

Abstract
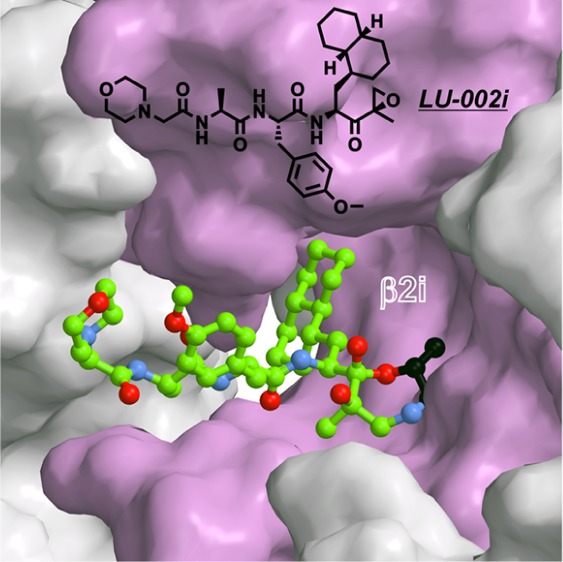
Subunit-selective proteasome inhibitors are valuable tools to assess the biological and medicinal relevance of individual proteasome active sites. Whereas the inhibitors for the β1c, β1i, β5c, and β5i subunits exploit the differences in the substrate-binding channels identified by X-ray crystallography, compounds selectively targeting β2c or β2i could not yet be rationally designed because of the high structural similarity of these two subunits. Here, we report the development, chemical synthesis, and biological screening of a compound library that led to the identification of the β2c- and β2i-selective compounds LU-002c (4; IC50 β2c: 8 nM, IC50 β2i/β2c: 40-fold) and LU-002i (5; IC50 β2i: 220 nM, IC50 β2c/β2i: 45-fold), respectively. Co-crystal structures with β2 humanized yeast proteasomes visualize protein–ligand interactions crucial for subunit specificity. Altogether, organic syntheses, activity-based protein profiling, yeast mutagenesis, and structural biology allowed us to decipher significant differences of β2 substrate-binding channels and to complete the set of subunit-selective proteasome inhibitors.
Introduction
Proteasomes are proteolytic machines responsible for the degradation of misfolded proteins localized in the cytosol and nucleus of eukaryotic cells.1 Their 20S core particles (CPs) are C2-symmetrical barrel-shaped complexes assembled of 28 subunits that are arranged in four stacked seven-membered rings.2 The two outer rings are made of seven α subunits (α1–7) and the two inner rings consist of seven homologous yet distinct β subunits (β1–7). In ubiquitously expressed constitutive proteasomes, the proteolytic activities reside within the subunits β1c (caspase-like activity), β2c (trypsin-like activity), and β5c (chymotrypsin-like activity).3 In lymphoid tissues, these subunits are replaced by their interferon-γ-inducible counterparts, β1i (LMP2), β2i (MECL-1), and β5i (LMP7),4 yielding the so-called immunoproteasome particles (iCPs) that preferentially generate antigenic peptides with high affinity for major histocompatibility complex (MHC) class I receptors.5
Proteasomes are validated drug targets in oncology, and numerous structurally diverse inhibitors of natural and nonnatural origin have been reported so far.6 Most synthetic compounds are N-terminally capped peptides of two to four residues with a C-terminal electrophilic warhead that forms a covalent linkage with the nucleophilic hydroxyl group and possibly the free N terminus of threonine-1 (Thr1) of the catalytically active proteasomal β subunits.7 Subunit specificity of peptidic ligands is largely determined by the sequence of the peptide fragment, although the nature of the warhead can confer selectivity as well.8 The first-generation boronic acid bortezomib and the second-generation epoxyketone carfilzomib target more than one subunit at a time and therefore are considered broad-spectrum proteasome inhibitors.6a Bortezomib and carfilzomib are now approved drugs for the treatment of multiple myeloma.9,10 Current industrial and academic drug design efforts focus on the development of subunit-selective proteasome inhibitors and their potential therapeutic use in chronic inflammatory diseases. For instance, the first immunoproteasome-selective compound KZR-616,11 an analog of ONX 0914,12 has recently entered phase 1b/2 clinical trials for the treatment of lupus erythematosus. Besides medical issues, selective inhibition of individual proteasome subunits may aid investigations on the involvement of these sites in different cellular pathways including MHC class I antigen presentation and control of cytokine levels. Although there is an overlap in the substrate preferences of the cCP and iCP subunits, distinct structural features and amino acid linings of the substrate-binding channels β1c and β1i as well as β5c and β5i could be identified and subsequently allowed for the development of specific inhibitors.12,13 The design of inhibitors targeting exclusively β2c or β2i however remained a challenge because of the high structural similarity between the trypsin-like active sites13d In 2018, Liskamp and co-workers reported a set of β2-selective inhibitors. However, these compounds, which are characterized by a sulfonyl fluoride as the C-terminal electrophile, a basic P1 residue, and a free N terminus, display limited preference for either β2c or β2i.14 In addition, Kezar Life Sciences developed an epoxyketone inhibitor with moderate selectivity for human β2i.11
Recently, we published a set of activity-based protein-profiling (ABPP) probes and inhibitors selective for each of the six catalytic activities of human cCP and iCP, including compounds LU-002c (β2c) and LU-002i (β2i; Figure 1).15 Here, we describe the design, synthesis, and screening of focused compound libraries that allowed us to identify these β2c and β2i inhibitors, respectively. Crystallographic data on humanized yeast proteasomes in complex with selective ligands provide insights into their mode of binding and reveal so far unnoticed differences in substrate and inhibitor specificity for the trypsin-like active sites of cCP and iCP.
Figure 1.

Chemical structures and IC50 values for the lead structures LU-102 (1),16 LU-112 (2),16 and ONX 0914 (3)12 that guided the development of the β2c- and β2i-selective compounds LU-002c (4) and LU-002i (5), respectively. IC50 values were measured by competitive ABPP.
Results
Development of Selective Inhibitors for Subunit β2c
The previously identified vinyl sulfone inhibitor LU-102 (Figure 1), which inhibits β2c and β2i with similar potency,16 was used as a starting point for creating selective β2c ligands. We generated a compound library based on the vinyl sulfone warhead and the 4-aminomethylphenyl side chain on P1 of LU-102, as these moieties proved to be crucial for β2 selectivity in general.16 In a first step, we replaced the N cap of LU-102 by a set of groups often found in peptide-based proteasome inhibitors (6–12). Next, we synthesized compounds with relatively small amino acid side chains in the P2 position (4, 13–20) and finally incorporated bulky aliphatic side chains at P2 and P3 (21–36). In total, 32 compounds were prepared using established protocols for the chemical synthesis of the 4-aminomethylphenylalanine vinyl sulfone warhead and solution-phase coupling of the peptide vinyl sulfones to the corresponding alpha-amino acids (see Supporting Information).27
All compounds were evaluated for β2c/β2i inhibition by our competitive ABPP assay at the final concentrations of 0.01, 0.1, 1.0, and 10.0 μM, and the apparent IC50 values were determined (Table 1). Among the N-cap series 6–12, compound 7 (pyrazine N cap) showed the highest selectivity for β2c over β2i (40-fold), but also decreased potency for β2c compared to LU-102 (23-fold). Screening of small P2 residues (compounds 4, 13–20) identified several ligands with both good selectivity and potency for β2c: 4 (P2 alanine; 10 nM, 32-fold selectivity over β2i), 13 (P2 serine; 11 nM, 47-fold), 15 (P2 methoxyserine; 8 nM, 25-fold), 16 (P2 threonine; 8 nM, 41-fold), and especially 18 (P2 glycine; 26 nM, 224-fold). Combining 2-methylthiazole N caps (20) with bulky P2 or P3 residues (21–36) revealed several potent and selective β2c compounds as well: see for instance, compounds 20 (P2 methoxyserine, P3 leucine; 72 nM, 14-fold), 22 (P2 leucine, P3 cyclohexyl; 18 nM, 30-fold), 30 (P2 cyclohexyl-homoalanine, P3 leucine; 11 nM, 25-fold), and 36 (P2 and P3 cyclohexyl; 40 nM, 10.5-fold). Altogether, based on the data shown in Table 1, we conclude that (1) subunit β2c accepts small as well as bulky P2 residues but disfavors oversized P3 side chains and that (2) β2i disfavors small P2 side chains and large P3 groups.
Table 1. Chemical Structures of Compounds 4, 6–36 and Their Inhibitory Activity (Apparent IC50 Values) against β2c and β2i (Determined by Competitive ABPP)a.



A high β2i/β2c ratio indicates selectivity for β2c. Raw data used for the calculations of IC50 values are in the Supporting Information.
To establish the apparent IC50 values more accurately and to obtain insights into the coinhibition of β1c, β1i, β5c, and β5i activities, we selected the compounds 4, 7, 13, 16, 18, 20, 22, and 25 for further analysis. In our competitive ABPP assay using Raji cell extracts (containing both cCPs and iCPs), a wider range of final concentrations were tested. All compounds inhibited β2c at low nanomolar concentrations (Table 2). The inhibitors 4, 13, 18, and 20, featuring small side chains on P2, displayed considerably enhanced selectivity for β2c over β2i (≥27-fold) compared to LU-102 (1.6-fold; Table 2), with 18 being the most selective (54-fold).
