

Abstract
A structure-activity/structure-property relationship study based on the physicochemical as well as in vitro pharmacokinetic properties of a first generation matrix metalloproteinase (MMP)-13 inhibitor (2) was undertaken. After systematic variation of inhibitor 2, compound 31 was identified which exhibited microsomal half-life higher than 20 min, kinetic solubility higher than 20 μM, and a permeability coefficient greater than 20 × 10−6 cm/s. Compound 31 also showed excellent in vivo PK properties after IV dosing (Cmax = 56.8 μM, T1/2(plasma) = 3.0 h, Cl = 0.23 mL/min/kg) and thus is a suitable candidate for in vivo efficacy studies in an OA animal model.
Keywords: Matrix metalloproteinase 13, Structure-property relationship, Structure-activity relationship, Microsomal stability, Solubility, Permeability, In vitro and in vivo pharmacokinetics
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
1. Introduction
Osteoarthritis (OA), the degeneration of cartilage, is the most common disabling condition in the Western world.1 Several risk factors such as genetic predisposition, obesity, and joint malalignment are associated with OA while the pathogenesis of cartilage degeneration remains largely unexplained.2 In principle, any synovial joint in the human body can be affected by OA, but commonly the larger, weight-bearing joints such as the hips, knees, and the lumbar region of the spine are mostly targeted. Clinically, pain and stiffness of affected joints with up to complete loss of mobility are the main symptoms of OA. So far, medical treatment options are rather limited and are mainly focused on relieving pain to maintain motoric and functional capabilities.3 Therefore, oral analgesics, especially paracetamol, ibuprofen, and celecoxib, which are all non-steroidal anti-inflammatory drugs (NSAID) and selective COX-2 inhibitors, are the first-choice drugs for the treatment of OA.4 These treatment options are ultimately ineffective because the biochemical mechanisms responsible for cartilage degeneration are not addressed. Therefore, there is an urgent need for the development of disease modifying agents to increase the quality of life for OA patients. Despite intensive academic and industrial research over the past years the problem still remains unsolved.5
Several zinc-dependent matrix metalloproteinases (MMPs) are known to play crucial roles in the turnover of extracellular matrix (ECM) and the associated destruction of articular cartilage in OA6 In particular, the role of MMP-13, which is also known as human collagenase-3, has been extensively analyzed and the enzyme shown to be primarily responsible for the cleavage of type II collagen in OA.7
Multiple attempts at developing an MMP-13 inhibitor-based therapeutic have failed mostly due to dose limiting side effects collectively described as musculoskeletal syndrome (MSS).8 The exact cause of MSS is not known. It is believed that the zinc- chelating properties of the first generation MMP inhibitors evoked a lack of selectivity toward other metalloproteases and/or non-related proteins.8 As a consequence, compounds lacking zinc chelating groups have been screened to identify inhibitors that did not suffer from non-specific binding. Several selective, nonzinc chelating MMP-13 inhibitors have been reported and biologically evaluated.9 Although all of these compounds showed very impressive selectivity profiles toward MMP-13, none has yet completed clinical trials. Co-crystal structural analysis and molecular modeling studies indicate that most of the non-zinc chelating compounds modulated the activity of MMP-13 by binding within a “specificity pocket” (subsite S1’) and its surrounding loop (S1’ specificity loop or Ω-loop).9d, 10
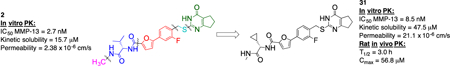
An initial high-throughput screening (HTS) effort at the Scripps Research Institute, as part of the Molecular Libraries Probe Production Centers Network (MLPCN) program, followed by a first round of medicinal chemistry efforts identified compound 1 (Figure 1A), a 2.4 μM inhibitor of MMP-13.11 The X-ray co-crystal structure of MMP-13 in complex with 1 revealed that the inhibitor bound within the known S1’ subsite.12 The p-methylphenyl ring points toward the catalytic center and shows a π-π stacking interaction with one of the imidazole rings, which coordinates the MMP-13 active site zinc ion (Figure 1B). Furthermore, the cyclopentyl moiety of the pyrimidinone unit of 1 is oriented toward the S1’ specificity loop.9d Using this cocrystal structure for design purposes, we recently reported a three-step approach for the development of highly potent and selective MMP-13 inhibitors.13 In steps one and two we applied comparative structural analyses followed by molecular design to obtain non-selective but highly potent Zn2+-chelating compounds. In the third step the chelating moiety was removed to increase selectivity. This approach resulted in the synthesis of a set of compounds, including 2, which exhibit a 1000-fold improvement of inhibitory potency compared to 1. Due to the assumption that off-target inhibition of zinc containing enzymes plays a pivotal role in MSS, 2 was screened against a panel of more than 25 proteases, including MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP, which are able to cleave collagen14 and are the closest relatives of MMP-13 with sequence homologies higher than 60%. At a concentration of 5 μM, inhibitor 2 did not inhibit any protease outside the MMP-family. Even within the MMP- family, 2 showed ca. 1000-fold selectivity versus the other MMPs screened (Figure 1A).13
Figure 1.
