

Abstract
Absolute quantification in targeted proteomics is challenging due to a variety of factors, including low specificity in complex backgrounds, limited analytical throughput, and wide dynamic range. To address these problems, we developed a hybrid offset-triggered multiplex absolute quantification (HOTMAQ) strategy that combines cost-effective mass difference and isobaric tags to enable simultaneous construction of an internal standard curve in the MS1 precursor scan, real-time identification of peptides at the MS2 level, and mass offset-triggered accurate quantification of target proteins in synchronous precursor selection (SPS)-MS3 spectra. This approach increases the analytical throughput of targeted quantitative proteomics by up to 12-fold. The HOTMAQ strategy was employed to verify candidate protein biomarkers in preclinical Alzheimer’s disease with high accuracy. The greatly enhanced throughput and quantitative performance, paired with sample flexibility, makes HOTMAQ broadly applicable to targeted peptidomics, proteomics, and phosphoproteomics.
Graphical Abstract:
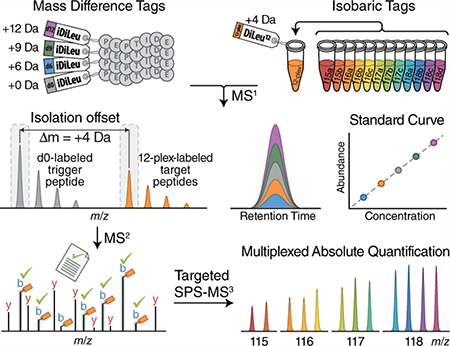
Targeted mass spectrometry (MS) is widely used to measure the absolute abundance of subset of proteins of interest with high sensitivity, reproducibility, and quantitative accuracy.1–5 Selected reaction monitoring (SRM) or parallel reaction monitoring (PRM) coupled with stable isotope-mass spectrometry, in which stable-isotope encoded peptide standards are spiked into samples in known amounts to determine absolute abundances of target peptides via signal intensity ratios (termed as AQUA), is a gold standard for absolute quantification in targeted proteomics.6–8 To improve the acquisition efficiency of SRM/PRM, internal standard triggered-parallel reaction monitoring (IS-PRM) has used spiked-in isotopic internal standards to prompt real-time measurement of analytes and on-the-fly adjustment of acquisition parameters.9 Similarly, a method termed TOMAHAQ (triggered by offset, multiplexed, accurate-mass, high-resolution, and absolute quantification) has utilized synthetic peptides to trigger quantification based on a known mass offset.10 This method has greatly increased analytical throughput of target proteomics by sample multiplexing. However, the dependence on single-point calibration for absolute quantification in AQUA and TOMAHAQ may provide inaccurate estimates when the amounts of target peptides span a wide dynamic range,10 particularly in preclinical and clinical biofluids.11 Furthermore, the high cost of heavy isotope-encoded peptide standards and isotope labels used in current targeted proteomics methods also limits their accessibility. It is therefore highly desirable to develop a method that is able to simultaneously address these remaining issues, including low specificity in complex sample backgrounds, limited analytical throughput, and limited dynamic range.
Stable isotope labels can be categorized into two types: (i) mass difference labels that introduce mass shifts of several daltons onto precursor ions, permitting their direct relative and absolute quantification in full MS (MS1) spectra, such as stable isotope labeling by amino acids in cell culture (SILAC) and mass differential tags for relative and absolute quantification (mTRAQ);12–16 and (ii) isobaric labels that impart a single nominal mass shift onto precursors in MS1 spectra but produce discrete reporter ions for relative quantification of peptides in tandem MS (MS/MS) spectra, such as iTRAQ and TMT.17–20 We have developed our own cost-effective amine-reactive N,N-dimethyl leucine (DiLeu) tags that offer the flexibility to employ either approach for multiplexed quantification of many samples in a single LC−MS/MS experiment (Figure S1). Isotopic DiLeu (iDiLeu) tags enable 5-plex mass difference quantification through the use of 3 Da mass differences between tags,21 while DiLeu isobaric tags enable up to 12-plex quantification via reporter ions using high-resolution MS/MS acquisition.22 Both variants share the same chemical structure, differing only in their composition and number of heavy stable isotopes (13C, 2H, 15N, and 18O). Here, we describe a novel hybrid offset-triggered multiplex absolute quantification (HOTMAQ) strategy to combine mass difference tags (iDiLeu) and isobaric tags (DiLeu) to enable accurate absolute quantification of targeted peptides across multiple complex samples. Numerous figures of merit, including limit of quantification, quantitative accuracy, and dynamic range are systematically assessed.
