsic apoptosis, and miR‐23 has also pro‐apoptotic properties. (C) Necroptosis. Necroptosis is favoured by low levels of caspase‐8, combined with sufficient concentrations of receptor‐interacting protein 3 (RIP3) and mixed lineage kinase domain like pseudokinase (MLKL). Following tumour necrosis factor receptor 1 (TNFR1) stimulation by tumour necrosis factor (TNF), RIP1 interacts with RIP3, and its phosphorylation is activated MLKL. They form a complex called the necrosome, which causes cell membrane rupture. miR‐155 inhibits necroptosis by silencing RIP1
Table 1.
miRNAs modulating apoptosis and necroptosis pathways in ALS
| miRNA | Target molecule | Regulatory effect | Apoptosis/Necroptosis pathways | Data in ALS |
|---|---|---|---|---|
| miR‐125b | BCL2 | Pro‐apoptotic35 | Intrinsic apoptosis | Up‐regulated in the CNS of SOD1G93A mice38, 39 |
| 7sl lncRNA | Pro‐apoptotic37 | |||
| Anti‐apoptotic36 | ||||
| p53 | Extrinsic apoptosis | |||
| miR‐155 | BCL2 | Pro‐apoptotic35 | Intrinsic apoptosis |
Up‐regulated in microglia of SOD1G93A mice39
Increased in the ALS spinal cord: fivefold in mice and twofold in humans40 Anti‐miR‐155 prolonged survival in SOD1 mice40, 41 |
| Anti‐necroptotic77 | ||||
| RIP1 | Necroptosis | |||
| miR‐365 and miR‐24 | BCL2 | Pro‐apoptotic43 | Intrinsic aptoptosis | Dysregulated in microglia ALS mice39 |
| miR‐23a/b | Apaf‐1 | Anti‐apoptotic44 | Intrinsic aptoptosis | Increased in skeletal muscle of ALS patients; influence on mitochondrial function28 |
| Pro‐apoptotic45 | ||||
| XIAP | Intrinsic aptoptosis | |||
| miR‐27a/b | Apaf‐1 | Anti‐apoptotic44 | Intrinsic apoptosis | Reduced in serum of ALS patients46 |
| Pro‐apoptotic68 | ||||
| FADD | Extrinsic apoptosis | |||
| miR‐133a | Caspase‐9 | Anti‐apoptotic47 | Intrinsic aptoptosis | Reduced in in SOD1G93A mice muscle26 |
| miR‐1 | Unknown | Anti‐apoptotic47 | Intrinsic aptoptosis | Reduced in in SOD1G93A mice muscle26 |
| BCL2, HSP60, HSP70 | Pro‐apoptotic48 | Intrinsic aptoptosis | ||
| miR‐29a | MCL‐1, P85a, CDC42 | Pro‐apoptotic48, 50 | Intrinsic aptoptosis |
Reduced in SOD1G93A mice muscle26
Increased in the lumbar spinal cord of ALS SOD1G3A mice49 |
| miR‐375 |
cIAP? cFLIP‐L? p53, ELAVL4 |
Pro‐apoptotic69 | Extrinsic apoptosis | Reduced in ALS iPSCs‐derived MNs carrying FUS mutations71 |
| Anti‐apoptotic70 | ||||
| miR‐34a | BCL2 | Pro‐apoptotic56, 57 | Intrinsic apoptosis | Decreased in SOD1G93A mice brainstem and spinal cord62 |
| SIRT1 | ||||
| miR‐26 | MTDH EZH2 | Pro‐apoptotic59 | Intrinsic and extrinsic apoptosis | Decreased in SOD1G93A mice brainstem and spinal cord62 |
| miR‐21 | BCL2, TGFBR2, Pdcd4 | Anti‐apoptotic65 | Intrinsic apoptosis | Increased in spinal cord and decreased in brainstem of SOD1G93A mice62 |
| PPARα, FASL | Extrinsic apoptosis | |||
| PTEN | Intrinisc and extrinsic apoptosis |
ALS: Amyotrophic lateral sclerosis; Apaf‐1: apoptotic protease activating factor‐1; CDC42: cell division control protein 42 homolog; cFLIP‐L: cellular FLICE (FADD‐like IL‐1β‐converting enzyme)‐inhibitory protein; cIAP: Cellular inhibitor of apoptosis 1; CNS: central nervous system; FADD: Fas‐associated protein with death domain; FASL: Fas Ligand; ELAVL4: ELAV‐like protein 4; EZH2: enhancer of zeste homolog 2; iPSCs: induced pluripotent stem cells; lncRNA: long noncoding RNA; miRNA: micro‐RNA; MCL‐1: induced myeloid leukaemia cell differentiation; MNs: motor neurons; MTDH: Metadherin; Pdcd4: programmed cell death 4; PPARα: peroxisome proliferator activated receptor alpha; PTEN: phosphatase and tensin homolog; RIP1: receptor‐interacting protein 1; SIRT1: silent information regulator 1; TGFBR2: transforming growth factor beta receptor II; XIAP: X‐linked inhibitor of apoptosis protein.
2. miRNAs AND INTRINSIC APOPTOSIS
Apoptosis is classically defined as PCD, or intracellular processes‐mediated cell death.