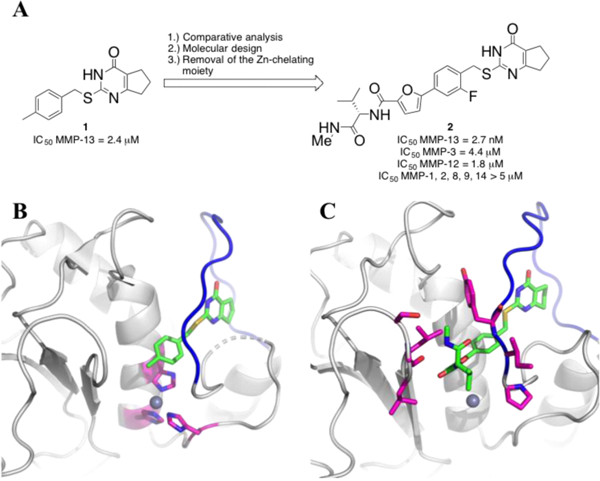
(A) Hit to lead optimization of 2. (B) The co-crystal structure of 1 with MMP-13 (PDB 4L19). Compound 1 is represented in green carbon sticks (red oxygen, blue nitrogen, and yellow sulfur), and hydrogen atoms are omitted for clarity. Three Zn-chelating His residues (His 222, His 226, and His 232) are represented in magenta carbon sticks. The S1’ specificity loop is shown as a blue tube. (C) The co-crystal structure of (S)-213 with MMP-13 (PDB 5UWL). Compound 2 is represented in green carbon sticks. The S1’ specificity loop is shown as a blue tube. The amino acid side chains that can form direct interaction with parts of (S)-2 (N-methylvaline-biaryl units) are shown as magenta sticks.
Based on the physicochemcial as well as in vitro pharmacokinetic (PK) properties of 2 we have performed and report herein a systematic variation of this inhibitor scaffold and investigated the influence of the structural modifications within a structure-activity relationship (SAR)/structure-property relationship (SPR) study. Our final goal was to obtain a set of compounds suitable for in vivo testing in an OA animal model.
2. Results and Discussion
The kinetic solubility, permeability through an artificial membrane (PAMPA assay)15, clogD7.4 and clogP values, and in vitro stability in human, rat, and mouse liver microsomes for 2 were determined (Table 1). These data were used to focus a SAR/SPR study to design selective MMP-13 inhibitors with improved pharmacokinetic (PK) properties. The goal was to reach the following in vitro property benchmarks before evaluating compounds further in in vivo PK experiments: (A) at least 20 min in vitro half-life (t1/2) in liver microsomes;16 (B) kinetic solubility higher than 15 μM as well as permeability coefficients greater than 20 × 10−6 cm/s;16 and (C) clogP and clogD7.4 values in the 0–3 and 1–4 range, respectively, values viewed as suitable for passive diffusion after oral dosing.17
Table 1.
Physicochemical and in vitro PK properties of 2.
| Property | Compound 2 |
|---|---|
| clogD7.4a | 4.2 |
| clogPb | 3.0 |
| Kinetic solubility | 15.7 μM |
| Permeability | 2.38 × 10−6 cm/s |
| t1/2 (human/rat/mouse) | 12/9/20 min |
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw.
The kinetic solubility of 2 (15.7 μM) as well as the clogP and clogD7.4 values (3.0 and 4.2) represented the lower and upper limits of the desired ranges, respectively. Compound 2 showed a moderate permeability value (2.38 × 10−6 cm/s) and a retention in the lipid bilayer of 47%. The in vitro half-life of 2 in human, mouse, and rat liver microsomes was 12, 20, and 9 min, respectively. (Sutent was used as a reference for all microsome stability studies, and its half-life was 75, 13, and 34 min in human, mouse, and rat microsomes, respectively18).
