

Abstract
Interleukin 34 (IL-34) constitutes a cytokine that shares a common receptor, colony-stimulating factor-1 receptor (CSF-1R), with CSF-1. We recently identified a novel type of monocytic cell termed follicular dendritic cell-induced monocytic cells (FDMCs), whose differentiation depended on CSF-1R signaling through the IL-34 produced from a follicular dendritic cell line, FL-Y. Here, we report the functional mechanisms of the IL-34–mediated CSF-1R signaling underlying FDMC differentiation. CRIPSR/Cas9-mediated knockout of the Il34 gene confirmed that the ability of FL-Y cells to induce FDMCs completely depends on the IL-34 expressed by FL-Y cells. Transwell culture experiments revealed that FDMC differentiation requires a signal from a membrane-anchored form of IL-34 on the FL-Y cell surface, but not from a secreted form, in a direct interaction between FDMC precursor cells and FL-Y cells. Furthermore, flow cytometric analysis using an anti–IL-34 antibody indicated that IL-34 was also expressed on the FL-Y cell surface. Thus, we explored proteins interacting with IL-34 in FL-Y cells. Mass spectrometry analysis and pulldown assay identified that IL-34 was associated with the molecular chaperone 78-kDa glucose-regulated protein (GRP78) in the plasma membrane fraction of FL-Y cells. Consistent with this finding, GRP78-heterozygous FL-Y cells expressed a lower level of IL-34 protein on their cell surface and exhibited a reduced competency to induce FDMC differentiation compared with the original FL-Y cells. These results indicated a novel GRP78-dependent localization and specific function of IL-34 in FL-Y cells related to monocytic cell differentiation.
Keywords: monocyte, cell differentiation, interleukin, cytokine, chaperone, GRP78, cell surface protein, plasma membrane, CRISPR/Cas, Western blot, colony-stimulating factor-1 receptor, follicular dendritic cell, interleukin 34
Introduction
Colony-stimulating factor-1 receptor (CSF-1R)2 activation promotes the proliferation, survival, and differentiation of mononuclear phagocytes, such as macrophages, Langerhans cells, and osteoclasts (1–4). Specifically, binding of CSF-1 to CSF-1R can induce the activation of the tyrosine kinase activity of CSF-1R through the phosphorylation of seven tyrosine residues (Tyr559, Tyr697, Tyr706, Tyr721, Tyr807, Tyr921, and Tyr974) of the cytoplasmic domain, followed by activation of signal transduction cascades including Src/Pyk2 and PI3K pathways (5, 6). Accordingly, CSF1op/op mice bearing a null mutation in the Csf1 gene exhibit osteoperitrotic phenotypes including toothlessness, skeletal defects, and impaired development of macrophages and osteoclasts (7). However, the phenotype of CSF-1R-deficient mice is more severe than that of the CSF1op/op mice; for example, Langerhans cells and microglia are completely absent in CSF-1R–deficient mice (8, 9).
Interleukin 34 (IL-34) has been identified as an alternative ligand for CSF-1R (10). IL-34 exhibits a similar stimulating activity for CSF-1R as the primary identified ligand, CSF-1 (11), although these two proteins share no sequence homology. Moreover, Il34 gene expression driven by the Csf1 promoter was shown to rescue the bone, osteoclast, tissue macrophage, and fertility defects of CSF1op/op mice; thus, IL-34 is considered to have redundant function with CSF-1 (2). Two groups have established IL-34–knockout (KO) mice, in which a LacZ reporter gene was inserted in exon 3 of the Il34 gene, resulting in a defect in the expression of functional IL-34 protein (12, 13). In these reports, disruption of IL-34 resulted in a selective defect of maintenance and differentiation of Langerhans cells and microglia. These studies therefore concluded that the IL-34 dependence of Langerhans and microglial cell differentiation was a result of the spatiotemporal difference between IL-34 and CSF-1 expression in the microenvironments during development of these cell lineages. However, it remains controversial whether IL-34 and CSF-1 exhibit identical activity with regard to CSF-1R signaling, because the binding affinity of IL-34 for CSF-1R is higher than that of CSF-1 (11), different phosphorylation patterns at intracellular tyrosine residues of CSF-1R are induced by IL-34 and CSF-1 binding to CSF-1R (14), and these cytokines induce different types of macrophage differentiation (14, 15).
Germinal centers (GCs) comprise transiently formed microenvironments in secondary lymphoid tissues after immunization, which are mainly composed of antigen (Ag)-activated B cells, follicular helper T cells, and follicular dendritic cells (FDCs). In GCs, Ag-activated B cells introduce somatic hypermutation in their Ig variable gene, and subsequently high-affinity B cells for a given antigen are clonally selected by the interaction with FDC and/or follicular helper T cells (16–18). Although several lines of evidence have shown a crucial role of FDCs for GC reactions, the detailed molecular mechanisms leading Ag-activated B cells to form GCs and the role of FDCs in plasma or memory phenotype B cell differentiation remain unclear (19–21). Toward this end, we previously established a mouse FDC line, FL-Y, from the lymph nodes of Ag-primed mice (22). FL-Y cells express major FDC markers, including FDC-M1, FcγRIIβ, and complement receptor, and proliferate in response to tumor necrosis factor (TNF)-α and an agonistic anti-lymphotoxin β receptor (LTβR) antibody. FL-Y cells are also capable of supporting the viability of GC B cells, thereby constituting a useful tool for analyzing GC reactions in vitro (23). Furthermore, we established a manipulated FL-Y line, termed FL-YB, which was transfected with an expression vector for B cell–activating factor belonging to the TNF family (BAFF), and exhibits enhanced activity for supporting GC-phenotype B cell viability (24). Moreover, Zhang et al. (25) demonstrated that cultured B cells stimulated with anti-IgM plus IL-4/IL-21 strongly proliferated and maintained the expression of BCL6 in the presence of FL-YB cells, indicating that FL-YB cells are capable of reconstituting GC environments.
Previously, we also found that FL-Y cells induced the differentiation of a novel class of monocytic cells, termed FDC-induced monocytic cells (FDMCs), from the population of T cell– and B cell–depleted c-kit+CD11b− splenocytes in vitro (26). Notably, FDMCs accelerated the proliferation of GC-phenotype B cells in anti-CD40 mAb-stimulated B cells. Furthermore, using RNAi-mediated knockdown of Il34 mRNA expression and Ab-mediated blockage of IL-34 and CSF-1R functions, we identified that IL-34, but not CSF-1, was a critical cytokine for stimulating FDMC differentiation, although FL-Y produced both IL-34 and CSF-1 (26). However, the specific function of IL-34 underlying FDMC differentiation remained unclear. Therefore, in the present study we explored the functional mechanisms of the IL-34 produced by FL-Y cells for FDMC differentiation.
Results
IL-34 plays a critical role in FDMC differentiation
To further confirm the IL-34–dependent differentiation of FDMCs, we first established IL-34 KO FL-Y cells by using the CRISPER/Cas9 system to eliminate IL-34 expression. A small transacting guide RNA (gRNA) and a targeting vector for disrupting the Il34 gene were designed against exon 3 of the Il34 gene, in which the LacZ gene was inserted for generating IL-34 KO mice as in the previous reports (Fig. 1A) (12, 13). FL-Y cells were transfected simultaneously with expression vectors for the gRNA and Cas9, along with IL-34–targeting vectors, and the successful replacement of exon 3 with the targeting vector was determined using genomic PCR. We established three independent clones in which a puromycin-resistance (clone 1) or blasticidin S–resistance gene (clone 2), respectively, was inserted into a single Il34 allele by a single transfection and wherein these two drug-resistance marker genes were inserted in both Il34 alleles by sequential transfection (clone 3). As shown in Fig. 1B, genomic PCR products indicated successful knockout of the Il34 allele by drug-resistance genes in IL-34 KO FL-Y cells. Sequencing analysis of genomic PCR products (F/R) in clones 1 and 2 showed that another allele was also inactivated by a 197- or 57-bp deletion in the vicinity of the gRNA-binding site, respectively (Fig. S1A). RT-PCR analysis using a primer pair (F/R) for amplifying the coding region of Il34 showed that expression of functional Il34 mRNA was undetectable in IL-34 KO FL-Y lines, whereas smaller DNA fragments were amplified in all clones (clones 1–3) (Fig. 1C). Sequencing analysis of smaller fragments amplified from clones 1, 2, and 3 cDNAs showed that Il34 mRNA in clones 1, 2, and 3 did not include exon 3, resulting in an encoded nonfunctional protein (Fig. S1B). Consistent with this, no PCR products were amplified by using the exon 3–specific primer (ex3F) in IL-34 KO clones (clones 1–3). In contrast, Csf1 mRNA expression levels in IL-34 KO FL-Y lines were comparable with that in the original FL-Y cells (Fig. 1C).
