

Abstract
We report a systematic study examining two synthetic routes, reductive amination and Mitsunobu coupling, for preparation of chiral γ-peptide nucleic acid (γPNA) monomers and oligomers. We found that the reductive amination route is prone to epimerization, even under mild experimental conditions. The extent of epimerization could be minimized by utilizing a bulky protecting group such as PhFl; however, it is difficult to remove in the subsequent oligomer synthesis stage. On the other hand, we found that the Mitsunobu route produced optically superior products using standard carbamate protecting groups.
Keywords: γPNA, Conformational-preorganization, Tight and specific binding
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
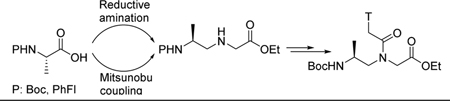
1. Introduction
Achieving tight and specific binding has been a major goal of molecular design for recognition and self-assembly. However, it is generally difficult to realize one without compromising the other due to their inverse relationship.1 Nature has devised a creative solution to this problem by stitching individual chemical building blocks, such as amino acids, nucleotides and nucleosides, into linear chains which then self-organize into compacted 3-dimensional structures. Such conformational preorganization, whereby the substrates adopt the conformations of the bound state prior to recognition, would not only improve the binding free energy, by minimizing the entropic cost, but also increase the rigidity of the system, by making it less accommodating to structural changes—hence the enhancement in recognition specificity. The concept of conformational preorganization has long been recognized by Pedersen,2 Cram,3 and Lehn,4 and successfully applied in the design of small-molecules for guest-host interactions. However, such a concept has been less successful with the design of larger macromolecular and oligomeric systems due to their many degrees of freedom—although some progress has been made.5
A particular class of oligonucleotide molecule whose conformation one would like to be able to define is peptide nucleic acid (PNA). PNA is synthetic analogue of DNA and RNA, comprised of a pseudopeptide backbone and nucleobases connected through a flexible carboxymethylene linker.6 PNA has many appealing features, including chemical and enzymatic stability, tight and sequence specific binding with DNA as well as RNA, and an ability to invade certain sequences of double-stranded DNA (dsDNA).7 The latter property is especially intriguing because of its potential utility in transcriptional regulation. However, with the original achiral backbone design,6 invasion of dsDNA by PNA is limited to mostly homopurine and homopyrimidine targets.8 Invasion of mixed-sequence dsDNA has been reported,9 but it often requires elaborate nucleobase substitutions and is generally restricted to a narrow temperature range.10 It has been shown by Nielsen,11 as well as our own group12 that the failure of PNA to productively invade mixed-sequence dsDNA at physiological temperature is not due to the lack of base-pair accessibility, but rather due to the lack of binding free energy. Since PNA does not have a well-defined conformation, one way to gain additional binding free energy without changing the oligo length or sequence would be to transform the conformation of PNA from a random fold into a right-handed helical motif, one that closely mimics that of the bound PNA-DNA hybrid duplex. We showed in a recent series of studies that such conformational preorganization can be accomplished by installing an appropriate stereogenic center at the γ-backbone.13,14 Those γPNA oligomers prepared from chiral monomers, starting with L-amino acids via the reductive amination13 or Mitsunobu15,16 route, adopt a righthanded helix, while those prepared from D-amino acids adopt a left-handed helix; however, only those right-handed helical γPNAs hybridize to DNA or RNA with high affinity and sequence specificity, and are able to invade dsDNA (or dsRNA).17 The efficiency of strand invasion, however, is strongly dependent on the optical purity of the monomers. The presence of even a minute amount of the epimerized mixture can have a profound adverse effect on strand invasion. It is therefore necessary to have optically pure monomers, if the corresponding γPNA oligomers were to be able to invade dsDNA (or dsRNA) and be successfully employed in antigene applications. Herein we report a systematic study examining two synthetic routes for preparing optically-pure γPNA monomers.
2. Results and Discussion
So far most of the chiral γPNA monomers were prepared through the reductive amination route, starting with either Boc- or Fmoc-protected amino acids.13,18–21 Though commonly employed in peptide synthesis, Boc- and Fmoc-protected amino aldehydes, the intermediates employed in reductive amination, are known to undergo epimerization, even under mild experimental conditions such as rapid chromatography on silica gel.22–25 Attempts to minimize epimerization have generally relied on the immediate use of cold, unpurified amino aldehyde solutions. Although such a precaution was exercised in many of these instances, epimerization could still occur. To address this issue, we explored two synthetic routes in the preparation of γPNA backbone: (i) reductive amination with the α-amino group separately protected with Boc- and N(9-(9-phenylfluorenyl)) (PhFl), and (ii) Mitsunobu reaction with Boc-protection. The sterically bulky PhFl-protecting group was chosen because it has been shown by Rapoport26,27 to be configurationally stable under reductive amination conditions. Similarly, the Mitsunobu reaction was ascertained because of its superior enantioselectivity.16 In the latter case, instead of going through the configurationally unstable aminoaldehyde, the protected amino acid is immediately converted to aminoalcohol, the enantioselective chemical transformation for which has already been established.13
The two routes employed the same batch of commercially available L-alanine 1 as a starting material (99.6% ee), as shown in Scheme 1. In route (i), L-alanine 1 was protected with Boc and PhFl, separately. Conversion of 2a and 2b to Weinreb amides 3a and 3b, followed by reduction with LiAlH4 yielded the aldehydes. Condensation of 4a-i (denoting the Weinreb amide reduction route) and 4b with glycine ethyl ester via the reductive amination route produced the desired alanyl-γPNA backbones 5a-i and 5b, respectively. In route (ii), Boc-L-alanine 2a was converted to aminoalcohol 6 under a mild condition.28 Treatment of 6 with NBS-Gly-OEt under the Mitsunobu condition followed by deprotection with thiophenol/K2CO3 yielded the backbone intermediate 5a-ii (denoting the Mitsunobu route). All three alanyl-γPNA backbones 5a-i, 5a-ii and 5b were coupled to thymine nucleobase using DCC/DhbtOH and then hydrolyzed with 2N NaOH to give the desired monomers 10a-i, 10a-ii and 10b, respectively, (Scheme 2).
Scheme 1. Synthesis of alanyl-γPNA backbones.
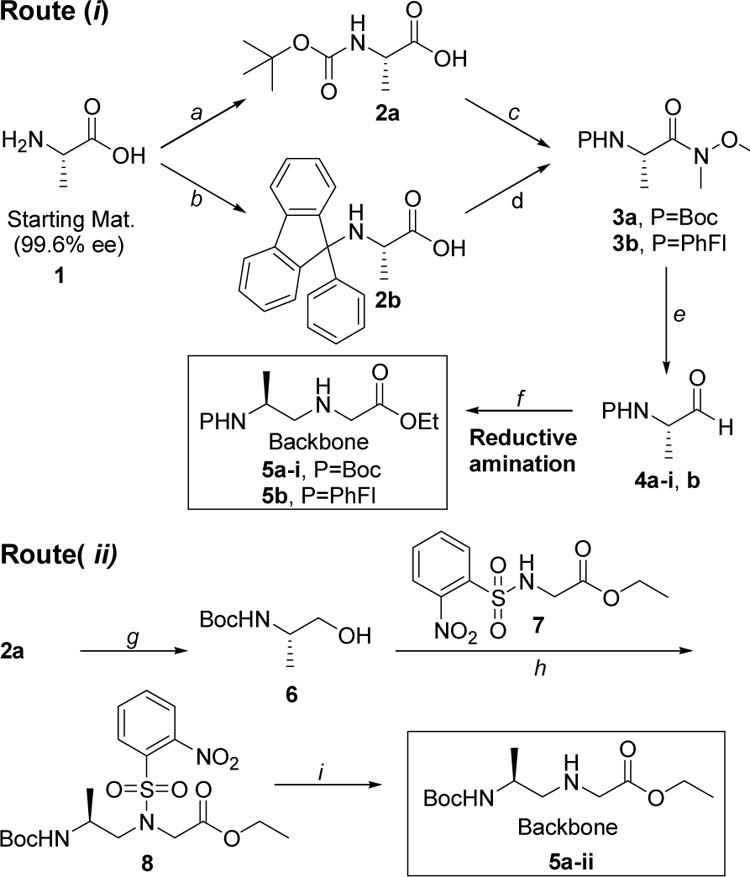
Reagents and conditions: (a) (Boc)2O, NaHCO3, 1,4-dioxane/water, rt; (b) (CH3)3SiCl, CHCl3/CH3CN, reflux, then Et3N, Pb(NO3)2, 9bromo-9-phenylfluorene, rt; (c) NMM, ClCO2iBu, DCM, −15 °C, CH3NHOCH3•HCl, rt; (d) DCC, DhbtOH, CH3NHOCH3•HCl, DMF, rt; (e) LiAlH4, THF, 0 °C; (f) glycine ethyl ester, MeOH, 4 °C, then acetic acid, NaBH3CN; (g) i) NMM, ClCO2iBu, DME, 0 °C, ii) NaBH4, H2O, 0 ºC; (h) o-NBS-Gly-OEt, TPP, DIAD, THF, 0 °C→rt; (i) PhSH, K2CO3, ACN, rt.