The term ‘intrinsic apoptosis’ refers to mitochondrial‐related apoptosis. It can be triggered by intracellular pathogenic injuries, such as oxidative stress or DNA damage, and it is regulated by the BCL2 protein.32, 33, 34 Under normal conditions, the anti‐apoptotic protein BCL2 inhibits apoptosis by blocking dimerization of the pro‐apoptotic molecules BAX and BAK.33, 34 In the presence of pathological stimuli, the BCL2 protein is hindered by the pro‐apoptotic BH3‐only proteins, favouring BAX and BAK dimerization on the outer mitochondrial membrane. BAX and BAK binding promotes permeabilization of the mitochondrial outer membrane and cytochrome c (cyt c) release into the cytoplasm. Cytosolic cyt c binds apoptotic protease activating factor‐1 (Apaf‐1) to facilitate formation of the multiprotein complex the apoptosome, which stimulates caspase‐9. Caspase‐9 activation further facilitates the caspase cascade, including caspase‐3, and the completion of apoptosis.33, 34
Intrinsic apoptosis can be controlled by several miRNAs, and a group of miRNAs are also dysregulated in ALS. Among them, miR‐125b and miR‐155 were found to display both pro‐apoptotic and anti‐apoptotic properties. Specifically, they participate in BCL2 inhibition in response to CD154 (CD40 ligand) in human leukemic B‐cells,35 thus activating the apoptotic process.
Nevertheless, miR‐125b is known as a negative regulator of p53,36 which in turn is a mediator of apoptotic pathways during cell stress. Finally, miR‐125b was reported to target the 7sl long noncoding RNA (lncRNA) as well,37 and because lncRNA is a p53 repressor, this may also contribute to its pro‐apoptotic properties.
miR‐125b was demonstrated to be up‐regulated in the CNS of SOD1‐G93A mice, correlating with neurodegeneration and astrocytosis.38 miR‐125b and miR‐155 levels were also augmented in the microglia of SOD1G93A mice, where they promote neuroinflammation.39 Increased miR‐155 expression was demonstrated in the ALS spinal cord, with a fivefold increase in rodent and twofold increase in patients samples.40 Anti‐miR‐155 prompted generalized de‐repression of mRNA targets in peritoneal macrophages and was found to disseminate in the mouse brain and spinal cord after intraventricular delivery. Treatment of SOD1‐G93A mice with anti‐miR‐155 significantly increased disease duration by 15 days and life length by 10 days.40 These data were confirmed by another set of experiments demonstrating that intraventricular delivery of anti‐miR‐155 reversed the expression of microglial miR‐155 targeted genes and that peripheral anti‐miR‐155 administration extended the lifespan.41 Moreover, abnormal microglia and monocyte signatures were recovered after genetic deletion of microglial miR‐155, and the lifespan of SOD1 mice increased by 51 days in females and 27 days in male animals.41 miR‐155 appears to be consistently over‐expressed in ALS in different studies and may be a biomarker for early disease identification.42
Several other miRNAs, including miR‐195, miR‐24, and miR‐365‐2, regulate the anti‐apoptotic protein BCL2 by targeting the 3′‐UTR.43 Up‐regulation of these molecules promotes PCD in breast cancer cells otherwise resistant to apoptosis. Additionally, miR‐365 and miR‐24 are dysregulated in microglia ALS.39
Apaf‐1, a key element of the apoptosome, is regulated by four RNA molecules (miR‐23a/b and miR‐27a/b), which assemble, forming two miRNA clusters, miR‐23a‐27a‐24 and miR‐23b‐27b‐24.44 MiR‐23a/b and miR‐27a/b inhibited Apaf‐1 expression in vitro, and their levels were inversely correlated with Apaf‐1 levels in the mouse cortex. Indeed, whereas hypoxic injuries induce the down‐regulation of miR‐23b and miR‐27b and increase Apaf‐1 expression in mouse neurons, overexpression of the two miRNAs in transgenic mice suppressed the apoptosis of neural cells induced by hypoxia.44 These findings indicate that these miRNAs may represent potential therapeutic strategies in neuronal apoptosis‐related diseases.