Figure 1.

Role of IL-34 in FDMC differentiation. A, gene targeting strategy for establishing IL-34 KO FL-Y cells. Exon 3 of the Il34 gene was replaced with the puromycin-resistance marker gene (puror) and the blasticidin S–resistance gene (BSR). Open arrowheads show primer pairs used for detecting successful recombination. B, genomic PCR analysis of the Il34 allele in FL-Y cells. Genome DNA was prepared from IL-34 KO FL-Y cells and analyzed by PCR. Il34 KO alleles were identified using indicated primer pairs (F/puro or F/BSR2). Closed arrowheads on the right show PCR products with the expected sizes. C, RT-PCR analysis of Il34 mRNA expression in IL-34 KO FL-Y cells. mRNA was isolated from WT FL-Y and IL-34 KO FL-Y cells (clones 1–3), and Il34 transcript was amplified by RT-PCR. Alternatively, an exon 3-specific primer (ex3F) was also used to amplify the Il34 transcripts. Closed arrowheads on the right show a PCR product with the expected size. The open arrowhead shows smaller PCR products than the expected one. D, T cell– and adherent cell–depleted splenocytes (1 × 106) from BALB/c mice were cultured with 1 × 104 cells of FL-Y or IL-34 KO FL-Y or without FL-Y (−) for 10 days. Cultured cells were collected and analyzed by flow cytometry after staining for CD11b. The percentage of CD11b+ cells is indicated. E, the number of generated FDMCs in each culture was calculated from the percentage of CD11b+ cells in the total number of viable cells in D. F, T cell– and adherent cell–depleted splenocytes (1 × 106) from BALB/c mice were cultured with 1 × 104 cells of FL-Y in the presence or absence of GW2580 (1 μm). The number of CD11b+ cells was determined as shown in D and E. The data are presented as the means ± S.D. of triplicate cultures. The data are representative of at least three independent experiments. Statistical differences are marked: **, p < 0.01 versus FL-Y; ***, p < 0.005 versus the DMSO control.
To explore the ability of IL-34 KO FL-Y cell lines to induce FDMCs, we cultured T cell–depleted splenocytes on an IL-34 KO FL-Y cell layer. After 9 days, the number of CD11b+ cells induced on the IL-34 KO FL-Y cell lines was significantly decreased compared with that on the original FL-Y cell line (Fig. 1, D and E). Furthermore, because receptor-type protein-tyrosine phosphatase ζ and syndecan-1 were found to function as additional IL-34 receptors (27, 28), we used a selective ATP-competitive CSF-1R inhibitor (GW2580) to evaluate the significance of CSF-1R signaling through IL-34 in FDMC differentiation, as previously reported (26). Notably, when GW2580 was added to the culture for FDMC induction, the generation of CD11b+ cells was completely suppressed (Fig. 1F). These data strongly supported that CSF-1R signaling through the IL-34 expressed by FL-Y plays a critical role in FDMC differentiation and that IL-34 KO FL-Y lines represent useful tools to study the functional mechanisms of IL-34 underlying FDMC differentiation.
In addition, the IL-34–dependent differentiation and immunological significance of FDMCs in vivo were confirmed by immunizing IL-34 KO mice with 4-hydroxy-3-nitrophenyl acetyl-conjugated chicken γ globulin plus alum, and the number of CD11b+CD115+ cells in the CD3e−B220−I-Ad−CD11c− population was examined because this population contained the in vivo counterpart of FDMCs in the spleen of immunized mice as previously described (26). Flow cytometric analysis showed that the number of CD11b+CD115+ cells among the splenocytes of IL-34 KO mice was significantly decreased compared with that in WT mice (Fig. S2, A and B). Furthermore, anti-NP IgG titer in the sera of IL-34 KO mice was lower than that of WT mice, although IgM titer was comparable between WT and IL-34 KO mice (Fig. S2C). These results suggested that IL-34 might be involved in the antibody response associated with the class switch recombination.
Cell surface–anchored IL-34 is involved in FDMC differentiation
To explore the IL-34–dependent mechanisms underlying FDMC differentiation on the FL-Y cell layer, we first used a Transwell culture system to block the cell–cell contact of FDMC precursor cells with FL-Y cells. When FL-Y cells were physically separated from the FDMC precursor cells by a Transwell membrane, the number of CD11b+ cells generated was significantly reduced to a level comparable with that on IL-34 KO FL-Y cells (Fig. 2A). We next treated FL-Y and IL-34 KO FL-Y cells with 0.1% paraformaldehyde (PFA). PFA treatment of living cells can block the transport of newly translated intracellular proteins to extracellular compartments by cross-linking the molecules expressed on the cell surface (29). Notably, CD11b+ cells were still generated on the PFA-treated FL-Y cell line, indicating that the ability of FL-Y to induce FDMCs was maintained in the absence of secreted IL-34 molecules produced from FL-Y cells (Fig. 2B). Moreover, culture supernatants of PFA-treated FL-Y cells exhibited no stimulating activity for the M-NFS 60 cell line, whose cell growth depended on CSF-1R signaling (Fig. S3A). To further ascertain the potential role of soluble IL-34 secreted from FL-Y cells in FDMC differentiation, secreted IL-34 protein purified from the culture supernatant of FL-Y cells overexpressing IL-34 was added to the culture for FDMC induction. However, generation of CD11b+ cells cultured on the original FL-Y cells was not increased by the addition of purified IL-34, suggesting no additive effects of soluble IL-34 on FDMC differentiation (Fig. S3B). These results indicated that there is little or no contribution of secreted IL-34 to FDMC differentiation and that another form of IL-34 in FL-Y cells might function in this regard.
Figure 2.

Involvement of cell-surface IL-34 in FDMC differentiation. A, in the Transwell culture, 1 × 104 FL-Y cells were seeded to the lower chamber, and T cell– and adherent cell–depleted splenocytes (1 × 106 cells) from BALB/c mice were added to the upper chamber separated by a 0.45-μm-pore size membrane. After 9 days, the number of CD11b+ cells in the upper chamber was estimated by flow cytometry. The data are presented as the means ± S.D. of triplicate cultures. Statistical differences are marked: *, p < 0.05 versus intact culture condition for FDMC induction of FL-Y cells. B, T cell– and adherent cell–depleted splenocytes (1 × 106) from BALB/c mice were cultured with 2 × 105 cells of the original FL-Y (FL-Y) or IL-34 KO FL-Y (IL-34 KO) cells, each of which was pretreated with (+) or without (−) 0.1% PFA. After 10 days, the cultured cells were analyzed by flow cytometry after staining for CD11b. The data are presented as the means ± S.D. of triplicate cultures. Statistical differences are marked: **, p < 0.01 versus the original FL-Y treated with PFA. C, IL-34 expression on the cell surface of the FL-Y cell line was determined by flow cytometry (left panels) after staining with isotype IgG (shaded) or anti–IL-34 antibody (red line). The level of IL-34 surface expression is indicated as the ΔMFI value, which was calculated by subtracting the MFI value of the isotype-matched control from that of each sample (right panel). The data are representative of at least three independent experiments. Max, maximum.
Next, we analyzed IL-34 localization on the FL-Y cell surface because FDMC differentiation was observed on PFA-treated FL-Y cells that were deficient in IL-34 secretion. When FL-Y cells were stained with an anti-mouse IL-34 Ab, an IL-34 signal was slightly detected on the cell surface of FL-Y cells, whereas signal was completely abolished on IL-34 KO FL-Y cells (Fig. 2C).