Scheme 2. Synthesis of alanyl-γPNA thymine monomers.
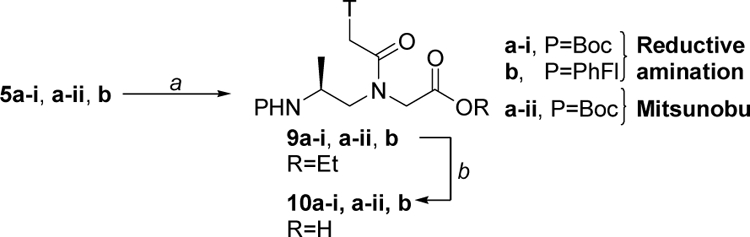
Reagents and conditions: (a) T-CH2CO2H, DCC, DhbtOH, DMF, 50 ºC; (b) 2N NaOH:THF (1:1), 0 ºC.
Next, we determined the optical purities of the alanyl-γPNA backbones 5a-i, 5a-ii and 5b and the corresponding monomers 10a-i, 10a-ii and 10b using fluorine NMR, after coupling to Mosher’s reagent. In this instant, the protecting groups (Boc and PhFl) were removed and the resulting amines were coupled to R-(+)-α-Methoxy α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl) (Scheme 3). The other diastereomers, D-alanine-derived γPNA backbone and thymine monomer, were also prepared for comparison using the Mitsunobu protocol.
Scheme 3. Synthesis of MTPA derivatives.
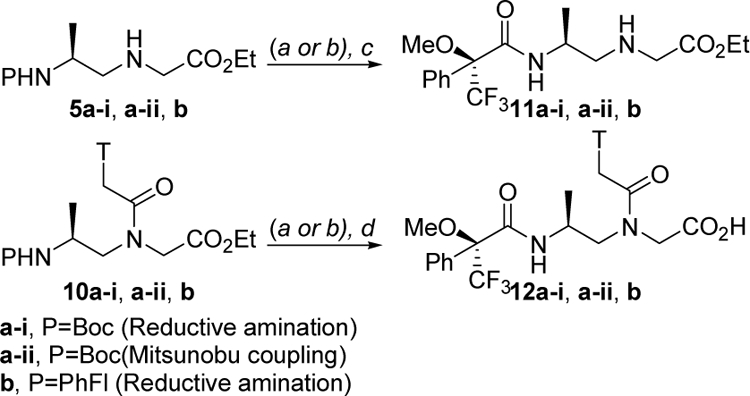
Reagents and conditions: (a) 5% m-cresol, TFA, DCM, rt (Boc removal); (b) H2-Pd/C, MeOH, rt (PhFl removal); (c) MTPA-Cl, DIPEA, DCM 0 °C→rt; (d) MTPA-Cl, NaHCO3, acetone/H2O (1/1).
19F-NMR analysis of the reductive amination backbone 11a-i (Boc-protected) showed roughly 5% epimerization (Figure 1), whereas that protected with PhFl (11b) and Bocprotected 11a-ii prepared via the Mitsunobu route showed excellent ee values (>99.5%, the minimum detection limit of 19F-NMR)29. It was noted that the degree of epimerization (~ 5%) for 11a-i was carried from the backbone to the final monomer (12a-i, Figure 2). This indicates that epimerization occurred during the reductive amination step. On the other hand, the reductive amination product containing PhFlprotecting group gave enantiopure backbone because the steric bulkiness of PhFl prevents deprotonation of the αproton in aldehyde 4b. 19F-NMR of MTPA-derived monomers 12a-i, 12a-ii and 12b showed two peaks due to the existence of two rotamers (major and minor), which occur as the result of rotational restriction around the amide nitrogen that connects the backbone to the nucleobase.
Figure 1.
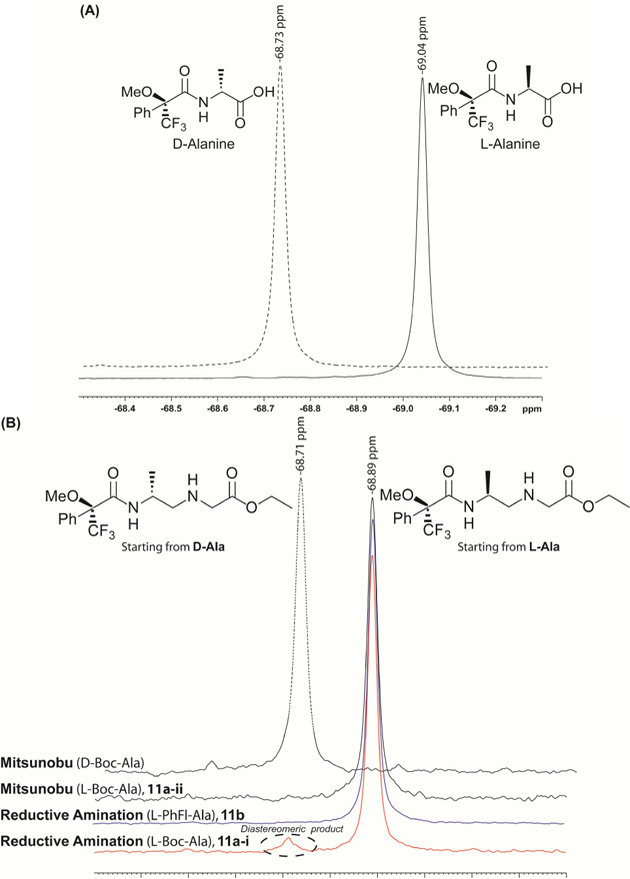
19F-NMR of MTPA-derivatized (A) D- and Lalanine starting materials, and (B) alanyl-γPNA backbones prepared from the reductive amination (11a-i and 11b) and Mitsunobu (11a-ii) routes.
Figure 2.
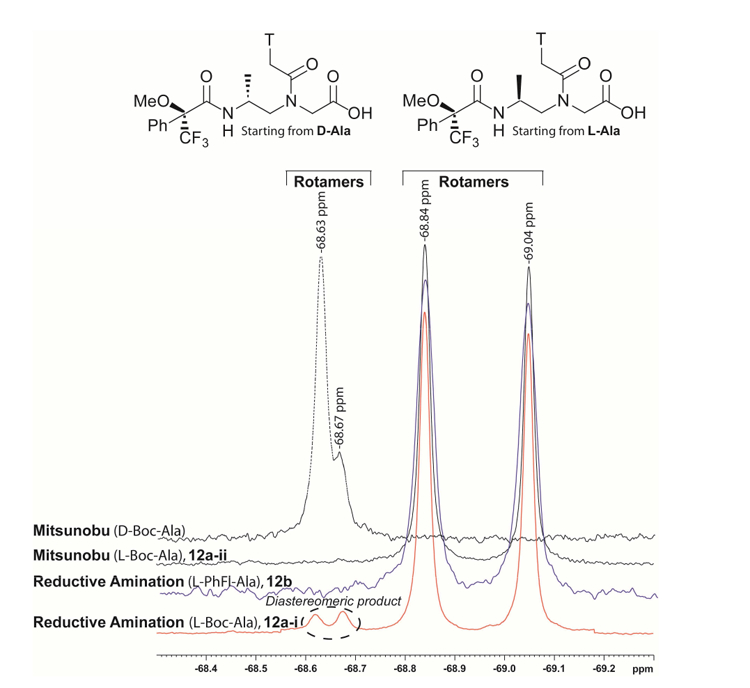
19F-NMR of MTPA-derivatized alanyl-γPNA Tmonomers prepared from the reductive amination (12a-i and 12b) and Mitsunobu (12a-ii) routes.