Table 2. Apparent IC50 Values of Compounds 1 (LU-102), 4, 7, 13, 16, 18, 20, 22, and 25 for the Six Catalytic Sites from Human cCPs and iCPs in Raji Cell Lysates, as Established by Competitive ABPP.
| apparent IC50 (μM) |
ratio |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | β2c | β2i | β5c | β5i | β1c | β1i | β2i/β2c | β1i/β2c | β1c/β2c | β5i/β2c | β5c/β2c |
| 1 (LU-102) | 0.013 | 0.020 | 1.33 | 1.17 | >100 | >100 | 2 | >7700 | >7700 | 90 | 102 |
| 4 (LU-002c) | 0.0050 | 0.14 | 1.3 | 2.8 | >100 | >100 | 27 | >19 000 | >19 000 | 540 | 250 |
| 7 | 0.17 | 2.9 | >100 | >100 | >100 | >100 | 17 | >600 | >600 | >600 | >600 |
| 13 | 0.0060 | 0.23 | 1.4 | 2.2 | >100 | >100 | 40 | >17 000 | >17 000 | 380 | 241 |
| 16 | 0.0070 | 0.11 | 0.75 | 2.1 | >100 | >100 | 16 | >14 000 | >14 000 | 300 | 107 |
| 18 | 0.046 | 2.5 | 8.6 | 12.7 | >100 | >100 | 54 | >2200 | >2200 | 187 | 276 |
| 20 | 0.077 | 4.0 | 45.3 | 57.1 | >100 | >100 | 52 | >1300 | >1300 | 740 | 590 |
| 22 | 0.065 | 0.42 | >100 | >100 | >100 | >100 | 6 | >1500 | >1500 | >1500 | >1500 |
| 25 | 0.44 | 3.1 | >100 | >100 | >100 | >100 | 7 | >220 | >220 | >220 | >220 |
Next, we assessed the inhibitory effects in living RPMI-8226 cells (Table 3). Initial screenings identified compound 4 as the most active, and we included this compound as LU-002c in our suite of subunit-selective proteasome inhibitors.15 In subsequent studies, we identified compound 16 to be even more potent and selective, and we dubbed this compound LU-012c.
Table 3. Inhibition of Proteasome Activities by Compounds 1 (LU-102), 4 (LU-002c), 7, 13, 16 (LU-012c), 18, 20, 22, and 25 in Intact RPMI-8226 Cells.
| apparent IC50 (μM) |
ratio | ||||||
|---|---|---|---|---|---|---|---|
| compound | β2c | β2i | β5c | β5i | β1c | β1i | β2i/β2c |
| 1 (LU-102)a | 0.29 | 0.41 | >10 | >10 | >10 | >10 | 1.4 |
| 4 (LU-002c)a | 1.80 | >10 | >10 | >10 | >10 | >10 | >5.6 |
| 7 | >10 | >10 | >10 | >10 | >10 | >10 | n.d. |
| 13 | 2.00 | >10 | >10 | >10 | >10 | >10 | >5 |
| 16 (LU-012c) | 1.250 | >10 | >10 | >10 | >10 | >10 | >8 |
| 18 | >10 | >10 | >10 | >10 | >10 | >10 | n.d. |
| 20 | >10 | >10 | >10 | >10 | >10 | >10 | n.d. |
| 22 | >10 | >10 | >10 | >10 | >10 | >10 | n.d. |
| 25 | >10 | >10 | >10 | >10 | >10 | >10 | n.d. |
Data cited from the literature; n.d., not determined.
Development of β2i-Selective Inhibitors
For the development of β2i-selective compounds, we used ONX 0914 (3)12 as the starting point (Figure 1). Though ONX 0914 is a β5i-selective inhibitor, it also targets other proteasome subunits12,13b (Figure 1) and shows slight selectivity for β2i over β2c (IC50 (β2i) 0.59 μM; IC50 (β2c) 1.1 μM, 1.9-fold).13b During our efforts to create β5i-selective compounds, we noted that the substitution of P1 phenylalanine in ONX 0914 for cyclohexylalanine enhances the selectivity for both β5i and β2i over the respective constitutive subunits (ratio β2c/β2i = 6) and that any additional modifications of the P2 and P3 positions as well as the N cap led to the loss of activities for the trypsin-like sites.13b On the basis of these observations, we reasoned that large aliphatic amino acid residues at P1 might lead to β2i-selective inhibitors. To probe this hypothesis, a set of epoxyketone inhibitors with large hydrophobic P1 residues (compounds 5, 37–53, Table 4) was synthesized (for details, see Supporting Information).
Table 4. Structures of Compounds 5, 37–53 and Their Inhibitory Activity (Apparent IC50 Values) against β2c and β2i (Determined by the Competitive ABPP Assay)a.


A high β2c/β2i ratio indicates selectivity for β2i.
n.d., not determined.*Compounds 44a and 44b are diastereomers; for details on stereochemistry, see Supporting Information.
The compounds were tested at the final concentrations of 0.01, 0.1, 1.0, and 10.0 μM by our competitive ABPP assay, and the apparent IC50 values for the inhibition of β2c and β2i were determined (Table 4). In this first evaluation step, compounds 5 (P1 1-decalanine; 320 nM, >31-fold), 39 (P1 cyclohexyl-homoalanine; 215 nM, >46-fold), 41b (methylcyclohexylalanine; 265 nM, 24-fold), and 44b (bicyclohexylalanine; 100 nM, 9-fold) showed the highest selectivity for β2i over β2c.
Next, the inhibition of all six sites by compounds 5 and 39 were tested at a wider range of final concentrations (Table 5). In this setup, compound 5 proved to be the most selective β2i ligand (ratio β2c/β2i: 67) as it did not inhibit any of the β1 and β5 proteasome subunits. By contrast, epoxyketone 39 proved to be a dual inhibitor of both β2i and β5i with high selectivity over the corresponding constitutive subunits (ratio β2c/β2i: 44; ratio β5c/β5i: 109).
Table 5. Apparent IC50 (μM) Values of Compounds 5 and 39 against the Six Catalytic Active Sites from Human cCPs and iCPs, as Determined in Raji Cell Lysates by Competitive ABPP.
| compound | β2i | β2c | β5i | β5c | β1i | β1c | ratio β2c/β2i | ratio β5c/β5i |
|---|---|---|---|---|---|---|---|---|
| 5 (LU-002i) | 0.18 | 12.1 | >100 | >100 | >100 | >100 | 67 | ∼1 |
| 39 | 0.057 | 2.5 | 0.046 | 5.0 | >100 | >100 | 44 | 109 |
Epoxyketone 5, the most selective β2i inhibitor of the series, was termed LU-002i and published as part of a set of compounds and ABPP probes to visualize all the six catalytic activities of human constitutive and immunoproteasomes.15 However, the decalin moiety of 5 was synthesized as a mixture of stereoisomers that could not be separated. To address the question whether one or both of the possible stereomers are active, the following attempts were undertaken to synthesize a stereomerically pure analogue of 5 (LU-002i). First, compounds with partially reduced naphthyl rings containing only one chiral carbon center within the bicyclic system were synthesized: 68 (R) and 71 (S) (Scheme 1; Supporting Information). In the competitive ABPP assay in Raji cell lysates (Table 6), 68 was inactive, whereas 71 selectively targeted β2i, though with a dramatic loss of potency (IC50 2.5 μM) compared to 5 (IC50 0.18 μM).
Scheme 1. Synthesis of Compounds 68 and 71.

Reagents and conditions: (a) (i) LiAlH4/Et2O, 99%; (ii) TsCl/triethylamine (TEA)/dichloromethane (DCM), 97%; (iii) NaCN/dimethylformamide (DMF), 95%; (b) (i) KOH/ethylene glycol; (ii) N,O-dimethylhydroxylamine hydrochloride, 2-(6-chloro-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU)/N,N-diisopropylethylamine (DiPEA)/DCM, 49% over two steps; (c) LiAlH4/Et2O; (d) 58/CuSO4/DCM, 84% two-step yield; (e) Et2AlCN/i-PrOH/tetrahydrofuran (THF), 58%; (f) (i) 6 M HCl, reflux; (ii) Boc2O/TEA/THF/H2O, 58% over two steps; (g) N,O-dimethylhydroxylamine hydrochloride, HCTU/DiPEA/DCM, 77%; (h) tBuLi/2-bromopropene/Et2O, −78 °C, 78%; (i) NaBH4/CeCl3·7H2O/MeOH, 59%; (j) (1) VO(acac)2/tBuOOH/DCM; (2) Dess–Martin periodinane/DCM, 33% over two steps; (k) trifluoroacetic acid (TFA), quantitative yield; (l) (1) 67, tBuONO/HCl (4N in dioxane) DCM/DMF, −30 °C; (2) 66, DiPEA, DMF, 40% over two steps.
Table 6. Apparent IC50 (μM) Values of Compounds 68, 71, 74, 77, 86, and 87 against the Six Catalytic Active Sites from Human cCPs and iCPs, Determined in Raji Cell Lysates by Competitive ABPP.
| compound | β2i | β2c | β5i | β5c | β1i | β1c |
|---|---|---|---|---|---|---|
| 68 | >100 | >100 | >100 | >100 | >100 | >100 |
| 71 | 2.5 | >100 | >100 | >100 | >100 | >100 |
| 74 | 12.0 | >100 | >100 | >100 | >100 | >100 |
| 77 | 0.38 | 28 | >100 | >100 | >100 | >100 |
| 86 | 34.0 | >100 | >100 | >100 | >100 | >100 |
| 87 | 0.19 | 19 | 28.40 | >100 | >100 | 53 |
In a second approach to unravel the active stereomer of 5, fully reduced decalin systems were produced, yielding the peptide epoxyketones 74 and 77, respectively (Scheme 2; Supporting Information). Competitive ABPP revealed that 74 inhibits β2i with an IC50 of 12.0 μM without touching the other five active sites of cCP and iCP particles (Table 6). Compound 77 in turn proved to be a potent β2i inhibitor (IC50 0.38 μM) with some cross-reactivity against β2c (IC50 28 μM). Notably, the absolute stereochemistry of the P1 side chain in 77 matches that of the corresponding carbon center in ligand 71, but it appears that decalin at P1 (77) is more effective for β2i inhibition than the corresponding partially oxidized bicyclic system (71).