We further demonstrate the utility of the HOTMAQ approach by analyzing cerebrospinal fluids (CSF) collected from individuals in preclinical stage of Alzheimer’s disease (AD) and healthy controls to verify candidate protein biomarkers. As the most common form of dementia, Alzheimer’s disease has three main neuropathological features, namely, brain atrophy, neurofibrillary tangles composed of hyperphosphorylated tau protein, and amyloid plaques composed of aggregated amyloid β (Aβ).23 The preclinical phase of AD is characterized by the presence of neuropathological abnormalities but without presentation of cognitive impairment.24–26 CSF is in direct contact with the extracellular space of the brain, making CSF an optimal source of biomarkers for AD, of which pathology is restricted to the brain.27 Three CSF biomarkers, Aβ1–42, total tau, and phosphorylated tau, have been well-established to reflect molecular pathologies of the hallmarks of Alzheimer’s disease.28,29 Even though these biomarkers have been found to aid in AD diagnosis, it is still beneficial to discover additional biomarkers for diagnosis of AD at an earlier asymptomatic stage, which begins many years before clinical dementia. To serve as diagnostic tools in clinical practice or monitoring therapeutic intervention, biomarkers must be measured by reliable and validated methods with high accuracy.30,31
EXPERIMENTAL SECTION
Participants.
Twenty-two enrollees (11 individuals in preclinical AD stage and equal number of healthy controls) from Wisconsin Alzheimer’s Disease Research Center (ADRC) participated in this study. Classification of participants as cognitively normal was based on a comprehensive neuropsychological test battery.32 Global cognitive function was evaluated by Mini-Mental State Examination (MMSE), on which a score of ≤24 is an indication for cognition impairment.33 All participants had MMSE scores of ≥27, indicating cognition normal. Classification of preclinical AD was based on evidence of amyloid beta accumulation on 11C Pittsburgh Compound B positron emission tomography (PET) imaging and hypometabolism on 18F Fluoro-2-deoxy-glucose (FDG)-PET, which is associated with synaptic function.24 The University of Wisconsin Institutional Review Board approved all study procedures. All participants provided signed informed consent.
Peptide and Protein Standards Preparation.
Stock solutions of synthesized human peptides (Biomatik, Ontario, Canada), GSPAINVAVHVFR (Transthyretin, TTR), THLGEALAPLSK (Neurosecretory protein VGF, VGF), and NVTELNEPLSNEER were prepared at 1 μg/μL. Sixteen aliquots were labeled with 4-plex iDiLeu and 12-plex DiLeu reagents, which were synthesized as previously described.21,22 In each aliquot, 20 μg of each peptide standard was combined, and the mixture was resuspended in 20 μL of 0.5 M triethylammonium bicarbonate buffer. Peptide standards were labeled at a ratio of 15:1 (tags−peptide, by weight). A volume of 50 μL of activated reagents was added to each aliquot to 75:25 organic−aqueous solution ratio. The reaction was incubated for 1 h under room temperature. Hydroxylamine (50%) was added to a final concentration of 0.25% to quench the reaction. In total, 200 μg of apolipoprotein E4 (ApoE4) protein standard (Sigma-Aldrich, Saint Louis, MO) was digested with trypsin (Promega, Madison, WI) and labeled with tags based on the digestion procedure described above. To obtain correction factors for iDiLeu-labeled target peptides, 2.5 μg of labeled peptide standards (d0, d6, d9, d12, and 12-plex DiLeu) were cleaned-up with SCX SpinTips (Protea Biosciences, Morgantown, WV) according to the manufacturer’s protocol, respectively. The eluate was dried in vacuo and desalted with ZipTip C18 pipette tips (Merck Millipore, Darmstadt, Germany). The isotopic peak fractions of 12-plex DiLeu were determined by labeling each channel with yeast tryptic digests, respectively. Trigger precursor mass inclusion list and trigger product mass inclusion list were constructed by analyzing d0-labeled peptide standards in yeast (Promega, Madison, WI) digests background alone to determine the most intense charge state of each target peptide and product ions within greater than 20% relative abundance. A target product mass inclusion list was determined by analyzing a mix of 12-plex DiLeu-labeled peptide standards in yeast digest background. Only b-type ions were included for peptides with arginine at C-termini.
Cerebrospinal Fluid Sample Preparation.