As compounds exhibiting low in vitro metabolic stabilities (t1/2) tend to have high in vivo clearance rates and poor in vivo PK properties,19 increasing the metabolic stability of 2 by removing metabolically labile groups was the first focus of the present study. The monooxygenases of the P450 family in the liver and intestine are the most important enzymes for drug metabolism.20 The binding sites of these enzymes are generally lipophilic and interact preferably with lipophilic molecules.21 To improve the metabolic stability of 2 we wanted to reduce the overall lipophilicity (lower clogP and clogD7.4) and remove or block labile groups and metabolically vulnerable sites at the same time.21 Therefore, the determination of in vitro half-life in human, mouse, and rat liver microsomes was used to assess the in vivo metabolic stability of new compounds. In principle, the reduction of clogP and clogD7.4 values by adding more polar atoms or groups would also help to increase the solubility of the inihibitor analogs.
Compound 2 was divided into five parts: (1) the terminal methylamine group (Figure 2, pink), (2) the amino acid part (Figure 2, blue), (3) the phenyl-furan group (Figure 2, red), (4) the carbon-sulfur linker (Figure 2, turquoise), and (5) the pyrimidinone scaffold (Figure 2, green). We designed and synthesized analogs of 2 by varying these five subunits in order to systematically study SAR and SPR.
Figure 2.
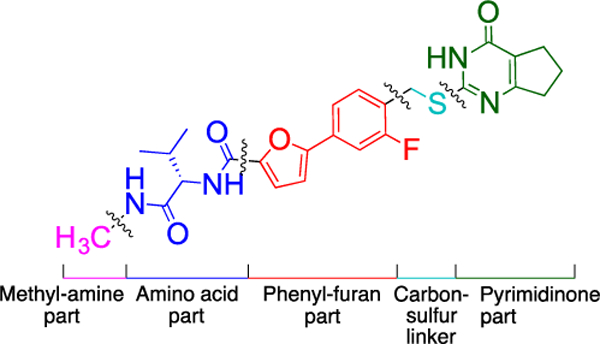
The five subunits of compound 2.
All synthesized compounds were first analyzed for their inhibitory activity against MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, and MT1-MMP catalyzed hydrolysis of the triple-helical collagen mimic substrate fTHP-1512, 22 ‘ followed by the determination of the inhbition of type II collagen cleavage at an inhibitor concentration of 20 μM. Compounds exhibiting at least 5-fold selectivity for MMP-13 and >90% inhibition of collagen cleavage at 20 μM were investigated for their metabolic stability in human, mouse, and rat liver microsomes as a guidline to assess their in vivo hepatic stability.
Variation of the terminal methylamine unit.
According to the co-crystal structure of MMP13•(S)-3 (Figure 1C), the terminal methylamine group of the inhibitor reaches into a substrate binding area of MMP-13 without direct interaction with the target structure. Thus, the terminal part of 2 is a desired location to install polar groups to improve solubility and microsomal stability. A series of compounds was prepared (Scheme 1 and Table 2). Briefly, the Suzuki reaction of bromofuran 4 and boronic acid 5 was followed by a benzylic bromination reaction. Substitution of the bromide with thiopyrimidone 6 gave methyl ester 7, which was hydrolyzed under basic conditions. Subsequent amino acid coupling with different valine derivatives resulted in the synthesis of 15 analogs (Table 2).13
Scheme 1.
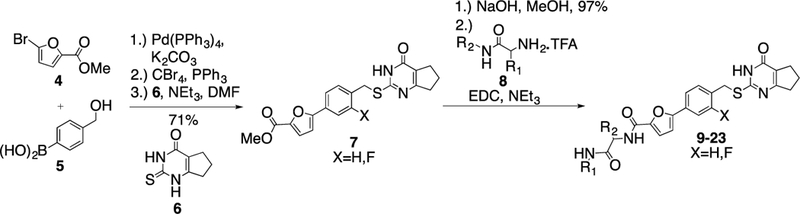
Synthesis of compounds with variation of the terminal methylamine unit of 2.
Table 2.
Inhibition potency, selectivity profile, and stability in rat, mouse, and human liver microsomes of compounds derived from Scheme 1.
