

Abstract
Retinal degeneration 3 (RD3) protein promotes accumulation of retinal membrane guanylyl cyclase (RetGC) in the photoreceptor outer segment and suppresses RetGC activation by guanylyl cyclase–activating proteins (GCAPs). Mutations truncating RD3 cause severe congenital blindness by preventing the inhibitory binding of RD3 to the cyclase. The high propensity of RD3 to aggregate in solution has prevented structural analysis. Here, we produced a highly soluble variant of human RD3 (residues 18–160) that is monomeric and can still bind and negatively regulate RetGC. The NMR solution structure of RD3 revealed an elongated backbone structure (70 Å long and 30 Å wide) consisting of a four-helix bundle with a long unstructured loop between helices 1 and 2. The structure reveals that RD3 residues previously implicated in the RetGC binding map to a localized and contiguous area on the structure, involving a loop between helices 2 and 3 and adjacent parts of helices 3 and 4. The NMR structure of RD3 was validated by mutagenesis. Introducing Trp85 or Phe29 to replace Cys or Leu, respectively, disrupts packing in the hydrophobic core and lowers RD3's apparent affinity for RetGC1. Introducing a positive charge at the interface (Glu32 to Lys) also lowered the affinity. Conversely, introducing Val in place of Cys93 stabilized the hydrophobic core and increased the RD3 affinity for the cyclase. The NMR structure of RD3 presented here provides a structural basis for elucidating RD3–RetGC interactions relevant for normal vision or blindness.
Keywords: guanylate cyclase (guanylyl cyclase), nuclear magnetic resonance (NMR), retinal degeneration, phototransduction, photoreceptor
Introduction
Phototransduction in vertebrate rods and cones occurs as a result of cGMP hydrolysis by light-activated phosphodiesterase PDE6, thus causing closure of cGMP gated channels in outer segments and hyperpolarization of plasma membrane (1–3). Recovery from excitation requires rapid resynthesis of cGMP by membrane guanylyl cyclase (RetGC),4 which occurs after illumination and causes a sharp decline in free Ca2+ concentrations in the outer segment (3, 4). There are two homologous isozymes of the cyclase: RetGC1 (coded in different species by GUCY2D or Gucy2e genes) and RetGC2 (GUCY2F) (5–7). Two types of regulatory proteins control RetGC activity: guanylyl cyclase–activating proteins (GCAPs), which are calcium sensors mediating the negative calcium feedback on RetGC (4, 8), and a non–calcium-binding protein, retinal degeneration 3 (RD3) (9). RD3 was first discovered as a 23-kDa protein whose deficiency caused severe photoreceptor degeneration and blindness in human patients of recessive Leber's congenital amaurosis (LCA) and in rd3 mouse strain (10). Lack of RD3 strongly reduces RetGC content in photoreceptors, likely because of the RD3 involvement in a process of RetGC delivery to the outer segment (11, 12). RD3 also acts as a potent negative regulator of RetGC activity that binds to the cyclase with high affinity and blocks its activation by GCAPs (13, 14). The ability of RD3 to bind and suppress RetGC is likely required for preventing aberrant activation of the cyclase by GCAPs and is critical for the survival of photoreceptors (14). The RetGC binding domain in RD3 was previously hypothesized to occupy the central region of the RD3 primary structure (14); however, the three-dimensional structure of RD3 is currently unknown and could not be reliably predicted because of a lack of sequence homology with other known protein structures. In this study, we report the NMR solution structure of a human RD3 and functionally validate the RD3 structure by site-directed mutagenesis.
Results
A soluble and functional RD3 mutant
A major challenge with the study of RD3 has to do with its low abundance in photoreceptors (11), making it unfeasible to purify native RD3 from retinal tissue in quantities sufficient for structural studies. Another obstacle is the high propensity of recombinant functional RD3 to aggregate and precipitate when expressed in bacterial or eukaryotic cells (13–15). To overcome this obstacle, we designed a variant of RD3, in which the nonessential residues for RetGC regulation (14) were deleted, leaving residues 18–160 that encompass the RetGC-binding interface. Three basic residues in RD3 (Lys154, Arg156, and Arg158) were replaced with Glu, and Arg68 and Ser160 were replaced with Asp, to lower the unusually high pI of RD3. These modifications prevented aggregation of the variant RD3 (named RD3d), which remained soluble at concentrations exceeding 300 μm and remained monomeric at such high concentrations (Fig. 1, B and C). The apparent SEC elution time indicated that RD3d is either a dimer or possibly a monomer, having a nonspherical shape with a gyration radius larger than a spherical globular protein of the same mass. To distinguish between these two possibilities, multiangle light scattering (SEC-MALS) analysis (16, 17) was performed, which can measure molar mass independent of shape. The SEC-MALS analysis determined a molar mass of 17 kDa for RD3d (Fig. 1C). Thus, RD3d is a monomer in solution, and the anomalous diffusion measured by SEC is caused by the nonspherical shape of the protein (16).
Figure 1.

A, primary structure of the hRD3. The residues that were deleted from the soluble RD3d form are shown in gray; residues 18–160 are highlighted in bold; the asterisks mark residues replaced by negatively charged side chains; α1–α4 cylinders mark α-helical structures; and straight lines mark the unstructured regions, either predicted (gray) or found in the NMR structure (black). Residues in the regions previously mapped as parts of the cyclase-binding interface in RD3 (14) are highlighted in red. B, size-exclusion chromatography of the purified RD3d suggests dimerization or nonspherical shape of the protein. RD3 purified from E. coli as described under “Experimental procedures” was chromatographed on a Superdex 200 HR column. The vertical arrows mark the peak elution volumes for molecular mass standards (Bio-Rad chromatography standards: thyroglobulin, IgG, ovalbumin, myoglobin, and vitamin B12). Note that the 18-kDa RD3d elutes in a volume corresponding to a 32-kDa globular protein. Inset, 15% SDS-PAGE of the purified RD3d, Coomassie Blue R-250 stain. C, SEC-MALS analysis of RD3. The molar mass of RD3 in solution (circles) was calculated from a Zimm plot analysis of the observed light scattering intensity using a refractive index increment, dn/dc = 0.185 liter g−1 (16, 50). The protein concentration was 200 μm. D, CD spectra of RD3d (red trace) and full-length WT RD3 (black trace). Molar ellipticity is plotted along the y axis in units of mdeg cm2 dmol−1. A quantitative spectral analysis indicates that RD3d secondary structure is comprised of 65% α-helix and 35% random coil compared with 55% α-helix and 45% random coil for WT RD3. The larger percentage of random coil for WT RD3 is consistent with the truncated regions (residues 1–17 and 161–195) adopting an unstructured random coil.
RD3d and full-length RD3 have a similar fold
Far-UV CD experiments demonstrated that RD3d is properly folded, and the RD3d CD spectrum resembles that of the full-length WT RD3 (Fig. 1D). The CD spectra of RD3d (Fig. 1D, red trace) and WT RD3 (Fig. 1D, black trace) in both cases revealed a protein secondary structure comprised of predominantly α-helix and zero β-sheet, suggesting that RD3d and WT RD3 both have similar overall secondary structure.
RD3d shows the ability to bind and inhibit RetGC1 in vitro and in cyto
We verified the ability of RD3d to inhibit RetGC–GCAP complex activity using a standardized in vitro assay (14), in which recombinant RetGC1 expressed in HEK293 cells was reconstituted with recombinant myristoylated GCAP1 purified from Escherichia coli (Fig. 2A). RD3d retained the ability to completely suppress activation of the cyclase in vitro in a dose-dependent manner, albeit with higher than WT EC50 (36 ± 10 nm versus 3 ± 0.6 nm, n = 3, p = 0.005, Student's t test). However, we also found it important that RD3d, like WT RD3, still suppressed the cyclase activity within the submicromolar range, in stark contrast to a full-length RD3 mutant in which the sequence of the five central amino acid residues at positions 93–98, a part of the “hot spot” for the cyclase regulation, was scrambled (14) (EC50 2386 ± 690 nm, n = 3, p = 0.004).