Scheme 2. Synthesis of Compounds 74 and 77.

Reagents and conditions: (a) H2, PtO2, AcOH, 99%.
With this information in hand, an enantiomerically pure diastereomeric set of peptide epoxyketones 86 and 87 was synthesized (Scheme 3; Supporting Information). Compound 86 appeared to be a weak (IC50 34 μM) but selective β2i inhibitor, whereas epoxyketone 87 strongly inhibits β2i (IC50 0.19 μM) with β2c, β1c, and β5i as off-targets at high micromolar concentrations (Table 6). On the basis of the assumption that carbon 1 in the decalin system of compound 87 has the (S) configuration as in 71 and 77, and assuming that the catalytic hydrogenation proceeded to deliver decalin with cis stereochemistry, the observed results strongly suggest that the stereochemistry of the most active and selective β2i inhibitor is as shown in structure 87 (Scheme 3).
Scheme 3. Synthesis of Compounds 86 and 87.

Reagents and conditions: (a) H2, PtO2, AcOH, quantitative yield; (b) (i) LiAlH4/Et2O, 92%; (ii) TsCl/TEA/DCM, 95%; (3) NaCN/DMF, 83%; (c) (i) KOH/ethylene glycol; (ii) N,O-dimethylhydroxylamine hydrochloride, HCTU/DiPEA/DCM, 88% over two steps; (d) LiAlH4/Et2O; (e) 58/CuSO4/DCM, 85% over two steps; (f) Et2AlCN/i-PrOH/THF, 75%.
To test whether compound 87 is the major active component of the stereomeric mixture that makes up compound 5 (the previously described β2i-selective inhibitor, LU-002i15), both were assessed in a competitive ABPP assay in Raji cell extracts at final inhibitor concentrations ranging from 0 to 3 μM (Figure 2). As both preparations are about equally active and selective, diastereomer 87 appears to be indeed the main active component in the stereomeric mixture that has previously been reported as LU-002i.15
Figure 2.
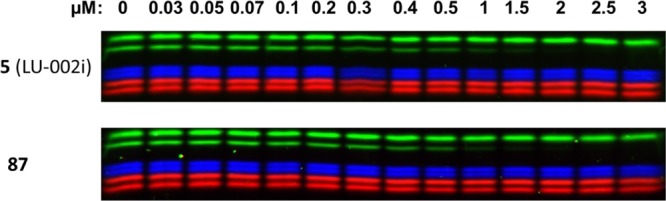
Comparative ABPP assay of compounds 5 (LU-002i) and 87, determined in Raji cell lysates.
Next, compound 87 was tested in intact RPMI-8226 cell lines, in comparison with the dual β2i/β5i inhibitor 39. The cells were first treated with the inhibitor at various concentrations, then lysed, incubated with the ABPP mixture, denatured, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), as described before. Like in Raji cell lysates, compound 87 selectively targeted only β2i (IC50 0.159 μM) without affecting the remaining proteolytically active proteasome subunits, whereas epoxyketone 39 inhibited both β2i (IC50 0.124 μM) and β5i (IC50 0.183 μM) (Figure 3). Thus, inhibitor 39 represents a co-inhibitor of β2i and β5i with potential medicinal relevance, especially because targeting of β2 has previously been shown to sensitize cells to β5 inhibitors,17 and dual subunit inhibition is required for suppressing autoinflammatory reactions.11
Figure 3.

Inhibition profiles of compounds 39 and 87, determined in intact RPMI-8226 cell lines.
As the next research objective, we decided to investigate whether a β2i-selective activity-based probe (ABP) could be derived from LU-002i (5). As the attachment of a fluorescent tag at the N terminus of subunit-selective inhibitors may be detrimental to selectivity, we decided to graft the reporter group onto the tyrosine residue at P2 by substituting the methyl group for an appropriately functionalized alkyl group (Scheme 4). The resulting ABP 97 was tested in Raji cell lysates to profile the proteasome activities. At a final concentration of 3 μM, β2i labeling was selective and could be easily distinguished (Figure 4A). In a competitive ABPP assay with probe 97, labeling of β2i could be completely abolished by preincubation with LU-002i (5, β2i) at 3 μM. The β2i signal was partially reduced after treatment with LU-002c (4, β2c) at high concentrations and completely abolished after preincubation with LU-102 (1, β2c/β2i) (Figure 4B). Finally, a competitive ABPP assay with probe 97 side-by-side with the three-probe mixture used previously in competitive ABPP experiments was carried out. This time, treatment with LU-002i (5, β2i) selectively blocked β2i labeling by the three probes at 3 μM, whereas LU-002c (4, β2c) completely prevented β2c identification (final concentration of 0.3 μM) and partially inhibited β2i labeling. Furthermore, LU-102 (1, β2c/β2i) blocked both β2c and β2i labeling at 1 μM (Figure 4B). These results match those published earlier on these compounds against the same set of probes.15 Altogether, these data demonstrate that ABP 97 is a potent and highly selective ABP for visualizing β2i activities of human immunoproteasomes.
Scheme 4. Synthesis of Probe 97.
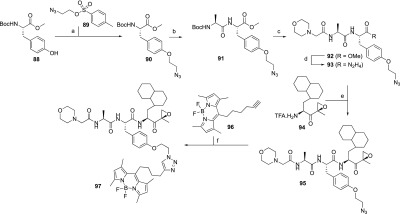
Reagents and conditions: (a) 89, K2CO3/DMF, 80%; (b) (i) TFA, 99%; (ii) Boc-Ala-OH, HCTU/DiPEA/DCM, 93%; (c) (i) TFA, 99%; (ii) 2-morpholino acetic acid, HCTU/DiPEA/DCM, 32%; (d) N2H4·H2O, MeOH, 99%; (e) tBuONO/HCl (4N in dioxane), DCM/DMF (1/1, v/v), −30 °C, 56%; (f) CuSO4, sodium ascorbate, DMF, 18%.
Figure 4.

(A) Activity-based proteasome profiling using probe 97 at different concentrations. Cocktail ABPs were added as control. (B) Left: competitive ABPP assay using ABP 97 and the inhibitors 1 (LU-102, 0.1 μM), 4 (LU-002c, 0.3 μM), and 5 (LU-002i, 3 μM). Right: competitive ABPP assay with probe 97 side-by-side with the three-probe mixture used previously in competitive ABPP experiments and the inhibitors 1 (LU-102, 0.1 μM), 4 (LU-002c, 0.03 μM), and 5 (LU-002i, 3 μM).
X-ray Structures of Selected Inhibitors in Complex with Yeast and Humanized CPs.
To obtain more insights into the structural features that drive either β2c or β2i selectivity of ligands, we aimed at determining the X-ray structures of selected compounds in complex with CPs. As structural data on human apo iCP are not available, we recently developed chimeric yeast proteasomes, which feature the key elements of human β5 subunits, as structural tools.18 On the basis of this work, we created here β2 humanized yeast proteasomes.
Although the yeast proteasome (yCP) α subunits can be easily exchanged by human counterparts, the replacement of most β entities, that is, β1, β2, β5, β6, and β7 is lethal to yeast.13d,18,19 Strikingly, however, the single-point mutation S171G suffices to rescue the lethal phenotype that is caused by the substitution of the endogenous yeast (y) β2 subunit with the human (h) β2c counterpart.19 We created the respective β2c chimeric yeast strain (Figures 5A, S8), purified, and crystallized its mutant proteasome. The X-ray structure (Table S13) revealed that the β2 propeptide was released from the active site Thr1 and that the overall fold of the subunit was intact (Figure 6A). Although the S171G mutation had no obvious impact on the structure of the matured mutant proteasome, it likely supports subunit folding and proteasome assembly. Any pronounced effects of Gly171 on β2 activity are excluded, as yeast viability does not depend on peptide bond hydrolysis by β2.20
Figure 5.

Schematic representation of yeast (y) and human (h) β2 subunits and their propeptides. Secondary structure elements, helices (H), and sheets (S) are numbered. (A) The full-length hβ2c (green) and hβ2i (pink) subunits cannot substitute the endogenous yβ2 subunit (gray), neither with their natural propeptides (pp; colored) nor with the yβ2 one (gray) (for details, see the experimental procedures). Strikingly, the human β2c subunit can replace the yeast counterpart when featuring the single-point mutation S171G.19 (B) Schematic illustration of human–yeast chimeric β2i constructs according to panel (A). Sequences highlighted in pink were taken from human β2i, whereas the gray ones originate from the yeast β2 entity. All tested variants, except for the construct encoding the residues 1–53 from human β2i, caused lethality when expressed in a pup1Δ yeast strain.
Figure 6.