By contrast, it was shown that miR‐23a can exert a pro‐apoptotic role by inhibiting X‐linked inhibitor of apoptosis protein (XIAP), which binds and inhibits caspase 3, 7, and 9. Specifically, various miRNAs, such as miR‐23a, miR‐24, and the miR‐130 cluster, bind to the XIAP 3′‐UTR, inducing apoptosis.45
Compared to healthy controls, miR‐23a, along with miR‐29b, miR‐206, and miR‐455, was up‐regulated in the skeletal muscle of ALS subjects.28 In this context, miR‐23a caused a decrease in peroxisome proliferator‐activated receptor γ coactivator‐1α (PGC‐1α) protein expression in ALS tissue. This potentially has a negative effect on skeletal muscle mitochondrial function.28 miR‐27a was found diminished in the serum of ALS patients compared to healthy individuals.46
miR‐133 and miR‐1 are normally present in adult skeletal muscle and cardiomyocytes. miR‐133 binds caspase‐9 at its 3′‐UTR binding site, and it's up‐regulation by ischaemic postconditioning or through miRNA mimics prevents apoptosis induced by ischaemia‐reperfusion in rat hearts.47 miR‐1 is proposed to promote intrinsic apoptosis by regulating BCL2, HSP60 and HSP7048; however, it is reported that along with miR‐133a, it plays a key role in protecting myocardial cells in case of damage, suppressing apoptosis‐associated genes.47
Several physiological or pathological events impact miRNA expression, in turn activating or inhibiting the intrinsic apoptotic pathway. For example, in ischaemia‐reperfusion heart damage, altered levels of several miRNAs, such as miR‐1, miR‐21, miR‐29, and miR‐133, modify the expression of several miRNA‐targeted genes, including Bcl‐2, PTEN, Mcl‐1, HSP20, HSP60, HSP70, LRRFIP1, Pdcd4, and Sirt‐1, which can independently or jointly prompt intrinsic apoptosis and influence tissue damage.48
Several of the above cited miRNAs were found altered in SOD1G93A rodent muscle tissue; in particular, miR‐1, miR‐29, and miR‐133 were reduced compared to wild‐type. This local expression can influence the CNS mRNA patterns, particularly those linked with the myelination events in the spinal cord.26 Otherwise, higher expression of miR‐29a, which is specific for the CNS, has been observed from postnatal day 70 in the lumbar tract of the SOD1G3A mouse spinal cord compared to controls.49 After miR‐29a knockdown obtained by a one‐time intraventricular administration of a miR‐29a‐specific antagomir in the CNS, miR‐29 down‐regulation did not show amelioration in terms of disease progression and motor performance, despite a slight increase in lifespan in male mice.49 Indeed, up‐regulation of miR‐29 caused a reduction in the induced myeloid leukaemia cell differentiation (MCL‐1) protein, which, when expressed, has an anti‐apoptotic role similar to BCL2.50
The miR‐34 family comprises three miRNAs encoded by two different loci: the miR‐34a locus is located on chromosome 1, whereas the miR‐34b/34c cluster is located on chromosome 11. Although the latter is expressed in lungs, miR‐34 is ubiquitous, with the greatest level in the brain.51 mir‐34a has a role in cell death, particularly in caspase‐dependent apoptosis, which has been extensively recognized.52 At first, ectopic expression of miR‐34a in neuroblastoma cell lines (in which its levels are normally decreased) was found to induce apoptosis.53 Then, Chang and colleagues reported that p53 induced the up‐regulation of miR‐34a, which in turn regulates genes related to cell proliferation, DNA repair and apoptosis, thus playing a crucial role in p53‐dependent and independent apoptosis.54 These data were confirmed by Bommer et al55, who demonstrated that miR‐34a is one of the effectors of the p53 tumour suppressor function and that BCL2 is one of its target genes. Moreover, BCL2 was found to be down‐regulated by miR‐34a in a mouse model of Alzheimer's disease, and miR‐34a repression in cell lines resulted in up‐regulation of the BCL2 protein and a decrease in caspase 3 levels.56 Silent information regulator 1 (SIRT1) is a deacetylase that regulates apoptosis induced by oxidative stress and DNA damage by inactivating several molecular targets, including p53. miR‐34a was shown to inhibit SIRT1, thus promoting activation of acetylated p53 and leading to increased expression of its transcriptional target p21 and PUMA.57 Therefore, miR‐34a and p53 may promote apoptosis by regulating each other through a positive feedback loop.
Similar to miR‐34, miR‐26 is an apoptosis‐involved miRNA that exhibits different expression profiles in different biological and pathologic processes, such as growth, development, and tumourigenesis.58 It was found down‐regulated in several types of tumours, where it likely exerts a tumour‐suppressor function, and it is over‐expressed in gliomas, where it promotes cell growth and proliferation by regulating one of its target, PTEN. In particular, miR‐26 prompted activation of caspase‐9 and caspase‐8, inducing both intrinsic and extrinsic apoptosis, respectively, in human breast cancer cells by targeting EZH2 and MTDH.59 The latter gene, which encodes for a histone‐methyltransferase enzyme involved in transcriptional repression, is also silenced during myogenesis, suggesting a role for miR‐26 in the regulation of proliferation and differentiation.60 Finally, through autophagy inhibition, miR‐26 increased apoptosis in hepatocellular carcinoma cells.61
A decrease in miR‐34a and miR‐26b levels was found in the spinal cord and brainstem nuclei of SOD1‐G93A rodent models.62
Finally, given its constant over‐expression in numerous cancers, miR‐21 has been widely studied in oncology for its anti‐apoptotic properties. At first, miR‐21 knockdown was shown to increase apoptotic cell death in murine models and in human glioblastoma cells.63, 64 Then, a number of tumour‐suppressor genes implicated in apoptosis were identified as targets of miR‐21 regulation. Among them are molecules involved in intrinsic apoptosis, such as the anti‐apoptotic protein BCL2, transforming growth factor beta receptor II (TGFBR2), which participates in the pro‐apoptotic signalling of TGF‐beta, programmed cell death 4 (Pdcd4), a pro‐apoptotic gene found up‐regulated in hepatocellular cancer, and other proteins participating in the extrinsic pathway, such Fas‐ligand (FASL) and peroxisome proliferator activated receptor alpha (PPARα).65 Phosphatase and tensin homolog (PTEN), one of the most important miR‐21 targets, is a mediator of both intrinsic and extrinsic apoptosis because it has a role in both mitochondrial and tumour necrosis factor (TNF) signalling.