Figure 2.

RD3d retains the ability to regulate RetGC1. A, the RD3d expressed in E. coli inhibits RetGC1–GCAP1 complex in submicromolar range in vitro. The activity of a recombinant RetGC1 reconstituted with 1.5 μm purified GCAP1 was assayed at different concentrations of WT RD3 (●), RD3d (○), and full-size RD3 with the scrambled residues 93–98 (14) (■) and normalized per maximal activity of the cyclase assayed in the absence of RD3; data (mean ± S.D., n = 3) are fitted using a Synergy Kaleidagraph software assuming a sigmoidal function, A% = 100/(1 + ([RD3]/EC50)−h), where h is a Hill coefficient. For WT, RD3d, and RD393–98, the EC50 values were 3, 36, and 2386 nm, and the Hill coefficients were 0.78 ± 0.09, 0.71 ± 0.07, and 0.69 ± 0.2, respectively. B and C, RD3d-GFP co-localiziation with mOrange RetGC1 in HEK293 cells. B, confocal images of RD3d-GFP (top) and WT RD3-GFP (middle) co-expressed with mOrange RetGC1 in HEK293 cells, and fluorescence distribution profiles for both tags (bottom) across the cells marked with asterisks. Note that both RD3 and RetGC1 in each case co-localize predominantly in the endoplasmic reticulum (ER) membranes and are void from the nucleus (marked n in B and C). C, neither RD3d nor WT RD3 bind W708R RetGC1 mutant. Confocal images (top) of WT RD3-GFP (left) and RD3d-GFP (right) co-expressed with W708R mOrange RetGC1 and the respective fluorescence profiles (bottom). Note that both forms of RD3-GFP are uniformly distributed throughout the cytoplasm and the nucleus of the cells and does not co-localize with the mutant cyclase.
We further tested whether RD3d could form a complex with RetGC1 in living cells (Fig. 2, B and C). The RetGC loses its ability to maintain active complex with regulatory proteins when extracted from the membrane (18). However, its binding with GCAPs and RD3 can be directly observed in HEK293 cells when fluorescently tagged RetGC1 anchors these cytosolic proteins to the endoplasmic reticulum membranes (15, 19, 20) and prevents their diffusion over the cytoplasm and the nucleus (Fig. 2B), a characteristic “tennis racquet” pattern (15, 19, 20). RD3d-GFP, in a manner similar to that of the WT RD3 (Fig. 2B), co-localized with WT mOrange RetGC1 but did not bind the R708W RetGC1, a mutation linked to congenital blindness that inactivates the cyclase (20). The respective Pearson's correlation coefficients (PCC) for the two fluorescent tags distribution over the entire cell was 0.83 ± 0.07 (mean ± S.D.), 24 cells, and 0.069 ± 0.11, 18 cells (p < 0.0001, unpaired t test assuming unequal variance); note that PCC ≤ 0.5 indicates the lack of co-localization, whereas PCC = 1.0 is the theoretical limit value for complete co-localization (21).
To summarize, despite the lower affinity for the cyclase, the essential regulatory properties of RD3—to bind and inhibit RetGC at submicromolar range—were preserved in RD3d. Therefore, we reasoned that the main RD3 biological function was retained in the RD3d structure.
NMR structure of RD3d
NMR spectral assignments for RD3d were reported previously (Biological Magnetic Resonance Data Bank accession number 27305) (22). These previous assignments were used in the current study to obtain NMR-derived structural restraints from NOESY and residual dipolar coupling (RDC) data. NMR structures were then calculated on the basis of distance restraints derived from analysis of NOESY (23) and long-range orientational restraints derived from RDC data (24) as described under “Experimental procedures.” The final NMR-derived structures of RD3d were resolved for 141 amino acids, starting at Arg19 and ending at Asp160. The last 20 residues at the C terminus (Arg140–Asp160) are unstructured and dynamically disordered. The 10 lowest-energy NMR structures are overlaid in Fig. 3A, and the structural statistics are summarized in Table 1. The overall precision of the NMR ensemble is expressed by a root-mean-square deviation (RMSD) of 0.55 Å calculated from the coordinates of the main-chain atoms. The quality of the NMR structures were assessed using PROCHECK-NMR (25), which shows that 96.8% of the residues occur in the allowed or favorable regions from the Ramachandran plot. RD3d forms an elongated overall structure (70 Å long by 30 Å wide) with a four-helix bundle (helix α1, Pro21–Val51; helix α2, Pro75–Lys87; helix α3, Pro90–Gln107; and helix α4, Val111–Thr139) shown in Fig. 3A. Helices α1 and α2 are connected by a long unstructured loop (residues Arg52–Ser74). Helices α1 and α4 are each quite long (27 Å) and interact with one another in an anti-parallel fashion that gives rise to a very long end-to-end distance in the structure. The elongated shape of the RD3d NMR structure can explain the unusually large radius of gyration observed for RD3 by SEC (Fig. 1C).
Figure 3.
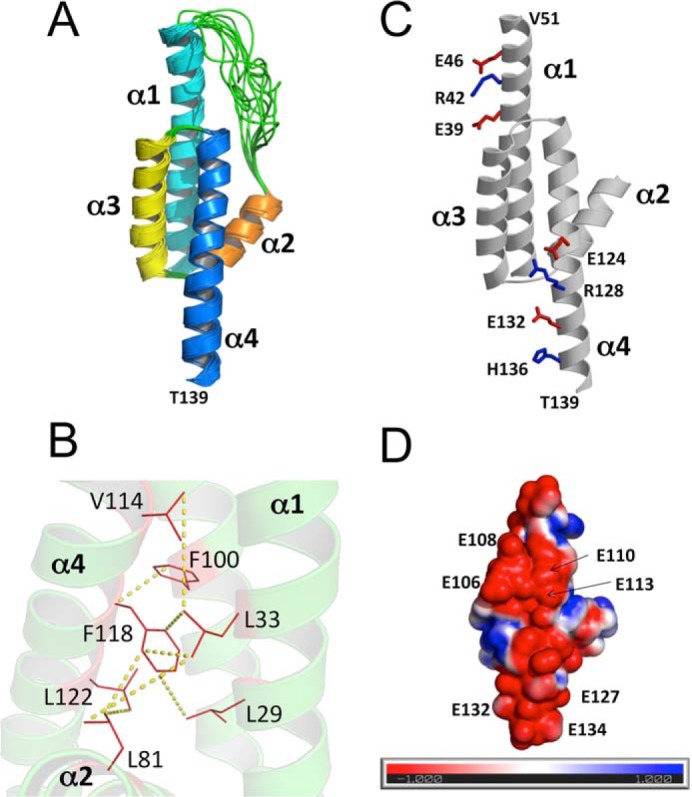
NMR-derived structures of RD3d. A, ensemble of 10 lowest energy NMR structures of RD3d (Protein Data Bank code 6DRF). Main-chain structures are depicted by a ribbon diagram. Structural statistics are given in Table 1. B, energy-minimized average structure of RD3d, showing the side-chain atoms of residues in the hydrophobic core. Yellow dashed lines show representative long-range NOE distances measured between residues in the hydrophobic core. C, RD3d salt-bridge interactions in helices α1 and α4 rigidify the elongated RD3 structure. Positively and negatively charged side-chain atoms are depicted by sticks and are colored blue and red, respectively. D, surface representation of RD3d showing the electrostatic potential of solvent-accessible surface residues with nearly the same view as in C. Negatively charged surface is highlighted red, and positively charged surface is blue. Exposed glutamate residues are indicated that form an extended negatively charged patch.