Establishment of FL-Y cells expressing Strep-tagged IL-34
It has been reported that the differentiation and growth of mononuclear phagocytes are stimulated by the soluble form of IL-34 because of the absence of the membrane-spanning domain in the IL-34 amino acid sequence (10). In contrast, flow cytometric analysis of FL-Y cells showed a detectable level of cell-surface localization of IL-34, although it is difficult to analyze how IL-34 is anchored on the cell surface because only an extremely small amount of IL-34 is expressed in FL-Y cells. Therefore, we generated an FL-Y cell line expressing twin-Strep-tagged IL-34 to identify the molecule(s) regulating IL-34 expression on the FL-Y cell surface. The twin-Strep-tag, composed of two Strep-tag peptides (WSHPQFEK) connected by a short linker (GGGSGGGSGGSA), is useful for detecting and purifying target molecules by using anti–Strep-tag mAb and Strep-Tactin–conjugated resin, respectively (30). The twin-Strep-tag sequence was inserted between the signal sequence and the mature protein sequence of IL-34, and the N-terminal twin-Strep-tagged IL-34 was introduced to FL-Y cells by retroviral transduction (FL-Y–IL-34–Nst). Western blotting analyses using an anti–IL-34 Ab and an anti–Strep-tag mAb showed that Strep-tagged IL-34 protein was detectable in the cell lysate of FL-Y–IL-34–Nst cells but not in the lysates of the original FL-Y and IL-34 KO FL-Y cells (Fig. 3A). In comparison, the endogenous IL-34 signal was not detected by Western blotting with the anti–IL-34 Ab, likely because FL-Y cells express intracellular and cell surface IL-34 at extremely low levels (Fig. 3A). Flow cytometric analysis showed that the IL-34–Nst protein was detectable on the cell surface of FL-Y–IL-34–Nst cells using either the anti–IL-34 Ab or anti–Strep-tag mAb and that the expression level of IL-34 on FL-Y–IL-34–Nst cells was significantly higher than that on the original FL-Y cells (Fig. 3B).
Figure 3.
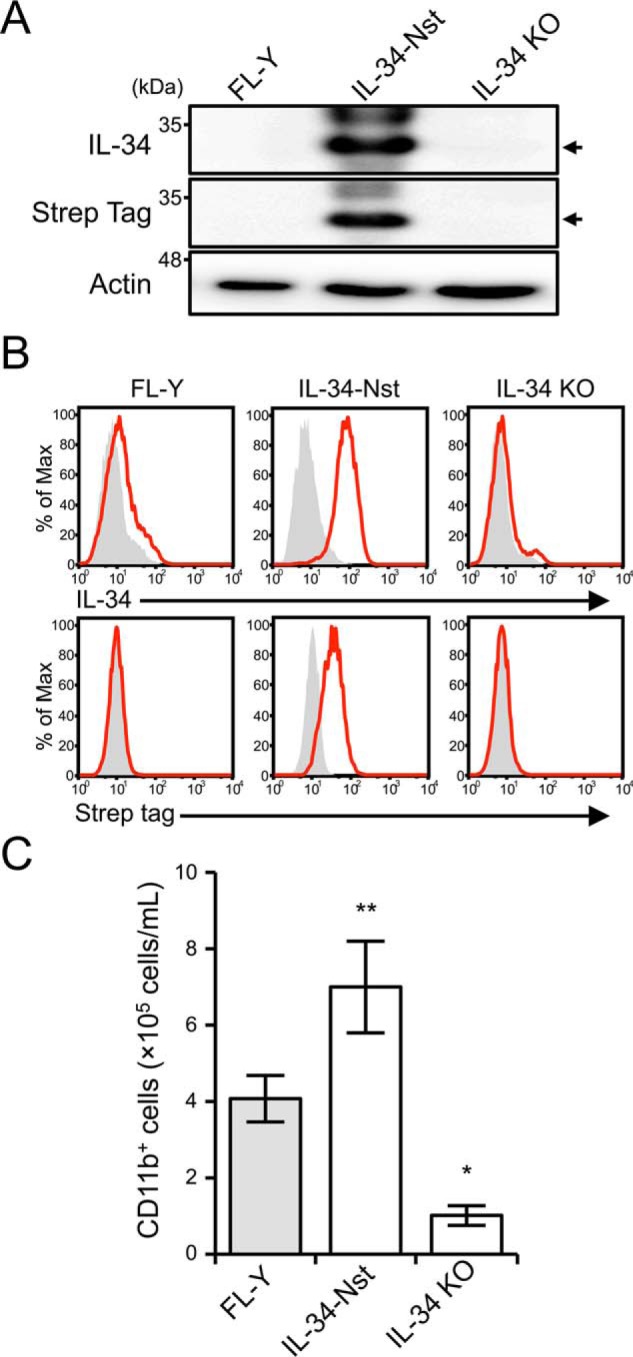
Establishment of FL-Y cells expressing Strep-tagged IL-34. A, immunoblot analysis of IL-34 in FL-Y, FL-Y–IL-34–Nst (IL-34-Nst), and IL-34 KO FL-Y (IL-34 KO) cells. Whole cell lysates were subjected to Western blotting analysis using anti–IL-34, anti–Strep-tag, and anti–β-actin Abs. β-Actin was used as an internal control. The blots are representative of at least three independent experiments. Arrows indicate IL-34–Nst. B, flow cytometric analysis of FL-Y cells after staining with anti–IL-34 or anti–Strep-tag Abs (red line). Negative controls that were stained with an isotype-matched control Ab are shown in gray histograms. C, T cell– and adherent cell–depleted splenocytes (1 × 106) from BALB/c mice were cultured with 2 × 105 cells of WT FL-Y, FL-Y–IL-34–Nst (IL-34-Nst), and IL-34 KO FL-Y (IL-34 KO) cells that were pretreated with 0.1% paraformaldehyde. After 8 days, the cultured cells were analyzed by flow cytometry after staining for CD11b. The data are representative of at least three independent experiments. The data are presented as the means ± S.D. of triplicate cultures. Statistical differences are marked: **, p < 0.01 versus FL-Y; *, p < 0.05 versus FL-Y.
To determine the biological activity of IL-34–Nst protein, IL-34–Nst in the conditioned medium collected from FL-Y–IL-34–Nst culture was purified by using Strep-Tactin–conjugated Sepharose and applied for stimulation of the M-NFS60 cell line (Fig. S4A). Purified IL-34–Nst protein prepared from the FL-Y–IL-34–Nst culture exhibited a biological activity for stimulating CSF-1R signaling in a dose-dependent manner (Fig. S4B). Furthermore, soluble IL-34–Nst protein was highly glycosylated at levels comparable with those of the commercially available rIL-34 protein (Fig. S4, C and D). Next, to examine whether FL-Y–IL-34–Nst cells exhibited an enhanced activity to induce FDMCs, we performed culture for FDMC induction under PFA-treated conditions. As shown in Fig. 3C, the FDMC-inducing activity of FL-Y–IL-34–Nst cells was significantly higher than that of the original FL-Y cells, indicating that cell surface IL-34–Nst was involved in FDMC differentiation.
Identification of GRP78 as an IL-34–binding molecule in the plasma membrane of FL-Y cells
To determine how IL-34 was anchored on the cell surface of FL-Y cells, we purified the plasma membrane fraction from FL-Y–IL-34–Nst cells. Consistent with the data from flow cytometric analysis, Western blotting analysis showed that IL-34–Nst protein was detectable in the plasma membrane fraction of FL-Y–IL-34–Nst cells but not in that of IL-34 KO FL-Y cells (Fig. 4A). Notably, the molecular mass of the IL-34–Nst protein in the plasma membrane fraction of FL-Y was mainly about 35 kDa, whereas higher molecular mass forms of IL-34–Nst were also detected in the whole cell lysate (Fig. 4A). Next, we performed protein identification of IL-34–associated molecules in the plasma membrane fraction prepared from FL-Y–IL-34–Nst cells. The purified plasma membrane fraction was reacted with Strep-Tactin resin to obtain Strep-tagged IL-34 and molecules bound to IL-34–Nst. After extensive washing, molecules bound to the resin were sequentially eluted with the elution buffer containing desthiobiotin, a competitor to the Strep-tag, and the SDS-PAGE sample buffer. Molecules pulled down with IL-34–Nst in the plasma membrane fraction of FL-Y–IL-34–Nst cells were identified by using LC-MS/MS analysis (Fig. 4B). Among some of the specific signals detected in FL-Y–IL-34–Nst cells, but not in FL-Y–IL-34 cells that were transduced with an untagged IL-34 expression vector, a 35-kDa protein was identified by LC-MS/MS analysis as the IL-34 that was used as bait, suggesting that the pulldown experiment was efficient. The ∼75-kDa band specifically bound to IL-34–Nst was subjected to LC-MS/MS analysis and identified as GRP78 (Fig. 4B, Table 1, and Fig. S5). GRP78, also known as binding immunoglobulin protein or heat shock protein A5, is involved in protein folding and unfolded protein responses at intracellular compartments (31).
Figure 4.