Although its role in ALS has not been completely recognized, over‐expression in the spinal cord and down‐regulation in the brainstem were described in mouse models.62 Indeed, it may play a function in glial interplay and the inflammatory response because it regulates astrocyte hypertrophy and scar formation after a spinal cord injury.66
3. miRNA AND EXTRINSIC APOPTOSIS
Extrinsic apoptosis is triggered by the binding of extracellular ligands to specific surface receptors, called death receptors (DRs). The most well‐characterized DRs belong to the TNF family, such as TNF receptor 1 (TNFR1) and Fas. Ligand binding and activation cause the assembly of the death‐inducing signalling complex (DISC), which in turn cooperates with the adaptor protein Fas‐associated protein with death domain (FADD), mediated by their death effector domains (DEDs). This interplay leads to the release and cleavage of pro‐caspase‐8 that once activated, prompts a caspase signalling cascade that ultimately elicits apoptosis. Several miRNAs can selectively recognize and inhibit death ligands and receptors.
As previously noted, miR‐125b targets p53, preventing cellular death, likely through a nonintrinsic‐cell mechanism.67
In human embryonic kidney cells, the miRNA cluster miR‐23a‐27a‐24 may separately promote either caspase‐dependent or independent apoptosis by silencing FADD expression.68 In particular, FADD is expected to be the target of miR‐27a based on a bioinformatics target prediction algorithm; its levels are increased proportionally to a low level of miR‐27a, and its expression was found to be directly inhibited by miR‐27a binding to its target sequence.68 Moreover, the miR‐23a‐27a‐24 cluster enhances TNFα cytotoxicity by increasing the expression of TNF receptor associated factor 2 (TRAF‐2) protein, which is an effector of TNFR1 signalling. As discussed above, serum miR‐27a was reduced in ALS patients.46
miR‐375 also increases TNFα‐induced apoptosis, although the mechanisms are unclear, although reductions in both cellular inhibitor of apoptosis 1 (cIAP) and cellular FLICE (FADD‐like IL‐1β‐converting enzyme)‐inhibitory protein (cFLIP‐L) by miR‐375 have been described.69
miR‐375 is implicated in the motor neuron disease phenotype, likely playing a protective role.70 miR‐375 is implicated in neurogenesis, inducing cell proliferation and protecting MNs from DNA damage‐induced apoptosis.70 Moreover, MNs derived from an SMA patient presented low levels of miR‐375, increased expression of p53 protein, and increased susceptibility to apoptosis in response to DNA damage.70 Remarkably, it was recently shown that miR‐375 levels were also diminished in ALS induced pluripotent stem cells (iPSCs)‐derived MNs harbouring FUS pathogenic mutations.71 Accordingly, FUS mutant MNs displayed an over‐expression of p53 and other pro‐apoptotic miR‐375 predicted targets, such as ELAV‐like protein 4 (ELAVL4). Indeed, ALS spinal MNs present increased levels of p53 protein, which may be the main driver of neuronal death mediated by apoptotic mechanisms.72
Thus, miR‐375 dysregulation is a condition shared by two different motor neuron disorders, SMA70 and FUS‐ALS71 MNs, and miR‐375 was reduced by 40% and 40%‐50%, respectively. The molecular mechanisms of miR‐375 dysregulation in SMA are unknown at present; however, it was hypothesized that FUS loss‐of‐function mutations in ALS are responsible for FUS protein discharge from the nucleus and interruption of miR‐375 synthesis.
Moreover, in addition to p53‐mediated apoptosis, miR‐375 reduction may have effects on other relevant pathways. It was associated with an up‐regulation of its target ELAVL4, an RNA‐binding protein implicated in CNS development, functioning, and plasticity.73 For this reason, it was proposed that FUS/miR‐375/ELAVL4 signalling could be a regulatory pathway involved in the RNA metabolism of MNs.71
4. miRNA AND PROGRAMMED NECROSIS
Necrosis was traditionally considered an unprogrammed and premature cell death caused by damage from pathologic external stress, resulting in cell disruption and outflow of cell content from the nucleus and cytoplasm, which in turn can represent pro‐inflammatory signals.32 Nevertheless, this hypothesis has been questioned by recent evidence suggesting that necrosis can also be a regulated process, sharing some features with apoptosis. One type of RN is called necroptosis. Similar to apoptosis, necroptosis is a type of PCD and involves DRs of the TNF family. By contrast, it is a caspase‐independent mechanism, induces membrane disruption and the release of cellular contents into the extracellular matrix, and activates the immune response.32
This process can be triggered by ligation of TNF‐α to its receptor TNFR1, and it is promoted when caspase‐8 is inhibited or reduced.74, 75 It is mediated by receptor‐interacting protein 1 (RIP1) and RIP3, which, along with the mixed lineage kinase domain like pseudokinase (MLKL), form a complex called the necrosome that causes the formation of pores in the cell membrane and finally its rupture.76
Re et al30 suggested that necroptosis participates in MN death in sALS. They demonstrated that in a coculture of MNs derived from human embryonic stem cells with astrocytes obtained from postmortem sALS patient CNS, unlike controls, the sALS astrocytes induced MN toxicity after 7 days. Treatment with a RIP1 antagonist, necrostatin‐1, and Rip1 shRNA knockdown increased the number of surviving MNs. Furthermore, treatment with necrosulfonamide, an MLKL inhibitor, led to similar improvement. Thus, these results provided evidence that a caspase‐independent form of PCD, necroptosis, has a leading role in MN death in sALS, both in in vitro and in vivo models.30 Additional findings in support of necroptosis in motor neuron disorders came from Ito and colleagues, who described the presence of this form of RN, both in cellular and animal models and in postmortem human samples, demonstrating the presence of increased levels of RIP1, RIP3, and MLKL.31 miR‐155 recognizes RIP1 as a target and was demonstrated to be over‐expressed after hydrogen‐peroxide administration in cardiomyocyte progenitor cells. Thus, miR‐155 can inhibit necroptosis in a similar manner as necrostatin.77 As mentioned above, increased miR‐155 levels were detected in the ALS CNS,40 with an unclear effect on necroptosis.