Table 1.
NMR structural statistics for RD3
| NMR restraints | Value (restraint violation) |
|---|---|
| Short-range NOEs | 727 (0.0 ± 0.0) |
| Long-range NOEs | 216 (0.0 ± 0.0) |
| Hydrogen bonds | 144 (not used in water refinement) |
| Dihedral angles | 172 (0.1 ± 0.3) |
| 1DHN RDC | 72 (0.0 ± 0.0) |
| RDC Q-factor | 0.289 |
| Coordinate precision (Å)a | |
| RMSD backbone atoms | 0.548 |
| RMSD all heavy atoms | 1.177 |
| Deviation from idealized geometry | |
| Bonds (Å) | 0.006 ± 0.000 |
| Angles (°) | 0.633 ± 0.012 |
| Impropers (°) | 0.827 ± 0.029 |
| Ramachandran plot (%) | |
| Favored region | 85.1 |
| Allowed region | 11.7 |
| Outlier region | 3.2 |
| Structure qualityb | |
| Clash score | 6 |
| Ramachandran outliers | 0.5% |
| Side-chain outliers | 4.8% |
a Coordinate precision was calculated for residues 4–34 and 58–122.
b Structure quality metrics assessed by MolProbity (49).
A four-helix bundle in RD3d is stabilized by a core of hydrophobic residues (Fig. 3B). Residues on the inner surface of helices α1 (residues Leu29 and Leu33), α3 (Phe100), and α4 (Val114, Phe118, and Leu122) each point inward toward the hydrophobic core (see yellow dotted lines in Fig. 2B). The Leu81 side chain from helix α2 also makes contacts in the hydrophobic core (Fig. 2B). The Phe118 side chain is located in the center of the core and connects the four helices together. Multiple NOE contacts to the Phe118 side chain (yellow dotted lines) reveal hydrophobic contacts that may stabilize the four-helix bundle. The N-terminal end of helix α2 (residues Pro75–Gln80) projects away from the hydrophobic core and is mostly solvent-exposed.
The solvent-exposed ends of helices α1 (residues Glu39–Val51) and α4 (residues Glu124–Thr139) are stabilized by a series of salt-bridge interactions (Fig. 3C). These salt bridges in α1 (see residues Glu39, Lys42, and Glu46 in Fig. 3C) and in α4 (Glu124, Arg128, Glu132, and His136 in Fig. 3C) help to rigidify the solvent-exposed ends of these helices, which creates the long end-to-end distance in the elongated structure. A space-filling representation of the RD3 structure reveals a highly negatively charged surface on one side (Fig. 3D), where a number of negatively charged glutamate residues (Glu106, Glu108, Glu110, Glu113, Glu127, Glu132, and Glu134) are all clustered in an extended localized patch on the protein surface. Whether or not any of them serve to make extensive electrostatic contacts with RetGC remains to be determined in future studies.
Hot-spot residues on the surface of RD3d
Previous mutational analysis on RD3 has identified several mutations that weaken RD3 binding to RetGC (14). These RetGC-sensitive residues in RD3 are located in discontinuous stretches throughout the amino acid sequence (red letters in Fig. 1A) but are localized spatially in close proximity to one another near the center of the NMR structure (highlighted red in Fig. 4). RD3 residues at each end of the elongated NMR structure (highlighted blue in Fig. 4) are not essential for the RetGC inhibitory binding (14). Many of the residues affecting RetGC regulation are solvent-exposed (see His89, Cys93, Pro95, Ile97, Arg99, Arg101, Gln102, and Ser120 in Fig. 4), consistent with these residues being able to make direct contact with RetGC. By contrast, a few of the RetGC-sensitive residues are buried in the hydrophobic core (Ile88, Ala96, Phe100,and Leu122), suggesting that these residues are inaccessible and must not make any regulatory contact with RetGC. Future studies are needed to pinpoint and map all the particular residues on the surface of RD3 that are clustered to form a binding site for RetGC.
Figure 4.
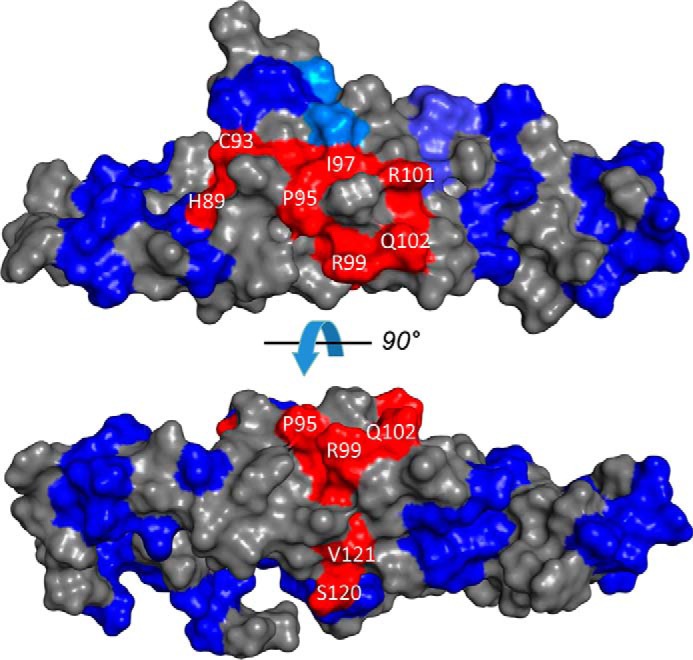
The RetGC interface localizes to the central part of the RD3 surface with a similar view as in Fig. 3C rotated 90° counterclockwise. Fragments of RD3 primary structure implicated in the inhibitory binding of RD3 to RetGC1 (red) and those that sustain mutagenesis without loss of the inhibitory binding to the cyclase (blue) (14) are superimposed on the RD3 NMR model.
Validation of the RD3 NMR structure by site-directed mutagenesis
To further test in a functional assay the NMR structure derived from the RD3d, we introduced mutations in unmodified full-length RD3, aiming at altering the interactions within the α-helical bundle portion containing the interface for binding to RetGC. It has been previously shown that the sequences of a majority of short regions in RD3 primary structure (marked in blue in Fig. 4) could be scrambled without loss of RD3 ability to bind and inhibit RetGC1 (14). To affect the interactions between the parts of the α-helical bundle, rather than the surface-exposed side chains involved in interaction with RetGC, we selected two hydrophilic residues, Cys85 and Cys93, which according to the NMR structure are embedded in a hydrophobic core formed by interacting residues from helices α3 and α4 and only minimally exposed to solvent (Figs. 4 and 5A). We surmised that the replacement of Cys85 with Trp would make less favorable the formation of a tight structure involving helices α2 and α3 and the residues in the loop 2/3, thus affecting the interface with the cyclase. We reasoned that Trp85, although preserving the hydrophobic interactions in that location, would be nonetheless overly large to fit into the size of the pocket containing the surrounding residues and thus tend to push them further apart from each other, likely reducing the RD3 affinity for the target enzyme. Indeed, the dose dependence of the RetGC1–GCAP1 complex inhibition by the C85W RD3 (Fig. 5B) was right-shifted, with its EC50 value increased to 184 ± 21 nm (mean ± S.D.), drastically higher than the 3 ± 0.6 nm of the WT RD3 (n = 3, p = 0.005). Replacement of Leu29 with Phe was also predicted to affect the cyclase-binding interface, because the distance between the residues in that pocket were also unfavorable for accommodating the larger hydrophobic residue, albeit with a less drastic disturbance of the interactions between the interface-containing helices as in case of C85W substitution. We found that this substitution did reduce the EC50 of the cyclase inhibition by RD3, just less drastically than C85W (17 ± 3 nm, n = 3, p = 0.013).