A Sprotte 24-gauge or 25-gauge spinal needle was used for cerebrospinal fluid (CSF) collection by lumbar puncture at L3/4 or L4/5. Each CSF sample was gently mixed and centrifuged at 2 000g × 10 min. The supernatant was collected as 0.5 mL aliquots in a polypropylene tube and stored at −80 °C. The 1 mL CSF samples were dried down in vacuum centrifugation with a Savant SC 110 SpeedVac concentrator (Thermo Scientific, Waltham, MA). Sample powder was resuspended in 100 μL of lysis buffer, which contained 8 M urea, 50 mM tris base (adjust pH to 8 with hydrochloric acid), 5 mM CaCl2, 20 mM NaCl, and 1 tablet of EDTA-free protease inhibitor cocktail. Protein concentration was measured with bicinchoninic acid (BCA) protein assay (Thermo Scientific Pierce, Rockford, IL). Proteins from each sample were reduced by adding dithiothreitol (DTT) to a final concentration of 5 mM and incubated at room temperature for 1 h. Reduced cysteines were alkylated by adding iodoacetamide to a final concentration of 15 mM and incubating for 30 min in the dark. The protein mixture was diluted with 50 mM Tris buffer (pH 8) to a final urea concentration of 1 M, followed by digestion with trypsin at a 1:50 enzyme to protein ratio by incubating at 37 °C for 18 h. The digestion reaction was quenched by acidification with 10% TFA to pH 3, followed by desalting with Bond Elut OMIX C18 pipet tips (Agilent Technologies, Santa Clara, CA). CSF protein digests from participants were randomly labeled with 12-plex DiLeu tags as described above. In total, 2 μg of DiLeu-labeled protein digests were combined with 4-plex iDiLeu-labeled peptide standards. Each combined sample was cleaned up with SCX SpinTips and desalted with Bond Elut OMIX C18 pipet tips. All the labeled samples were then dried in vacuo and reconstituted in 3% acetonitrile (ACN) and 0.1% formic acid (FA) in water. The sample group allocation was hidden during the experiment and data analysis to maintain the blinded study.
NanoLC–MS Acquisition.
Online nano LC was performed on a Dionex Ultimate 3000 nanoLC system (Thermo Scientific). Capillary column (16 cm length, 75 μm i.d.) was self-fabricated and packed with reversed-phase BEH C18 material (1.7 μm, 130 Å, Waters Corporation). Samples were loaded onto the column in 100% solvent A (water, 0.1% FA) at a flow rate of 0.3 μL min−1. Separation was performed using a linear gradient from 4% to 35% solvent B (ACN, 0.1% FA) for 90 min. Eluting peptides were electrosprayed into an Orbitrap Fusion Lumos Tribrid quadrupole-ion trap-Orbitrap mass spectrometer (Thermo Scientific). A trigger precursor mass inclusion list containing iDiLeu d0-labeled trigger peptide precursors (m/z, z, scheduled retention time, and scan event index) was used in MS1 survey scans with ±15 ppm of the m/z and ±1.5 min elution time window (Orbitrap mass analyzer; resolution = 60 000, automatic gain control [AGC] = 1 × 106, mass range = 450–950 m/z). Only precursors appearing in the trigger precursor inclusion list were selected for MS2 acquisition. iDiLeu d0-labeled trigger peptides were isolated in the quadrupole within a window of 0.4 m/z, activated by collision induced dissociation (CID), and detected in the Orbitrap mass analyzer (normalized collision energy [NCE] = 35, resolution = 15 000, AGC = 1 × 105, max. injection time = 100 ms). Real-time identification of each trigger peptide was conducted by matching product ions in their MS2 spectra to those specified in a trigger product mass inclusion list.10 If at least five trigger product ions from the list were detected within ±15 ppm, MS2 acquisition of the 12-plex DiLeu-labeled target peptide precursor was triggered. The 12-plex DiLeu-labeled target peptides were isolated within a window of 0.4 m/z at specific offset from the trigger peptide, which is based on charge state and number of DiLeu tags on the peptide and activated by CID using an NCE of 34 so that trigger and target scans can be discrete for downstream data analysis (Orbitrap mass analyzer; resolution = 60 000, AGC = 2 × 105, max. injection time = 500 ms). Product ions corresponding to b- or y-type ions of target peptides were specified in a target product mass inclusion list for targeted SPS-MS3 analysis. Six product ions were selected for SPS-MS3 with an isolation window of 0.4 m/z, activated by higher energy collisional dissociation (HCD) (Orbitrap mass analyzer; resolution = 60 000, NCE = 55, AGC = 1 × 106, max. injection time = 1000 ms, mass range = 100– 500 m/z).
Data Analysis.
All peptide and protein identification were performed using Peaks Studio 7.5 software (Bioinformatics Solutions Inc., Waterloo, ON, Canada). The data refinement was applied to correct precursor mass by default. All raw files were searched against Uniprot Homo sapiens reviewed database (http://www.uniprot.org) with trypsin as the digestion enzyme. The error tolerance for precursor mass was 25 ppm using monoisotopic mass and 0.02 Th for fragment ions. The maximum missed cleavages per peptide was set to 2, which was allowed to be cleaved at both ends of the peptides. Fixed modification was set as carbamidomethylation of cysteine residues (+57.0215 Da). Labels (+141.1154 Da for d0, 145.1208 Da for 12-plex DiLeu, 147.1409 Da for d6, 150.1631 Da for d9, and 153.1644 Da for d12) of peptide N-termini and lysine residues and oxidation of methionine (+15.9949 Da) were selected as variable modifications. Peptides were considered to be unambiguous identifications with FDR below 1%.