Interaction of IL-34 with GRP78 in the plasma membrane of FL-Y cells. A, immunoblot analysis of the plasma membrane fraction prepared from FL-Y–IL-34–Nst (IL-34-Nst) and IL-34 KO FL-Y (IL-34 KO) cells. Plasma membrane fractions extracted from the indicated cells were subjected to Western blotting analysis using anti–IL-34 and anti–Strep-tag Abs. VCAM was used as an internal control. Arrows indicate IL-34–Nst. B, silver staining of IL-34 and proteins associated with IL-34. IL-34–Nst proteins in the plasma membrane fraction prepared from FL-Y–IL-34 (IL-34) and FL-Y–IL-34–Nst (IL-34-Nst) cells were pulled down by using the Strep-Tactin Sepharose resin and eluted by 2.5 mm desthiobiotin. Plasma membrane fractions (Input), eluted fractions with desthiobiotin (EF), and residual fractions on the resin (RF) were subjected to SDS-PAGE. C, GRP78 was bound to IL-34–Nst in the plasma membrane fraction. The plasma membrane fraction of FL-Y–IL-34–Nst cells was mixed with control (Cont) or Strep-Tactin (ST) Sepharose, and bound molecules were eluted with SDS-PAGE sample buffer for Western blotting with anti-GRP78 Ab. D, plasma membrane fraction of FL-Y–IL-34 (IL-34) and FL-Y–IL-34–Nst (IL-34-Nst) cells was reacted with Strep-Tactin Sepharose, and binding molecules were eluted with SDS-PAGE sample buffer for Western blotting with anti-GRP78 Ab. E, recombinant GST or GST–GRP78 was mixed with the whole cell lysate of FL-Y–IL-34–Nst cells and precipitated with glutathione-Sepharose. Eluted fractions were subjected to SDS-PAGE and Western blotting analysis.
Table 1.
Amino acid sequences of the GRP78 peptides identified by LC-MS/MS analysis
| Residues | Peptide sequence | Ion score |
|---|---|---|
| 83–97 | NQLTSNPENTVFDAK | 74 |
| 83–98 | NQLTSNPENTVFDAKR | 16 |
| 125–139 | TKPYIQVDIGGGQTK | 10 |
| 140–153 | TFAPEEISAMVLTK | 28 |
| 187–198 | DAGTIAGLNVMR | 51 |
| 346–353 | VLEDSDLK | 16 |
| 355–368 | SDIDEIVLVGGSTR | 39 |
| 449–465 | SQIFSTASDNQPTVTIK | 50 |
| 525–533 | ITITNDQNR | 60 |
| 534–541 | LTPEEIER | 37 |
| 564–574 | NELESYAYSLK | 71 |
| 623–634 | ELEEIVQPIISK | 36 |
Considering that there is no report of binding between IL-34 and GRP78 on the cell surface, we first performed a pulldown assay of the plasma membrane fraction prepared from FL-Y–IL-34–Nst cells using Strep-Tactin Sepharose. The pulldown assay showed that IL-34–Nst and GRP78 were co-precipitated with the Strep-Tactin Sepharose resin but not the control Sepharose (Fig. 4C). To rule out the possibility of nonspecific binding of GRP78 and IL-34 on the Strep-Tactin Sepharose, we used the plasma membrane fraction prepared from FL-Y–IL-34 cells. To further assess whether IL-34 was able to interact with GRP78, we used recombinant glutathione S-transferase (GST)–GRP78 that was expressed in Escherichia coli cells and purified with glutathione-Sepharose. When GST–GRP78 was reacted with total cell lysates prepared from FL-Y–IL-34–Nst cells, GST–GRP78 and IL-34–Nst were co-precipitated by glutathione-Sepharose, which further supported the specific binding of GRP78 to IL-34 (Fig. 4E).
GRP78 regulates IL-34 expression on the cell surface and FDMC differentiation
To determine the role for GRP78 in IL-34 expression on the cell surface and subsequent FDMC differentiation, we established GRP78-heterozygous FL-Y cells by using the CRISPER/Cas9 system. A targeting vector for the GRP78 locus was constructed against exon 2 of the Grp78 gene that contains the translational start codon (Fig. 5A). FL-Y cells were transfected with expression vectors for a gRNA specific to exon 2 of the Grp78 gene and for Cas9, along with the targeting vectors, from which we obtained two-independent GRP78-heterozygous clones (Fig. 5B). RT-PCR analysis showed that Grp78 mRNA expression was decreased by 20–70% in GRP78-heterozygus cells compared with that in the original cell line (Fig. 5C). Furthermore, GRP78 protein expression in the GRP78-heterozygous FL-Y line was also reduced to ∼50% of that in the original FL-Y cells (Fig. 5D). Notably, we observed that the expression level of cell surface–expressing IL-34 in two independent GRP78-heterozygous clones was reduced to ∼20% relative to that in the original FL-Y cells (Fig. 5E). The reduction of IL-34 cell-surface expression was also observed in GRP78-heterozygous FL-Y–IL-34–Nst cells, even though excess IL-34 was expressed (Fig. S6). To examine whether the reduced expression of GRP78 abrogates FL-Y–induced FDMC differentiation, we cultured T cell–depleted splenocytes on PFA-treated FL-Y cells for 12 days to induce FDMCs. The number of CD11b+ cells generated on GRP78-heterozygous FL-Y cells was reduced to approximately half of that on WT FL-Y cells (Fig. 5F). These results clearly indicated that GRP78 promotes IL-34 expression on the FL-Y cell surface and contributes to inducing FDMC differentiation.
Figure 5.

GRP78-heterozygous FL-Y cells exhibit reduced FDMC-inducing activity. A, gene targeting strategy for establishing GRP78-heterozygous FL-Y cells. Open arrowheads show primer pairs used for detecting successful recombination. B, genomic PCR analysis of the Grp78 gene in WT (+/+) and GRP78-heterozygous (+/−) FL-Y cells. Genome DNA was prepared from GRP78-heterozygous FL-Y cells (clones 1 and 2) and amplified by PCR. The Grp78 KO allele was identified using indicated primer pairs (F/puro and r-plox/R). C, RT-PCR analysis of Grp78 mRNA expression in GRP78-heterozygous FL-Y cells (clones 1 and 2). RNA was isolated from WT and GRP78-heterozygous FL-Y cells (clones 1 and 2), and the Grp78 transcript was amplified by RT-PCR. D, Western blotting analysis of GRP78 expressed in FL-Y and GRP78-heterozygous FL-Y cells (clones 1 and 2). Whole cell lysates prepared from FL-Y and GRP78-heterozygous FL-Y cells (clones 1 and 2) were separated by SDS-PAGE and subjected to Western blotting. E, IL-34 expression on the cell surface of the GRP78-heterozygous FL-Y cell line was determined by flow cytometry after staining with an anti–IL-34 antibody as shown in Fig. 2C. The level of IL-34 cell-surface expression is indicated as described for Fig. 2C (ΔMFI). F, competency of GRP78-heterozygous FL-Y cells for inducing FDMC differentiation. T cell– and adherent cell–depleted splenocytes (1 × 106) from BALB/c mice were cultured with FL-Y and GRP78-heterozygous (±) FL-Y (clones 1 and 2) that were pretreated with 0.1% paraformaldehyde as shown in Fig. 2B. Cultured cells were collected and analyzed by flow cytometry after staining for CD11b. The data are presented as the means ± S.D. of triplicate cultures. The data are representative of at least three independent experiments. Statistical differences are marked: *, p < 0.05 versus original FL-Y (+/+).
Discussion
In this study, we found that a membrane-anchored form, but not a secreted from, of IL-34 produced from FL-Y cells was involved in monocytic cell differentiation, although the secreted form of IL-34 detected in culture medium could also exhibit a stimulating activity through CSF-1R. IL-34 has been thought to constitute a secreted molecule, because of the absence of a membrane-spanning domain in the primary structure of IL-34. However, flow cytometric analysis showed that WT but not IL-34 KO FL-Y cells expressed a detectable level of cell-surface IL-34 (Figs. 2C and 3B). Moreover, overexpression of IL-34 via an IL-34–Nst retroviral expression vector enhanced not only IL-34 expression levels on the FL-Y cell surface, but also monocytic cell differentiation, further supporting the conclusion that cell-surface IL-34 contributes to the induction of monocytic cell differentiation in a dose-dependent manner.