Figure 5.
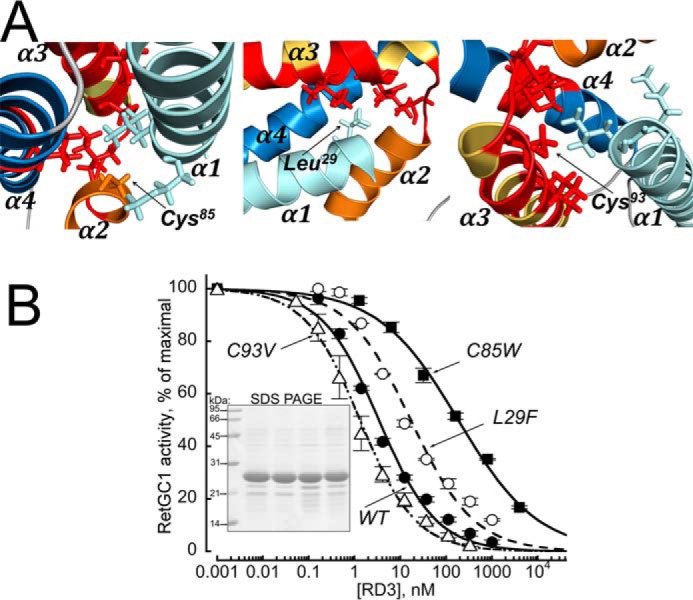
A, residues Cys85 (left), Leu29 (middle), and Cys93 (right) were replaced in a full-length RD3 with the respective Trp, Phe, or Val to modify the interactions between residues proximal to the RetGC-binding domain in the NMR structure of the RD3d (marked in red). The C85W and L29F, inserting overly large residues, are expected to be unfavorable for the packing of the cyclase-binding domain, whereas the C93V is expected to stabilize the interactions within the domain. B, the activity of a recombinant RetGC1 reconstituted with 1.5 μm purified GCAP1 was assayed at different concentrations of the WT (●), C85W (■), L29F (○), and C93V (▵) full-length RD3 and normalized per maximal activity of the cyclase assayed in the absence of RD3; data were normalized and fitted as described in Fig. 2A. The EC50 values for the WT, C85W, L29F, and C93V RD3 from the fit were 3.4, 184, 17.3, and 1.3 nm, with a negative (0.5 < h ≤ 0.7) cooperativity in all cases. Inset, 15% SDS-PAGE of the RD3 variants used in the experiment. Lanes left to right show molecular mass standards, WT RD3, C85W, C93V, and L29F; Coomassie Blue stain.
Conversely, we reasoned that replacement of a less hydrophobic Cys93 with a similarly sized but more hydrophobic residue, Val93, would stabilize the interactions in the RetGC binding interface portion of helices α3 and α4 and likely improve the affinity of RD3 for the cyclase. Fig. 5B shows that the dose dependence of the RetGC1–GCAP1 complex inhibition by the C93V RD3 became shifted toward lower concentrations (EC50 = 1.3 ± 0.1 nm, n = 3, p = 0.024), demonstrating the expected increase of the RD3 affinity for the cyclase.
We further surmised that substitution of Glu32 with Lys, which places a positive charge in close proximity to the positively charged Arg101 located in the hot-spot region near helix α3 (Fig. 6A), may repel the cyclase and weaken the RD3 affinity for RetGC1. In contrast, we did not anticipate a reduction of the RD3 affinity for the cyclase if such a positively charged residue replaced Glu108, which is located outside of the hot spot. As expected, these two substitutions (E32K and E108K) have quite different effects on RetGC1 binding (Fig. 6B): although the E108K RD3 retained high affinity for the cyclase (EC50 = 1.5 ± 0.3 nm, n = 3), the E32K mutant exhibited a more than 300-fold lower affinity compared with the WT (EC50 = 624 ± 216 nm, n = 3, p = 0.0075).
Figure 6.
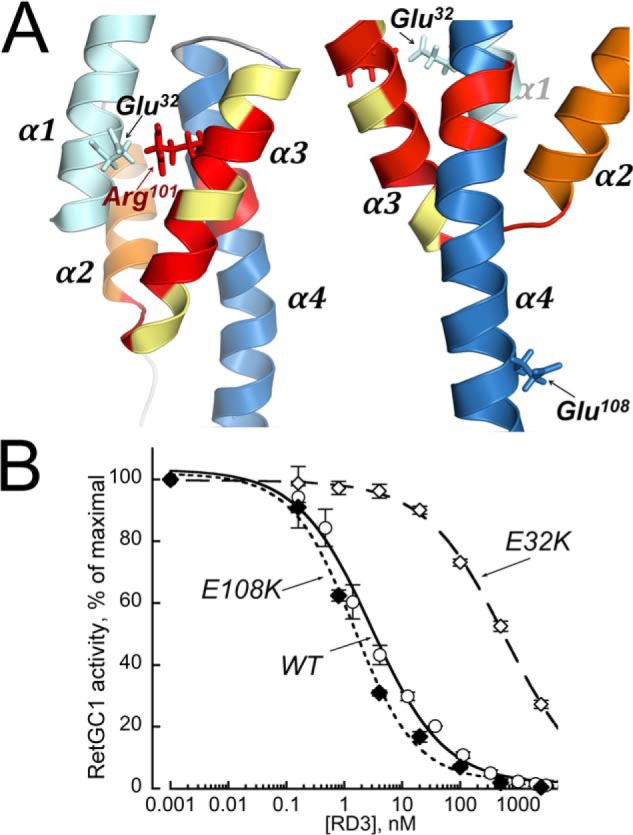
Differential effects of replacing charges in two surface exposed residues in a full-length RD3. A, Glu32 in helix α1 (left), proximal to Arg101 located in the putative RetGC-binding interface part (14) of α3, or Glu108 in helix α4, facing away from the putative cyclase-binding interface (right), was replaced by positively charged Lys. B, the activity of a recombinant RetGC1 reconstituted with 1.5 μm purified GCAP1 was assayed at different concentrations of the full-length WT (○), E108K (♦), or E32K (♢) RD3 as described in Fig. 2A. The respective EC50 values for the WT, E108K, and E32K were 2.8, 1.5, and 594 nm with a negative (0.6 < h < 0.8) cooperativity in all cases.
Discussion
RD3 is essential for vertebrate photoreceptor function and the survival of rods and cones in the retina. Premature truncation of RD3 that occurs in LCA12 patients and, in a similar manner, in rd3 mice (10) eliminates RD3 and reduces the content of RetGC in the outer segment (11, 12). The latter becomes restored upon transgenic expression of RD3 in homozygous rd3 mice (26). There are strong indications that RD3 is directly involved in facilitating the intracellular transport of RetGC from the inner to the outer segment (12). However, the lack of effective transport of RetGC to the outer segment in RD3-deficient photoreceptors cannot solely account for the severe and rapid death of the rd3 rods and cones, because these photoreceptors still possess low but detectable levels of RetGC activity and can produce a diminished yet clearly detectable photoresponse (10, 14). Despite the partial preservation of cyclase activity in homozygous rd3 mice, the RD3-deficient photoreceptors degenerate much faster than RetGC1/RetGC2 double-knockout mice, which completely lack RetGC activity and the ability to respond to light, altogether (13, 14, 27, 28). The degeneration pattern of rd3 retinas favors a hypothesis (13, 14) that RD3 is also required for silencing RetGC by blocking a premature GCAP-dependent activation in the inner segment prior to its reaching the outer segment. Although this hypothesis and the proposed RD3-related regulatory in vivo mechanisms (9) are yet to be delineated, the high-affinity binding of RD3 to RetGC (11, 13) must be an essential part of RD3-related mechanisms. The essential ability of RD3 to bind and inhibit RetGC at submicromolar concentrations remains preserved in the RD3d variant (Fig. 2). Deletion of the unstructured terminal regions of RD3 reduces the RD3 affinity for the cyclase 12-fold (EC50 36 nm versus 3 nm; Fig. 2A) but does not shift it outside the physiological range required for co-localization with the cyclase in vivo (Fig. 2B). Although the overall reduction of the affinity of RD3 for the cyclase indicates that the structure of RD3d has some limitations and that the truncated regions may contribute to the cyclase interaction, the four-helix bundle structure preserved in RD3d plays a fundamental role in recognition and inhibition of the target enzyme. Indeed, scrambling the amino acid sequence in the central part of RD3 drastically reduces the binding affinity more so than trimming the N- and C-terminal regions of the protein (Fig. 2) (14).