Since reductive amination is a two-step process, involving reduction of Weinreb amide (or oxidation of alcohol) to aldehyde and reductive coupling of aldehyde with amine, we decoupled the two reaction steps and determined the extent of their epimerization separately. Optically pure Boc-protected aminoalcohols, for both D- and L-isomers, were prepared using a known procedure.28 19F-NMR data showed that they were optically-pure (Figure 1S, Supporting Information). Due to the concern that utilization of LiAlH4 in the reduction of the Weinreb amide to aldehyde might be a predominant source of epimerization, as the result of prolonged exposure of the aldehyde to high pH aqueous solution in the workup, we examined two additional reactions, Swern30 and ParikhDoering,31 that have been shown to be milder and less prone to epimerization in the oxidation of alcohol to aldehyde (Scheme 4). For a meaningful comparison, we optimized the conditions of these reactions further with respect to reaction time, temperature, molar ratio of base (Et3N) and solvent that would give greater than 90% chemical yield within one-hour and the highest optical purity (Supporting Information). Under such conditions, the optical purities of the aldehydes produced by the three methods, reduction of Weinreb amide by LiAlH4, Swern and Parikh-Doering oxidation, were similar (~ 1.5%), as shown in Figure 3. This was determined by FNMR following an immediate conversion of the aldehydes back to alcohols and subsequent Boc removal and coupling to Mosher’s reagent. Following a similar characterization protocol, we next selected one of the aldehyde products with a known ee value, in this case produced by Swern oxidation (4a-ii), due to the convenience in scaling, and subjected it to an optimized reductive amination condition. F-NMR spectrum of the resulting backbone product following Boc-removal and coupling to Mosher’s reagent showed nearly doubling the degree of epimerization. These results together show that under these optimized conditions, each reaction step (alcohol to aldehyde and aldehyde to secondary amine) contributed a similar degree of epimerization (~ 1.5% each).
Scheme 4. Comparison of the oxidation methods.
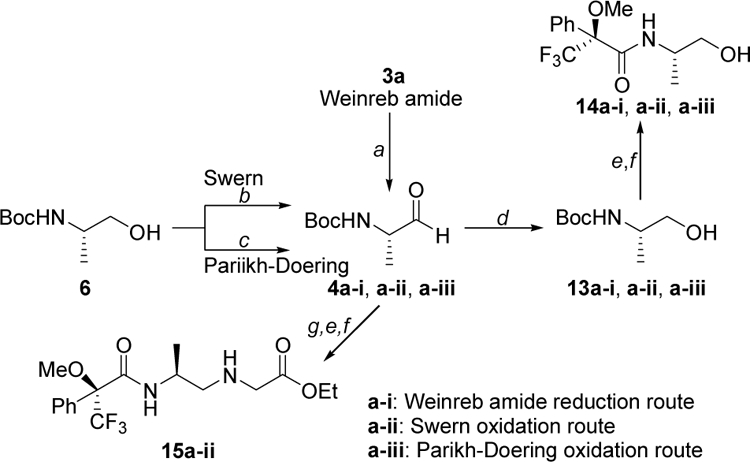
Reagents and conditions: (a) LiAlH4, THF, 0 °C; (b) Swern [(COCl)2, DMSO, Et3N, DCM, −78 °C]; (c) Parikh-Doering [Py•SO3, DMSO/DCM (1/1), Et3N, 0 °C→rt]; (d) NaBH3CN, MeOH, 0 °C→rt; (e) 5% m-cresol, TFA, DCM, rt; (f) MTPA-Cl, DIPEA, DCM 0 °C→rt; (g) glycine ethyl ester, MeOH, 4 °C, then acetic acid, NaBH3CN.
Figure 3.
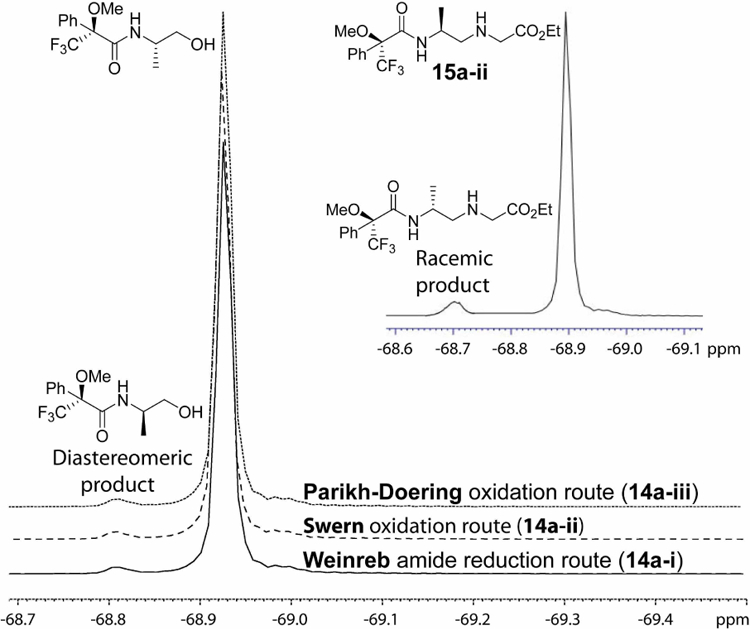
19F-NMR spectra of MTPA-derivatized alcohols prepared via the Weinreb amide reduction (14a-i) and Swern (14a-ii) and Parikh-Doering (14a-iii) oxidation routes, followed by reduction of aldehydes to alcohols, Boc deprotection and coupling to MTPA-Cl. Insert: 19F-NMR of the alanyl-γPNA backbone (15a-ii) prepared by reductive amination of the Swern oxidation product followed by Boc deprotection and coupling to MTPA-Cl.
To determine the effect of epimerization on the conformation of PNAs, we measured the CD spectra of a series of oligomers prepared from two separate batches of monomers: those obtained via the reductive amination (10a-i) and Mitsunobu (10a-ii) routes, Table 1. CD spectra were recorded at room temperature for all oligomers as single strands (Figure 4). As expected, no noticeable CD signals were observed for the unmodified PNA1 in the nucleobase absorption regions (200–350 nm).13 This is because PNA, as individual strands, do not have well-defined conformations. On the other hand, a pronounced biphasic exciton coupling pattern characteristic of a right-handed helix was observed for PNA2a, 2b, 3a and 3b, with maxima at 220 and 260 nm and minima at 240 and 280 nm.13 An interesting finding was that the amplitudes of the CD signals of PNA2a at 220 and 260 nm are smaller than that of PNA2b. This is expected because PNA2a was prepared from the epimerized building block (10a-i). Since the epimerized monomer contained only ~ 95% of the desired (S)-diastereomer, which adopts a right-handed helix, and the other 5% being an (R)-diastereomer, which adopts a left-handed helix, its incorporation into PNA2a would cancel each other to an extent that leads to a decrease in the overall amplitude of the CD signal. A similar pattern was observed for PNA3a, with four epimerized building blocks, as compared to that of PNA3b which contained optically pure monomers, with the latter exhibiting greater signal strengths. We ruled out the differences in the CD amplitudes as the result of variations in the concentrations, because the UV-absorption profiles of the oligomers were identical at 90 °C, the temperature at which all the strands were completely denatured, indicating that they were of the same concentration.
Table 1.
Sequence of PNA oligomers
| PNA | Sequence | M.W. |
|---|---|---|
| 1 | H-GCATGTTTGA-LLys-NH2 | 2886 |
| 2a (10a–i) | H-GCATGTTTGA- LLys-NH2 | 2900 |
| 2b (10a–ii) | H-GCATGTTTGA- LLys-NH2 | 2900 |
| 3a (10a–i) | H-GCATGTTTGA- LLys-NH2 | 2942 |
| 3b (10a–ii) | H-GCATGTTTGA- LLys-NH2 | 2942 |
T = Alanyl-γPNA T-monomeric unit; 10a–i: prepared via reductive amination route, 10a-ii: Mitsunobu route.
Figure 4.
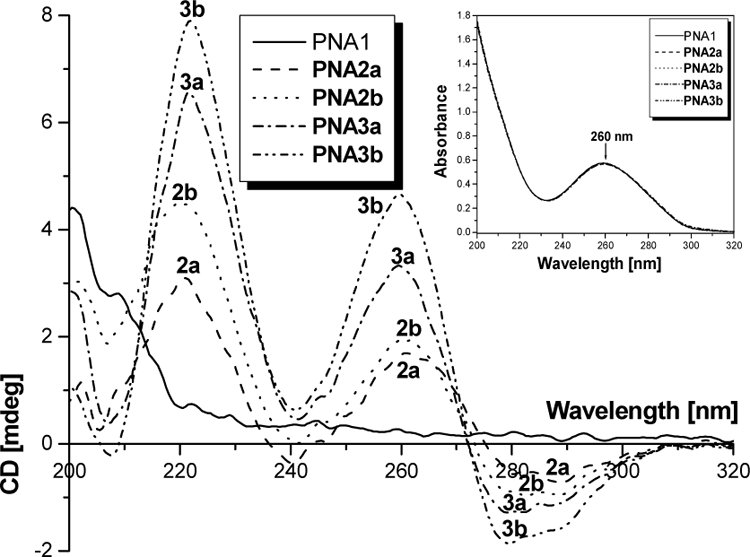
CD spectra of single-strand unmodified PNA (PNA1) and alanyl-γPNAs (PNA2a, 2b, 3a and 3b). All samples were prepared at 5 µM strand concentration each in 10 mM NaPi buffer. CD spectra were recorded at 22 °C. Inset: UV-Vis spectra of the indicated oligomers at 90 °C showing that their concentrations were the same at 260 nm.