We also identified for the first time that the cell-surface localization of IL-34 is regulated in a GRP78-dependent manner. GRP78, a member of the heat shock protein 70 family, is mainly localized to the endoplasmic reticulum (ER) and comprises the most abundant molecular chaperone regulating protein folding, preventing protein aggregation, and facilitating the unfolded protein response (31). Although GRP78 is mainly found in the ER lumen, it has been reported that GRP78 is redistributed to the nucleus, mitochondria, and cytoplasm under some circumstances (32–34). In addition, several reports have revealed that GRP78 is also located on the cell surface of several cell lines including leukemias, lymphomas, and cancer cells derived from prostate, breast, ovarian, and brain tissues (35–37), as well as on normal cells, such as CD4+CD25+ T cells and hematopoietic stem cells (38, 39). Furthermore, GRP78 expression level on the cell surface was enhanced by ER stress and hypoxia conditions to decrease ER stress-induced cell death (40, 41). Accumulating data have revealed that cell-surface GRP78 can form complexes with several proteins and functions as a multifunctional receptor for a wide variety of ligands. Binding of a ligand or an agonistic Ab to cell-surface GRP78 also triggers the activation of intracellular signal transduction cascades through the PI3K/Akt pathway as well as NF-κB-dependent mechanisms, resulting in the induction of protein synthesis, DNA synthesis, and proliferation. For example, cell-surface GRP78 serves as a receptor for activated α2-macroglobulin and triggers intracellular signal transduction (42, 43), leading to the cell proliferation and metastasis of prostate cancer cells (44). Cripto, also known as teratocarcinoma-derived growth factor 1, forms complexes with GRP78 at the cell surface to inhibit transforming growth factor β (TGF-β) signaling and enhance cell proliferation (45). High expression levels of cell-surface GRP78 have thereby been implicated in cancer growth, metastasis, and resistance to chemotherapy. Alternatively, GRP78 is also shown to act as a receptor for several types of viruses and plays an important role for viral entry into animal cells (46, 47). It might thus be important for understanding FDC functions to determine whether IL-34 is able to trigger signal transduction through binding to GRP78.
Previously, it was reported that GRP78 binds to TGF-β/LAP and regulates its cell surface expression. Notably, the TGF-β in complex with GRP78 contains a high-glycosylated and furin-processed latent form, which differs from the secreted and the intracellular form of TGF-β (38). Thus, cell-surface IL-34 molecules that are anchored and/or regulated by GRP78 might have an unconventional structure or glycosylation pattern. Consistent with this conjecture, in the present study we found that the molecular mass of cell-surface IL-34 (∼35 kDa) is different from that of the secreted form of IL-34 (high molecular mass) or a recombinant IL-34 expressed in E. coli (low molecular mass) (Fig. S4D).
We further found that GRP78 could be detected in the plasma membrane fraction of the mouse FDC line, FL-Y (Fig. 4, C and D), and plays a critical role in regulating IL-34 expression on the cell surface. Although it is still not clear whether the GRP78 expressed in FL-Y cells functions as an anchor protein at the plasma membrane to present IL-34 and/or as receptor that can trigger autocrine intracellular signal transduction, a paracrine CSF-1R signaling via cell-surface IL-34 appears to be important for FDMC differentiation because this process was completely blocked by treatment with the CSF-1R inhibitor GW2580.
In a prior study, we reported that the level of Il34 mRNA in FL-Y cells was enhanced by stimulation with TNF-α plus an agonistic anti-LTβR mAb or during FDMC induction culture, indicating that IL-34 expression was enhanced by activation signals for FDC (26). Although little has been reported regarding the effects of IL-34 on acquired immunity including GC reactions, the IL-34 produced from FDC might regulate Ab responses in vivo because TNF receptor and LTβR-mediated signals have been shown to be important for secondary lymphoid organ development, FDC differentiation, and host immune responses including GC formation and the affinity maturation of Abs (48–51). Several groups have reported that the level of IL-34 expression correlates with the severity of autoimmune diseases including rheumatoid arthritis (52, 53), systemic lupus erythematosus (SLE) (54), and Sjogren's syndrome (55). In patients with rheumatoid arthritis, abundant IL-34 expression that was mainly produced by synovial fibroblasts can be observed in the sera and synovia (52) and is associated with pathogenesis (53). Serum IL-34 levels also correlate with anti-dsDNA Ab titer and SLE disease activity index in patients with SLE. Notably, serum IL-34 levels were significantly decreased after successful treatment for SLE (54), suggesting that IL-34 may be useful as a biomarker for autoimmune diseases, as well as a target for treatments. Thus, it may be important to determine the regulatory mechanism of cell-surface forms, as well as secreted forms, of IL-34 in inflammatory conditions for understanding the development and severity of autoimmune diseases.
In conclusion, in this study we revealed that CSF-1R signaling triggered by cell-surface IL-34 is involved in the development of a specific type of monocytic cells. However, the detailed functions of the cell-surface IL-34 underlying FDMC differentiation remain unclear. Thus, further studies are required to confirm the specific functions of IL-34 on the cell surface. Nevertheless, these findings provide a foundation for understanding the new regulatory mechanisms of monocyte differentiation by GRP78-dependent cell-surface IL-34.
Experimental procedures
Animals
Male BALB/c mice were purchased from Japan Charles River Breeding Laboratories (Kanagawa, Japan). All mice were treated in accordance with the guidelines approved by the Committee of Laboratory Animal Care of Okayama University and usually used at 7–12 weeks of age.
Cell line
The mouse FDC line, FL-Y, was established from the long-term culture of primary FDCs derived from immunized BALB/c mice as described previously (22). The FL-Y cell line was maintained in culture medium consisting of a 1:1 mixture of Dulbecco's modified Eagle's medium (Nissui Pharmaceuticals, Tokyo, Japan) and RPMI 1640 medium (Nissui Pharmaceuticals) supplemented with 10% fetal bovine serum (Nichirei Biosciences, Tokyo, Japan), 2 mm glutamine, 1 mm pyruvic acid, 100 μg/ml penicillin G, 50 μg/ml streptomycin, and 50 μm 2-mercaptoethanol, in the presence of 5 ng/ml TNF-α (PeproTech, Rocky Hill, NJ). The M-NFS-60 cell line was kindly provided by Prof. Shinya Suzu at Kumamoto University (56) and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum in the presence of 100 ng/ml human macrophage colony-stimulating factor (57).
Establishment of IL-34 and GRP78 KO FL-Y lines
To generate targeting vectors for Il34 gene knockout, 5′ and 3′ DNA fragments flanking exon 3 of the Il34 gene were amplified with primer pairs, IL-34–1st-F with IL-34–1st-R and IL-34–2nd-F with IL-34–2nd-R, respectively (Table S1). PCR was carried out using KOD FX neo DNA polymerase (Toyobo, Osaka, Japan). Each DNA fragment was cloned in the pCR-Blunt vector (Invitrogen) and confirmed by sequencing analysis using BigDye 1.1 and the ABI310 genetic analyzer (Applied Biosystems, Foster City, CA) according to the manufacturer's instruction. 5′ and 3′ DNA fragments were subcloned stepwise between NotI and ClaI sites in the pBluescript SK (+) vector. Blasticidin S–resistance (BSR) or puromycin-resistance cassettes excised from pLox-BSR or pLox-puro vector, respectively, were inserted into the BamHI site between two DNA fragments (58). For constructing gRNA expression vectors, we designed two guide sequences (T1 and T2) targeting exon 3 of the Il34 gene. Two annealed complementary oligonucleotides as listed in Table S1 were cloned into the gRNA expression vector (catalog no. 41824; Addgene, Cambridge, MA).
To generate a targeting vector for the GRP78 knockout, 5′ and 3′ DNA fragments flanking exon 2 of the Grp78 gene were amplified with primer pairs GRP78–1st-F and GRP78–1st-R for the 5′ DNA fragment and GRP78–2nd-F and GRP78–2nd-R for the 3′ DNA fragment. Primers used for construction are listed in Table S1.
FL-Y cells were treated for 24 h with a targeting vector (1 μg), a gRNA vector (1 μg), and Cas9 expression vector (1 μg) (catalog no. 41815; Addgene) that were mixed with X-tremeGENE HP DNA transfection reagents (2 μl) (Roche Diagnostics, Mannheim, Germany) in 200 μl of Opti-MEM (Invitrogen) according to the manufacturer's instructions. Stably transfected clones were selected in culture medium containing 2 μg/ml puromycin or 10 μg/ml blasticidin S hydrochloride (Kaken Pharmaceutical, Tokyo, Japan) for 2–3 weeks. After limiting dilution of drug-resistant cells, isolated clones were examined for successful knockout of Il34 and Grp78 genes by genomic PCR, RT-PCR, and Western blotting. Primers used for successful recombination are listed in Table S1.