| IC50 (nM)* | Microsome stability (min) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | X | MMP-13 | MMP-2 | MMP-8 | clogD7.4a | clogPb | rat | mouse | human |
| 2 | Me | 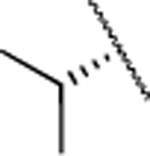 |
F | 2.7±0.6 | >5000 | >5000 | 4.2 | 3.0 | 9 | 20 | 12 |
| 9 | 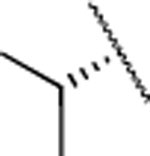 |
H | 12.9±1. 1 | 500±100 | 140±25.0 | 2.2 | 2.6 | 61 | 39 | >120 | |
| 10 | 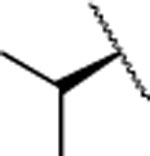 |
H | 14.4±1.7 | 362±82.0 | >5000 | 2.2 | 2.6 | nd | nd | nd | |
| 11 | 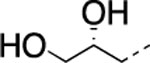 |
 |
H | 7.1±0.6 | 172±23.0 | 83±26.0 | 2.97 | 2.6 | 45 | >120 | >120 |
| 12 | 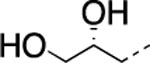 |
 |
H | 6.9±1.2 | 232±32.0 | 117±51.0 | 2.97 | 2.6 | nd | nd | nd |
| 13 | 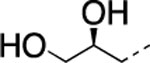 |
 |
H | 5.5±0.6 | 114± 18.0 | 38±19.0 | 2.92 | 2.6 | nd | nd | nd |
| 14 |  |
 |
H | 6.4±0.9 | 219±28.0 | 110±46.0 | 2.97 | 2.6 | 42 | >120 | >120 |
| 15 |  |
H | 7.3±0.8 | 187±38.0 | 35±16.0 | 3.5 | 2.8 | 19 | 65 | 64 | |
| 16 |  |
H | 7.8±1.0 | 189±36.0 | 29±13.0 | 3.5 | 2.8 | nd | nd | nd | |
| 17 |  |
 |
F | 9.1±0.4 | > 5000 | 1600±470 | 4.7 | 3.6 | 9 | 38 | 18 |
| 18 |  |
 |
H | 1.6±0.6 | > 5000 | > 5000 | 4.9 | 3.9 | 34 | 26 | 14 |
| 19 |  |
 |
H | > 5000 | nd | nd | 4.9 | 3.9 | nd | nd | nd |
| 20 |  |
 |
F | > 5000 | nd | nd | 5.1 | 4.1 | nd | nd | nd |
| 21 |  |
 |
F | 5.1±0.3 | > 5000 | > 5000 | 5.1 | 4.1 | 42 | 22 | 13 |
| 22 |  |
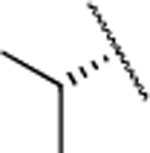 |
F | 4.4±1.0 | > 5000 | > 5000 | 3.9 | 3.6 | 29 | 9 | 27 |
| 23 | 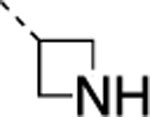 |
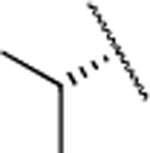 |
F | 20±1.2 | >5000 | >5000 | 2.4 | 3.6 | 25 | >120 | >120 |
The IC50 values for MMP-1, MMP-9 and MT1-MMP for all compounds are > 5 μM. nd = not determined.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw.
The IC50 values for inhibition of MMP-13 catalyzed hydrolysis of fTHP-1512‘ 22, the selectivity profile among the remaining collagenases (MMP-1, MMP-2, mMp-8, MMP-9, and MT1-MMP), and the in vitro microsomal stability in human, mouse, and rat liver microsomes of the synthesized compounds were evaluated (Table 2). All compounds exhibited IC50 values for MMP-13 in the low nanomolar range and higher than 10-fold selectivities among the collagenases compared to MMP-13. Interestingly, substituting the methylamine group with polar residues and changing the stereochemistry in the amino acid component (as in the enantiomeric pairs 9/10 and 15/16 as well as the diastereomeric pairs 11/12 and 13/14) did not influence the potency of these compounds toward MMP-13 but did impact the selectivity profile negatively compared to 2. Compounds 21, 22, and 23 showed no inhibition of fTHP-15 hydrolysis by MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP at 5 μM, which was the highest concentration tested. Furthermore, all compounds except 19 and 20 (IC50 > 5 μM in the fTHP-15 assay) were able to inhibit collagen cleavage by >95% at a concentration of 20 μM. Compounds 18 and 21 exhibited acceptable stability in mouse and rat liver microsomes (t1/2 for 18 = 26 and 34 min in mouse and rat, respectively, while t1/2 for 21 = 22 and 42 min in mouse and rat, respectively), but the stability in human microsomes was lower (t1/2 = 13 and 14 min for 18 and 21, respectively). Compounds 22 and 23 showed excellent selectivity within the MMP family and very desirable in vitro stabilities in rat and human microsomes (Table 2).
Variation of the amino acid unit of compound 2.