Peak intensity generated by Genesis peak detection algorithm was processed in Thermo Xcalibur 2.2. Precursor ion integration tolerance was set to 15 ppm. The peak intensity of target peptides was used for quantification only when the retention time of iDiLeu and 12-plex DiLeu-labeled peptides extracted ion chromatogram was within 2 min. An in-house software program was developed for isotopic interference correction of MS1 precursor and reporter ion-based peptide quantification based on a series of established equations.21,22 The custom software program is open source and freely available at GitHub. The fractional intensities of 12-plex DiLeu and 4-plex iDiLeu primary reporter ion peaks and isotopic peaks are shown in Tables S1 and S2. Channel-normalization was performed to correct systematic biases of 12-plex DiLeu tags by summed reporter ion ratios in Excel. The Student’s t-test (two-tailed) was performed for comparisons between two groups of independent samples. A p-value <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Rationale of High-Throughput HOTMAQ Strategy.
An overview of this quantification method is outlined in Figure 1. Synthetic peptides are labeled with iDiLeu tags (d0, d6, d9, d12) to impart mass additions of 141.1, 147.1, 150.2, 153.2 Da to peptides, respectively. Individual peptide samples of interest are concurrently labeled with 12-plex isobaric DiLeu tags, introducing a nominal mass addition of 145.1 Da per tag, as a substitution for the d3 iDiLeu tag (mass addition of 144.1 Da). The 4-plex iDiLeu-labeled synthetic peptides are diluted in a series of known concentrations and spiked into 12-plex DiLeu-labeled samples to construct standard curves for simultaneous absolute quantification in a single LC−MS run. Because iDiLeu and DiLeu tags are identical in chemical structure, the multiplexed synthetic peptides and target peptides have the same chromatographic elution profiles, while their differences in stable isotope configurations make them distinct in mass from one another by 4, 6, 9, and 12 Da, enabling determination of total amounts of multiplexed isobaric DiLeu-labeled target peptides via the iDiLeu standard curve in the MS1 scan. In addition to generating iDiLeu standard curves, the d0-labeled synthetic peptides also function as real-time monitors by matching MS2 spectra to a product mass inclusion list to specifically trigger acquisition of 12-plex DiLeu-labeled target peptides based on their known offset mass of 4.01 Da per tag. To maximize effective time for measuring multiplexed target peptides in a scheduled time window, MS2 acquisition alternates between two scan modes: a fast low-resolution scan for d0-labeled synthetic peptides and a high-resolution scan for 12-plex DiLeu-labeled target peptides. While quantification accuracy and precision of reporter ion-based quantitation methods suffer due to ratio distortion from coisolated and cofragmented nearly isobaric peptides, preselection of fragment ions for targeted synchronous precursor selection (SPS)-MS3 analysis can mitigate this effect and enable accurate quantification.10 The relative abundance of each 12-plex DiLeu-labeled peptide is determined by SPS-MS3 acquisition, and the absolute amounts of target peptides in each sample are determined by employing the total amount obtained in the standard curve (eq 1).
| (1) |
AA12-plex is the total absolute amount of 12-plex DiLeu-labeled target peptides obtained from a standard curve at the MS1 level. SIi (i = 115a, 115b, 116a, 116b, 116c, 117a, 117b, 117c, 118a, 118b, 118c, 118d) is the signal intensity of DiLeu reporter ions acquired in MS3 spectra. AAi is the absolute amount for each DiLeu tag-labeled peptide.
Figure 1.

Schematic illustration for the HOTMAQ method. (A) Synthetic peptides are labeled with 4-plex iDiLeu at different concentrations and spiked into 12-plex DiLeu-labeled analytes. (B) Labeled peptides are detected with identical chromatographic elution profiles as five precursor ion clusters. The iDiLeu labeled-synthetic peptides are used to generate internal calibration curves to quantify the total amount of multiplexed target peptides. iDiLeu d0-labeled synthetic trigger peptides and multiplexed DiLeu-labeled target peptides are separated in MS1 spectra by a mass offset of 4.01 Da, which enables synthetic trigger peptides to initiate quantitative analysis of target peptides via MS2 regardless of target peptide precursor abundances. (C) Real-time MS2 analysis of d0-labeled synthetic peptides by matching MS2 spectrum to a product mass inclusion list unambiguously triggers fragmentation of 12-plex DiLeu-labeled target peptides in a predefined monitoring window. Acquisition parameters alternate between a low-resolution scan for monitoring d0-labeled trigger peptides and a high-resolution scan for quantifying 12-plex DiLeu-labeled target peptides. Fragment ions of 12-plex DiLeu-labeled target peptides are selected for synchronous precursor selection (SPS)-MS3 analysis. (D) The relative abundance of each 12-plex DiLeu-labeled peptide is accurately determined by targeted SPS-MS3 acquisition at a resolving power of 60K (at m/z 200). The absolute amounts of target peptides are quantified by integrating the total amount obtained using the standard curve.
Construction and Validation of HOTMAQ Strategy.