5. miR‐9 AND ‐206 AND CELL DEATH
As noted above, the most constant finding in ALS rodent and human tissues is the increase in miR‐9 in the CNS and miR‐206 in skeletal muscle and serum.23, 24, 25, 26
MiR‐9 is an miRNA highly expressed in the nervous system, which regulates neurogenesis by inhibiting neural stem cell self‐renewal and promoting neural differentiation and proliferation. As a master regulator of neurogenesis, a potential role for miR‐9 in apoptosis was supported by the observation that miR‐9 down‐regulation significantly increased cell death in the forebrain of Xenopus embryos. By contrast, apoptosis did not occur in Xenopus embryo hindbrains and in other model systems.78 In particular, it was described that an anti‐miR‐9 morpholino causes apoptosis in neural progenitor cells (NPCs) only in the forebrain, with mechanisms that involve the p53 pathway.79 Moreover, there is strong evidence for the function of miR‐9 in neurodegeneration, and up‐regulation of this miRNA was reported in postmortem brains of patients affected by Alzheimer's disease and reduced levels in the precocious stages of Huntington's disease. Finally, miR‐9 is down‐regulated in mouse embryonic cell‐derived MNs carrying the SMN1 mutation and modify the expression and dynamics of intermediate filaments.78 It is unclear whether the specific effect of miR‐9 up‐regulation in the CNS is positive or detrimental; however, it is uncontroversial that miR‐9 down‐regulation in the spinal cord causes neurofilament dysregulation and MN degeneration.13 Additionally, miR‐9 is implicated in cell proliferation and migration. As altered miR‐9 levels were described in different types of cancer, miR‐9 is also likely implicated in tumour formation and development. In consideration of the variety of its targets, miR‐9 may have different effects on several types of cells and tissue, either inducing proliferation or cell death.78 Overall, these findings suggest that miR‐9 has different roles in different contexts, and considering its dysregulation in MNs, proper modulation with drugs or gene therapy could represent an interesting challenge.
miR‐206 along with miR‐1 and miR‐133 are among the best characterized human and mouse muscle‐specific miRNAs. They are implicated in various steps of the muscle differentiation processes, including alternative splicing, DNA synthesis, and cell apoptosis.80 During development, miR‐1 and miR‐206 inhibit target paired‐box transcription factor 7 (Pax7) and its homolog Pax3, thus restricting satellite cell proliferative potential and promoting their differentiation in myogenic progenitor cells.81 Conversely, miR‐1 and miR‐206 down‐regulation provokes Pax7 and Pax3 overexpression and, subsequently, inhibition of myoblast differentiation. In this context, because Pax7 and Pax3 are pro‐survival factors, their down‐regulation by miR‐206 can cause apoptosis. It is well‐known that ALS is associated with higher expression of miR‐206 in muscle tissue, where it plays a protective role, and is necessary for the regeneration of neuromuscular junctions after acute nerve injury.23 ALS mouse models knocked‐out for the miR‐206 gene display later and incomplete muscle reinnervation compared to SOD1G93A mice wild‐type for this miRNA.82 It was recently reported that compared to controls, ALS patients showed higher miR‐206 levels in plasma, and interestingly, those values were correlated with muscle strength measured by the Medical Research Council (MRC) scale and were predictive of slower progression over 12 months.29 In later disease stages, the miR‐206 concentration decreased in muscle and serum, most likely reflecting a loss of muscle fibres. Considering this, miR‐206 could be a biomarker of lower MN involvement and muscle damage in ALS29 and an interesting therapeutic target.
However, even if preliminary data in ALS rodents showed a good therapeutic efficacy for its up‐regulation, a proper pharmacological/molecular modulation of miR‐206 may remain difficult in the clinic.
6. THERAPEUTIC PERSPECTIVES
In addition to providing interesting and reliable tools for understanding the pathogenic mechanisms underlying ALS and other neurodegenerative disorders, miRNAs represent promising therapeutic intervention strategies because the modulation of a single miRNA molecule interferes with different target genes, thus modifying broad cellular pathways and intervening at several levels in a complex disease such as ALS.
Approaches for miRNA modulation include (a) substitution of down‐regulated miRNAs with synthetic miRNA mimics, which act at the gene‐level inhibiting translation, (b) up‐regulated miRNA blockade and degradation through antisense oligonucleotides (anti‐miRs) and antagomirs, (c) de‐repression of miRNA target genes through miRNA masks, which bind to the 3′UTR of mRNA, hiding the miRNA binding site, and (d) alteration of miRNA expression at the transcription level using small drug inhibitors.83, 84
Concerns limiting these procedures include the confined biodistribution with systemic delivery and the capacity to pass the blood brain barrier, which can be overcome using intrathecal infusion, and the molecular stability over time. To improve miRNA cellular entry and allow molecular stabilization against degradation, delivery vehicles, such as liposomes, polymeric particles, and viral vectors, have been developed. In this scenario, the adenovirus‐associated virus is one of the most promising carriers.
Because in most cases miRNAs exert their function in multiple pathways, the major issue of miRNA targeted therapy is the risk of influencing the transcription of off‐target tissues, leading to the occurrence of possible side effects.