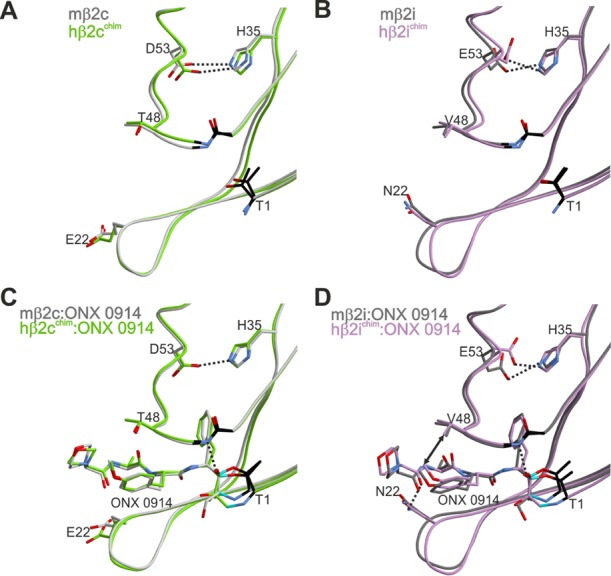
Structural superpositions of the natural mouse β2c (A,C) and β2i (B,D) subunits with their human–yeast chimeric counterparts in the ligand-free (A,B) and ONX 0914-bound (C,D) states. Amino acids are labeled by the one-letter code. Hydrogen bonds are depicted by black dashed lines. Hydrophobic interactions are highlighted by double arrows. Color coding is according to Figure 5. Note that ONX 0914 has been previously modeled into the mouse β2 subunits as a morpholine adduct with Thr1,13d whereas in the chimeric subunits it was built as a seven-membered ring structure according to the revised reaction mechanism of epoxyketones with Thr1.21 PDB IDs: 3UNE (mouse cCP), 3UNH (mouse iCP), 3UNB (mouse cCP:ONX 0914), 3UNF (mouse iCP:ONX 0914), 6HTB (hβ2c chimera), 6HV3 (hβ2i chimera), 6HTC (hβ2c chimera:ONX 0914), 6HV4 (hβ2i chimera:ONX 0914).
As no rescuing mutation for the hβ2i subunit is known to date, we created various chimeric hβ2i-yβ2 constructs and tested whether they can substitute wild-type (WT) yβ2. Surprisingly, only a construct featuring the β2i amino acids 1–53 was viable (Figure 5B). As this sequence covers the entire β2 substrate-binding channel, we used this construct for structural analyses (Table S13).
The superposition of ligand-free β2c/i chimeric structures with the natural mouse counterpart13d proved their structural similarity (Figure 6A,B). The subsequent crystal soakings with ONX 0914 as a reference compound confirmed that the β2 proteolytic centers were reactive (Figure S9) and visualized a similar binding mode for the inhibitor as in the respective mouse crystal structures13d (Figure 6C,D). The β2 subunits can accommodate bulky P1 residues without any pronounced conformational changes of the protein backbone (Figure S10A,B). The corresponding spacious P1 binding site is created by Gly45 at the bottom of the S1 pocket.13d Although the chemical nature and the orientation of amino acid 45 differ among most proteasome subunits, Gly45 has been preserved in β2 subunits throughout evolution.13d Though the mutation of Gly45 to Ala does neither impair yeast growth nor affect subunit folding and ligand binding, any additional increase of residue 45 is predicted to sterically interfere with the surrounding protein side chains (Figures S8, S11, and S12, Table S13).
On the basis of the structural similarity of human–yeast chimeric and mouse β2 active sites, a set of 29 ligand complex structures was determined with WT and β2 chimeric yeast proteasomes (Table S13).
The β2c-selective compound 4 (LU-002c) was found to be well-stabilized in the β2c and β2i active sites. The interactions of the 4-aminomethylphenyl group at P1 with the carboxylic amino acid side chains in position 53 are supposed to be the driving forces for the general β2 selectivity of 4 (LU-002c) as well as the related compounds LU-102 (1) and LU-112 (2) (Figure 1).16 The selectivity for subunit β2c might be gained by dual anchoring of the 4-aminomethylphenyl group to Asp53 in β2c versus a single interaction with Glu53 in β2i (Figure 7A,C). In addition, the shorter P2 Ala side chain of 4 (LU-002c) compared to Leu in LU-102 increases β2c selectivity by reducing the potency for β2i (Figure 1). Most likely, small P2 residues like Ala fail to undergo favorable van der Waals interactions with Val48 in β2i (Figure 7C) and thereby lead to the observed β2c selectivity of 4 (LU-002c).
Figure 7.

Human–yeast chimeric proteasomes in complex with β2c (4; green)- and β2i (39; purple)-selective inhibitors. (A,C,E,G) 2FO–FC electron density maps for the compounds bound to the β2c (green) and β2i (purple) chimeric subunits, respectively, are shown as blue meshes contoured to 1σ. (B,D,F,H) Structural superposition of ligand-free and ligand-bound chimeric β2c and β2i subunits. Polar and hydrophobic interactions are depicted according to Figure 6. PDB IDs: 6HTB (hβ2c chimera), 6HTD (hβ2c chimera:4), 6HUV (hβ2c chimera:39), 6HV3 (hβ2i chimera), 6HV5 (hβ2c chimera:4), 6HVV (hβ2i chimera:39).
For the most selective β2i inhibitor, compound 5 (LU-002i), crystallographic data could only be obtained with WT yCP (Figure S13, Table S14). We assume that the ligand could not be trapped at the mutant β2 active site, as the reactivity of chimeric subunits is impaired18 and as compound 5 is poorly soluble in aqueous solutions because of its apolar decalin moiety. Chimeric proteasome structures in complex with 39 however could be achieved. Compounds 5 (LU-002i) and 39 are derived from the epoxyketone inhibitor ONX 0914. Epoxyketones have recently been shown to form seven-membered,21 instead of six-membered,22 ring structures with the nucleophilic Thr1 residue of the proteasomal β subunits. Although the 1,4-oxazepane (seven-membered) ring structure fits our experimental electron densities in most cases, we also have structural data which match better the six-membered 1,4-morpholine system (e.g., see Figure S13A,B). However, the kind of irreversible covalent structure inhibitors formed with Thr1 has no further implications for drug development, as subunit selectivity of epoxyketone inhibitors is mostly gained by the interactions of the ligands’ side chains with the protein surroundings.
ONX 0914 slightly favors β2i over β2c,12 which may be supported by an advantageous hydrophobic interaction of its P2-methoxy group with Val48 of β2i, a contact that is not provided in subunit β2c (Figure 6C,D). Furthermore, Asn22 forms hydrogen bonds with the amide oxygen atom of the morpholine cap of ONX 0914, whereas Glu22 in subunit β2c fails to provide this additional stabilization (Figure 6C,D). The interaction with Asn22 in β2i is also observed with other tripeptide ligands like 39 (Figure 7G,H), implying that peptide substrates in general might be better stabilized in the β2i substrate-binding channel than in the β2c one. Notably, a similar observation has previously been reported for Thr22 in subunit yβ1/β1c.13a
The co-crystal structure of the β2i chimera with compound 39 shows a well-defined 2FO–FC electron density map for the ligand (Figure 7G). A comparison of the ligand-free and ligand-bound states of the β2i chimera indicates a movement of His35 upon inhibitor binding (Figure 7H). Despite this structural flexibility and plasticity of the S1 pocket, the hydrogen bond between His35 and Glu53 remains intact. Compared to β2i, the β2c active site appears to be more rigid, as binding of 39 does not trigger any structural changes of His35 (Figure 7F). Presumably, the P1 side chain of 39 is less well-defined in the β2c active site because of the tight anchoring of and the resulting steric hindrance with His35 (Figure 7E,F). Thus, although the β2 subunits in general accept large P1 side chains, it appears that the plasticity of the β2i active site tolerates bulky residues even more readily than β2c.
Discussion and Conclusions
Here, we describe the development and evaluation of a set of potent and selective inhibitors of human β2c and β2i proteasome activities. Because of the structural similarities of the mammalian β2c and β2i subunits, no key guidelines for compound design strategies could be derived from the crystal structures so far.13d Thus, we used the previously described inhibitors LU-102 (1),16 LU-112 (2),16 and ONX 0914 (3)12 as the starting points, which have no or only moderate preference for one of the two human β2 subunits over the other. By changing the P sites of the ligands, we disfavored the most closely related subunit, either β2i or β2c, and gained selectivity.
Substantial organic synthesis efforts and thorough empiric screening of compound libraries derived from these lead structures finally led to the identification of selective compounds and to the development of suitable probes for ABPP assays. Furthermore, previously unaddressed stereochemistry issues on LU-002i (5) have now been resolved and the exact configuration of the bioactive compound has been determined.
Selected β2c and β2i inhibitors were analyzed by X-ray crystallography in complex with the WT yeast CP and with chimeric human–yeast proteasomes, incorporating key elements of the human β2c and β2i substrate-binding channels, respectively. Despite the artificial character of chimeras, they were previously shown to serve as excellent structural tools18 and now again prove valuable for explaining the selectivity patterns observed for the β2 compound libraries described here. Both β2c and β2i can incorporate large P1 residues in their spacious S1 pocket. Because of the favorable hydrogen bond interactions with Asp/Glu53, LU-102 derivatives with their 4-aminomethylphenyl side chain at P1 are in general more potent β2 inhibitors than ONX 0914-based compounds, featuring apolar P1 residues.16 Selectivity for β2c was gained by installing small P2 residues on LU-102. Epoxyketones with bulky hydrophobic P1 residues and small P3 side chains were found to show β2i selectivity. Because of the plasticity of the S1 pocket and the flexibility of His35 in subunit β2i, large apolar P1 side chains can be better accommodated in β2i than in β2c.