Measuring 12-plex DiLeu-labeled peptides at an appropriate point along the iDiLeu standard curve is a prerequisite for accurate quantitative measurements by the HOTMAQ method, so we first evaluated its quantitative performance paired with iDiLeu d0- and 12-plex DiLeu-labeled synthetic peptide standards in a background of iDiLeu d0- and 12-plex DiLeu-labeled yeast tryptic peptides, which were combined at unity ratios in all proof-of-principle experiments. Three synthetic peptides were labeled with iDiLeu d0 and 12-plex DiLeu and spiked at known concentrations into the labeled yeast background. Because trigger peptides in low abundance are not able to adequately initiate acquisition of target peptide precursors and those present in extremely high abundance could reduce quantitative accuracy due to greater isotopic interference at any MS level, the optimal ratio between trigger peptides and target peptides were determined initially by testing samples at 10:1, 20:1, 50:1, and 100:1 mixing ratios, with unity ratios maintained for 12-plex DiLeu-labeled target peptides. The ratio of 20:1 was determined to be optimal with a relative error within 10% and coefficient of variation (CV) below 7% (Figure S2).
To evaluate the quantification accuracy of HOTMAQ, 12-plex DiLeu-labeled peptide standards were prepared by combining at ratios of 1:1:1:1:1:1:1:1:1:1:1:1 and 1:1:2:2:5:10:10:5:2:2:1:1 (115a–118d) with 100 amol on column. As an example, a d0 iDiLeu- and 12-plex DiLeu-labeled peptide (THLGEALAPLSK), observed as two distinct peak clusters with a 2.67 m/z difference (at 3+) (Figure 2A), had nearly identical retention time at 62.2 min. Upon real-time identification of the d0-labeled peptide, acquisition of the 12-plex DiLeu-labeled peptide follows (Figure 2B). A target product mass inclusion list was constructed for targeted SPS-MS3 analysis as interference product ions may be more abundant than target product ions and lead to ratio distortion in standard SPS-MS3. After isotopic interference correction, the 12-plex DiLeu ratios (in triplicate) were plotted against each other. Across all channels, the mean ratios were within 10% and 18% of the expected values with average CVs of 6.3% and 13.1% for 1:1 (Figure 2C) and 10:1 (Figure 2D) ratios, respectively.
Figure 2.

HOTMAQ feasibility in a combined interference model. Synthetic human peptide standards were spiked into the iDiLeu d0- and 12-plex DiLeu-labeled yeast tryptic peptides, which were combined at unity ratios. (A) iDiLeu d0 and 12-plex DiLeu-labeled peptides with the sequence THLGEALAPLSK coelute with an identical retention time of 62.2 min. The two precursor ion clusters (z, 3+) are separated by 2.67 m/z, as the peptide carries two tags at both N-terminal and lysine side chain. (B) An example of an iDiLeu d0-labeled peptide successfully triggering fragmentation of 12-plex DiLeu-labeled targeted peptides. 12-plex DiLeu-labeled peptide standards were combined at ratios of 1:1:1:1:1:1:1:1:1:1:1:1 (C) and 1:1:2:2:5:10:10:5:2:2:1:1 (D) (115a–118d). The average signal intensity of all the 12 channels was used for normalization in part C. The average signal intensity of 115a, 115b, 118c, and 118d at the unit ratio was used for normalization in part D. Error bars represent the standard deviations.
We also compared the quantitative accuracy of this method with conventional PRM and standard data-dependent SPS-MS3 using labeled peptides mixed at a ratio of 1:1:2:2:5:10:10:5:2:2:1:1 spanning from 100 amol to 1 fmol in the labeled yeast background (Figure 3A). The CVs for all three quantification methods were below 20%. As shown in the radar plot, the relative errors at ratios of 2, 5, and 10 were within 10% for the HOTMAQ method but were up to 22% for the other two quantification methods (Figure 3B). The accuracy of the PRM-MS2 method suffered due to interference of nearly isobaric yeast contaminant ions that were isolated and fragmented together with target ions. SPS-MS3 improved quantitative precision, but the remaining fragment ion interference caused an underestimation of the mixing ratios.
Figure 3.

Comparison of quantification accuracy for PRM, standard SPS-MS3, and HOTMAQ. (A) 12-plex DiLeu-labeled peptide standards were combined at ratios of 1:1:2:2:5:10:10:5:2:2:1:1 (115a–118d). The sample was quantified by PRM, standard SPS-MS3, and HOTMAQ, respectively. (B) Radar plot of relative errors of (A) at the average ratio of 2, 5, and 10. The greater the distance of each tested condition from a relative error of 0, the lower the quantification accuracy of the corresponding quantification method.