Retroviral transduction of the FL-Y line for IL-34 overexpression
The Il34 cDNA fragment was amplified by RT-PCR using the primer pair as listed in Table S1 using KOD-FX Neo DNA polymerase (Toyobo) and cloned into the pCR-Blunt vector (pCR-Blunt-IL-34). An Il34 DNA fragment encoding IL-34 tagged with a twin-Strep-tag between the signal sequence (residue 1–20) and the mature peptide chain was constructed by two-step PCR amplification and subcloned into the pCR-Blunt vector (pCR–Blunt–IL-34–Nst). For constructing IL-34 and IL-34–Nst expression shuttle vectors, the EcoRI DNA fragments of the Il34 and Il34–Nst gene was elicited from pCR–Blunt–IL-34 and pCR–Blunt–IL-34–Nst, respectively, and cloned in the EcoRI site of the pQCXIP vector (Clontech).
Culture supernatants containing a recombinant retrovirus were prepared using the GP2–293 packaging cell line transfected transiently with each shuttle vector and the pVSV-G plasmid vector (Clontech). FL-Y cells were transduced with each retrovirus in the presence of 4 μg/ml Polybrene and 5 mm HEPES (pH 7.4). Stably puromycin-resistant FL-Y cells were selected with puromycin (4 μg/ml) (Sigma–Aldrich).
FDMC induction culture
Culture for the induction of FDMC differentiation was performed as described previously (26). Briefly, each well in 24-well culture plates was coated with 5 × 103 or 1 × 104 FL-Y cell lines by incubating the cells overnight in 1 ml of the culture medium. After removing the culture medium, 1 × 106 spleen cells that were depleted of T cells and adherent cells in 1 ml of the culture medium were added into each FL-Y–coated well. The cells were cultured at 37 °C under a humidified atmosphere of 95% air and 5% CO2. T cell– and adherent cell–depleted cells were prepared from BALB/c spleen cells by lysing red blood cells and removing adherent cells by panning with plastic culture plates and depleting T cells using Dynabeads Mouse PanT (Thy1.2) (Invitrogen) or CD90.2 (Thy1.2) Magnetic Particles-DM (BD Biosciences, San Jose, CA). After culture for 7–12 days, viable cells were enumerated by trypan blue dye exclusion, and the frequency of CD11b+ cells was estimated by flow cytometry. To block CSF-1R signaling during FDMC differentiation, 1 μm GW2580 (Abcam) was added on day 0.
Flow cytometric analysis
The cells were treated with normal rat IgG (10 μg/ml) for 30 min to block nonspecific binding of labeled Abs and subsequently stained with phycoerythrin-labeled anti-CD11b mAb (M1/70; BioLegend, San Diego, CA) in a staining buffer (PBS containing 0.2% (w/v) BSA and 0.1% (w/v) NaN3) at 4 °C for 30 min. After washing three times with the staining buffer, labeled cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (Tree Star, Ashland, OR). For IL-34 staining, FL-Y cells were fixed with 4% formaldehyde for 10 min and stained with sheep anti–IL-34 Ab (AF5195; R&D Systems, Minneapolis, MN) at 4 °C for 1 h. After washing, these cells were incubated with biotinylated anti-sheep IgG (Vector Laboratories, Burlingame, CA), followed by streptavidin-conjugated phycoerythrin–Cy5 (BioLegend).
Western blotting
Cell lysates were prepared in SDS-PAGE sample buffer and subjected to SDS-PAGE and Western blotting. Membranes were probed with sheep anti-mouse IL-34 Ab (AF5195; R&D Systems), mouse anti–Strep-tag mAb (Strep Mab Classic) (IBA, Göttingen, Germany), or rabbit anti-GRP78 Ab (N-20, Santa Cruz Biotechnology, Dallas, TX; and GL-19, Sigma–Aldrich). The antibody binding was detected using a combination with rabbit HRP–anti-sheep IgG (Invitrogen), HRP-anti-mouse IgG1 (Southern Biotechnology Associates, Birmingham, AL), or HRP–anti-rabbit IgG (GE Healthcare, London, UK) with ECL Plus or ECL Pro (PerkinElmer Life Sciences), respectively. Anti–β-actin Ab (sc-47778; Santa Cruz Biotechnology) was used as a loading control.
Extraction of membrane-bound protein and pulldown assay
Cell membrane fractions in FL-Y, FL-Y–IL-34, FL-Y–IL-34–Nst, and IL-34 KO FL-Y cells were purified using a plasma membrane extraction kit (BioVision, Mountain View, CA) according to the manufacturer's instruction. Cultured FL-Y cells were washed with PBS and detached with a cell scraper from culture plates. Plasma membrane fractions prepared from several FL-Y lines were solubilized with radioimmune precipitation assay buffer (150 mm NaCl, 20 mm HEPES, pH 7.4, 1% NP-40, 0.1% SDS) at 4 °C overnight and subsequently centrifuged at 21,880 × g for 10 min at 4 °C. The supernatants were used as the plasma membrane fractions. For pulldown assay, the plasma membrane fractions were diluted with a 10-fold volume of NP buffer (50 mm NaH2PO4, 300 mm NaCl, pH 8.0) to apply the Strep-Tactin Sepharose resin (IBA). After washing with NP buffer, proteins bound to the resin were eluted with elution buffer (NP buffer containing 2.5 mm desthiobiotin (Sigma–Aldrich)) and analyzed by SDS-PAGE and Western blotting.
Mass spectrometry analysis
Purified plasma membrane fractions prepared from FL-Y–IL-34–Nst cells (1 × 108 cells) were suspended in 40 ml of NP buffer and incubated with 40 μl of the Strep-Tactin Sepharose resin at 4 °C overnight. After washing with NP buffer, proteins bound to the resin were eluted with 20 μl of elution buffer. Eluate was separated by SDS-PAGE and stained using a silver staining kit (Invitrogen), followed by in-gel digestion with 10 μg/ml sequencing grade modified trypsin (Promega, Madison, WI) overnight at 37 °C (59). The digested peptides were eluted with 0.1% formic acid and subjected to LC-MS/MS analysis on an LC-MS–IT–TOF instrument (Shimadzu, Kyoto, Japan) interfaced with a nano reverse-phase LC system (Shimadzu), as described previously (60). MS/MS data were acquired in the datum-dependent mode using LC-MS solution software (Shimadzu) and converted to a single text file (containing the observed precursor peptide m/z, fragment ion m/z, and intensity values) using Mascot Distiller (Matrix Science, London, UK). MS/MS data were obtained independently and merged for the Mascot analysis. The file was analyzed using Mascot (Matrixscience) MS/MS ion search to search and assign the obtained peptides to the SwissProt database. The search parameter was as follows: database, SwissProt 2017_05; taxonomy, all; enzyme, trypsin; variable modifications, carbamidomethyl (C), oxidation (M), propionamid (C); peptide tol., ± 0.05 Da; and MS/MS tol., ± 0.05 Da.
Preparation of recombinant GST–GRP78 protein and pulldown assay
GST-tagged GRP78 was constructed using a pGEX-KG-PreS vector (61) and expressed in E. coli BL21 Star (DE3), followed by purification via glutathione-Sepharose chromatography (GE Healthcare). To obtain whole cell lysate, FL-Y–IL-34–Nst cells (6 × 106 cells) were lysed in 200 μl of radioimmune precipitation assay buffer and centrifuged at 1560 × g for 5 min after sonication. GST and GST–GRP78 protein (40 μg) was mixed with 100 μl of whole cell lysate. Subsequently, 20 μl of glutathione-Sepharose was reacted with the sample overnight at 4 °C. After extensive washing, proteins bound to Sepharose were eluted with 30 μl of SDS sample buffer. The eluate was subjected to SDS-PAGE followed by Western blotting.
Author contributions
S. O., N. K., N. H., H. T., and M. M. supervision; S. O. and M. M. funding acquisition; S. O., Y. M., M. T., K. M., F. Y., E. K., S. Y., K. H., N. K., N. H., H. T., and M. M. investigation; S. O. and M. M. writing-original draft; S. O. and M. M. project administration; S. O., N. K., N. H., H. T., and M. M. writing-review and editing.
Supplementary Material
This work was supported in part by Grant-in-aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan 25460590 and funds from the Ryobi Teien Foundation and the Takeda Science Foundation (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S6.