In the next step, we modified the amino acid component of 2 by coupling 24 with different amino acid derivatives (Scheme 2, Table 3). Hydrolysis by proteases or peptidases can contribute to drug degradation in the plasma as well as the gastrointestinal tract.23 Avoiding amide bonds or shielding them with adjacent bulky groups are common structural modification strategies to lower the hydrolysis rate of a compound. Furthermore, if an amino acid is part of a lead structure, changing the stereochemistry or incorporating unnatural amino acids can help to decrease enzymatic hydrolysis.16 The switch from the naturally occurring amino acid L-valine to D-valine (25) caused a 100-fold drop of the inhibition potency toward MMP-13 (Table 3). The same effect could be observed for the enantiomeric pairs 26 vs. 27 and 28 vs. 29, and changing the stereochemistry of the cyclohexyl and tert-butyl containing amino acid resulted in a decrease of IC50 values for MMP-13 (4.4 vs. 159 nM for cyclohexyl and 2.4 vs. 289 nM for tert-butyl). The selectivity amongst MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP was the same for 25-27 and none of these compounds inhibited the remaining collagenases at the highest concentration tested (5 μM). In contrast, compound 28 showed low nanomolar inhibition of MMP-8 (IC50 = 17 nM). The tetrahydropyran containing compound 30, possessing high inhibition potency for MMP-13 (IC50 = 1.9 nM), showed increased stability in human liver microsomes compared to the cyclohexyl containing compound (26). However, the selectivity of 30 for MMP-13 over MMP-8 (IC50 =113 nM) is only ca. 50-fold. Compound 31, which incorporated a cyclopropyl moiety, showed a highly increased in vitro half-life in mouse and human liver microsomes (31 and 74 min, respectively) compared to 2 and ca. 500- and 100-fold selectivity for MMP-13 vs. MMP-2 and MMP-8, respectively. All compounds except 25 (<40%) and 33 (<10%) inhibited collagen cleavage with >90% at an inhibitor concentration of 20 μM (Table 3).
Scheme 2.
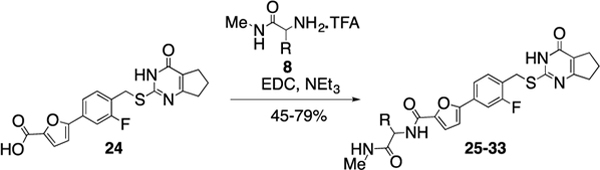
Synthesis of compounds with variation of the amino acid component of 2.
Table 3.
In-vitro biological evaluation of compounds derived by Scheme 2
| IC50 (nM)* | Microsome stability (min) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | R | MMP-13 | MMP-2 | MMP-8 | clogD7.4a | clogPb | rat | mouse | human | Collagen cleavage (20 μM) |
| 2 | 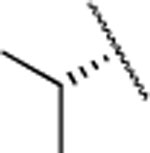 |
2.7±0.6 | >5000 | >5000 | 4.2 | 3.0 | 9 | 20 | 12 | >90% |
| 25 | 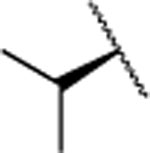 |
257±35.0 | >5000 | >5000 | 4.2 | 3.0 | 8 | 11 | 6 | <40% |
| 26 | 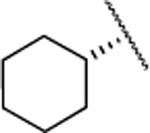 |
4.4±1.5 | >5000 | >5000 | 5.2 | 4.2 | 4 | 22 | 7 | >90% |
| 27 | 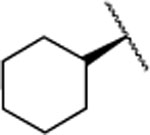 |
159±59.0 | >5000 | >5000 | 5.2 | 4.2 | nd | nd | nd | >90% |
| 28 | 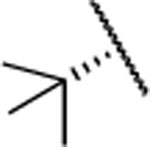 |
2.4±0.1 | 128±23.0 | 17±1.9 | 4.5 | 3.4 | nd | nd | 8 | >90% |
| 29 | 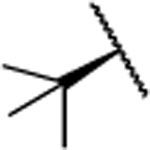 |
289±24.0 | nd | nd | 4.5 | 3.4 | nd | nd | nd | >90% |
| 30 | 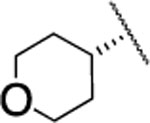 |
1.9±0.3 | 936±190 | 113±23.0 | 3.5 | 1.8 | 4 | 52 | 29 | >90% |
| 31 | 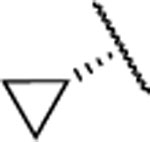 |
8.5±1.4 | >5000 | 832±53.0 | 3.9 | 2.5 | 13 | 31 | 74 | >90% |
| 32 | 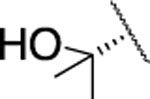 |
2.5±0.5 | 584±21.0 | 128±47.0 | 3.1 | 1.8 | 15 | 33 | 59 | >90% |
| 33 | 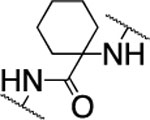 |
43.2±4.8 | >5000 | >5000 | 4.8 | 3.7 | nd | nd | nd | <10% |
The IC50 values for MMP-1, MMP-9, and MT1-MMP are >5 μM for all compounds. nd = not determined.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw.