Next, we evaluated the absolute quantification accuracy of the HOTMAQ method using 4-plex iDiLeu- and 12-plex DiLeu-labeled peptide standards. 12-plex DiLeu-labeled peptides were combined at a ratio of 1:1:2:2:5:10:10:5:2:2:1:1 with the lowest amount at 100 amol in the DiLeu-labeled yeast background, which is the limit of quantification for SRM/PRM but not in a multiplexed assay.34 Each of the three peptide standards were quantified with excellent linearity (R2 = 0.999). Figure 4A presents an example of standard curve for peptide (THLGEALAPLSK). By incorporating 12-plex DiLeu ratios measured by targeted SPS-MS3, the final amounts for each channel were 0.08, 0.09, 0.21, 0.2, 0.38, 0.88, 0.95, 0.38, 0.18, 0.20, 0.08, and 0.09 fmol (115a–118d) with an average relative error of 11% and CV of 8.4% (Figure 4B). These results illustrate that the overall accuracy and precision for absolute quantification by HOTMAQ is excellent in a multiplexed experiment.
Figure 4.

Assessing accurate absolute quantification of target peptides at low attomoles. (A) Calibration curve constructed for peptide (THLGEALAPLSK) with exceptional linearity of R2 = 0.9992. (B) The HOTMAQ method demonstrated exceptional absolute quantification accuracy at ratios of 1:1:2:2:5:10:10:5:2:2:1:1 (115a–118d) with the minimal loading amount at 100 amol. Error bars represent the standard deviations.
Applying HOTMAQ Method to Quantify Candidate Biomarkers in Preclinical AD.
We further applied the HOTMAQ method to quantify and validate three reported candidate protein biomarkers, transthyretin (TTR), neurosecretory protein VGF (VGF), and apolipoprotein E (ApoE) in CSF samples collected from 11 individuals in preclinical AD and an equal number of healthy controls in triplicate.29,35 Detailed characteristics of study participants are provided in Table S3. The mean (SD) age of healthy controls and preclinical AD patients was 59.8 (6.6) and 59.4 (4.6) years, respectively. All the samples were analyzed in less than 3 h, owing to the 12-fold increase in analytical throughput by HOTMAQ. The linear dynamic range of each target protein was optimized to include the endogenous abundance in the standard curve (Figure 5A–C). We observed that each of the three targeted proteins exhibited down-regulation in preclinical AD stage (Figure 5D–F), which is consistent with the results previously reported for AD dementia,36–38 though some groups have reported inconsistent findings.39 The protein amounts of all samples were located within the 5th–95th percentile, except for one outlier of TTR. Student’s t-test yielded a p-value of 0.03 for ApoE, indicating a significant difference in ApoE between healthy controls and preclinical AD patients (decreased 17.4%). Statistical significance was not found for TTR (p = 0.32) and VGF (p = 0.26).
Figure 5.

Quantification of candidate protein biomarkers in preclinical AD. A total of 22 CSF samples from preclinical AD and control subjects were labeled with 12-plex DiLeu. The spiked-in 4-plex iDiLeu-labeled synthetic peptides displayed excellent linearity for TTR (A), VGF (B), and ApoE (C). Compared to healthy controls (gray), TTR (D), VGF (E), and ApoE (F) were observed to be down-regulated in preclinical AD patients (orange). Student t-test was performed for comparison between preclinical AD and control subjects. The demarcated line on the plots for parts D–F shows the average amount.
The ApoE-encoding gene APOE has three major polymorphic alleles, ε2, ε3, and ε4, all of which differently modulate amyloid beta aggregation and clearance in AD pathogenesis.40 The APOE ε4 allele is strongly associated with an earlier age of AD onset and increased risk of late-onset AD.41 For comparison regarding to APOE ε4 genotype, the subjects were categorized as ε4 noncarriers (ε2/ε3, ε3/ε3) and ε4 carriers (ε2/ε4, ε3/ε4, ε4/ε4) in the healthy controls and preclinical AD group, respectively. ε4 carriers exhibited lower amount of ApoE than ε4 noncarriers in healthy subjects (Figure S3A). Consistent with the known increased susceptibility to AD dementia among women,42 we observed that the average CSF ApoE level of healthy controls was lower in women than in men, while remaining the same in both preclinical AD patients (Figure S3B).
CONCLUSIONS
Targeted mass spectrometry is a valuable technique for sensitive and quantitative detection of a subset of proteins of interest in academia and industry. The novel HOTMAQ strategy presented in this study increases analytical throughput of absolute quantification by up to 12-fold with exceptional accuracy. The unparalleled advantage of HOTMAQ is that a single run can achieve three aims: (1) an internal standard curve can be constructed specifically for each target peptide through mass difference labeling; (2) real-time identification of trigger peptide prompts unambiguous detection and quantification of target peptide in a scheduled time window; (3) targeted SPS-MS3 analysis enables accurate determination of 12-plex DiLeu reporter ion abundances. HOTMAQ has a similar limit of quantification with conventional SRM/PRM at the low attomole level but in a 12-plex experiment and with improved quantification accuracy. Compared to other targeted proteomics methods, the HOTMAQ approach employs cost-effective iDiLeu and DiLeu tags, eliminating the need for synthesizing expensive heavy isotope-encoded peptide standards. The combination of precursor isotopic labeling and isobaric labeling has been used for 24-plex quantification by dimethyl labeling at acidic pH and DiLeu labeling at basic pH, allowing for relative quantification in discovery-based proteomics studies,43 while HOTMAQ is developed for multiplexed absolute quantification in targeted mass spectrometry. The limitation of HOTMAQ is that it can only be applied using an advanced mass spectrometer, such as the Fusion Lumos Orbitrap instrument. The DiLeu-labeled peptide standards need to be analyzed individually to construct the trigger product mass inclusion list and target product mass inclusion list prior to sample analysis. The initial costs for starting materials for 12-plex DiLeu and iDiLeu will still amount to less than the cost of an appropriately sized commercial 10-plex TMT kit and the amounts available for the cost will allow for larger experiments and more replicates. The cost saving of the HOTMAQ strategy is a great advantage of many, but the synthesis of 12-plex DiLeu and 4-plex iDiLeu does require time and effort.