Taken together, we here present the most selective β2c and β2i ligands reported so far. As part of a set of inhibitors and ABPs that is capable of disabling and visualizing the individual activities of human constitutive and immunoproteasomes,15 these compounds might become valuable tools for fundamental as well as applied biochemical and biomedical research on proteasomes and hopefully elucidate more details on the biological role and impact of the trypsin-like active sites of human proteasomes.
Experimental Section
General Procedures
All reagents were of commercial grade and used as received unless indicated otherwise. The purity of all tested compounds is >95% on the basis of liquid chromatography–mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR). 1H- and 13C NMR spectra were recorded on a Bruker AV-400 (400 MHz), AV-600 (600 MHz), or AV-850 (850 MHz) spectrometer. Chemical shifts are given in ppm (δ) relative to CD3OD or CDCl3 as an internal standard. Coupling constants are given in Hz, and peak assignments are based on 2D 1H correlation spectroscopy and 13C heteronuclear single quantum coherence NMR experiments. All 13C attached proton test spectra are proton-decoupled. LC-MS analysis was performed on a Finnigan Surveyor high-performance liquid chromatography (HPLC) system with a Gemini C18 50 × 4.60 mm column (detection at 200–600 nm) coupled to a Finnigan LCQ Advantage Max mass spectrometer with electrospray ionization (ESI). Methods used are: 15 min (0–0.5 min: 10% MeCN; 0.5–10.5 min: 10–90% MeCN; 10.5–12.5 min: 90% MeCN; 12.5–15 min: 90–10% MeCN) or 12.5 min (0–0.5 min: 10% MeCN; 0.5–8.5 min: 10–90% MeCN; 8.5–10.5 min: 90% MeCN; 10.5–12.5 min: 90––10% MeCN). HRMS was recorded on an LTQ Orbitrap (ThermoFinnigan). For reverse-phase HPLC purification, an automated Gilson HPLC system equipped with a C18 semiprep column (Phenomenex Gemini C18, 5 μm 250 × 10 mm) and a GX281 fraction collector was used.
General Procedure for Boc Removal
The appropriate Boc-protected C-terminally modified leucine derivative was dissolved in TFA and stirred for 20 min. Co-evaporation with toluene (3×) afforded the TFA salt, which was used without further purification.
General Procedure for Azide Couplings
Compounds 6–53, 68, 71, 74, 77, 86, 87, and 97 were prepared via azide coupling of the appropriate protected tripeptide hydrazide and either an epoxyketone amine or a vinyl sulfone amine. Peptide hydrazides were prepared by hydrazinolysis of peptide methyl esters synthesized as described in the Supporting Information. The hydrazide was dissolved in 1:1 DMF/DCM (v/v) and cooled to −30 °C. tBuONO (1.1 equiv) and HCl (4 N solution in 1,4-dioxane, 2.8 equiv) were added, and the mixture was stirred for 3 h at −30 °C, after which thin-layer chromatography analysis (10% MeOH/DCM, v/v) showed the complete consumption of the starting material. The epoxyketone or vinyl sulfone amine was added as a free amine to the reaction mixture as a solution in DMF with 5.0 equiv of DiPEA. The mixture was allowed to warm to room temperature overnight. The mixture was diluted with ethyl acetate (EtOAc) and extracted with H2O (3×) and brine. The organic layer was dried over MgSO4 and purified by reverse-phase HPLC. For compounds featuring Boc-protecting groups, TFA was added, and the reaction mixture was stirred for 30 min. The crude was purified by reverse-phase HPLC.
N3Phe-Leu-Ser-Phe(4-CH2NH2)VS TFA salt (13). The synthesis of tripeptide hydrazide N3Phe-Leu-Ser(tBu)-NHNH2 is described in the Supporting Information. The title compound was prepared according to the general procedure for azide coupling on a 50 μmol scale and purified by HPLC (30–40% MeCN–H2O) to yield 2.8 mg (3.8 μmol, 8%). 1H NMR (600 MHz, MeOD): δ 7.46–7.23 (m, 9H), 6.85–6.81 (m, 1H), 6.76–6.73 (m, 1H), 4.39–4.27 (m, 2H), 4.20–4.17 (m, 1H), 4.11 (s, 2H), 3.84–3.81 (m, 1H), 3.76–3.73 (m, 1H), 3.25–3.22 (m, 1H), 3.07–2.98 (m, 3H), 2.95 (s, 3H), 1.69–1.52 (m, 3H), 1.02–0.88 (m, 6H). 13C NMR (150 MHz, MeOD): δ 174.33, 172.23, 171.81, 146.65, 139.69, 137.84, 133.00, 131.84, 131.26, 130.42, 130.24, 129.65, 128.10, 65.38, 62.75, 56.70, 53.80, 52.56, 44.09, 42.77, 41.38, 40.25, 38.72, 25.82, 23.46, 21.84. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 15.0 min): Rt (min): 6.27 (ESI–MS (m/z): 628.20 (M + H)+). HRMS calcd for C30H41N7O6S, 628.29118 [M + H]+; found, 628.29123.
Morp-Ala-Tyr(Me)-HomoCha-EK TFA salt (39). The synthesis of Boc-HomoCha-EK is described in the Supporting Information, and the Boc-protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 50 μmol scale and purified by HPLC (30–45% MeCN–H2O) to yield 12.3 mg (17.2 μmol, 34%). 1H NMR (600 MHz, MeOD): δ 7.25–7.01 (m, 2H), 6.91–6.67 (m, 2H), 4.60–4.57 (m, 1H), 4.48–4.28 (m, 2H), 3.77 (s, 3H), 3.71–3.70 (m, 4H), 3.21 (d, J = 4.9 Hz, 1H), 3.09–2.88 (m, 4H), 2.84–2.79 (m, 1H), 2.56–2.37 (m, 4H), 1.83–1.63 (m, 6H), 1.52–1.39 (m, 4H), 1.38–1.16 (m, 9H), 0.97–0.83 (m, 2H). 13C NMR (150 MHz, MeOD): δ 209.19, 174.20, 173.30, 171.99, 159.94, 131.41, 130.02, 114.75, 67.85, 62.40, 60.01, 55.75, 55.60, 54.71, 53.06, 52.92, 49.65, 38.48, 38.15, 34.61, 34.02, 29.11, 27.72, 27.44, 27.38, 18.65, 16.84. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.23 (ESI–MS (m/z): 601.33 (M + H)+). HRMS calcd for C32H48N4O7, 601.35958 [M + H]+; found, 601.35945.
Morp-Ala-Tyr(Me)-1-(R)-TetraNal-EK TFA salt (68). The synthesis of Boc-1-TatraNal-EK is described in the Supporting Information, and the Boc protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 56 μmol scale and purified by HPLC (30–45% MeCN–H2O) to yield 14.2 mg (22.4 μmol, 40%). 1H NMR (400 MHz, MeOD): δ 7.22–7.13 (m, 2H), 7.12–7.01 (m, 4H), 6.88–6.77 (m, 2H), 4.63–4.59 (m, 2H), 4.41–4.36 (m, 1H), 4.06–3.84 (m, 6H), 3.78 (s, 3H), 3.17–2.98 (m, 2H), 2.93–2.67 (m, 5H), 2.14–1.54 (m, 6H), 1.45 (s, 3H), 1.35 (d, J = 7.1 Hz, 3H). 13C NMR (100 MHz, MeOD): δ 209.40, 174.19, 173.10, 165.04, 160.01, 140.66, 137.91, 131.40, 130.18, 129.98, 129.94, 127.07, 126.54, 114.84, 64.84, 59.89, 58.37, 55.83, 55.67, 53.93, 52.88, 51.64, 50.57, 39.22, 37.99, 36.17, 30.02, 29.64, 20.16, 18.11, 16.76. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.10 (ESI–MS (m/z): 635.00 (M + H)+). HRMS calcd for C35H46N4O7, 635.34393 [M + H]+; found, 635.34371.
Morp-Ala-Tyr(Me)-1-(S)-TetraNal-EK TFA salt (71). The synthesis of Boc-1-TatraNal-EK is described in the Supporting Information, and the Boc-protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 50 μmol scale and purified by HPLC (30–45% MeCN–H2O) to yield 16.5 mg (26.0 μmol, 52%). 1H NMR (400 MHz, MeOD): δ 7.24–7.13 (m, 3H), 7.13–6.98 (m, 3H), 6.86–6.78 (m, 2H), 4.77–4.59 (m, 2H), 4.41–4.36 (m, 1H), 4.00–3.86 (m, 6H), 3.21 (d, J = 5.0 Hz, 1H), 3.10 (dd, J = 14.0, 6.0 Hz, 1H), 3.03–2.64 (m, 5H), 2.02–1.60 (m, 6H), 1.45 (s, 3H), 1.33 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, MeOD): δ 208.97, 174.18, 173.57, 164.98, 160.01, 141.21, 138.13, 131.41, 130.09, 130.01, 129.75, 126.83, 126.76, 114.84, 114.75, 64.82, 60.04, 58.35, 55.89, 55.66, 53.91, 53.05, 51.05, 50.54, 38.67, 38.02, 35.13, 30.57, 27.13, 20.23, 18.10, 16.86. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.18 (ESI–MS (m/z): 635.07 (M + H)+). HRMS calcd for C35H46N4O7, 635.34393 [M + H]+; found, 635.34370.