Variation of the phenyl-furan unit of compound 2.
Next studied was the variation of the phenyl-furan biaryl unit in 2. First, the synthesis of suitable building blocks containing different heterocycles was needed (Scheme 3). The phenyl- thiazole (37) and phenyl-oxazole (41) linkers were synthesized in the same way: a Suzuki reaction was followed by a radical benzylic bromination reaction using N-bromosuccinimide and benzoyl peroxide as the radical initiator to give the desired products. The imidazol-phenyl linker 44 was isolated after a three-step sequence: the methyl-imidazole derivative 42 was brominated, coupled with (4-(hydroxymethyl)phenyl)-boronic acid, and finally a benzylic bromination gave the desired product 44. The three-step sequence for the formation of the pyridine containing linkers 48 and 50 was initiated by the conversion of 45 into the corresponding boronic acid pinacol ester 46 using Pd(dppf)2 as the catalyst. Again, a Suzuki reaction was followed by a benzylic bromination reaction to yield 48 and 50.
Scheme 3.
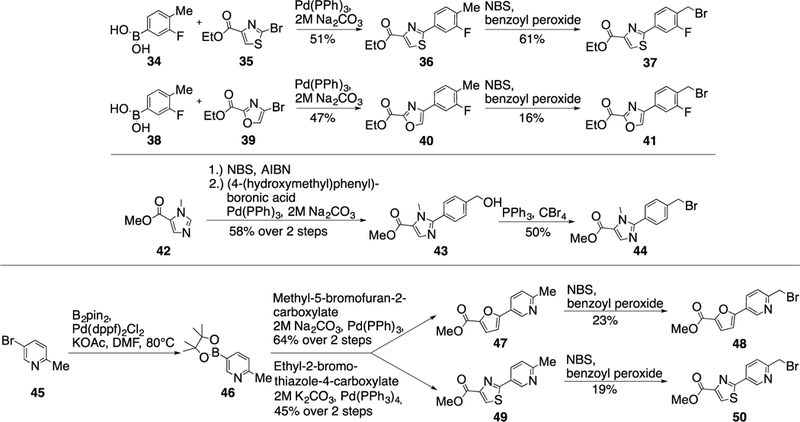
Synthesis of building blocks containing different heterocycles.
The five compounds 37, 41, 44, 48, and 50 were coupled with thiopyrimidone 6 under basic conditions before the ester moiety was hydrolyzed and the valine derivative 51 was added via amide bond formation using EDC as the coupling reagent to give compounds 52-56 (Scheme 4 and Table 4).
Scheme 4.
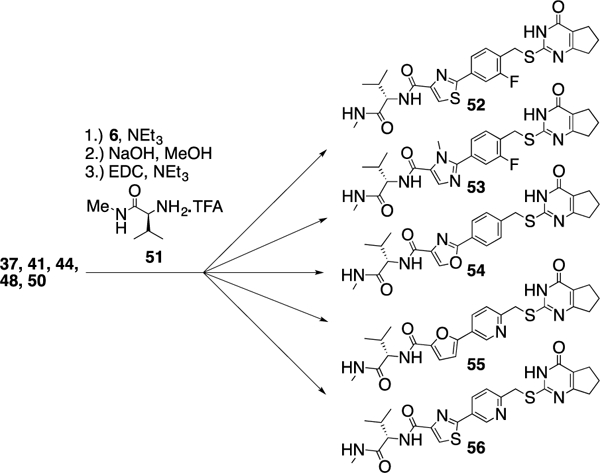
Replacement of the phenyl-furan linker in 2.
Table 4.
In-vitro biological evaluation of compounds derived by Scheme 4.