Next, we determined the thermal stability of the same set of oligomers upon hybridization to a complementary (antiparallel) DNA strand using variable-temperature UVspectroscopy (Figure 5). Roughly a 3 °C decrease in the Tm was observed for the PNA2a-DNA hybrid duplex as compared to that of PNA2b-DNA, due to epimerization in the monomer (10a-i) employed in the synthesis of PNA2a. These results are in agreement with our recent findings in which we showed that the left-handed γPNAs are unable to hybridize to DNA or RNA due to conformational mismatch, despite the sequence complementarity—which explains the destabilizing effect observed here. Similarly, the epimerized PNA3a-DNA duplex showed a 6 °C decrease in the Tm as compared to the enantiomerically-pure PNA3b-DNA. The differences in the thermal stability of the PNA-DNA duplexes are consistent with the CD data.
Figure 5.
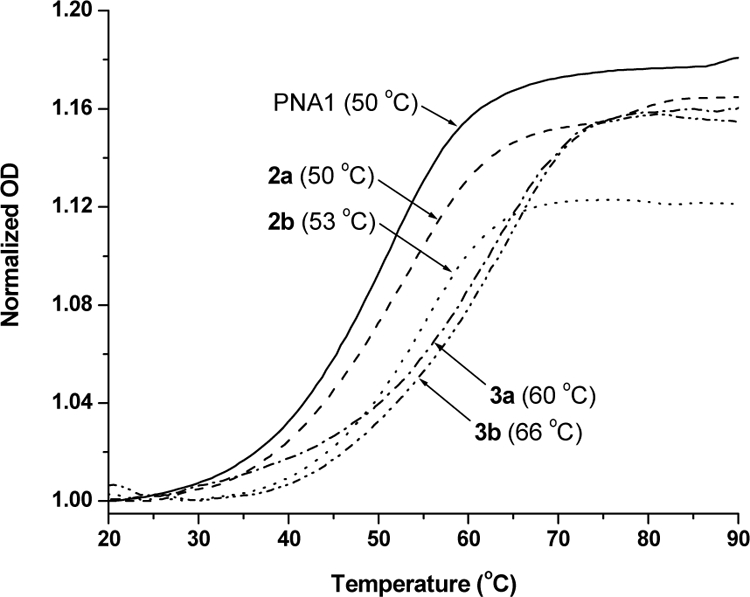
UV-melting profiles of PNA-DNA hybrid duplexes. The concentration of each strand was 5 µM prepared in 10 mM NaPi buffer (pH 7.4). Both the heating and the cooling runs were recorded, with both runs showing nearly identical profiles. The Tms were determined by taking the first derivative of the melting curves.
3. Conclusion
In summary, we have shown that depending on the size of the protecting group (Boc vs. PhFl) and the synthetic route (reductive amination vs. Mitsunobu) through which the backbone intermediates were prepared, optically-pure γPNA monomers could be obtained. Utilization of PhFl-protection via reductive amination and Boc-protection via the Mitsunobu route yielded optically-pure backbone intermediates and the corresponding γPNA monomers, while Boc-protection via reductive amination resulted in considerable epimerization. Further study revealed that reduction of the Weinreb amide or oxidation of the alcohol to aldehyde via the Swern or ParikhDoering method contributes about half to the overall epimerization, while the other half comes from the reductive amination of aldehyde with glycine to the backbone.
Incorporation of the epimerized monomers into oligomers resulted in conformational heterogeneity and reduced the degree of conformational preorganization and binding affinity of PNA towards complementary DNA strand, as demonstrated in the CD and UV-thermal melting measurements. When the oligomer was comprised entirely of the epimerized building blocks (A, C, G, T), a dramatic reduction in the Tm (15–20 °C, unpublished data) was observed. Obtaining optically-pure γPNA monomers is therefore critical to achieving ultrahigh binding affinity and effectively being able to invade double helical B-DNA or RNA (data not shown).
4. Experimental Section
Material and Methods.
All commercial reagents were used without further purification. Solvents were dried by standard methods and distilled freshly prior to use. All chemicals were purchased from Aldrich except for L-alanine, which was purchased from Novabiochem. All Boc/Z-protected PNA monomers were purchased from Applied Biosystems. 1H-,13C- and 19F-NMR spectra were recorded on a Bruker Avance AV-300 NMR spectrometer using standard Bruker software. Flash chromatography was performed using standard grade silica gel from Sorbent Technologies. TLC was performed with silica gel 60 F-254 precoated plates from Sorbent Technologies. MALDI-TOF experiments were performed on a PerSeptive Biosystems Voyager STR MALDI-TOF mass spectrometer using a 10mg/ml solution of α-hydroxycinnamic acid in ACN-water (1:1) with 0.1% TFA. Mass spectra were recorded on a Finnigan LCQ ESI/APCI ion trap mass spectrometer by electrospray ionization. CD experiments were performed on a Jasco J-715 spectropolarimeter equipped with a thermoelectrically-controlled single-cell holder. UV-Vis measurements were taken on a Varian Cary 300 Bio spectrophotometer equipped with a thermoelectrically controlled multi-cell holder.
General procedures
Oligomer synthesis
All PNA oligomers were synthesized on methylbenzhydryl amine (MBHA) resin according to the published procedures. All oligomers were cleaved from the resin using m-cresol: thioanisole:TFMSA:TFA (1:1:2:6) cocktail. The resulting mixtures were precipitated with ether, purified by RP-HPLC and characterized by MALDI-TOF mass spectrometry. Concentrations of PNA oligomers were determined from the OD at 260 nm recorded at 90 ºC, using the following extinction coefficient: T = 8,600 M−1cm−1, A = 13,700 M−1cm−1, C = 6,600 M−1cm−1, and G = 11,700 M−1cm-1. The typical yields for these oligomers are in the 27–37% range after purification.
Circular Dichroism (CD)
All samples were prepared in buffer containing 10 mM sodium phosphate (pH 7.4) at 5 µM strand concentration each. All samples were annealed prior to recording the CD spectra, by heating to 90 ºC for 5 min followed by gradual cooling to room temperature. All spectra represent an average of at least ten scans collected at the rate of 100 nm/min between 200–320 nm in a 1-cm path-length cuvette at 22 ºC. CD spectrum from buffer solution was subtracted from the sample spectra, which were then smoothed via an eight-point adjacent averaging algorithm.
UV-Melting of PNA-DNA hybrid duplexes
All samples were prepared in buffer containing 10 mM sodium phosphate (pH 7.4). UV-Vis absorbance at 260 nm was recorded at the rate of 1°C/min for both the heating (2095 °C) and cooling (95–20 °C) runs. The Tms were determined by taking the first derivative of the heating profiles (both the heating and cooling profiles yielded similar results, with little hysteresis).
Monomer Synthesis
N-tert-Butoxycarbonyl alanine (2a):
L-alanine 1 (10 g, 112.2 mmol) was dissolved in water and sodium bicarbonate (2 equiv., 224.5 mmol, 18.9 g) was added with stirring. The resulting solution was cooled to 5 °C and Boc2O (1.5 equiv., 168.4 mmol, 36.8 g) was added slowly along with 1,4-dioxane (also cooled). The resulting mixture was stirred at 0 °C for 1 h and allowed to warm to room temperature overnight. Water was added and the aqueous layer was extracted twice with EtOAc (200 mL). The organic layer was back extracted twice with saturated NaHCO3 solution. The combined aqueous layers were acidified to pH 1 with 10% HCl and extracted 3 times with EtOAc (100 mL). The combined organic layers were dried (sodium sulfate) and concentrated in vacuo. The resulting residue was purified by silica gel chromatography. 1H-NMR (300 MHz, DMSO-d6): δ 1.21 (d, J = 7.32 Hz, 3H), δ 1.37 (s, 9H), 3.92 (q, J = 7.42 Hz, 1H), 7.05 (d, J = 7.54 Hz, 1H), 12.03 (brs, 1H). ESI-MS (positive mode): m/z 190.20 (Mcalc for C8H15NO4: 189.21).