Morp-Ala-Tyr(Me)-1-DecAla-EK TFA salt (74). The synthesis of Boc-1-DecAla-EK is described in the Supporting Information, and the Boc-protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 50 μmol scale and purified by HPLC (40–50% MeCN–H2O) to yield 14.6 mg (22.8 μmol, 46%). 1H NMR (400 MHz, MeOD): δ 7.20–7.11 (m, 2H), 6.86–6.78 (m, 2H), 4.62–4.51 (m, 2H), 4.38–4.33 (m, 1H), 4.07–3.83 (m, 6H), 3.77 (d, J = 3.7 Hz, 3H), 3.22 (d, J = 12 Hz, 1H), 3.06–3.01 (m, 1H), 2.95 (d, J = 12 Hz, 1H), 2.85–2.79 (m, 1H), 1.84–1.13 (m, 25H). 13C NMR (100 MHz, MeOD): δ 209.81, 174.11, 173.35, 164.81, 159.95, 131.42, 130.00, 114.77, 64.78, 60.08, 58.26, 55.64, 53.89, 52.81, 50.50, 50.29, 39.17, 38.74, 38.20, 33.84, 29.05, 27.91, 26.60, 22.38, 20.22, 18.11, 16.88. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.75 (ESI–MS (m/z): 641.13 (M + H)+). HRMS calcd for C35H52N4O7, 641.39088 [M + H]+; found, 641.39081.
Morp-Ala-Tyr(Me)-1-DecAla-EK TFA salt (77). The synthesis of Boc-1-DecAla-EK is described in the Supporting Information, and the Boc- protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 23 μmol scale and purified by HPLC (40–50% MeCN–H2O) to yield 6.8 mg (10.6 μmol, 46%s). 1H NMR (400 MHz, MeOD): δ 7.19–7.13 (m, 2H), 6.84–6.80 (m, 2H), 4.63–4.60 (m, 1H), 4.53–4.50 (m, 1H), 4.40–4.34 (m, 1H), 4.01–3.90 (m, 6H), 3.78 (d, J = 1.9 Hz, 3H), 3.17 (d, J = 5.1 Hz, 1H), 3.07–3.02 (m, 1H), 2.94 (d, J = 5.1 Hz, 1H), 2.88–2.80 (m, 1H), 1.85–1.07 (m, 25H). 13C NMR (100 MHz, MeOD): δ 209.46, 174.13, 173.41, 164.82, 159.97, 131.43, 129.96, 114.77, 64.78, 59.91, 58.27, 55.62, 53.90, 52.92, 50.97, 50.52, 43.08, 39.49, 39.11, 38.25, 36.48, 33.73, 27.99, 27.69, 26.76, 26.54, 22.32, 21.21, 18.10, 16.87. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.79 (ESI–MS (m/z): 641.07 (M + H)+). HRMS calcd for C35H52N4O7, 641.39088 [M + H]+; found, 641.39070.
Morp-Ala-Tyr(Me)-1-DecAla-EK TFA salt (86). The synthesis of Boc-1-DecAla-EK is described in the Supporting Information, and the Boc-protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 25 μmol scale and purified by HPLC (30–45% MeCN–H2O) to yield 8.6 mg (11.4 μmol, 46%). 1H NMR (500 MHz, MeOD): δ 7.15 (d, J = 8.7 Hz, 2H), 6.85–6.80 (m, 2H), 4.62–4.59 (m, 1H), 4.55–4.52 (m, 1H), 4.40–4.35 (m, 1H), 4.01–3.91 (m, 6H), 3.78 (s, 3H), 3.21 (d, J = 5.1 Hz, 1H), 3.07–3.03 (m, 1H), 2.95 (d, J = 5.1 Hz, 1H), 2.85–2.81 (m, 1H), 1.83–1.52 (m, 10H), 1.47–1.41 (m, 5H), 1.37–1.18 (m, 10H). 13C NMR (125 MHz, MeOD): δ 209.83, 174.13, 173.36, 164.80, 159.96, 131.42, 129.99, 114.77, 64.78, 60.09, 58.26, 55.64, 53.90, 52.81, 50.49, 50.29, 39.21, 39.17, 38.75, 38.20, 34.85, 33.84, 29.05, 27.92, 27.91, 26.60, 22.38, 20.22, 18.12, 16.87. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.92 (ESI–MS (m/z): 641.13 (M + H)+). HRMS calcd for C35H52N4O7, 641.39088 [M + H]+; found, 641.39065.
Morp-Ala-Tyr(Me)-1-DecAla-EK TFA salt (87). The synthesis of Boc-1-DecAla-EK is described in the Supporting Information, and the Boc-protecting group was removed according to the general procedure. The title compound was prepared according to the general procedure for azide coupling on a 44 μmol scale and purified by HPLC (30–45% MeCN–H2O) to yield 12.7 mg (16.8 μmol, 38%). 1H NMR (500 MHz, MeOD): δ 7.15 (d, J = 8.6 Hz, 2H), 6.84–6.79 (m, 2H), 4.66–4.58 (m, 1H), 4.53–4.50 (m, 1H), 4.39–4.35 (m, 1H), 4.02–3.92 (m, 6H), 3.17 (d, J = 5.1 Hz, 1H), 3.07–3.03 (m, 1H), 2.94 (d, J = 5.1 Hz, 1H), 2.86–2.82 (m, 1H), 1.84–1.18 (m, 25H). 13C NMR (125 MHz, MeOD): δ 209.47, 174.13, 173.41, 164.81, 159.96, 131.43, 129.96, 114.77, 64.76, 59.91, 58.24, 55.64, 53.89, 52.93, 50.96, 50.55, 43.06, 39.48, 39.10, 38.25, 36.48, 33.72, 27.98, 27.68, 26.75, 26.53, 22.31, 21.21, 18.08, 16.87. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 6.88 (ESI–MS (m/z): 641.13 (M + H)+). HRMS calcd for C35H52N4O7, 641.39088 [M + H]+; found, 641.39077.
Morp-Ala-Tyr(O–C2H4-BODIPY(FL))-1-DecAla-EK (97). The synthesis of compound 95 is described in the Supporting Information. Compound 95 (23 mg, 33 μmol) and BODIPY-FL-alkyne 96(23) (13 mg, 40 μmol, 1.2 equiv) were dissolved in DMF. An aqueous solution of sodium ascorbate (100 μL, 25 μmol, 0.75 equiv) and an aqueous solution of CuSO4 (100 μL, 17 μmol, 0.5 equiv) were added. The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo, and purification by HPLC (50–70% MeCN–H2O) yielded the title compound (6.7 mg, 5.9 μmol, 18%). 1H NMR (600 MHz, MeOD): δ 7.91 (s, 1H), 7.17 (d, J = 8.2 Hz, 2H), 6.84 (dd, J = 12.1, 8.3 Hz, 2H), 6.14 (s, 2H), 4.82 (t, J = 5.0 Hz, 2H), 4.64 (dt, J = 8.3, 5.5 Hz, 1H), 4.57 (ddd, J = 13.6, 10.7, 3.3 Hz, 1H), 4.46–4.34 (m, 4H), 3.99 (dd, J = 24.6, 16.0 Hz, 8H), 3.34–3.16 (m, 3H), 3.16–3.03 (m, 4H), 2.98 (dt, J = 11.4, 5.6 Hz, 1H), 2.87 (t, J = 7.2 Hz, 4H), 2.51 (s, 8H), 2.44 (s, 8H), 1.99 (q, J = 7.4 Hz, 3H), 1.93–1.80 (m, 3H), 1.75 (ddd, J = 16.6, 8.4, 3.4 Hz, 6H), 1.69–1.54 (m, 6H), 1.52 (d, J = 2.8 Hz, 1H), 1.49 (d, J = 2.9 Hz, 5H), 1.45–1.37 (m, 5H), 1.37–1.18 (m, 7H). 13C NMR (150 MHz, MeOD): δ 209.76, 209.41, 174.09, 173.28, 173.22, 172.91, 164.76, 161.88, 161.64, 158.47, 154.93, 148.55, 147.84, 142.18, 132.57, 131.57, 130.91, 130.89, 130.77, 124.23, 122.62, 115.63, 115.58, 67.69, 64.80, 64.75, 60.04, 59.87, 58.23, 55.64, 53.89, 52.90, 52.77, 50.96, 50.52, 50.20, 49.43, 49.28, 49.14, 49.00, 48.86, 48.72, 48.57, 43.06, 39.48, 39.22, 39.11, 38.74, 38.28, 38.23, 36.50, 35.66, 34.99, 34.89, 33.84, 33.73, 32.20, 32.15, 30.72, 30.63, 29.08, 29.04, 27.99, 27.91, 27.69, 27.58, 27.00, 26.76, 26.59, 26.53, 25.88, 22.38, 22.31, 21.21, 18.14, 16.88, 16.49, 14.48. LC–MS (linear gradient 10 → 90% MeCN/H2O, 0.1% TFA, 12.5 min): Rt (min): 8.07 (ESI–MS (m/z): 1024.33 (M + H)+). HRMS calcd for C55H78BF2N9O7, 1024.60108 [M + H]+; found, 1024.60174.
Biological and Structural Analysis
Competition Assays in Cell Lysates
Lysates of Raji cells were prepared by sonication in three volumes of lysis buffer containing 50 mM Tris pH 7.5, 1 mM DTT, 5 mM MgCl2, 250 mM sucrose, 2 mM ATP, and 0.05% (w/v) digitonin. The protein concentration was determined by the Bradford assay. Cell lysates (diluted to 5 μg of total protein in buffer containing 50 mM Tris pH 7.5, 2 mM DTT, 5 mM MgCl2, 10% (v/v) glycerol, and 2 mM ATP) were exposed to the inhibitors for 1 h at 37 °C prior to incubation with cocktail ABPs for another 1 h, followed by 3 min boiling with a reducing gel-loading buffer and fractionation on 12.5% SDS-PAGE. In-gel detection of residual proteasome activity was performed in the wet gel slabs directly on a ChemiDoc MP system using Cy2 settings to detect BODIPY(FL)-LU-112, Cy3 settings to detect BODIPY(TMR)–NC–005-VS, and Cy5 settings to detect Cy5-NC-001. The intensities of bands were measured by fluorescent densitometry and normalized to the intensity of bands in the mock-treated extracts. The average values of three independent experiments were plotted against the inhibitor concentrations (in the initial screening, experiments were only carried out one time). The IC50 (ligand concentrations giving 50% inhibition) values were calculated using GraphPad Prism software.