| IC50 (nM)* | Microsome stability (min) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | R1 | MMP-13 | MMP-2 | MMP-8 | clogD7.4a | clogPb | rat | mouse | human | Collagen cleavage (20 μM) |
| 2 | 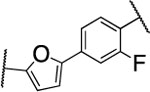 |
2.7±0.6 | >5000 | >5000 | 4.2 | 3.0 | 9 | 20 | 12 | >90% |
| 52 | 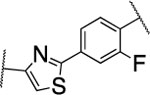 |
7.1±0.8 | >5000 | 1100±236 | 3.6 | 3.3 | 9 | 26 | 12 | >90% |
| 53 | 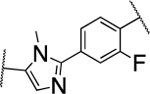 |
>5000 | nd | nd | 3.3 | 2.7 | 14 | 110 | 59 | nd |
| 54 | 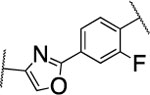 |
18±4.9 | nd | nd | 3.2 | 2.5 | 10 | 31 | 21 | 40% |
| 55 | 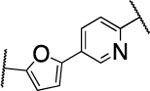 |
38±7.5 | >5000 | >5000 | 3.3 | 1.5 | 4 | 12 | 96 | 65% |
| 56 | 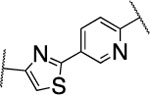 |
148±8.5 | nd | nd | 3.8 | 3.3 | 5 | 20 | 82 | nd |
The IC50 values for MMP-1, MMP-9, and MT1-MMP are >5 μM for all compounds. nd = not determined.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw.
The furan ring in 2 was replaced by different five-membered heterocyles to minimize the potential toxicity and metabolizing liability of the electron rich furan heterocycle,24 which can be easily oxidized and degraded by enzymes of the CYP450 family.25 Compounds 52, 53, and 54 showed low nanomolar IC50s (7.1, 18.1, and 37.5 nM, respectively) as well as high selectivity among the collagenases MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP (Table 4). Among the three compounds 54 exhibited an excellent half-life (96 min) in human liver microsomes, but unfortunately only 52 inhibited collagen cleavage with >90% at an inhibitor concentration of 20 μM. Further assessment via a dose-responsive assay showed that 52 inhibited collagen cleavage with a 14.9 nM IC50.
Substitution of the carbon-sulfur linker of compound 2.
As the sulfur atom of the thiopyrimidone component of 2 could be easily oxidized in vivo, we designed a new set of compounds focusing on blocking the possible metabolically labile site by replacing the sulfur with either with a nitrogen, oxygen, or a carbon atom (Scheme 5A-C). 4-Bromo-2-fluorobenzeneacetic acid (57) was reduced to the corresponding homo-benzylic alcohol and the boronic acid pinacol ester was introduced via a palladium catalyzed cross-coupling reaction to give compound 58. A Suzuki coupling was followed by a three-step sequence to install the amidine moiety in 60 before a condensation reaction with ethyl-2-oxocyclopentane-1-carboxylate completed the synthesis of methyl-ester 61 (Scheme 5A).
Scheme 5.
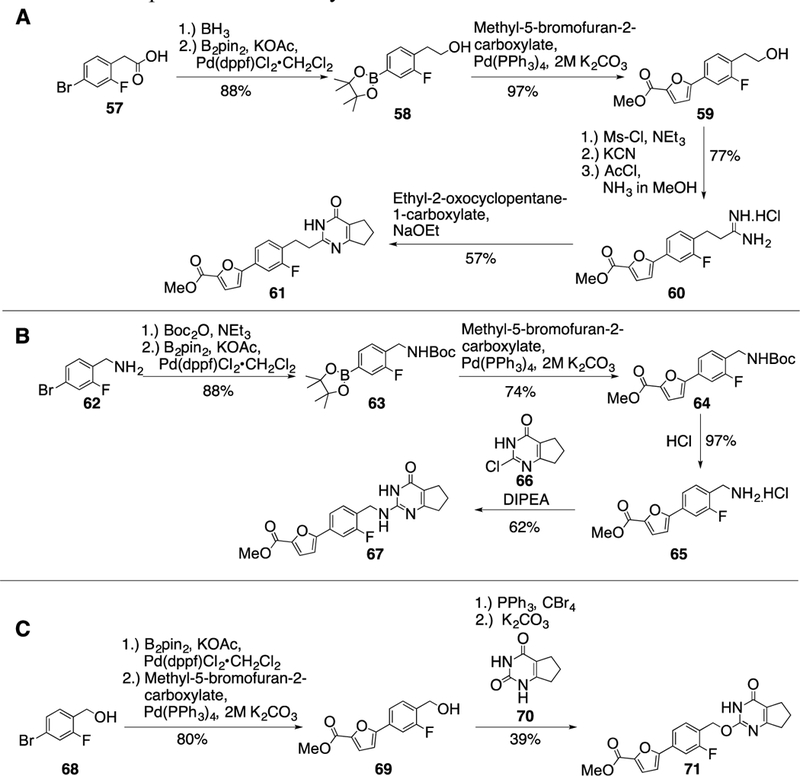
Synthesis of intermediates replacing the sulfur atom of compound 2.