Thus far, two miRNAs implicated in cell death were tested as possible therapeutic targets in ALS: miR‐155 and miR‐29a. In both cases, as described above, miRNA overexpression was inhibited with an antagomir and a modest amelioration of the phenotype was observed.40, 49 No major side effects were recorded, although their detection is limited by the reduced lifespan of SOD1G93A mice.
Considering that miR‐375 can preserve MNs from DNA damage‐induced apoptosis via inhibition of p53 and given its down‐regulation in ALS carrying the FUS mutation,71 miR‐375 can represent an interesting therapeutic target. In this case, the up‐regulation of miR‐375 should be obtained.
Finally, because a global reduction in miRNA levels is a shared molecular characteristic for different forms of motor neuron diseases, strategies to generally up‐regulate their production, such as enhancing DICER activity through small molecules, such as enoxacin, can be explored.17
7. CONCLUSION
Cell death, with various mechanisms spanning from apoptosis to necroptosis, appears to be the final manner of MN loss in ALS. In this context, it could represent a key aspect by which therapeutic efforts can be rationally focused, in addition to a perfect knowledge of the primum movens of the disease. As important regulators of cellular processes, including cell death, miRNAs can be potential targets for therapy development for motor neuron disorders. However, because miRNAs can influence several targets within different molecular pathways, their modification with pharmacological agents or gene therapy strategies can be particularly complex. In this apparently difficult scenario, several themes, such as the opportunity to counteract the general depression of miRNAs in ALS or the specific role of several miRNAs (ie, miR‐375, which can inhibit p53‐mediated cell death) can emerge. These aspects, and hopefully new ones, warrant further investigation for the progress of therapeutic strategies for ALS.
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interests.
ACKNOWLEDGEMENTS
Joint Programme Neurodegenerative Disease (JPND) Research grant DAMNDPATHS (2014) and ARISLA grant smallRNALS (2014) to SC and the Italian Ministry of Health RF‐2013023555764 and Regione Lombardia TRANS‐ALS to GPC are gratefully acknowledged. The authors thank the Associazione Centro Dino Ferrari for its support.
Gagliardi D, Comi GP, Bresolin N, Corti S. MicroRNAs as regulators of cell death mechanisms in amyotrophic lateral sclerosis. J Cell Mol Med. 2019;23:1647–1656. 10.1111/jcmm.13976
REFERENCES
- 1. Hardiman O, Al‐Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 2. Bucchia M, Ramirez A, Parente V, et al. Therapeutic development in amyotrophic lateral sclerosis. Clin Ther. 2015;37:668‐680. [DOI] [PubMed] [Google Scholar]
- 3. Rizzo F, Riboldi G, Salani S, et al. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci. 2014;71:999‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al‐Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96‐104. [DOI] [PubMed] [Google Scholar]
- 5. Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59‐62. [DOI] [PubMed] [Google Scholar]
- 6. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron. 2011;72:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130‐133. [DOI] [PubMed] [Google Scholar]
- 8. Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. May 2008;40:572‐574. [DOI] [PubMed] [Google Scholar]
- 9. Sreedharan J, Blair IP, Tripathi VB, et al. TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522‐531. [DOI] [PubMed] [Google Scholar]
- 11. Krokidis MG, Vlamos P. Transcriptomics in amyotrophic lateral sclerosis. Front Biosci (Elite Ed). 2018;10:103‐121. [DOI] [PubMed] [Google Scholar]
- 12. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell. 1993;75:843‐854. [DOI] [PubMed] [Google Scholar]
- 13. Haramati S, Chapnik E, Sztainberg Y, et al. miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci USA. 2010;107:13111‐13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rinchetti P, Rizzuti M, Faravelli I, Corti S. MicroRNA metabolism and dysregulation in amyotrophic lateral sclerosis. Mol Neurobiol. 2018;55:2617‐2630. [DOI] [PubMed] [Google Scholar]
- 15. Magri F, Vanoli F, Corti S. miRNA in spinal muscular atrophy pathogenesis and therapy. J Cell Mol Med. 2018;22:755‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Campos‐Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ. Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain. 2013;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emde A, Eitan C, Liou LL, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS‐causing mutations: a new mechanism for ALS. EMBO J. 2015;34:2633‐2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wakabayashi K, Mori F, Kakita A, Takahashi H, Utsumi J, Sasaki H. Analysis of microRNA from archived formalin‐fixed paraffin‐embedded specimens of amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2014;2:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Freischmidt A, Müller K, Zondler L, et al. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36:2660 e15‐20. [DOI] [PubMed] [Google Scholar]
- 20. Raman R, Allen SP, Goodall EF, et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia‐response and RNA processing functions. Neuropathol Appl Neurobiol. 2015;41:201‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahashi I, Hama Y, Matsushima M, et al. Identification of plasma microRNAs as a biomarker of sporadic Amyotrophic Lateral Sclerosis. Mol Brain. 2015;8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benigni M, Ricci C, Jones AR, Giannini F, Al‐Chalabi A, Battistini S. Identification of miRNAs as potential biomarkers in cerebrospinal fluid from amyotrophic lateral sclerosis patients. Neuromolecular Med. 2016;18:551‐560. [DOI] [PubMed] [Google Scholar]