Based on a pilot mutational analysis (14), the RetGC binding interface in RD3 involves several short regions, Lys87–Pro90, Cys93–Ile97, Arg99–Gln102, and Arg119–Leu122. In the RD3 primary structure, these sites are spread over the central part of the molecule in a discontinuous fashion (red letters in Fig. 1A). The RD3 NMR structure, however, places all the implicated residues of the RetGC interface into close spatial proximity (highlighted red in Fig. 4). Because extraction of the cyclase from the membrane drastically disables its regulatory properties (18), we used a functional assay of the cyclase in HEK293 membranes (13, 14) rather than studying a detergent-solubilized form. All tested mutants were able to bind to the regulated form of the membrane-embedded cyclase but displayed different apparent affinities. Our mutational analysis of two cysteine residues (Cys85 and Cys93) elucidates why this region has such a sensitive effect on cyclase binding. The C85W RD3 mutant reduces the affinity of RD3/RetGC binding (Fig. 5B), because the bulky Trp substitution disrupts the hydrophobic core by prying apart helices α3 and α4 (Fig. 5A). Conversely, the C93V RD3 mutant increases the binding affinity to RetGC, because the bulky Val substitution at this position helps to fill a void in the interior of the four-helix bundle (Fig. 5A). In contrast to the Cys → Val substitution, various hydrophilic residues at position 93 drastically reduced RD3 affinity for the cyclase (14). The reduced affinity caused by the hydrophilic substitutions at residue 93 is not due to its contact with RetGC1 but rather due to its position between the four helices in the hydrophobic core. In essence, residue 93 serves as a linchpin inside the hydrophobic core that can sensitively tune the folding stability of RD3 and hence control its binding affinity for the cyclase.
One of the surprising observations from the NMR structure is a lack of a coiled-coil interaction involving helix α1, predicted by computational analyses of RD3 primary structure (9, 14). Such a coiled-coil interaction would be expected to connect two separate RD3 subunits and would predict a dimer structure. RD3d, which retains the intact helix α1 and an adjacent short N-terminal fragment, indeed exhibits a SEC elution time consistent with a dimer (Fig. 1C). However, a more rigorous measurement of molar mass by SEC-MALS unequivocally demonstrates that RD3d is a monomer (Fig. 1D). The anomalous diffusion of RD3 evidently results from its highly elongated shape (70 Å long and 30 Å wide) that increases the effective gyration radius of the molecule, mimicking that of a spherical dimer. The helix α1 does not contribute strongly to the RD3 docking with the cyclase (14), but we presently cannot exclude that it participates in a coiled-coil interaction with other cellular protein(s), such as those involved in a cellular transport, in enzymes other than RetGC (29, 30), or even in secondary interactions with the RetGC cyclase itself. Additional studies will be required to resolve this issue.
RD3 has little homology to other proteins, and our study presents the first three-dimensional structure for this new type of regulatory protein and its orthologs in other species. Although a four-helix bundle domain is frequently found in other proteins, the highly elongated shape of the four-helix bundle of RD3 has not been observed in any of the other known protein structures from the SCOP or DALI three-dimensional databases, suggesting that the elongated structure of RD3 may represent a new fold. The NMR structure also provides support for more detailed analysis of the cyclase-binding interface in RD3 and further studies of the RD3/RetGC1 complex structure. Knowledge of the RD3 three-dimensional structure will also guide future functional studies of the mechanisms through which RD3 is involved in cellular transport of RetGC to the outer segment and/or regulation of cyclase expression levels. Lastly, mutations in RD3 that lead to a rare and severe form of congenital blindness, LCA12, or other vision disorders began to emerge over the past decade (10, 31). Presently, the first RD3 mutations causing LCA12 have been identified as a truncation of RD3 (10, 31) upstream from or in the middle of the hot spot (14), which completely inactivates the ability of the deficient RD3 to bind the cyclase (13). However, we expect that missense point mutations will likely be found in the future as the number of genotyped cases of LCA12 increases with time, in a manner similar to the LCA1 CUCY2D blindness, where mutations were found first as truncations of RetGC1 but later led to dozens of missense point mutations linked to the disease (32, 33). We expect that the present study will provide a structural basis for delineating molecular mechanisms of blindness resulting from deficiency or abnormal function of RD3 in photoreceptors.
The structure of RetGC remains a major challenge for understanding its interaction with regulatory proteins (GCAPs and RD3), because only a part of its catalytic domain harboring the active site has been reliably modeled (34) and validated by biochemical analyses (35, 36). The structures of the remaining domains in RetGC (extracellular, kinase homology, and dimerization domains) are not known. We expect that the three-dimensional structure of RD3 may help establish the structure of the regulatory complex with RetGC. RD3 and GCAPs competitively bind to RetGC (13, 14). Hence, the RetGC-binding sites for RD3 and GCAPs could be at least partially overlapped. RetGC1 binding to both GCAP1 and GCAP2 involves the kinase homology and dimerization domains (15, 20). Consistent with a partial overlapping of the GCAP1- and RD3-binding sites, RetGC binding to RD3 (and the GCAPs (20)) are both disabled by W708R mutation in the kinase homology domain of RetGC1 (Fig. 2) (20). However, RD3 binding to RetGC1 is disrupted by removal of a C-terminal fragment of the cyclase (11), which is not essential for binding of GCAPs (20). Furthermore, some mutations in the RetGC1 dimerization domain that completely block its binding with GCAPs have little effect on RD3 binding (15). Therefore, the mutually exclusive regulation of the cyclase by GCAPs and RD3 is likely to occur via different allosteric mechanisms rather than by a direct competition for the same binding site.
Future structural studies on the RetGC–RD3 complex will first need to determine the stoichiometry of RD3 binding. Contrary to the GCAPs, the RD3-dependent inhibition of RetGC appears to have negative cooperativity (Figs. 2A, 5B, and 6B) (13, 14). Although the apparent negative cooperativity could possibly be explained by RD3 aggregation at higher protein concentrations, the same cannot be said for RD3d, because it remains monomeric even at high protein concentrations. It is therefore possible that at least two RD3 molecules bind to the cyclase in a negatively cooperative manner.