N-tert-Butoxycarbonyl alanine Weinreb amide (3a):
To a stirred solution of 2a (10 g, 52.9 mmol ) in anhydrous DCM (100 mL) was added N-methylmorpholine (2 equiv., 105.8 mmol, 10.7 g) at −15 °C (dry ice/methanol) under argon. Isobutylchloroformate (1 equiv., 55.5 mmol, 7.2 mL) was added drop-wise. After stirring for 15 min at the same temperature N,O-dimethylhydroxylamine hydrochloride (1.1 equiv., 58.2 mmol, 5.6 g) was added and the ice bath was removed. The reaction mixture was allowed to stir at rt for 14 h. The solution was diluted with DCM (200 mL) and washed with 10% aq. KHSO4 (100 mL), saturated NaHCO3 solution (2 × 50 mL) and saturated NaCl solution. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by silica gel column chromatography to afford 3a as a white solid. Yield 90%, Rf: 0.6 (3:2, EtOAc: hexane). 1H-NMR (300 MHz, CDCl3): δ 1.28 (d, J = 6.91 Hz, 3H), 1.41 (s, 9H), 3.18 (s, 3H), 3.74 (s, 3H), 4.65 (m, 1H), 5.22 (brs, 1H). 13C-NMR (75 MHz, CDCl3): δ 18.0, 27.8, 31.6, 45.9, 61.0, 78.8, 154.6, 173.1. ESI-MS (positive mode): m/z 233.7 (Mcalc for C10H20N2O4: 232.8).
N-tert-Butoxycarbonyl alanal (4a-i):
A stirred solution of 3a (5 g, 21.4 mmol) in dry THF (100 mL) was cooled to 0 °C under argon. Lithium aluminum hydride (2.0 equiv, 42.8 mmol, 1.6 g) was added portion-wise and after 1 h, the reaction mixture was quenched with 10% KHSO4 solution (100 mL) and allowed to stir for 10 minutes. The solution was then extracted with ethyl acetate (3 ×100 mL). The combined organic layers were washed with 1M HCl (2 × 50 mL), saturated NaHCO3 (2 × 50 mL) and saturated NaCl (2 × 50 mL). The organic layer was dried on Na2SO4 and evaporated under reduced pressure to afford white solid which was used in the next step without further purification. Yield 90%, Rf: 0.8 (3:2, EtOAc: Hexane). 1H-NMR (300 MHz, CDCl3): δ 1.33 (d, J = 7.42 Hz, 3H), 1.45 (s, 9H), 4.22 (m, 1H), 5.09 (brs, 1H), 9.56(s, 1H). 13C NMR (75 MHz, CDCl3): δ 14.3, 27.8, 55.0, 79.5, 154.8, 199.2. ESI-MS (positive mode): m/z 174.2(Mcalc for C8H15NO3: 173.2).
N-tert-butoxycarbonyl-1-(methyl)-aminoethylglycine ethyl ester (5a-i):
A stirred solution of 4a-i (1.1 g, 6.5 mmol) in dry MeOH (50 mL) was cooled to 0 °C under argon. In a separate round bottom flask, ethyl glycinate hydrochloride (2.1 equiv., 13.5 mmol, 1.9 g) was dissolved in MeOH (5 mL) and DIPEA (2.1 equiv, 13.6 mmol, 2.4 mL). The flasks were then mixed and stirred at 4 °C for 4 h at which point acetic acid (1.2 mL, 20 mmol) was added, followed by NaBH3CN (0.6 g, 10 mmol) and the solution was allowed to stir for another 30 min. 10% NaHCO3 solution (100 mL) was then added to the reaction mixture and extracted it with EtOAc (3 × 30 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4 and filtered. The solvent was evaporated under reduced pressure and the resulting residue was purified by column chromatography to afford 5a-i as viscous liquid. Yield 68%, Rf = 0.75 (20:1, EtOAc: EtOH). 1H-NMR (300 MHz, CDCl3): δ 1.15 (d, J = 6.6 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.46 (s, 9H), 2.64 (d, J = 5.9 Hz, 2H), 3.41(ABq, J = 17.4 Hz, 2H), 3.74 (m, 1H), 4.21 (q, J = 7.1 Hz, 2H), 4.79 (brs,1H). 13C NMR (75 MHz, CDCl3): δ 13.6, 18.4, 27.8, 45.6, 50.3, 53.9, 60.1, 78.4, 155.0, 171.9. ESI-MS (positive mode): m/z 261 (Mcalc for C12H24N2O4: 260.33).
N-(9-(9-Phenylfluorenyl))-alanine (2b).
To a stirred suspension of L-alanine (18 g, 0.2 mol) in 600 mL of CHCl3/CH3CN (5/1) in a Morton flask was added TMS-Cl (1 equiv., 0.2 mol, 26 mL) at room temperature under nitrogen. The mixture was heated under reflux for 2 h and then allowed to cool to room temperature. Addition of Et3N (20 equiv., 4 mol, 56 mL) at a rate sufficient to maintain gentle reflux was followed by the addition of Pb(NO3)2 (0.12 mol, 40 g) and a solution of 9-bromo-9-phenylfluorene (1 equiv., 0.2 mol, 64.2 g) in CHCl3, (200 mL). The resulting mixture was vigorously stirred for 48 h at room temperature, and then excess MeOH (0.5 mol) was added. Filtration followed by evaporation gave a residue which was partitioned between Et2O (1 L) and precooled 5% aqueous citric acid (1 L). The organic phase was washed with 1N NaOH (2 × 400 mL) and H2O (2 × 200 mL), and the combined aqueous layers were washed with Et2O (400 mL), cooled to 0 °C and neutralized with glacial AcOH. The precipitated product was extracted with Et2O (4 × 300 mL). The combined organic layers were washed with water (200 mL), dried over anhydrous Na2SO4 and evaporated to give compound 2b as a light yellow foam (55g, 84%), which was used without further purification. Careful crystallization of 2b from EtOAc/hexane produced a white solid: 1H-NMR (300 MHz, CDCl3): δ 1.18 (d, J = 7.16 Hz, 3H), 2.72 (q, J = 7.16 Hz, 1H), 6.14 (brs, 1H), 7.21–7.50 (m, 11H), 7.70–7.79 (m, 2H). ESI-MS (positive mode): m/z 329.9 (Mcalc for C22H19NO2: 329.14).
N-(9-(9-Phenylfluorenyl))-alanine Weinreb amide (3b):
To a stirred solution of 2b (10 g, 30.4 mmol) in dry DCM:DMF (3:1, 20 mL) were added DCC (1.1 equiv., 33.4 mmol, 6.9 g) and DhbtOH (1.1 equiv., 33.4 mmol, 5.4 g) under nitrogen at 0 °C. The stirring was continued for 1 h at the same temperature. In a separate flask the mixture of N,Odimethylhydroxylamine hydrochloride (1.1 equiv., 33.4 mmol, 3.2 g) and DIPEA (1.1 equiv., 33.4 mmol, 5.8 mL) in 5 mL DCM: DMF (3:1), cooled to 0 °C was added and the ice bath was removed. The reaction mixture was stirred at rt for 14 h. Following evaporation of the solvent, the residue was partitioned between ethyl acetate (100 mL) and saturated NaHCO3 solution (100 mL). The organic layer was washed with 10 % KHSO4 (3 × 50 mL), 10 % NaHCO3 (3 × 50 mL), brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure and purified by column chromatography to afford 3b as pale yellow solid. Yield 73%, Rf: 0.6 (3:2, EtOAc:hexane). 1H-NMR (300 MHz, CDCl3): δ 1.06 (d, J = 6.93 Hz, 3H), 2.7–3.1 (m, 1H), 2.85 (s, 3H), 2.88 (s, 3H), 3.3 (brs, 1H), 7.1–7.6 (m, 11H), 7.63–7.75 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 21.3, 31.4, 47.6, 59.7, 72.7, 118.9, 119, 124, 125.5, 126.2, 127.5, 127.6, 139.3, 140.6, 144.3, 148.9, 149.7, 176.5. ESI-MS (positive mode): m/z 372.8 (Mcalc for C24H24N2O2: 372.1).
N-(9-(9-Phenylfluorenyl))-alanal (4b):
The target compound was made from compound 3b by following the synthesis procedure of 4a. Yield 80%, Rf: 0.2 (3:2, EtOAc:hexane). 1H-NMR (300 MHz, CDCl3): δ 0.9 (d, J = 7.27 Hz, 3H), 2.57 (qd, J = 1.68, 7.27 Hz, 1H), 2.7 (bs, 1H), 7.05–7.45 (m, 11H), 7.56–7.65 (m, 2H), 9.17 (d, J = 1.68 Hz, 1H).