HOTMAQ utilizes synthetic peptides to unambiguously prompt selection and quantification of target peptides, obviating the need for immunoaffinity enrichment of proteins of interest in complex biofluids. This is particularly important as the production of high-quality protein antibodies has historically been the bottleneck in candidate biomarkers verification. The HOTMAQ strategy ideally bridges the gap between the discovery and verification phases for candidate biomarkers from large cohorts of clinical specimens. Using the HOTMAQ strategy to investigate preclinical AD, we observed significant down-regulation of ApoE in agreement with previous studies of AD dementia, and these results may provide support to the development of early stage diagnostic tools and therapeutic interventions to delay the onset of dementia. The utility of this new strategy goes beyond AD CSF biomarker verification and validation; its greatly enhanced throughput and quantitative performance, paired with sample flexibility, should be useful in targeted peptidomics, proteomics, and phophoproteomics in general.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by the National Institutes of Health (NIH) Grants R01AG052324, 1P50AG033514, R01DK071801, and P41GM108538. Support for this research was also provided by the University of Wisconsin-Madison, Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (Grant NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. L.L. acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b04580.
Supplementary Figure S1, structure of N,N-dimethyl leucine (DiLeu) tags; Supplementary Figure S2, ratio optimization for trigger peptides and target peptides; Supplementary Figure S3, comparison of ApoE regarding to gender difference and APOE ε4 genotype in preclinical Alzheimer’s disease; Supplementary Table S1, primary and isotopic peak fractions for DiLeu reporter ions; Supplementary Table S2, primary and isotopic peak fractions for iDiLeu-labeled target peptides; and Supplementary Table S3, characteristics of study participants (PDF)
REFERENCES
- (1).Gillette MA; Carr SA Nat. Methods 2013, 10 (1), 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Doerr A Nat. Methods 2013, 10 (1), 23. [DOI] [PubMed] [Google Scholar]
- (3).Marx V Nat. Methods 2013, 10 (1), 19–22. [DOI] [PubMed] [Google Scholar]
- (4).Doerr A Nat. Methods 2012, 9 (10), 950. [DOI] [PubMed] [Google Scholar]
- (5).Picotti P; Aebersold R Nat. Methods 2012, 9 (6), 555–566. [DOI] [PubMed] [Google Scholar]
- (6).Gerber SA; Rush J; Stemman O; Kirschner MW; Gygi SP Proc. Natl. Acad. Sci. U. S. A 2003, 100 (12), 6940–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Keshishian H; Addona T; Burgess M; Kuhn E; Carr SA Mol. Cell. Proteomics 2007, 6 (12), 2212–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wu R; Haas W; Dephoure N; Huttlin EL; Zhai B; Sowa ME; Gygi SP Nat. Methods 2011, 8 (8), 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gallien S; Kim SY; Domon B Mol. Cell. Proteomics 2015, 14 (6), 1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Erickson BK; Rose CM; Braun CR; Erickson AR; Knott J; McAlister GC; Wü M; Paulo JA; Everley RA; Gygi SP Mol. Cell 2017, 65 (2), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen I-H; Xue L; Hsu C-C; Paez JSP; Pan L; Andaluz H; Wendt MK; Iliuk AB; Zhu J-K; Tao WA Proc. Natl. Acad. Sci. U. S. A 2017, 114 (12), 3175–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1 (5), 376–386. [DOI] [PubMed] [Google Scholar]
- (13).Desouza LV; Taylor AM; Li W; Minkoff MS; Romaschin AD; Colgan TJ; Siu KW M. J. Proteome Res 2008, 7 (8), 3525–3534. [DOI] [PubMed] [Google Scholar]
- (14).Zhu H; Pan S; Gu S; Bradbury EM; Chen X Rapid Commun. Mass Spectrom 2002, 16 (22), 2115–2123. [DOI] [PubMed] [Google Scholar]