Experimental procedures
GCAP1 expression and purification
Myristoylated bovine GCAP1 (D6S) was expressed from pET11d vector (Novagen/Calbiochem) in a BLR(DE3) E. coli strain (Novagen/Calbiochem) harboring a pBB131 plasmid coding for a yeast N-myristoyl transferase and purified using previously published procedure (37) modified as follows. Cells typically grown in 2.0 liters of a standard LB medium (Thermo Fisher Scientific) containing 50 μg/ml kanamycin and 100 μg/ml ampicillin to reach A600 0.6–0.7. Free myristic acid (Sigma–Aldrich) was added from a concentrated ethanol solution to the suspension of bacterial cells to a final concentration of 100 μg/ml, 30 min prior to the induction with 0.5 mm isopropyl-β-d-thiogalactopyranoside (Research Products International). Three hours after the induction, the bacterial pellet was harvested by centrifugation at 8,000 × g for 20 min at 4 °C and frozen in −70 °C. The thawed pellet was resuspended in 100 ml of 10 mm Tris-HCl (pH 7.5) containing 2 mm EGTA and 14 mm 2-mercaptoethanol, and the cells were disrupted by ultrasonication. The expressed GCAP1 in the insoluble fraction of the inclusion bodies was collected by centrifugation at 20,000 × g for 20 min at 4 °C; extracted from the pellet by homogenization in 30 mm Tris-HCl (pH 7.5) containing 2 mm EGTA, 14 mm 2-mercaptoethanol, 2 mm MgCl2, and 8 m Sigma Ultra urea for 30 min at 4 °C; first dialyzed at 4 °C for 3–4 h against 2.0 liters of 10 mm Tris-HCl buffer (pH 7.5) containing 0.5 mm EGTA, 2 mm MgCl2, and 14 mm 2-mercaptoethanol; and then dialyzed overnight against 2.0 liters of 10 mm Tris-HCl buffer (pH 7.5) containing 0.1 mm EGTA, 2 mm MgCl2, and 14 mm 2-mercaptoethanol. The insoluble material was removed by centrifugation at 20,000 × g for 20 min, 4 °C. The concentration of Tris-HCl buffer (pH 7.5) in the supernatant was adjusted to 50 mm, and CaCl2 was added to a final concentration of 10 mm and kept for 20 min at room temperature. The precipitate was removed by centrifugation at 20,000 × g for 20 min at 4 °C. Supernatant was collected and, after adding NaCl to 1 m and DTT to 5 mm, applied on a 1.6 × 5.0-cm butyl-Sepharose Fast Flow column (GE Healthcare) pre-equilibrated with 20 mm Tris-HCl (pH 7.5) containing 1.0 m NaCl. The column was washed with ∼10 volumes of the same buffer, and GCAP1 was eluted with 5 mm Tris-HCl (pH 7.5) and concentrated to 5 ml using Amicon Ultra-15 (10,000 MWCO) centrifugal filter (Thermo Fisher Scientific). Concentrated solution was centrifuged at 200,000 × g for 10 min at 4 °C in a Beckman Optima TLX centrifuge and chromatographed on a GE Healthcare Sephacryl S-100 column (2.6 × 60 cm) pre-equilibrated with 20 mm Tris-HCl (pH 7.5), 100 mm NaCl. The main peak containing GCAP1 was collected, and EDTA was added to 2 mm to remove Ca2+ bound to GCAP1. The excess of EDTA was then removed by four cycles of 20-fold concentration/dilution in 10 mm Tris-HCl (pH 7.5) containing 30 μm EDTA using Amicon Ultra-15 (10,000 MWCO) to 300–350 μm. Concentrated protein was frozen in small aliquots and stored at −70 °C. The purity of GCAP1 preparations estimated by SDS gel electrophoresis was ≥95%.
RetGC1 expression and activity assay
A human recombinant RetGC1 was expressed from a modified Invitrogen/Thermo Fisher pRCCMV vector in HEK293 cells transfected using calcium-phosphate precipitation method, and the membrane fraction containing the expressed cyclase was purified as previously described (38). The guanylyl cyclase activity was assayed as previously described in detail (14, 38). Briefly, the assay mixture (25 μl) containing HEK293 membranes, 1.5 μm GCAP1, 30 mm MOPS-KOH (pH 7.2), 60 mm KCl, 4 mm NaCl, 1 mm DTT, 2 mm Ca2+/Mg2+/EGTA buffers, 0.9 mm free Mg2+, 0.3 mm ATP, 4 mm cGMP, 1 mm GTP, and 1 μCi of [α-32P]GTP, 100 μm zaprinast and dipyridamole, 10 mm creatine phosphate, 0.5 unit of creatine phosphokinase (Sigma–Aldrich) was incubated at 30 °C for 30 min, and the reaction was stopped by heat inactivation at 95 °C for 2 min. The resultant [32P]cGMP product was separated by TLC using fluorescently backed polyethyleneimine cellulose plates (Merck) developed in 0.2 m LiCl and eluted with 2 m LiCl, and the radioactivity was counted using liquid scintillation. Ca2+/EGTA buffer (<10 nm free Ca2+ concentrations at 0.9 mm free Mg2+) was prepared using the Tsien and Pozzan method (39). The results were averaged in each case as means ± S.D. from the indicated number of independent measurements.
RetGC–RD3 co-expression and imaging
To test mOrange RetGC1 and RD3-GFP co-localization in vivo, the two proteins were co-expressed in HEK293 cells as previously described (20). In brief, ∼90% confluent HEK293 cell cultures in LabTeck 4-well cover glass chamber were transfected with a mixture of 1 μg of mOrange RetGC1 DNA/0.02 μg of RD3-GFP/well using 3 μl/μg DNA of a Promega FuGENE reagent. Confocal images of the cells were taken after 24 h of incubation at 37 °C in 5% CO2 utilizing an Olympus FV1000 Spectral instrument, at 543- and 488-nm excitation in sequential mode for the red and the green fluorochromes, respectively. No changes to the original images were made except for minor γ correction applied to whole image for clear presentation in print. Quantitative analysis was performed by Olympus FluoView FV10-ASW software using original images, without γ corrections. PCC values were calculated for whole-cell images of RD3-GFP and mOrange RetGC1 localization, and the statistically significant difference between the PCC values was tested using Student's t test in a Synergy Kaleidagraph 4.
RD3 mutagenesis, expression, and purification
The mutations were introduced into human RD3 cDNA by PCR following a conventional “splicing-by-overlap extension” procedure (40) utilizing a high-fidelity Thermo Scientific PhusionFlash polymerase, and the mutated cDNA was inserted into the NcoI/BamHI sites of a pET11d vector as described previously (13, 14) and verified by automated Sanger sequencing. The recombinant human RD3 was expressed from a pET11d vector in a BL21(DE3) Codon Plus E. coli strain (Stratagene), extracted from the inclusion bodies, and purified as previously described in detail (14). The soluble RD3d mutant was purified using the same method, except that NaCl precipitation step was omitted and replaced by size-exclusion FPLC chromatography on a GE Health Sciences Superdex 200 HR10 × 30 column in 20 mm Tris-HCl buffer containing 100 mm NaCl at 0.5 ml/min elution rate. The purified RD3d was concentrated using Amicon Ultra-15 (10,000 MWCO) centrifugal concentrators. For expression of RD3-GFP in HEK293 cells, the RD3 cDNA with the Kozak motif was inserted in the BamHI/EcoRI sites of a Clontech pQBIfN3 vector in-frame with eGFP cDNA and used for co-transfection with mOrange RetGC1 as described above.
NMR spectroscopy
All NMR measurements were performed at 298 K using a Bruker Avance III 600 MHz spectrometer equipped with a four-channel interface and triple-resonance cryoprobe (TCI). NMR sample preparation of RD3d was described previously (22). Two-dimensional NMR experiments (HSQC and HSQC-IPAP (inphase/antiphase)) were recorded on a sample of 15N-labeled RD3d (0.5 mm) dissolved in 20 mm 2-amino-2-hydroxymethyl-propane-1,3-diol-d11 (Tris-d11 at pH 7.5) with 5 mm DTT-d10 (DTT-d10) and 95% H2O, 5% D2O. Three-dimensional NMR experiments for assigning backbone and side-chain resonances were recorded on a double-labeled sample (15N,13C-labeled RD3d) as described previously (22). NMR data were processed using NMRPipe (41) and analyzed with SPARKY (T. D. Goddard and D. G. Kneller, University of California at San Francisco).