N-(9-(9-Phenylfluorenyl))-1-(methyl)-aminoethyl glycine ethyl ester (5b):
The corresponding backbone 5b was synthesized from the aldehyde 4b via reductive amination as described in the synthesis of 5a. Yield 76%, Rf: 0.25 (3:2, EtOAc:hexane). 1H-NMR (300 MHz, CDCl3): δ 0.61 (d, J = 5.9 Hz, 3H), 1.27 (t, J = 7.13 Hz, 3H), 2.10 (bs, 1H), 2.2–2.4 (m, 3H), 3.16 (ABq, J = 17.32 Hz, 2H), 4.18 (q, J = 7.13 Hz, 2H), 7.12–7.28 (m, 5H), 7.32–7.39 (4H, m), 7.39–7.47 (m, 2H), 7.66–7.72 (m, 2H). 13C-NMR (75 MHz, CDCl3): δ13.7, 20.9, 47.2, 50.3, 55.7, 60, 72.4, 119.3, 124.7, 125, 125.6, 126.4, 127.1, 127.5, 127.7, 139.7, 140, 145.2, 149.6, 150.4, 172.
N-tert-butyloxycarbonyl alanol (6):
To a cold (−20 °C) stirred solution of Boc-alanine 2a (5 g, 26.5 mmol) in anhydrous 1,2-dimethoxyethane (DME) (30 mL) were successively added N-methylmorpholine (1 equiv., 26.5 mmol, 2.9 mL) and isobutylchloroformate (1 equiv., 26.5 mmol, 3.5 mL) under nitrogen. After 10 min, the precipitated white solid was removed by filtration and washed with DME (3 × 10 mL). The filtrate and DME washings were combined in a 500 mL round bottom flask and cooled to −20 °C. A solution of sodium borohydride (1.5 equiv., 39.6 mmol, 1.5 g) in water (15 mL) was added portion wise, followed by water (500 mL). The reaction mixture was extracted with ethyl acetate (3 × 200 mL) and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, evaporated to dryness, and then purified by silica gel column chromatography. Yield 90%, Rf: 0.5 (3:2, EtOAc:hexane). 1H-NMR (300 MHz, CDCl3): δ 1.15 (d, J = 6.6 Hz, 3H), 1.46 (s, 9H), 2.59 (brs, 1H), 3.49 (dd, J = 6.1, 10.7 Hz, 1H), 3.62 (dd, J = 4.0, 10.7 Hz, 1H), 3.78 (m, 1H), 4.58 (brs, 1H). ESIMS (positive mode): m/z 176 (Mcalc for C8H17NO3: 175.23).
[(2-tert-Butoxycarbonylamino-propyl)-(2-nitrobenzene sulfonyl)amino]-acetic acid ethyl ester (8):
Compound 6 (3.2 g, 18 mmol), o-NBS-GlyOEt (0.95 equiv., 17.1 mmol, 4.7 g) and triphenylphosphine (1 equiv., 18 mmol, 4.7 g) were dissolved in freshly distilled anhydrous THF (100 mL). The solution was stirred at 0 °C under nitrogen, and diisopropylazodicarboxylate (DIAD 3.5 mL, 18 mmol) was added drop-wise over the course of 20 min. The reaction mixture was stirred overnight at room temperature. The solvent was evaporated to dryness and the oily residue was re-dissolved in diethyl ether (25 mL) and cooled to 0 °C. The next day the precipitate was filtered off and the solvent evaporated in vacuo. The remaining residue was purified by silica gel column chromatography to give pale yellow oil. Yield 78%, Rf: 0.5 (2:3, EtOAc:hexane). 1H-NMR (300 MHz, CDCl3): δ 1.14 (m, 6H), 1.40 & 1.42 (2xs, 9H), 3.3 & 3.37 (2xd, J = 5.4 Hz, 1H), 3.50 (m, 1H), 3.87 (m, 1H), 4.04 (m, 2H), 4.25 (ABq, J = 19 Hz, 2H), 4.69 (brs, 1H), 7.58 (m, 1H), 7.67 (m, 2H), 8.01 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 13.4, 18.0, 27.8, 43.2, 44.3, 47.4, 52.4, 60.8, 61.2, 78.9, 123.5, 125.0, 130.1, 131.2, 132.3, 132.7, 133.1, 147.4, 154.9, 168.0. ESI-MS (positive mode): m/z 447 (Mcalc for C18H27N3O8S: 445.49).
(2-tert-Butoxycarbonylamino-propylamino)-acetic acid ethyl ester (5a-ii):
To a stirred solution of 8 (3 g, 6.7 mmol) in acetonitrile (75 mL) were added potassium carbonate (1.5 equiv., 10.1 mmol, 1.4 g) and thiophenol (2 equiv., 13.5 mmol, 1.6 mL) at room temperature. The reaction mixture was stirred overnight. The solvent was evaporated and the residue was partitioned between ethyl acetate (100 mL) and water (50 mL). The ethyl acetate layer was extracted with 10% aqueous citric acid (3 × 100 mL). To the combined aqueous layers solid potassium carbonate was added portionwise in high excess (CAUTION! A strong evolution of gas was observed). The aqueous mixture was then extracted with ethyl acetate (3 × 75 mL). The combined ethyl acetate layers were washed with brine and dried over Na2SO4. The solvent was evaporated and the residue was purified by silica gel column chromatography. Yield 65%, Rf: 0.75 (1:20, EtOH:EtOAc). 1H-NMR (300 MHz, CDCl3): δ 1.15 (d, J = 6.6 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.46 (s, 9H), 2.64 (d, J = 5.9 Hz, 2H), 3.41(ABq, J = 17.4 Hz, 2H), 3.74 (m, 1H), 4.21 (q, J = 7.1 Hz, 2H), 4.79 (brs, 1H). ESI-MS (positive mode): m/z 261 (Mcalc for C12H24N2O4: 260.33).
Boc γ-ala PNA thymine monomer ethyl ester (9a-i and 9a-ii):
To a stirred solution of thymine acetic acid (TCH2CO2H, 1.0 g, 5.3 mmol) in dry DMF (20 ml) were added DCC (1 equiv., 5.3 mmol, 1.1 g) and DhbtOH (1 equiv., 5.3 mmol, 0.9 g) under argon. The stirring was continued for 1 h at room temperature. Compound 5a-i (1.1 g, 4.4 mmol) in dry DMF (5 mL) was added and the reaction was stirred at 50 °C for 14 h. Following evaporation of the solvent, the residue was partitioned between EtOAc (100 mL) and saturated NaHCO3 solution (100 mL). The organic layer was washed with 10 % KHSO4 (3 × 50 mL), 10 % NaHCO3 (3 × 50 mL), brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure and purified by silica gel column chromatography to afford 9a-i as off white foamy solid. The same procedure was adopted to get 9a-ii from 5a-ii. Yield 60%, Rf = 0.75 (20:1, EtOAc:EtOH). 1H-NMR (300 MHz, DMSO-d6, 2 rotamers): δ 0.95 & 1.08 (2xd, J = 6.67 Hz, 3H), 1.17 & 1.24 (2xt, J = 7.15 Hz, 3H), 1.36 & 1.37 (2xs, 9H), 1.75 (s, 3H), 3.02–3.31 (m, 3H), 3.46–3.78 (m, 1H), 3.93–4.21 (m, 3H), 4.3 & 4.46 (2xs, 1H), 4.67 (ABq, J = 16.4 Hz, 1H), 6.67 & 6.85 (brs, 1H), 7.23(s, 1H), 11.24 & 11.26 (2xs, 1H). 13 C NMR (75 MHz, DMSO-d6): δ 11.8, 13.9, 17.8, 18.2, 24.4, 25.2, 28.1, 33.2, 44.5, 47.6, 48.1, 49.1, 51.7, 52.4, 60.3, 61.0, 77.6, 77.9, 108.1, 148.1, 150.9, 155.1, 164.3, 167.4, 167.8, 168.7, 169.1. ESI-MS (nagative mode): m/z 869 (Mcalc for C43H50N8O12: 870.9).