Competition Assays in Living RPMI-8226 Cells
RPMI-8226 cells were cultured in RPMI-1640 media supplemented with 10% (v/v) fetal calf serum, GlutaMAX, and penicillin/streptomycin in a 5% CO2-humidified incubator. An amount of (5–8) × 105 cells/mL cells was exposed to inhibitors for 1 h at 37 °C. The cells were harvested and washed twice with phosphate-buffered saline. The cell pellets were treated with lysis buffer (50 mM Tris pH 7.5, 2 mM DTT, 5 mM MgCl2, 10% (v/v) glycerol, 2 mM ATP, 0.05% (w/v) digitonin) on ice for 1 h, followed by centrifugation at 14 000 rpm for 15 min. Proteasome inhibition in the obtained cell lysates was determined using the method described above. The intensities of bands were measured by fluorescent densitometry and divided by the intensity of bands in the mock-treated extracts. Gels were stained by Coomassie Brilliant Blue, which was used to correct for gel-loading differences. The average values of three independent experiments were plotted against the inhibitor concentrations. The IC50 (compound concentrations causing 50% inhibition) values were calculated using GraphPad Prism software.
ABPP Assays in Raji Cell Lysates
Raji cell lysates (diluted to 5 μg of total protein in buffer containing 50 mM Tris pH 7.5, 2 mM DTT, 5 mM MgCl2, 10% (v/v) glycerol, and 2 mM ATP) were exposed to the probe for 1 h at 37 °C, followed by 3 min boiling with a reducing gel-loading buffer and fractionation by 12.5% SDS-PAGE. Separation was obtained by electrophoresis for 15 min on 80 V, followed by 120 min on 130 V. In-gel detection of residual proteasome activity was performed in the wet gel slabs directly on a ChemiDoc MP system using Cy2 settings.
Yeast Mutagenesis
hPSMB7 and hPSMB10 encoding the human β2c and β2i proteasome subunits, respectively, were purchased as yeast codon-optimized, synthetic gene fragments, each with a 30 bp 5′ overhang corresponding to the yeast PUP1 (yβ2) promoter sequence preceding the start ATG and a 40 bp 3′ overhang corresponding to the PUP1 terminator sequence following the stop codon. An AgeI site at the codons for Gly-1/Thr1 was incorporated into both genes.
The human PSMB7/10 ORFs were fused to the PUP1 promoter and terminator by recombinant polymerase chain reaction (PCR): both genes were amplified with the primers PSMB-for and PSMB-rev (Table S15). The PUP1 promoter was amplified from the template plasmid p15-PUP1-new with the primers pBS-rev and PUP1-prom-rev and the terminator with the primers PUP1-ter-for and pBS-uni (Table S15). The promoter fragment and the ORF fragments were fused by recombinant PCR in the presence of pBS-rev and PSMB-rev. The resulting fragment was then fused by recombinant PCR with the terminator fragment in the presence of pBS-rev and pBS-uni.
The recombinant gene fragments were cut with SacI and HindIII and ligated with SacI/HindIII cut vector pUC19 and afterward transferred into the shuttle vector pRS315, yielding p15-fl-PSMB7 and p15-fl-PSMB10. The S171G mutant version of PSMB7 was created by recombinant PCR with the pUC19 construct as the template and mutagenic primers PSMB7-S171G-for and PSMB7-S171G-rev (Table S15) and cloning of the resulting SacI/HindIII cut product into pRS315, yielding p15-fl-PSMB7*.
For replacement of the genuine human propeptide-encoding sequences by the PUP1 propeptide sequence, the PUP1 promoter, together with the propeptide encoding region, was amplified from p15-PUP120b with the primers pBS-rev and PUP1-Age-rev (Table S15), which introduces an AgeI site at the corresponding Gly-1/Thr1-encoding position of PUP1. The PCR product was cut with HindIII and AgeI and ligated with the respective AgeI/SacI fragments from p15-fl-PSMB7, p15-fl-PSMB7*, and p15-fl-PSMB10 into HindIII/SacI cut pRS315 to obtain the plasmids p15-P1pp-PSMB7, p15-P1pp-PSMB7*, and p15-P1pp-PSMB10.
Genes encoding the hybrids of yβ2 and hβ2i were constructed by recombinant PCR. For the hβ2i1–129 construct, an N-terminal fragment resulting from a PCR with primers pBS-rev and beta2i-129-rev on template p15-P1pp-PSMB10 was fused with a C-terminal fragment made by PCR with primers beta2i-129-for and pBS-uni on template p15-PUP1-new (Table S15). Accordingly, the hybrids hβ2i1–93 and hβ2i1–52 were constructed employing primers beta2i-1-93-for/beta2i-1-93-rev and beta2i-1-52-for/beta2i-1-52-rev, respectively. For the hβ2i1–52/93–129 hybrid, the N-terminal fragment was obtained by PCR on the hβ2i1–52 template with primers pBS-rev and y93-rev, and for the C-terminal fragment, the hβ2i1–129 template was used with primers 2i93-for and pBS-uni (Table S15).
All pRS315-based constructs were introduced into the yeast strain YWH10,20b which has the chromosomal PUP1 gene deleted and a WT PUP1 copy in a URA3-marked plasmid. After selection against URA3 on 5′-fluorouracil, clones that were viable without the WT PUP1 gene were recovered.
Crystallographic Analysis
WT and mutant yCP crystals were grown by hanging drop vapor diffusion as previously described.24 Inhibitor complex structures were obtained by incubating crystals in 5 μL cryobuffer (20 mM magnesium acetate, 100 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.8, and 30% (v/v) 2-methyl-2,4-pentanediol) supplemented with 0.5 μL of inhibitor (50 mM in dimethyl sulfoxide) for up to 48 h. Diffraction data were collected at the Paul Scherrer Institute, SLS, Villigen, Switzerland and the ESRF, Grenoble, France (λ = 1.0 Å). The evaluation of reflection intensities and data reduction was performed with the program package XDS.25 Molecular replacement was carried out with the coordinates of the yeast 20S proteasome (PDB entry code: 5CZ4(26)) by rigid body refinements (REFMAC527). COOT28 was used to build models. Translation/libration/screw refinements finally yielded excellent R factors as well as root-mean-square deviation bond and angle values. The coordinates, proven to have good stereochemistry from the Ramachandran plots, were deposited in the RCSB Protein Data Bank. For accession codes, see Table S13.
Acknowledgments
This work was supported by the China Scholarship Council (PhD fellowship to B.-T.X.), The Netherlands Organization for Scientific Research (NWO, TOPPUNT grant, to H.S.O.), the Young Scholars’ Program of the Bavarian Academy of Sciences and Humanities (fellowship to E.M.H.), and by the Deutsche Forschungsgemeinschaft (DFG, grant GR1861/10-1 to M.G.). We thank the staff of the beamlines X06SA at the Paul Scherrer Institute, Swiss Light Source, Villigen (Switzerland) and ID23 at the European Synchrotron Radiation Facility, Grenoble (France) for assistance during data collection. Richard Feicht is greatly acknowledged for the purification and crystallization of proteasome mutants.
Glossary
Abbreviations
- ABPP
activity-based protein profiling
- ABP
activity-based probe
- BODIPY
boron dipyrromethene (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene)
- cCP
constitutive proteasome core particle
- iCP
immunoproteasome core particle
- DiPEA
N,N-diisopropylethylamine
- EtOAc
ethyl acetate
- HCTU
2-(6-chloro-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- yCP
yeast proteasome core particle
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01884.
Assays of compounds 6–53 and 68, 71, 74, 77, 86, 87, and 97 in Raji lysates; pIC50 values and standard errors of all compounds in cell lysates and intact cells; structures of ABPs, complete synthetic details, and characterization of all compounds and synthetic intermediates; NMR spectra and LC–MS traces of compounds 13, 39, 68, 71, 74, 77, 86, 87, and 97 as well as X-ray data tables and oligonucleotides; PDB IDs; Authors will release the atomic coordinates upon article publication (PDF)
Molecular formula string of the compounds (CSV)
Accession Codes
Structure factors and coordinates were deposited in the RCSB Protein Data Bank under the accession codes listed in Table S13.