To measure RDCs (24) of RD3d, the filamentous bacteriophage Pf1 (Asla Biotech Ltd.) was used as an orienting medium. Pf1 (17 mg/ml) was added to 15N-labeled RD3d (0.5 mm) to produce weak alignment. 1H-15N residual dipolar coupling constants (DNH) were measured using a two-dimensional IPAP (inphase/antiphase) 1H-15N HSQC experiment as described by Ottiger et al. (42). Briefly, the backbone N-H RDCs were calculated by measuring the difference in 15N splitting for each amide resonance both in the presence and in the absence of the orienting medium. The RDC Q-factor and analysis of RDC data were calculated by PALES (43). The Q-factor is calculated as Q = RMS(Dmeas − Dcalc)/RMS(Dmeas), where Dmeas is the measured RDC, Dcalc is the calculated RDC, and RMS is the root mean square difference. A Q-factor of 30% corresponds to 2Å resolution.
NMR structure calculation
NMR-derived structures of RD3d were calculated using restrained molecular dynamics simulations within Xplor-NIH (44). Residual dipolar couplings, NOE distances, dihedral angles from TALOS+ (45), and backbone hydrogen bonds were used as structural restraints. NOEs were obtained from 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC (aliphatic) as described by Tanaka et al. (46) and were assigned automatically using PONDEROSA (47). Backbone dihedral angles were calculated by TALOS+ (45) using backbone chemical shifts (Hα, Cα, Cβ, CO, 15N, and HN) as input. Hydrogen-bond restraints in helices were verified by measuring amide hydrogen-deuterium exchange rates as described by Ames et al. (48). The Xplor-NIH structure calculation was performed in three stages: annealing, refinement, and water refinement. Annealing started from an extended random structure. A total of 200 structures were calculated, and the one with lowest energy was used as a starting structure during the refinement. From a total of 200 structures, the 20 lowest energy structures were refined in an explicit water environment and resulted in the final 10 structures, which were deposited to the Protein Data Bank (code 6DRF). Ramachandran plot was generated by PROCHECK-NMR (25) and structure quality was assessed by MolProbity (49).
Light-scattering experiments
SEC was performed using a Superdex 200 HR 10/30 column (GE Healthcare) at 4 °C equilibrated in buffer containing 20 mm Tris (pH 7.4). A 0.1-ml aliquot of protein (200 μm) was loaded onto the column and eluted at a flow rate of 0.5 ml/min. The molar mass of RD3 was determined by analytical SEC performed in-line with a MALS miniDawn instrument with a 690-nm laser (Wyatt Technologies, Inc.) coupled to refractive index instrument (Optilab Rex, Wyatt Technologies, Inc.). The molar mass of chromatographed protein was calculated from the observed light scattering intensity and differential refractive index (50) using ASTRA software (Wyatt Technologies, Inc.) based on a Zimm plot analysis using a refractive index increment, dn/dc = 0.185 liter g−1 (16, 50).
CD spectroscopy
CD spectra (190 nm to 290 nm) were acquired using a Chirascan circular dichroism spectrometer (Applied Photophysics Limited). All experiments were performed at 23 °C using a 1-cm pathlength quartz cuvette with a 0.5-nm bandpass and 1-s dwell time. Protein samples (full-length WT RD3 and RD3d) were dissolved in water (0.015 mg/ml protein concentration). Background signals caused by water absorption were subtracted to generate final spectra shown in Fig. 1D.
Author contributions
I. V. P., Q. Y., S. L., and D. C. data curation; I. V. P., Q. Y., S. L., D. C., and J. B. A. formal analysis; I. V. P. and Q. Y. validation; I. V. P., S. L., A. M. D., and J. B. A. investigation; I. V. P., Q. Y., S. L., D. C., and A. M. D. methodology; I. V. P., Q. Y., A. M. D., and J. B. A. writing-review and editing; Q. Y. software; A. M. D. and J. B. A. conceptualization; A. M. D. and J. B. A. supervision; A. M. D. and J. B. A. funding acquisition; A. M. D. and J. B. A. writing-original draft; A. M. D. and J. B. A. project administration.
This work was supported by National Institutes of Health Grants EY11522 (to A. M. D.) and EY012347 (to J. B. A.) and a Pennsylvania Department of Health CURE Formula grant (to A. M. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 6DRF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RetGC
- retinal membrane guanylyl cyclase
- GCAP
- guanylyl cyclase–activating protein
- LCA
- Leber's congenital amaurosis
- MALS
- multiangle light scattering
- PCC
- Pearson's correlation coefficient
- RD3
- retinal degeneration 3 protein
- SEC
- size-exclusion chromatography
- RMSD
- root-mean-square deviation
- MWCO
- molecular weight cut-off
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Arshavsky V. Y., and Burns M. E. (2014) Current understanding of signal amplification in phototransduction. Cell. Logist. 4, e29390 10.4161/cl.29390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arshavsky V. Y., Lamb T. D., and Pugh E. N. (2002) G proteins and phototransduction. Annu. Rev. Physiol. 64, 153–187 10.1146/annurev.physiol.64.082701.102229 [DOI] [PubMed] [Google Scholar]
- 3. Koch K. W., and Dell'Orco D. (2015) Protein and signaling networks in vertebrate photoreceptor cells. Front. Mol. Neurosci. 8, 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dizhoor A. M., Olshevskaya E. V., and Peshenko I. V. (2010) Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell Biochem. 334, 117–124 10.1007/s11010-009-0328-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dizhoor A. M., Lowe D. G., Olshevskaya E. V., Laura R. P., and Hurley J. B. (1994) The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352 10.1016/0896-6273(94)90449-9 [DOI] [PubMed] [Google Scholar]
- 6. Lowe D. G., Dizhoor A. M., Liu K., Gu Q., Spencer M., Laura R., Lu L., and Hurley J. B. (1995) Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539 10.1073/pnas.92.12.5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang R. B., Foster D. C., Garbers D. L., and Fülle H. J. (1995) Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606 10.1073/pnas.92.2.602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim S., Dizhoor A. M., and Ames J. B. (2014) Structural diversity of neuronal calcium sensor proteins and insights for activation of retinal guanylyl cyclase by GCAP1. Front. Mol. Neurosci. 7, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Molday L. L., Jefferies T., and Molday R. S. (2014) Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedman J. S., Chang B., Kannabiran C., Chakarova C., Singh H. P., Jalali S., Hawes N. L., Branham K., Othman M., Filippova E., Thompson D. A., Webster A. R., Andréasson S., Jacobson S. G., Bhattacharya S. S., et al. (2006) Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070 10.1086/510021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azadi S., Molday L. L., and Molday R. S. (2010) RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163 10.1073/pnas.1010460107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zulliger R., Naash M. I., Rajala R. V., Molday R. S., and Azadi S. (2015) Impaired association of retinal degeneration-3 with guanylate cyclase-1 and guanylate cyclase-activating protein-1 leads to leber congenital amaurosis-1. J. Biol. Chem. 290, 3488–3499 10.1074/jbc.M114.616656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peshenko I. V., Olshevskaya E. V., Azadi S., Molday L. L., Molday R. S., and Dizhoor A. M. (2011) Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519 10.1021/bi201342b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2016) Functional study and mapping sites for interaction with the target enzyme in retinal degeneration 3 (RD3) protein. J. Biol. Chem. 291, 19713–19723 10.1074/jbc.M116.742288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Dimerization domain of retinal membrane guanylyl cyclase 1 (RetGC1) is an essential part of guanylyl cyclase-activating protein (GCAP) binding interface. J. Biol. Chem. 290, 19584–19596 10.1074/jbc.M115.661371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer M., and Morgenstern B. (2003) Characterization of gelatine and acid soluble collagen by size exclusion chromatography coupled with multi angle light scattering (SEC-MALS). Biomacromolecules 4, 1727–1732 10.1021/bm0341531 [DOI] [PubMed] [Google Scholar]