- CSF-1
- colony-stimulating factor-1
- CSF-1R
- colony-stimulating factor-1 receptor
- GC
- germinal center
- KO
- knockout
- GRP78
- 78-kDa glucose-regulated protein
- FDC
- follicular dendritic cell
- FDMC
- FDC-induced monocytic cell
- TNF
- tumor necrosis factor
- gRNA
- guide RNA
- ER
- endoplasmic reticulum
- TGF
- transforming growth factor
- IL
- interleukin
- Ag
- antigen
- LTβR
- lymphotoxin β receptor
- PFA
- paraformaldehyde
- GST
- glutathione S-transferase
- SLE
- systemic lupus erythematosus
- BSR
- blasticidin S–resistance
- HRP
- horseradish peroxidase
- MFI
- mean fluorescence intensity.
References
- 1. Hume D. A. (2008) Macrophages as APC and the dendritic cell myth. J. Immunol. 181, 5829–5835 10.4049/jimmunol.181.9.5829 [DOI] [PubMed] [Google Scholar]
- 2. Wei S., Nandi S., Chitu V., Yeung Y.-G., Yu W., Huang M., Williams L. T., Lin H., and Stanley E. R. (2010) Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J. Leukoc. Biol. 88, 495–505 10.1189/jlb.1209822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garceau V., Smith J., Paton I. R., Davey M., Fares M. A., Sester D. P., Burt D. W., and Hume D. A. (2010) Pivotal advance: avian colony-stimulating factor 1 (CSF-1), interleukin-34 (IL-34), and CSF-1 receptor genes and gene products. J. Leukoc. Biol. 87, 753–764 10.1189/jlb.0909624 [DOI] [PubMed] [Google Scholar]
- 4. Hamilton J. A., and Achuthan A. (2013) Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 34, 81–89 10.1016/j.it.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 5. Knowlton M. L., Selfors L. M., Wrobel C. N., Gu T. L., Ballif B. A., Gygi S. P., Polakiewicz R., and Brugge J. S. (2010) Profiling Y561-dependent and -independent substrates of CSF-1R in epithelial cells. PLoS One 5, e13587 10.1371/journal.pone.0013587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu W., Chen J., Xiong Y., Pixley F. J., Dai X.-M., Yeung Y.-G., and Stanley E. R. (2008) CSF-1 receptor structure/function in MacCsf1r−/− macrophages: regulation of proliferation, differentiation, and morphology. J. Leukoc. Biol. 84, 852–863 10.1189/jlb.0308171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witmer-Pack M. D., Hughes D. A., Schuler G., Lawson L., McWilliam A., Inaba K., Steinman R. M., and Gordon S. (1993) Identification of macrophages and dendritic cells in the osteopetrotic (op/op) mouse. J. Cell Sci. 104, 1021–1029 [DOI] [PubMed] [Google Scholar]
- 8. Dai X. M., Ryan G. R., Hapel A. J., Dominguez M. G., Russell R. G., Kapp S., Sylvestre V., and Stanley E. R. (2002) Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99, 111–120 10.1182/blood.V99.1.111 [DOI] [PubMed] [Google Scholar]
- 9. Ginhoux F., Tacke F., Angeli V., Bogunovic M., Loubeau M., Dai X. M., Stanley E. R., Randolph G. J., and Merad M. (2006) Langerhans cells arise from monocytes in vivo. Nat. Immunol. 7, 265–273 10.1038/nrm1890,10.1038/ni1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin H., Lee E., Hestir K., Leo C., Huang M., Bosch E., Halenbeck R., Wu G., Zhou A., Behrens D., Hollenbaugh D., Linnemann T., Qin M., Wong J., Chu K., Doberstein S. K., et al. (2008) Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320, 807–811 10.1126/science.1154370 [DOI] [PubMed] [Google Scholar]
- 11. Ma X., Lin W. Y., Chen Y., Stawicki S., Mukhyala K., Wu Y., Martin F., Bazan J. F., and Starovasnik M. A. (2012) Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure 20, 676–687 10.1016/j.str.2012.02.010 [DOI] [PubMed] [Google Scholar]
- 12. Greter M., Lelios I., Pelczar P., Hoeffel G., Price J., Leboeuf M., Kündig T. M., Frei K., Ginhoux F., Merad M., and Becher B. (2012) Stroma-derived interleukin-34 controls the development and maintenance of Langerhans cells and the maintenance of microglia. Immunity 37, 1050–1060 10.1016/j.immuni.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y., Szretter K. J., Vermi W., Gilfillan S., Rossini C., Cella M., Barrow A. D., Diamond M. S., and Colonna M. (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753–760 10.1038/nrm3481,10.1038/nrm3480,10.1038/ni.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chihara T., Suzu S., Hassan R., Chutiwitoonchai N., Hiyoshi M., Motoyoshi K., Kimura F., and Okada S. (2010) IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 17, 1917–1927 10.1038/cdd.2010.60 [DOI] [PubMed] [Google Scholar]
- 15. Boulakirba S., Pfeifer A., Mhaidly R., Obba S., Goulard M., Schmitt T., Chaintreuil P., Calleja A., Furstoss N., Orange F., Lacas-Gervais S., Boyer L., Marchetti S., Verhoeyen E., Luciano F., et al. (2018) IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 8, 256 10.1038/s41598-017-18433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu M., and Schatz D. G. (2009) Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 30, 173–181 10.1016/j.it.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 17. Vinuesa C. G., Sanz Ĩ, and Cook M. C. (2009) Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 9, 845–857 10.1038/nri2637 [DOI] [PubMed] [Google Scholar]
- 18. Rajewsky K. (1996) Clonal selection and learning in the antibody system. Nature 381, 751–758 10.1038/381751a0 [DOI] [PubMed] [Google Scholar]
- 19. Allen C. D., and Cyster J. G. (2008) Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin. Immunol. 20, 14–25 10.1016/j.smim.2007.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park C. S., and Choi Y. S. (2005) How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology 114, 2–10 10.1111/j.1365-2567.2004.02075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kosco-Vilbois M. H. (2003) Are follicular dendritic cells really good for nothing? Nat. Rev. Immunol. 3, 764–769 10.1038/nri1179 [DOI] [PubMed] [Google Scholar]
- 22. Nishikawa Y., Hikida M., Magari M., Kanayama N., Mori M., Kitamura H., Kurosaki T., and Ohmori H. (2006) Establishment of lymphotoxin β receptor signaling-dependent cell lines with follicular dendritic cell phenotypes from mouse lymph nodes. J. Immunol. 177, 5204–5214 10.4049/jimmunol.177.8.5204 [DOI] [PubMed] [Google Scholar]
- 23. Aungier S. R., Ohmori H., Clinton M., and Mabbott N. A. (2015) MicroRNA-100–5p indirectly modulates the expression of Il6, Ptgs1/2 and Tlr4 mRNA in the mouse follicular dendritic cell-like cell line, FL-Y. Immunology 144, 34–44 10.1111/imm.12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Magari M., Nishikawa Y., Fujii Y., Nishio Y., Watanabe K., Fujiwara M., Kanayama N., and Ohmori H. (2011) IL-21-dependent B cell death driven by prostaglandin E2, a product secreted from follicular dendritic cells. J. Immunol. 187, 4210–4218 10.4049/jimmunol.1100934 [DOI] [PubMed] [Google Scholar]
- 25. Zhang T. T., Gonzalez D. G., Cote C. M., Kerfoot S. M., Deng S., Cheng Y., Magari M., and Haberman A. M. (2017) Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. Elife 6, e19552 10.7554/eLife.19552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamane F., Nishikawa Y., Matsui K., Asakura M., Iwasaki E., Watanabe K., Tanimoto H., Sano H., Fujiwara Y., Stanley E. R., Kanayama N., Mabbott N. A., Magari M., and Ohmori H. (2014) CSF-1 receptor-mediated differentiation of a new type of monocytic cell with B cell-stimulating activity: its selective dependence on IL-34. J. Leukoc. Biol. 95, 19–31 10.1189/jlb.0613311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nandi S., Cioce M., Yeung Y. G., Nieves E., Tesfa L., Lin H., Hsu A. W., Halenbeck R., Cheng H. Y., Gokhan S., Mehler M. F., and Stanley E. R. (2013) Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J. Biol. Chem. 288, 21972–21986 10.1074/jbc.M112.442731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Segaliny A. I., Brion R., Mortier E., Maillasson M., Cherel M., Jacques Y., Le Goff B., and Heymann D. (2015) Syndecan-1 regulates the biological activities of interleukin-34. Biochim. Biophys. Acta 1853, 1010–1021 10.1016/j.bbamcr.2015.01.023 [DOI] [PubMed] [Google Scholar]
- 29. Zhang M., Tang H., Guo Z., An H., Zhu X., Song W., Guo J., Huang X., Chen T., Wang J., and Cao X. (2004) Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat. Immunol. 5, 1124–1133 10.1038/ni1130 [DOI] [PubMed] [Google Scholar]