Author Present Address
⊥ Department of Drug Discovery and Development, Harrison School of Pharmacy, Auburn University, Auburn Al 36849 United States
Author Contributions
# B.-T.X. and E.M.H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Hershko A.; Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]; b Rock K. L.; Gramm C.; Rothstein L.; Clark K.; Stein R.; Dick L.; Hwang D.; Goldberg A. L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- a Lowe J.; Stock D.; Jap B.; Zwickl P.; Baumeister W.; Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]; b Groll M.; Ditzel L.; Löwe J.; Stock D.; Bochtler M.; Bartunik H. D.; Huber R. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature 1997, 386, 463–471. 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Voges D.; Zwickl P.; Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- a Aki M.; Shimbara N.; Takashina M.; Akiyama K.; Kagawa S.; Tamura T.; Tanahashi N.; Yoshimura T.; Tanaka K.; Ichihara A. Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes1. J. Biochem. 1994, 115, 257–269. 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]; b Griffin T. A.; Nandi D.; Cruz M.; Fehling H. J.; Kaer L. V.; Monaco J. J.; Colbert R. A. Immunoproteasome Assembly: Cooperative Incorporation of Interferon γ (IFN-γ)-inducible Subunits. J. Exp. Med. 1998, 187, 97–104. 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Groettrup M.; Kraft R.; Kostka S.; Standera S.; Stohwasser R.; Kloetzel P.-M. A third interferon-γ-induced subunit exchange in the 20S proteasome. Eur. J. Immunol. 1996, 26, 863–869. 10.1002/eji.1830260421. [DOI] [PubMed] [Google Scholar]
- Basler M.; Kirk C. J.; Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. 10.1016/j.coi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- a Huber E. M.; Groll M. Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew. Chem., Int. Ed. 2012, 51, 8708–8720. 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]; b Beck P.; Dubiella C.; Groll M. Covalent and non-covalent reversible proteasome inhibition. Biol. Chem. 2012, 393, 1101–1120. 10.1515/hsz-2012-0212. [DOI] [PubMed] [Google Scholar]; c Kisselev A. F.; van der Linden W. A.; Overkleeft H. S. Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borissenko L.; Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717. 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- a Screen M.; Britton M.; Downey S. L.; Verdoes M.; Voges M. J.; Blom A. E. M.; Geurink P. P.; Risseeuw M. D. P.; Florea B. I.; van der Linden W. A.; Pletnev A. A.; Overkleeft H. S.; Kisselev A. F. Nature of pharmacophore influences active site specificity of proteasome inhibitors. J. Biol. Chem. 2010, 285, 40125–40134. 10.1074/jbc.m110.160606. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Verdoes M.; Willems L. I.; van der Linden W. A.; Duivenvoorden B. A.; van der Marel G. A.; Florea B. I.; Kisselev A. F.; Overkleeft H. S. A panel of subunit-selective activity-based proteasome probes. Org. Biomol. Chem. 2010, 8, 2719–2727. 10.1039/c001036g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fostier K.; De Becker A.; Schots R. Carfilzomib: a novel treatment in relapsed and refractory multiple myeloma. Onco Targets Ther 2012, 5, 237–44. 10.2147/OTT.S28911. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Field-Smith A.; Morgan G. J.; Davies F. E. Bortezomib (Velcade?) in the treatment of multiple myeloma. Ther. Clin. Risk Manage. 2006, 2, 271–279. 10.2147/tcrm.2006.2.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallumsetla N.; Paludo J.; Kapoor P. Bortezomib in mantle cell lymphoma: comparative therapeutic outcomes. Ther. Clin. Risk Manage. 2015, 11, 1663–1674. 10.2147/tcrm.s72943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson H. W. B.; Lowe E.; Anderl J. L.; Fan A.; Muchamuel T.; Bowers S.; Moebius D. C.; Kirk C.; McMinn D. L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. 10.1021/acs.jmedchem.8b01201. [DOI] [PubMed] [Google Scholar]
- Muchamuel T.; Basler M.; Aujay M. A.; Suzuki E.; Kalim K. W.; Lauer C.; Sylvain C.; Ring E. R.; Shields J.; Jiang J.; Shwonek P.; Parlati F.; Demo S. D.; Bennett M. K.; Kirk C. J.; Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- a Huber E. M.; de Bruin G.; Heinemeyer W.; Soriano G. P.; Overkleeft H. S.; Groll M. Systematic analyses of substrate preferences of 20S proteasomes using peptidic epoxyketone inhibitors. J. Am. Chem. Soc. 2015, 137, 7835–7842. 10.1021/jacs.5b03688. [DOI] [PubMed] [Google Scholar]; b de Bruin G.; Huber E. M.; Xin B.-T.; van Rooden E. J.; Al-Ayed K.; Kim K.-B.; Kisselev A. F.; Driessen C.; van der Stelt M.; van der Marel G. A.; Groll M.; Overkleeft H. S. Structure-Based Design of β1i or β5i Specific Inhibitors of Human Immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]; c Parlati F.; Lee S. J.; Aujay M.; Suzuki E.; Levitsky K.; Lorens J. B.; Micklem D. R.; Ruurs P.; Sylvain C.; Lu Y.; Shenk K. D.; Bennett M. K. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]; d Huber E. M.; Basler M.; Schwab R.; Heinemeyer W.; Kirk C. J.; Groettrup M.; Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]; e Xin B.-T.; de Bruin G.; Huber E. M.; Besse A.; Florea B. I.; Filippov D. V.; van der Marel G. A.; Kisselev A. F.; van der Stelt M.; Driessen C.; Groll M.; Overkleeft H. S. Structure-Based Design of β5c Selective Inhibitors of Human Constitutive Proteasomes. J. Med. Chem. 2016, 59, 7177–7187. 10.1021/acs.jmedchem.6b00705. [DOI] [PubMed] [Google Scholar]; f Johnson H. W. B.; Anderl J. L.; Bradley E. K.; Bui J.; Jones J.; Arastu-Kapur S.; Kelly L. M.; Lowe E.; Moebius D. C.; Muchamuel T.; Kirk C.; Wang Z.; McMinn D. Discovery of Highly Selective Inhibitors of the Immunoproteasome Low Molecular Mass Polypeptide 2 (LMP2) Subunit. ACS Med. Chem. Lett. 2017, 8, 413–417. 10.1021/acsmedchemlett.6b00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artschwager R.; Ward D. J.; Gannon S.; Brouwer A. J.; van de Langemheen H.; Kowalski H.; Liskamp R. M. J. Potent and highly selective inhibitors of the proteasome trypsin-like site by incorporation of basic side chain containing amino acid derived sulfonyl fluorides. J. Med. Chem. 2018, 61, 5395–5411. 10.1021/acs.jmedchem.8b00685. [DOI] [PubMed] [Google Scholar]
- de Bruin G.; Xin B. T.; Kraus M.; van der Stelt M.; van der Marel G. A.; Kisselev A. F.; Driessen C.; Florea B. I.; Overkleeft H. S. A set of activity-based probes to visualize human (immuno)proteasome activities. Angew. Chem., Int. Ed. 2016, 55, 4199–4203. 10.1002/anie.201509092. [DOI] [PubMed] [Google Scholar]
- Geurink P. P.; van der Linden W. A.; Mirabella A. C.; Gallastegui N.; de Bruin G.; Blom A. E. M.; Voges M. J.; Mock E. D.; Florea B. I.; van der Marel G. A.; Driessen C.; van der Stelt M.; Groll M.; Overkleeft H. S.; Kisselev A. F. Incorporation of non-natural amino acids improves cell permeability and potency of specific inhibitors of proteasome trypsin-like sites. J. Med. Chem. 2013, 56, 1262–1275. 10.1021/jm3016987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mirabella A. C.; Pletnev A. A.; Downey S. L.; Florea B. I.; Shabaneh T. B.; Britton M.; Verdoes M.; Filippov D. V.; Overkleeft H. S.; Kisselev A. F. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem. Biol. 2011, 18, 608–618. 10.1016/j.chembiol.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weyburne E. S.; Wilkins O. M.; Sha Z.; Williams D. A.; Pletnev A. A.; de Bruin G.; Overkleeft H. S.; Goldberg A. L.; Cole M. D.; Kisselev A. F. Inhibition of the Proteasome β2 Site Sensitizes Triple-Negative Breast Cancer Cells to β5 Inhibitors and Suppresses Nrf1 Activation. Cell Chem. Biol. 2017, 24, 218–230. 10.1016/j.chembiol.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M.; Heinemeyer W.; de Bruin G.; Overkleeft H. S.; Groll M. A humanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit β5i. EMBO J. 2016, 35, 2602–2613. 10.15252/embj.201695222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo A. H.; Laurent J. M.; Yellman C. M.; Meyer A. G.; Wilke C. O.; Marcotte E. M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. 10.1126/science.aaa0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Groll M.; Heinemeyer W.; Jager S.; Ullrich T.; Bochtler M.; Wolf D. H.; Huber R. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10976–10983. 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Heinemeyer W.; Fischer M.; Krimmer T.; Stachon U.; Wolf D. H. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 1997, 272, 25200–25209. 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- Schrader J.; Henneberg F.; Mata R. A.; Tittmann K.; Schneider T. R.; Stark H.; Bourenkov G.; Chari A. The inhibition mechanism of human 20Sproteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- Groll M.; Kim K. B.; Kairies N.; Huber R.; Crews C. M. Crystal Structure of Epoxomicin:20S Proteasome Reveals a Molecular Basis for Selectivity of α’,β’-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc. 2000, 122, 1237–1238. 10.1021/ja993588m. [DOI] [Google Scholar]
- Verdoes M.; Hillaert U.; Florea B. I.; Sae-Heng M.; Risseeuw M. D. P.; Filippov D. V.; van der Marel G. A.; Overkleeft H. S. Acetylene functionalized BODIPY dyes and their application in the synthesis of activity based proteasome probes. Bioorg. Med. Chem. Lett. 2007, 17, 6169–6171. 10.1016/j.bmcl.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Gallastegui N.; Groll M. Analysing properties of proteasome inhibitors using kinetic and X-ray crystallographic studies. Methods Mol. Biol. 2012, 832, 373–390. 10.1007/978-1-61779-474-2_26. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M.; Heinemeyer W.; Li X.; Arendt C. S.; Hochstrasser M.; Groll M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. 10.1038/ncomms10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2184–2195. 10.1107/s0907444904023510. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development ofCoot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.