Boc-γ-ala PNA thymine monomer (10a-i and 10a-ii):
To a solution of 9a-i (1.3 g, 1.5 mmol) in THF (20 mL) was added drop-wise 2N sodium hydroxide (20 mL) at 0 °C. After stirring for 30 min at the same temperature, water (100 mL) was added and the pH was adjusted to 4 at which point precipitate was formed. Filtered the precipitate, washed the solid with cold water and dried under vacuum to obtain the required title compound 10a-i (1.2 g) Yield: 95%, Rf = 0.1 (EtOAc:EtOH = 4:1). The same procedure was adopted for the synthesis of compound 10a-ii from 9a-ii. 1H-NMR (300 MHz, DMSO-d6, 2 rotamers): δ 0.96 & 1.08 (2xd, J = 6.9 Hz, 3H), 1.37 & 1.38 (2xs, 9H), 1.75 (s, 3H), 3.01–3.46 (m, 2H), 3.65–4.05 (m, 3H), 4.45 (s, 1H), 4.69 (ABq, J = 16.7 Hz, 1H), 6.7 & 6.88 (brs, 1H), 7.24 (s, 1H), 11.26 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ 11.8, 17.8, 18.2, 28.7, 44.4, 44.6, 47.6, 48.0, 50.1, 51.9, 52.2, 77.6, 77.9, 108.0, 141.8, 150.9, 155.1, 164.3, 167.1, 167.8, 170.5, 171.1. ESI-MS (positive mode): m/z 848.8(Mcalc for C41H46N8O12: 847.9).
PhFl-γ-ala PNA thymine monomer ethyl ester (9b):
The aforementioned procedure for compounds 9a-i and 9a-ii was used for the synthesis of 9b as white solid from the starting material 5b. Yield 75%, Rf: 0.7 (80:20, EtOAc:EtOH). 1HNMR (300 MHz, CDCl3, 2 rotamers): δ 0.75 & 0.86 (2xd, J = 6.48 Hz, 3H), 1.22 & 1.30 (2xt, J = 7.15 Hz, 3H), 1.89 (2xs, 3H), 2.15 & 2.7 (2xq, J = 6.48 Hz), 2.9–3.25 (m, 2H), 3.6 & 3.75 (ABq, J = 17.29 Hz, 2H), 4.10 & 4.16 (2xq, J = 7.15 Hz, 2H), 4.25 & 4.30 (2xABq, J = 16.33 Hz, 2H), 6.82–6.89 (2xq, J = 1.15 Hz), 7.16–7.4 (m, 11H), 7.64–7.76 (m, 2H), 8.08 (s, 1H). 13C-NMR (75 MHz, CDCl3): δ11.8, 13.9, 20, 20.4, 20.7, 47.1, 47.7, 53.8, 54.9, 59.6, 60.3, 61, 72.8, 107.9, 120, 125.3, 125.9, 126.7, 127.6, 128, 139.7, 140, 141.9, 145.3, 145.5, 149.5, 150.8, 164.3, 167.2, 167.4, 168.4, 168.9. ESI-MS (positive mode): m/z 567.1 (Mcalc for C33H34N4O5 :566.2)
PhFl-γ-ala PNA thymine monomer (10b):
The aforementioned procedure for compounds 10a-i and 10a-ii was used for the synthesis of 10b as white solid from 9b. Yield 80%, Rf: 0.6 (80:20, EtOAc:EtOH). 1H-NMR (300 MHz, DMSO-d6, 2 rotamers): δ 0.52 & 0.67 (2xd, J = 6.01 Hz, 3H), 1.72 &1.74 (2xs, 3H), 2.19 (2xq, J = 6.01 Hz, 1H), 2.8–3.3 (m, 2H), 3.3–4.0 (m, 2H), 7.10–7.55 (m, 12H), 7.7–7.9 (m, 2H), 11.20 (1bs, 1H). 13C-NMR (75 MHz, CDCl3): δ11.8, 14, 21, 19.9, 47.8, 49.1, 54.6, 72.8, 107.9, 120.2, 125.5, 125.9, 126.8, 128, 139.8, 141.9, 150.8, 164.3, 167.1, 170, 170.3, 171.9. ESI-MS (positive mode): m/z 539.1 (Mcalc for C31H30N4O5 :538.2)
Synthesis of MTPA-derivative of γ-alanine-PNA backbones 11a-i and 11a-ii:
To the stirred solution of Boc protected PNA backbones 5a-i and 5a-ii (0.5 g, 1.9 mmol) was added 5% m-cresol in TFA (7 mL) in DCM at 0 °C. The reaction mixture was stirred for 1 h at rt. The solvent was evaporated under reduced pressure and the resulting residue was triturated with diethyl ether (3 × 5 mL). The residue was dried under high vacuum for several hours and then dissolved in anhydrous DCM (5 mL) at room temperature under nitrogen. DIPEA (2.5 equiv., 4.6 mmol, 0.8 mL) and MTPACl (1.5 equiv., 2.8 mmol, 0.6 g) were added to the dissolved solution at 0 °C and the reaction mixture was stirred for 14 h at rt. The solution was diluted with DCM (50 mL) and washed with water (50 mL), saturated NaHCO3 (30 mL) and saturated NaCl (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo and the residue was purified by silica gel column chromatography to get the title compound 11a-i and 11a-ii. Yield 70%, Rf: 0.75 (1:20, EtOH: EtOAc). 1H-NMR (300 MHz, DMSO-d6): 1.10 (d, J = 6.7 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H), 2.55 & 2.59 (2xd, J = 7.0 Hz, 2H), 3.27(ABq, J = 17.2 Hz, 2H), 3.31 (brs, 1H), 3.40 & 3.41 (2xs, 3H), 3.98 (m, 1H), 4.08 (q, J = 7.1 & 14.3 Hz, 2H), 7.43 (m, 3H), 7.55 (m, 2H), 8.09 (d, J = 8.7 Hz, 1H). 13C-NMR (75 MHz, DMSO-d6): 13.9, 17.9, 44.4, 49.7, 52.7, 54.6, 60.1, 127.3, 128.2, 129.4, 132.9, 165.1, 172.
Synthesis of MTPA-derivative of γ-alanine-PNA backbone 11b:
Compound 5b (0.5 g, 1.9 mmol) was dissolved in anhydrous methanol (10 mL) at room temperature under nitrogen. 10% Pd/C (150 mg) was added carefully under nitrogen. The resulting reaction mixture was stirred for 2 h under hydrogen atmosphere. After the disappearance of starting material as judged by TLC, the reaction mixture was filtered through a celite pad and washed with methanol. The filtrate was concentrated in vacuo and the deprotected-free amine was directly taken to the next step without further purification. Amine (0.3 g, 1.9 mmol) was dissolved in anhydrous DCM (5 mL) at room temperature under nitrogen. DIPEA (1.5 equiv., 2.8 mmol, 0.5 mL) and MTPA-Cl (1.5 equiv., 2.8 mmol, 0.5 g) were added at 0 °C to the solution. The reaction mixture was stirred at room temperature for 14 h under nitrogen and quenched by adding water (30 mL). The reaction mixture was extracted with dichloromethane and the organic layer was washed with water (25 mL), saturated NaHCO3 (30 mL), saturated NaCl (30 mL) solutions and dried over anhydrous Na2SO4. The solution was filtered, and the solvent was removed under reduced pressure and the resulting material was subjected to silica gel column chromatography to obtain the required compound 12b. Yield: 70%, Rf: 0.75 (1:20, EtOH:EtOAc).
Synthesis of MTPA-derivative of γ-alanine PNA thymine monomers, 12a-i and 12a-ii:
Boc-group in compound 10a-i and 10a-ii was deprotected using 5% m-cresol in TFA. Deprotected-amine (0.5 g, 0.6 mmol) was dissolved in a mixture of water and acetone (1:1, 10 mL) at room temperature. Solid NaHCO3 (3 equiv., 1.8 mmol, 0.2 g) and MTPA-Cl (1.5 equiv., 0.9 mmol, 0.2 g) were added and the reaction mixture was stirred for 14 h at rt. Acetone was removed in vacuo and the resulting reaction mixture was diluted with water and extracted with ethyl acetate (2 × 20 mL). Combined organic layers were washed with saturated NaCl (20 mL) solution, dried over anhydrous Na2SO4 and filtered. The solvent was concentrated under reduced pressure and the residue was purified by column chromatography to afford MTPA derivatives 12a-i and 12a-ii. Yield 65%, Rf: 0.1 (1:20, EtOH:EtOAc). 1H-NMR (300 MHz, DMSO-d6): 1.06 (d, J = 6.5 Hz, 3H), 1.16–1.2 (2xt, J = 7.0 Hz, 3H), 1.72 & 1.74 (2xs, 3H), 3.11–3.36 (m, 2H), 3.34 & 3.45 (2xs, 3H), 3.78 (ABq, J = 18.8 Hz, 1H), 3.91 (ABq, J = 18.8 Hz, 1H), 4.09–4.28 (m, 1H), 4.29–4.54 (m, 1H), 4.59 (ABq, J = 16.8 Hz, 1H), 7.09 & 7.14 (2xs,1H), 7.38–7.57 (m, 5H), 8.23 & 8.38 (2xd, J = 8.5 Hz, 1H), 11.27 (brs, 1H). 13C-NMR (75 MHz, DMSO-d6): δ 11.8, 17.4, 44.1, 47.6, 51.6, 52.2, 54.9, 83.5, 107.9, 126.9, 128.2, 129.3, 133.4, 142.1, 151, 164.4, 164.9, 168.4, 171.3.