- 17. Xu X., Xu W., Rayo J., Ishida Y., Leal W. S., and Ames J. B. (2010) NMR structure of navel orangeworm moth pheromone-binding protein (AtraPBP1): implications for pH-sensitive pheromone detection. Biochemistry 49, 1469–1476 10.1021/bi9020132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch K. W. (1991) Purification and identification of photoreceptor guanylate cyclase. J. Biol. Chem. 266, 8634–8637 [PubMed] [Google Scholar]
- 19. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2008) Binding of guanylyl cyclase activating protein 1 (GCAP1) to retinal guanylyl cyclase (RetGC1): The role of individual EF-hands. J. Biol. Chem. 283, 21747–21757 10.1074/jbc.M801899200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Evaluating the role of retinal guanylyl cyclase 1 (RetGC1) domains in binding guanylyl cyclase activating proteins (GCAP). J. Biol. Chem. 290, 6913–6924 10.1074/jbc.M114.629642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zinchuk V., and Zinchuck O. (2008) Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr. Prot. Cell Biol. 39, 4191–4195 10.1002/0471143030.cb0419s39 [DOI] [PubMed] [Google Scholar]
- 22. Lim S., Cudia D., Yu Q., Peshenko I., Dizhoor A. M., and Ames J. B. (2018) Chemical shift assignments of retinal degeneration 3 protein (RD3). Biomol. NMR Assign. 12, 167–170 10.1007/s12104-018-9802-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clore G. M., and Gronenborn A. M. (1998) Determining the structures of large proteins and protein complexes by NMR. Curr. Opin. Chem. Biol. 2, 564–570 10.1016/S1367-5931(98)80084-7 [DOI] [PubMed] [Google Scholar]
- 24. Tjandra N., and Bax A. (1997) Direct measurement of disances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278, 1111–1114 10.1126/science.278.5340.1111 [DOI] [PubMed] [Google Scholar]
- 25. Laskowski R. A., Rullmann J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 26. Molday L. L., Djajadi H., Yan P., Szczygiel L., Boye S. L., Chiodo V. A., Gregory-Evans K., Sarunic M. V., Hauswirth W. W., and Molday R. S. (2013) RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet. 22, 3894–3905 10.1093/hmg/ddt244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baehr W., Karan S., Maeda T., Luo D. G., Li S., Bronson J. D., Watt C. B., Yau K. W., Frederick J. M., and Palczewski K. (2007) The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 282, 8837–8847 10.1074/jbc.M610369200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baehr W., and Palczewski K. (2007) Guanylate cyclase-activating proteins and retina disease. Subcell. Biochem. 45, 71–91 10.1007/978-1-4020-6191-2_4 [DOI] [PubMed] [Google Scholar]
- 29. Wimberg H., Janssen-Bienhold U., and Koch K. W. (2018) Control of the nucleotide cycle in photoreceptor cell extracts by retinal degeneration protein 3. Front. Mol. Neurosci. 11, 52 10.3389/fnmol.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharon D., Wimberg H., Kinarty Y., and Koch K. W. (2018) Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 63, 69–91 10.1016/j.preteyeres.2017.10.003 [DOI] [PubMed] [Google Scholar]
- 31. Preising M. N., Hausotter-Will N., Solbach M. C., Friedburg C., Rüschendorf F., and Lorenz B. (2012) Mutations in RD3 are associated with an extremely rare and severe form of early onset retinal dystrophy. Invest. Ophthalmol. Vis. Sci. 53, 3463–3472 10.1167/iovs.12-9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jacobson S. G., Cideciyan A. V., Peshenko I. V., Sumaroka A., Olshevskaya E. V., Cao L., Schwartz S. B., Roman A. J., Olivares M. B., Sadigh S., Yau K. W., Heon E., Stone E. M., and Dizhoor A. M. (2013) Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum. Mol. Genet. 22, 168–183 10.1093/hmg/dds421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stone E. M. (2007) Leber congenital amaurosis: a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 10.1016/j.ajo.2007.08.022 [DOI] [PubMed] [Google Scholar]
- 34. Liu Y., Ruoho A. E., Rao V. D., and Hurley J. H. (1997) Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc. Natl. Acad. Sci. U.S.A. 94, 13414–13419 10.1073/pnas.94.25.13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramamurthy V., Tucker C., Wilkie S. E., Daggett V., Hunt D. M., and Hurley J. B. (2001) Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J. Biol. Chem. 276, 26218–26229 10.1074/jbc.M010495200 [DOI] [PubMed] [Google Scholar]
- 36. Tucker C., Hurley J. H., Miller T. R., and Hurley J. B. (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 95, 5993–5997 10.1073/pnas.95.11.5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peshenko I. V., and Dizhoor A. M. (2006) Ca2+ and Mg2+ binding properties of GCAP-1: evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841 10.1074/jbc.M600257200 [DOI] [PubMed] [Google Scholar]
- 38. Peshenko I. V., and Dizhoor A. M. (2004) Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906 10.1074/jbc.C400065200 [DOI] [PubMed] [Google Scholar]
- 39. Tsien R. Y. (1989) A new generation of Ca2+ indicators with greatly improved fluorescence properties. Methods Cell Biol. 30, 127–156 10.1016/S0091-679X(08)60978-4 [DOI] [PubMed] [Google Scholar]
- 40. Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., and Pease L. R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68 10.1016/0378-1119(89)90359-4 [DOI] [PubMed] [Google Scholar]
- 41. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 42. Ottiger M., Delaglio F., Marquardt J. L., Tjandra N., and Bax A. (1998) Measurement of dipolar couplings for methylene and methyl sites in weakly oriented macromolecules and their use in structure determination. J. Magn. Reson. 134, 365–369 10.1006/jmre.1998.1546 [DOI] [PubMed] [Google Scholar]
- 43. Zweckstetter M. (2008) NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 3, 679–690 10.1038/nprot.2008.36 [DOI] [PubMed] [Google Scholar]
- 44. Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 10.1016/S1090-7807(02)00014-9 [DOI] [PubMed] [Google Scholar]
- 45. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanaka T., Ames J. B., Kainosho M., Stryer L., and Ikura M. (1998) Differential isotype labeling strategy for determining the structure of myristoylated recoverin by NMR spectroscopy. J. Biomol. NMR 11, 135–152 10.1023/A:1008212316986 [DOI] [PubMed] [Google Scholar]
- 47. Lee W., Petit C. M., Cornilescu G., Stark J. L., and Markley J. L. (2016) The AUDANA algorithm for automated protein 3D structure determination from NMR NOE data. J. Biomol. NMR 65, 51–57 10.1007/s10858-016-0036-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ames J. B., Tanaka T., Stryer L., and Ikura M. (1994) Secondary structure of myristoylated recoverin determined by three-dimensional heteronuclear NMR: implications for the calcium-myristoyl switch. Biochemistry 33, 10743–10753 10.1021/bi00201a023 [DOI] [PubMed] [Google Scholar]
- 49. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wyatt P. J. (1991) Combined differential light scattering with various liquid chromatography separation techniques. Biochem. Soc. Trans. 19, 485 10.1042/bst0190485 [DOI] [PubMed] [Google Scholar]