- 23. Williams AH, Valdez G, Moresi V, et al. MicroRNA‐206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549‐1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou F, Guan Y, Chen Y, et al. miRNA‐9 expression is upregulated in the spinal cord of G93A‐SOD1 transgenic mice. Int J Clin Exp Pathol. 2013;6:1826‐1838. [PMC free article] [PubMed] [Google Scholar]
- 25. Toivonen JM, Manzano R, Oliván S, Zaragoza P, García‐Redondo A, Osta R. MicroRNA‐206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS ONE. 2014;9:e89065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dobrowolny G, Bernardini C, Martini M, Baranzini M, Barba M, Musarò A. Muscle expression of SOD1(G93A) modulates microRNA and mRNA transcription pattern associated with the myelination process in the spinal cord of transgenic mice. Front Cell Neurosci. 2015;9:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valsecchi V, Boido M, De Amicis E, Piras A, Vercelli A. Expression of muscle‐specific MiRNA 206 in the progression of disease in a murine SMA model. PLoS ONE. 2015;10:e0128560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russell AP, Wada S, Vergani L, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis. 2013;49:107‐117. [DOI] [PubMed] [Google Scholar]
- 29. de Andrade HM, de Albuquerque M, Avansini SH, et al. MicroRNAs‐424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J Neurol Sci. 2016;368:19‐24. [DOI] [PubMed] [Google Scholar]
- 30. Re DB, Le Verche V, Yu C, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morrice JR, Gregory‐Evans CY, Shaw CA. Necroptosis in amyotrophic lateral sclerosis and other neurological disorders. Biochim Biophys Acta. 2017;1863:347‐353. [DOI] [PubMed] [Google Scholar]
- 33. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49‐63. [DOI] [PubMed] [Google Scholar]
- 34. Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19:555‐566. [DOI] [PubMed] [Google Scholar]
- 35. Willimott S, Wagner SD. miR‐125b and miR‐155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B‐cells. J Biol Chem. 2012;287:2608‐2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le MT, Teh C, Shyh‐Chang N, et al. MicroRNA‐125b is a novel negative regulator of p53. Genes Dev. 2009;23:862‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jalali S, Bhartiya D, Lalwani MK, Sivasubbu S, Scaria V. Systematic transcriptome wide analysis of lncRNA‐miRNA interactions. PLoS ONE. 2013;8:e53823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marcuzzo S, Bonanno S, Kapetis D, et al. Up‐regulation of neural and cell cycle‐related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Mol Brain. 2015;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parisi C, Arisi I, D'Ambrosi N, et al. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013;4:e959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koval ED, Shaner C, Zhang P, et al. Method for widespread microRNA‐155 inhibition prolongs survival in ALS‐model mice. Hum Mol Genet. 2013;22:4127‐4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butovsky O, Jedrychowski MP, Cialic R, et al. Targeting miR‐155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77:75‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cunha C, Santos C, Gomes C, et al. Downregulated glia interplay and increased miRNA‐155 as promising markers to track ALS at an early stage. Mol Neurobiol. 2017;55:4207‐4224. [DOI] [PubMed] [Google Scholar]
- 43. Su Y, Wu H, Pavlosky A, et al. Regulatory non‐coding RNA: new instruments in the orchestration of cell death. Cell Death Dis. 2016;7:e2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen Q, Xu J, Li L, et al. MicroRNA‐23a/b and microRNA‐27a/b suppress Apaf‐1 protein and alleviate hypoxia‐induced neuronal apoptosis. Cell Death Dis. 2014;5:e1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu W, Xu H, Zhu D, et al. miR‐200bc/429 cluster modulates multidrug resistance of human cancer cell lines by targeting BCL2 and XIAP. Cancer Chemother Pharmacol. 2012;69:723‐731. [DOI] [PubMed] [Google Scholar]
- 46. Tasca E, Pegoraro V, Merico A, Angelini C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin Neuropathol. 2016;35:22‐30. [DOI] [PubMed] [Google Scholar]
- 47. He B, Xiao J, Ren AJ, et al. Role of miR‐1 and miR‐133a in myocardial ischemic postconditioning. J Biomed Sci. 2011;18:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ye Y, Perez‐Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia‐reperfusion injury. Physiol Genomics. May 2011;43:534‐542. [DOI] [PubMed] [Google Scholar]
- 49. Nolan K, Mitchem MR, Jimenez‐Mateos EM, Henshall DC, Concannon CG, Prehn JHM. Increased expression of microRNA‐29a in ALS mice: functional analysis of its inhibition. J Mol Neurosci. 2014;53:231‐241. [DOI] [PubMed] [Google Scholar]
- 50. Nijhuis A, Curciarello R, Mehta S, et al. MCL‐1 is modulated in Crohn's disease fibrosis by miR‐29b via IL‐6 and IL‐8. Cell Tissue Res. 2017;368:325‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR‐34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591‐2600. [DOI] [PubMed] [Google Scholar]
- 52. Hermeking H. The miR‐34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193‐199. [DOI] [PubMed] [Google Scholar]
- 53. Welch C, Chen Y, Stallings RL. MicroRNA‐34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017‐5022. [DOI] [PubMed] [Google Scholar]