- 30. Skerra A., and Schmidt T. G. (2000) Use of the Strep-Tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol. 326, 271–304 10.1016/S0076-6879(00)26060-6 [DOI] [PubMed] [Google Scholar]
- 31. Kaufman R. J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233 10.1101/gad.13.10.1211 [DOI] [PubMed] [Google Scholar]
- 32. Reddy R. K., Mao C., Baumeister P., Austin R. C., Kaufman R. J., and Lee A. S. (2003) Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors. Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 278, 20915–20924 10.1074/jbc.M212328200 [DOI] [PubMed] [Google Scholar]
- 33. Sun F. C., Wei S., Li C. W., Chang Y. S., Chao C. C., and Lai Y. K. (2006) Localization of GRP78 to mitochondria under the unfolded protein response. Biochem. J. 396, 31–39 10.1042/BJ20051916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ni M., Zhou H., Wey S., Baumeister P., and Lee A. S. (2009) Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One 4, e6868 10.1371/journal.pone.0006868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arap M. A., Lahdenranta J., Mintz P. J., Hajitou A., Sarkis Á. S., Arap W., and Pasqualini R. (2004) Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 6, 275–284 10.1016/j.ccr.2004.08.018 [DOI] [PubMed] [Google Scholar]
- 36. Miao Y. R., Eckhardt B. L., Cao Y., Pasqualini R., Argani P., Arap W., Ramsay R. G., and Anderson R. L. (2013) Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin. Cancer Res. 19, 2107–2116 10.1158/1078-0432.CCR-12-2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez-Gronow M., Selim M. A., Papalas J., and Pizzo S. V. (2009) GRP78: a multifunctional receptor on the cell surface. Antioxid. Redox. Signal. 11, 2299–2306 10.1089/ars.2009.2568 [DOI] [PubMed] [Google Scholar]
- 38. Oida T., and Weiner H. L. (2010) Overexpression of TGF-β1 gene induces cell surface localized glucose-regulated protein 78-associated latency-associated peptide/TGF-β. J. Immunol. 185, 3529–3535 10.4049/jimmunol.0904121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miharada K., Karlsson G., Rehn M., Rörby E., Siva K., Cammenga J., and Karlsson S. (2011) Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell 9, 330–344 10.1016/j.stem.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y., Liu R., Ni M., Gill P., and Lee A. S. (2010) Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 285, 15065–15075 10.1074/jbc.M109.087445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai Y. L., Ha D. P., Zhao H., Carlos A. J., Wei S., Pun T. K., Wu K., Zandi E., Kelly K., and Lee A. S. (2018) Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc. Natl. Acad. Sci. U.S.A. 115, E4245–E4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Misra U. K., Gonzalez-Gronow M., Gawdi G., Wang F., and Pizzo S. V. (2004) A novel receptor function for the heat shock protein Grp78: silencing of Grp78 gene expression attenuates α2M*-induced signalling. Cell Signal. 16, 929–938 10.1016/j.cellsig.2004.01.003 [DOI] [PubMed] [Google Scholar]
- 43. Misra U. K., Deedwania R., and Pizzo S. V. (2006) Activation and cross-talk between Akt, NF-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J. Biol. Chem. 281, 13694–13707 10.1074/jbc.M511694200 [DOI] [PubMed] [Google Scholar]
- 44. Misra U. K., Deedwania R., and Pizzo S. V. (2005) Binding of activated α2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 280, 26278–26286 10.1074/jbc.M414467200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shani G., Fischer W. H., Justice N. J., Kelber J. A., Vale W., and Gray P. C. (2008) GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor β signaling and enhance cell growth. Mol. Cell Biol. 28, 666–677 10.1128/MCB.01716-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Triantafilou K., Fradelizi D., Wilson K., and Triantafilou M. (2002) GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J. Virol. 76, 633–643 10.1128/JVI.76.2.633-643.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nain M., Mukherjee S., Karmakar S. P., Paton A. W., Paton J. C., Abdin M. Z., Basu A., Kalia M., and Vrati S. (2017) GRP78 is an important host factor for Japanese encephalitis virus entry and replication in mammalian cells. J. Virol. 91, e02274–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mackay F., and Browning J. L. (1998) Turning off follicular dendritic cells. Nature 395, 26–27 10.1038/25630 [DOI] [PubMed] [Google Scholar]
- 49. Pasparakis M., Alexopoulou L., Episkopou V., and Kollias G. (1996) Immune and inflammatory responses in TNFα-deficient mice: a critical requirement for TNFα in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184, 1397–1411 10.1084/jem.184.4.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Endres R., Alimzhanov M. B., Plitz T., Fütterer A., Kosco-Vilbois M. H., Nedospasov S. A., Rajewsky K., and Pfeffer K. (1999) Mature follicular dendritic cell networks depend on expression of lymphotoxin β receptor by radioresistant stromal cells and of lymphotoxin β and tumor necrosis factor by B cells. J. Exp. Med. 189, 159–168 10.1084/jem.189.1.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fütterer A., Mink K., Luz A., Kosco-Vilbois M. H., and Pfeffer K. (1998) The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 9, 59–70 10.1016/S1074-7613(00)80588-9 [DOI] [PubMed] [Google Scholar]
- 52. Hwang S. J., Choi B., Kang S. S., Chang J. H., Kim Y. G., Chung Y. H., Sohn D. H., So M. W., Lee C. K., Robinson W. H., and Chang E. J. (2012) Interleukin-34 produced by human fibroblast-like synovial cells in rheumatoid arthritis supports osteoclastogenesis. Arthritis Res. Ther. 14, R14 10.1186/ar3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou R. P., Wu X. S., Xie Y. Y., Dai B. B., Hu W., Ge J. F., and Chen F. H. (2016) Functions of interleukin-34 and its emerging association with rheumatoid arthritis. Immunology 149, 362–373 10.1111/imm.12660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang H., Cao J., Lai X., and Wong C. K. (2016) Serum interleukin-34 levels are elevated in patients with systemic lupus erythematosus. Molecules 22, 35 10.3390/molecules22010035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciccia F., Alessandro R., Rodolico V., Guggino G., Raimondo S., Guarnotta C., Giardina A., Sireci G., Campisi G., De Leo G., and Triolo G. (2013) IL-34 is overexpressed in the inflamed salivary glands of patients with Sjögren's syndrome and is associated with the local expansion of pro-inflammatory CD14brightCD16+ monocytes. Rheumatology 52, 1009–1017 10.1093/rheumatology/kes435 [DOI] [PubMed] [Google Scholar]
- 56. Nakoinz I., Lee M. T., Weaver J. F., and Ralph P. (1990) Differentiation of the IL-3-dependent NFS-60 cell line and adaption to growth in macrophage colony-stimulating factor. J. Immunol. 145, 860–864 [PubMed] [Google Scholar]
- 57. Chihara T., Hashimoto M., Osman A., Hiyoshi-Yoshidomi Y., Suzu I., Chutiwitoonchai N., Hiyoshi M., Okada S., and Suzu S. (2012) HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J. Immunol. 188, 3620–3627 10.4049/jimmunol.1101593 [DOI] [PubMed] [Google Scholar]
- 58. Arakawa H., Lodygin D., and Buerstedde J. M. (2001) Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 1, 7 10.1186/1472-6750-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hatano N., and Hamada T. (2008) Proteome analysis of pitcher fluid of the carnivorous plant Nepenthes alata. J. Proteome Res. 7, 809–816 10.1021/pr700566d [DOI] [PubMed] [Google Scholar]
- 60. Tokumitsu H., Hatano N., Fujimoto T., Yurimoto S., and Kobayashi R. (2011) Generation of autonomous activity of Ca2+/calmodulin-dependent protein kinase kinase β by autophosphorylation. Biochemistry 50, 8193–8201 10.1021/bi201005g [DOI] [PubMed] [Google Scholar]
- 61. Tokumitsu H., Iwabu M., Ishikawa Y., and Kobayashi R. (2001) Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry 40, 13925–13932 10.1021/bi010863k [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.