Synthesis of MTPA-derivative of γ-alanine PNA thymine monomers, 12b:
Compound 10b was hydrogenated using the protocol described in the synthesis of compound 11b and the resulting amine was converted to compound 12b using the procedure mentioned in the synthesis of compounds 12a-i and 12a-ii.
N-tert-Butoxycarbonyl alanal (4a-ii):
Oxalyl chloride (2 equiv., 5.7 mmol, 0.5 mL) was added to dry DCM (5 ml) taken in a twoneck round bottom flask equipped with a stirrer under argon. The flask was cooled to −78 °C. To this flask was added DMSO (4 equiv., 11.4 mmol, 0.8 mL) drop-wise and stirring was continued for 30 min at −78 °C. Next, the alcohol 6 (0.5 g, 2.9 mmol) dissolved in dry DCM (5 mL) was added to the reaction mixture drop-wise and stirred for further 1h. Then Et3N (3 equiv., 8.6 mmol, 1.2 mL) was added with stirring at −78 °C. After 5 min the cooling bath was removed and the reaction mixture was allowed to warm to room temperature. Upon completion of the reaction as checked by TLC, the reaction was quenched with NH4Cl solution drop-wise. The organic layer was then separated. The aqueous phase was re-extracted with CH2Cl2 (20ml) and the organic layers were combined and washed with 10% citric acid solution, and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated to give the crude product. The crude product was carried forward to the next step immediately.
N-tert-Butoxycarbonyl alanal (4a-iii):
To a stirred solution of alcohol 6 (0.5 g, 2.9 mmol) in CH2Cl2/DMSO (1:1, 10 mL), SO3•py (4 equiv., 11.4 mmol, 1.8 g) and Et3N (3 equiv., 8.6 mmol, 1.2 mL) were added at 0 °C. The resulting mixture was then stirred for 1 h at room temperature before it was quenched with H2O (10 mL) and extracted with DCM (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated to afford the crude product which was carried forward to the next step immediately without any purification.
Synthesis of MTPA-derivative of alanol 14a-i, a-ii, a-iii:
To the stirred solution of Boc protected alanol 13a-i, 13a-ii and 13aiii (0.5g, 2.85 mmol) was added 5% m-cresol in TFA (7 mL) in DCM at 0 °C. The reaction mixture was stirred for 1 h at rt. The solvent was evaporated under reduced pressure and the resulting residue was column purified to get the corresponding amine product. This amine (0.05 g, 0.7 mmol) was then dissolved in anhydrous DCM (5 mL) at room temperature under nitrogen. DIPEA (2.5 equiv., 1.7 mmol, 0.3 mL) and MTPA-Cl (1.3 equiv., 0.9 mmol, 0.3 g) were added to the dissolved solution at 0 °C and the reaction mixture was stirred for 14 h at rt. The solution was diluted with DCM (10 mL) and washed with water (10 mL), saturated NaHCO3 (10 mL) and saturated NaCl (10 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo and the residue was purified by silica gel column chromatography to give the title compound 14a-i, 14a-ii and 14a-iii. Yield 80%, Rf: 0.5 (40:60, EtOAc: hexane). 1H-NMR (300 MHz, CDCl3): 1.23 (d, J = 6.8 Hz, 3H), 2.58 (brs, 1H), 3.38 (s, 3H), 3.52(m, 1H), 3.62 (m, 1H), 4.11 (m, 1H), 6.98 (d, J = 7.07 Hz, 1H), 7.39–7.56 (m, 5H). 13C-NMR (75 MHz, CDCl3): 16.7, 28.3, 47.6, 54.8, 66.3, 120.4, 122.7, 125.0, 127.3, 127.8, 128.6, 129.5,132.4,166.7.
Supplementary Material
Acknowledgment:
Financial supports for this work were provided in part by the National Institutes of Health (GM076251), National Science Foundation (CHE-1012467), and DSF Charitable Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information is available for 1H and 13C-NMR spectral data of compounds and MALDI-TOF spectra of PNA oligomers. The material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Lehn J-M Angew. Chem.-Int. Edit. Engl 1988, 27, 89. [Google Scholar]
- (2).Pedersen CJ J. Am. Chem. Soc 1967, 89, 2495. [Google Scholar]
- (3).Cram DJ Science 1988, 240, 760. [PubMed] [Google Scholar]
- (4).Dietrich B; Lehn J-M; Sauvage J-P Tetrahedron Lett 1969, 34, 2885. [Google Scholar]
- (5).Kool ET Chem. Rev 1997, 97, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nielsen PE; Egholm M; Berg RH; Buchardt O Science 1991, 254, 1497. [DOI] [PubMed] [Google Scholar]
- (7).Ray A; Norden B Faseb J 2000, 14, 1041. [DOI] [PubMed] [Google Scholar]
- (8).Nielsen PE Acc. Chem. Res 1999, 32, 624. [Google Scholar]
- (9).Lohse J; Dahl O; Nielsen PE Proc. Natl. Acad. Sci. U.S.A 1999, 96, 11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Demidov VV; Protozanova E; Izvolsky KI; Price C; Nielsen PE; Frank-Kamenetskii MD Proc. Natl. Acad. Sci. U.S.A 2002, 99, 5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nielsen PE; Christensen LJ Am. Chem. Soc 1996, 118, 2287. [Google Scholar]
- (12).Rapireddy S; Bahal R; Ly DH Biochemistry 2011, 50, 3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dragulescu-Andrasi A; Rapireddy S; Frezza BM; Gayathri C; Gil RR; Ly DH J. Am. Chem. Soc 2006, 128, 10258. [DOI] [PubMed] [Google Scholar]
- (14).Yeh JI; Shivachev B; Rapireddy S; Crawford MJ; Gil RR; Du S; Madrid M; Ly DH J. Am. Chem. Soc 2010, 132, 10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sahu B; Chenna V; Lathrop KL; Thomas SM; Zon G; Livak KJ; Ly DH J. Org. Chem 2009, 74, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sahu B; Sacui I; Rapireddy S; Zanotti KJ; Bahal R; Armitage BA; Ly DH J. Org. Chem 2011, 76, 5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bahal R; Sahu B; Rapireddy S; Lee C-M; Ly DH ChemBioChem 2012, 13, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kosynkina L; Wang W; Liang TC Tetrahedron Lett 1994, 35, 5173. [Google Scholar]
- (19).Englund EA; Appella DH Org. Lett 2005, 7, 3465. [DOI] [PubMed] [Google Scholar]
- (20).Tedeschi T; Sforza S; Corradini R; Marchelli R Tetrahedron Lett 2005, 46, 8395. [Google Scholar]
- (21).Dose C; Seitz O Org. Lett 2005, 7, 4365. [DOI] [PubMed] [Google Scholar]
- (22).Rittle KE; Homnick CF; Ponticello GS; Evans BE J. Org. Chem 1982, 47, 3016. [Google Scholar]
- (23).Stanfield CF; Parker JE; Kanellis PJ Org. Chem 1981, 46, 4797. [Google Scholar]
- (24).Miles NJ; Sammes PG; Kennewell PD; Westwood RJ Chem. Soc. Perkin. Trans. 1 1985, 2299. [Google Scholar]
- (25).Dellaria JFJ; Maki RG Tetrahedron Lett 1986, 27, 2337. [Google Scholar]
- (26).Lubell WD; Rapoport HJ Am. Chem. Soc 1987, 109, 236. [Google Scholar]
- (27).Lubell WD; Rapoport HJ Am. Chem. Soc 1989, 54, 3824. [Google Scholar]
- (28).Rodriguez M; Llinares M; Doulut S; Martinez J Tetrahedron Lett . 1991, 32, 923. [Google Scholar]
- (29).Seco JM; Quinoa E; Riguera R Chem. Rev 2004, 104, 17. [DOI] [PubMed] [Google Scholar]
- (30).Omura K; Swern D Tetrahedron 1978, 34, 1651. [Google Scholar]
- (31).Parikh JR; Doering WV E. J. Am. Chem. Soc. 1967, 89, 5505. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.