- 54. Chang T‐C, Wentzel EA, Kent OA, et al. Transactivation of miR‐34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bommer GT, Gerin I, Feng Y, et al. p53‐mediated activation of miRNA34 candidate tumor‐suppressor genes. Curr Biol. 2007;17:1298‐1307. [DOI] [PubMed] [Google Scholar]
- 56. Wang X, Liu P, Zhu H, et al. miR‐34a, a microRNA up‐regulated in a double transgenic mouse model of Alzheimer's disease, inhibits bcl2 translation. Brain Res Bull. 2009;80:268‐273. [DOI] [PubMed] [Google Scholar]
- 57. Yamakuchi M, Ferlito M, Lowenstein CJ. miR‐34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421‐13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao J, Liu Q‐G. The role of miR‐26 in tumors and normal tissues (Review). Oncol Lett. 2011;2:1019‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang B, Liu XX, He JR, et al. Pathologically decreased miR‐26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32:2‐9. [DOI] [PubMed] [Google Scholar]
- 60. Wong CF, Tellam RL. MicroRNA‐26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. J Biol Chem. 2008;283:9836‐9843. [DOI] [PubMed] [Google Scholar]
- 61. Jin F, Wang Y, Li M, et al. MiR‐26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017;8:e2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou F, Zhang C, Guan Y, et al. Screening the expression characteristics of several miRNAs in G93A‐SOD1 transgenic mouse: altered expression of miRNA‐124 is associated with astrocyte differentiation by targeting Sox2 and Sox9. J Neurochem. 2018;145:51‐67. [DOI] [PubMed] [Google Scholar]
- 63. Si M‐L, Zhu S, Wu H, Lu Z, Wu F, Mo Y‐Y. miR‐21‐mediated tumor growth. Oncogene. 2007;26:2799‐2803. [DOI] [PubMed] [Google Scholar]
- 64. Chan JA, Krichevsky AM, Kosik KS. MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029‐6033. [DOI] [PubMed] [Google Scholar]
- 65. Buscaglia LEB, Li Y. Apoptosis and the target genes of microRNA‐21. Chin J Cancer. 2011;30:371‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bhalala OG, Pan L, Sahni V, et al. microRNA‐21 regulates astrocytic response following spinal cord injury. J Neurosci. 2012;32:17935‐17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu J, Guo B, Chen Z, et al. miR‐125b promotes MLL‐AF9‐driven murine acute myeloid leukemia involving a VEGFA‐mediated non‐cell‐intrinsic mechanism. Blood. 2017;129:1491‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chhabra R, Adlakha YK, Hariharan M, Scaria V, Saini N. Upregulation of miR‐23a‐27a‐24‐2 cluster induces caspase‐dependent and ‐independent apoptosis in human embryonic kidney cells. PLoS ONE. 2009;4:e5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang J, Huang H, Wang C, Liu X, Hu F, Liu M. MicroRNA‐375 sensitizes tumour necrosis factor‐alpha (TNF‐α)‐induced apoptosis in head and neck squamous cell carcinoma in vitro. Int J Oral Maxillofac Surg. 2013;42:949‐955. [DOI] [PubMed] [Google Scholar]
- 70. Bhinge A, Namboori SC, Bithell A, Soldati C, Buckley NJ, Stanton LW. MiR‐375 is essential for human spinal motor neuron development and may be involved in motor neuron degeneration. Stem Cells. 2016;34:124‐134. [DOI] [PubMed] [Google Scholar]
- 71. De Santis R, Santini L, Colantoni A, et al. FUS mutant human motoneurons display altered transcriptome and microRNA pathways with implications for ALS pathogenesis. Stem Cell Reports. 2017;9:1450‐1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ranganathan S, Bowser R. p53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol J. 2010;4:11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bronicki LM, Jasmin BJ. Emerging complexity of the HuD/ELAVl4 gene; implications for neuronal development, function, and dysfunction. RNA. 2013;19:1019‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science. 2009;325:332‐336. [DOI] [PubMed] [Google Scholar]
- 75. He S, Wang L, Miao L, et al. Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell. 2009;137:1100‐1111. [DOI] [PubMed] [Google Scholar]
- 76. Murphy JM, Czabotar PE, Hildebrand JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443‐453. [DOI] [PubMed] [Google Scholar]
- 77. Liu J, van Mil A, Vrijsen K, et al. MicroRNA‐155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med. 2011;15:1474‐1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yuva‐Aydemir Y, Simkin A, Gascon E, Gao F‐B. MicroRNA‐9: functional evolution of a conserved small regulatory RNA. RNA Biol. 2011;8:557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bonev B, Pisco A, Papalopulu N. MicroRNA‐9 reveals regional diversity of neural progenitors along the anterior‐posterior axis. Dev Cell. 2011;20:19‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luo W, Nie Q, Zhang X. MicroRNAs involved in skeletal muscle differentiation. J Genet Genomics. 2013;40:107‐116. [DOI] [PubMed] [Google Scholar]
- 81. Chen JF, Tao Y, Li J, et al. microRNA‐1 and microRNA‐206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol. 2010;190:867‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. De Felice B, Guida M, Guida M, Coppola C, De Mieri G, Cotrufo R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene. 2012;508:35‐40. [DOI] [PubMed] [Google Scholar]
- 83. Basak I, Patil KS, Alves G, Larsen JP, Møller SG. microRNAs as neuroregulators, biomarkers and therapeutic agents in neurodegenerative diseases. Cell Mol Life Sci. 2016;73:811‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Baumann V, Winkler J. miRNA‐based therapies: strategies and delivery platforms for oligonucleotide and non‐oligonucleotide agents. Future Med Chem. 2014;6:1967‐1984. [DOI] [PMC free article] [PubMed] [Google Scholar]