- (15).Seyfried NT; Gozal YM; Dammer EB; Xia Q; Duong DM; Cheng D; Lah JJ; Levey AI; Peng J Mol. Cell. Proteomics 2010, 9 (4), 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hebert AS; Merrill AE; Bailey DJ; Still AJ; Westphall MS; Strieter ER; Pagliarini DJ; Coon JJ Nat. Methods 2013, 10 (4), 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Mol. Cell. Proteomics 2004, 3 (12), 1154–1169. [DOI] [PubMed] [Google Scholar]
- (18).Rauniyar N; Yates JR J. Proteome Res 2014, 13 (12), 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (20).Yu C; Huszagh A; Viner R; Novitsky EJ; Rychnovsky SD; Huang L Anal. Chem 2016, 88 (20), 10301–10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Greer T; Lietz CB; Xiang F; Li LJ Am. Soc. Mass Spectrom 2015, 26 (1), 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Frost DC; Greer T; Li L Anal. Chem 2015, 87 (3), 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zetterberg H Curr. Opin. Psychiatry 2015, 28 (5), 402–409. [DOI] [PubMed] [Google Scholar]
- (24).Sperling RA; Aisen PS; Beckett LA; Bennett DA; Craft S; Fagan AM; Iwatsubo T; Jack CR; Kaye J; Montine TJ; Park DC; Reiman EM; Rowe CC; Siemers E; Stern Y; Yaffe K; Carrillo MC; Thies B; Morrison-Bogorad M; Wagster MV; Phelps CH Alzheimer’s Dementia 2011, 7 (3), 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aluise CD; Robinson RAS; Beckett TL; Murphy MP; Cai J; Pierce WM; Markesbery WR; Butterfield DA Neurobiol. Dis 2010, 39 (2), 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Blennow K; Hampel H Lancet Neurol 2003, 2 (10), 605–613. [DOI] [PubMed] [Google Scholar]
- (27).Blennow K; Hampel H; Weiner M; Zetterberg H Nat. Rev. Neurol 2010, 6 (3), 131–144. [DOI] [PubMed] [Google Scholar]
- (28).Blennow K; de Leon MJ; Zetterberg H Lancet 2006, 368 (9533), 387–403. [DOI] [PubMed] [Google Scholar]
- (29).Hö M; Minthon L; Hansson O; Holmén-Larsson J; Pike I; Ward M; Kuhn K; Rüetschi U; Zetterberg H; Blennow K; Gobom JJ Proteome Res 2015, 14 (2), 654–663. [DOI] [PubMed] [Google Scholar]
- (30).Hampel H; Frank R; Broich K; Teipel SJ; Katz RG; Hardy J; Herholz K; Bokde ALW; Jessen F; Hoessler YC; Sanhai WR; Zetterberg H; Woodcock J; Blennow K Nat. Rev. Drug Discovery 2010, 9 (7), 560–574. [DOI] [PubMed] [Google Scholar]
- (31).Ludwig KR; Hummon AB Mol. BioSyst 2017, 13 (4), 648–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Almeida RP; Schultz SA; Austin BP; Boots EA; Dowling NM; Gleason CE; Bendlin BB; Sager MA; Hermann BP; Zetterberg H; Carlsson CM; Johnson SC; Asthana S; Okonkwo OC JAMA Neurol 2015, 72 (6), 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Folstein MF; Folstein SE; McHugh PR J. Psychiatr. Res 1975, 12 (3), 189–198. [DOI] [PubMed] [Google Scholar]
- (34).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Mol. Cell. Proteomics 2012, 11 (11), 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rifai N; Gillette MA; Carr SA Nat. Biotechnol 2006, 24 (8), 971–983. [DOI] [PubMed] [Google Scholar]
- (36).Carrette O; Demalte I; Scherl A; Yalkinoglu O; Corthals G; Burkhard P; Hochstrasser DF; Sanchez J-C Proteomics 2003, 3 (8), 1486–1494. [DOI] [PubMed] [Google Scholar]
- (37).Riisøen H Acta Neurol. Scand 1988, 78 (6), 455–459. [DOI] [PubMed] [Google Scholar]
- (38).Gupta VB; Laws SM; Villemagne VL; Ames D; Bush AI; Ellis KA; Lui JK; Masters C; Rowe CC; Szoeke C; Taddei K; Martins RN Neurology 2011, 76 (12), 1091–1098. [DOI] [PubMed] [Google Scholar]
- (39).Liu C-C; Kanekiyo T; Xu H; Bu G Nat. Rev. Neurol 2013, 9, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Han X Cell. Mol. Life Sci 2004, 61 (15), 1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kim J; Basak JM; Holtzman DM Neuron 2009, 63 (3), 287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Andersen K; Launer LJ; Dewey ME; Letenneur L; Ott A; Copeland JRM; Dartigues J-F; Kragh-Sorensen P; Baldereschi M; Brayne C; Lobo A; Martinez-Lage JM; Stijnen T; Hofman A Neurology 1999, 53 (9), 1992–1992. [DOI] [PubMed] [Google Scholar]
- (43).Frost DC; Rust CJ; Robinson RAS; Li L Anal. Chem 2018, 90 (18), 10664–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.