4-Bromo-2-fluorobenzylamine (62) was Boc-protected and the boronic acid pinacol ester was installed via a cross-coupling reaction using Pd(dppf)Cl2 as the metal catalyst. A Suzuki reaction followed by Boc-deprotection provided primary amine 65 and finally, the pyrimidone group was added under basic conditions to give methyl ester 67 (Scheme 5B).
4-Bromo-2-fluorobenzyl alcohol (68) was converted into the boronic acid pinacol ester and coupled with methyl-5- bromofuran-2-carboxylate to give intermediate 69, which was then exposed to triphenylphosphine and tetrabromomethane to provide the corresponding benzyl bromide, and the thiopyrimidone moiety was added via a substitution reaction using 70 as the nucleophile (Scheme 5C). After the methyl esters in 61, 67, and 71 were hydrolyzed, an amino acid coupling finished the syntheses of 72-74 (Scheme 6 and Table 5).
Scheme 6.
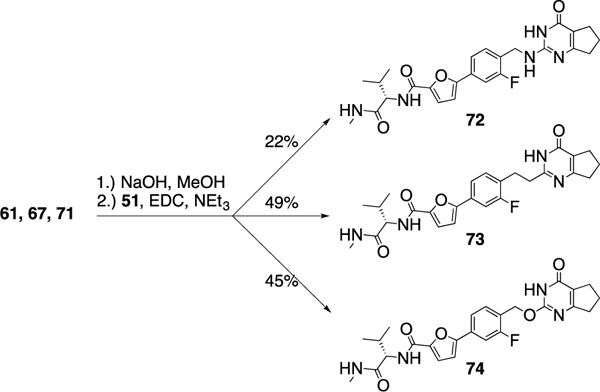
Synthesis of 72-74; replacing the sulfur atom of compound 2.
Table 5.
In-vitro biological evaluation of compounds derived from Scheme 6.
| IC50 (nM)* | Microsome stability (min) | |||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | MMP-13 | clogD7.4a | colgPb | rat | mouse | human | Collagen cleavage at 20μM |
|
| 2 | 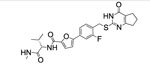 |
2.7±0.6 | 4.2 | 3.0 | 9 | 20 | 12 | >90% |
| 72 | 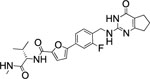 |
6.4±2.1 | 3.0 | 2.4 | 22 | 10 | 17 | >90% |
| 73 | 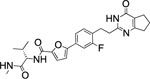 |
18.7±2.4 | 3.6 | 2.7 | 7 | 14 | 26 | >90% |
| 74 | 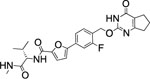 |
697±83.0 | 3.7 | 2.6 | 7 | 7 | 14 | <5% |
The IC50 values for MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP are >5 μM for all compounds.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw
Interestingly, the potency dropped by 3-fold from the nitrogen containing compound (72, IC50 = 5.5 nM) to the carbon atom containing inhibitor (73, IC50 = 18.7 nM) and even 100-fold for the replacement of the sulfur atom in 2 with an oxygen atom (76, IC50 = 697 nM) (Table 5). All three compounds showed an excellent selectivity profile among the remaining collagenases (MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP) with IC50 values higher than 5 μM for all enzymes tested. The newly introduced nitrogen atom in 72 reduced the clogP and clogD7.4 values (2.4 and 3.0, respectively) and increased the microsomal stability in human and rat liver microsomes to 17 and 22 min, respectively. Substitution of the sulfur atom by a CH2 group doubled the human microsomal half-life (26 min) compared to 2. Compounds 72 and 73 inhibited collagen cleavage greater than 95% at 20 μM, while 74 was not active in inhibiting collagen cleavage by MMP-13.
Variation of the pyrimidinone scaffold.
The next compound set was focused on variation of the cyclopentene ring of 2, binding in the S1’ subsite. Potentially, the CH2 groups could be easily accessible to metabolizing enzymes in the liver and could contribute to the moderate metabolic stability of 2. The thiopyrimidinone building blocks 75-79 were commercially available and six more (82, 84, 86, 89, 90 and 94) were synthesized (Scheme 7). A condensation reaction of thiourea and ethyl-2-cyano-4,4-diethoxybutyrate followed by hydrolysis of the diethyl acetal under acidic conditions gave 80 in excellent yield. Treatment of 83 and 85 with sodium hydroxide gave the corresponding chloro-pyrimidinones 84 and 86. Aminothiophene 87 was converted into the thiourea derivative 88 using amino thiocyanate and benzoyl chloride before refluxing in ethanolic KOH gave the desired thiopyrimidinone intermediates 89 and 90.26 Finally, the last thiopyrimidinone building block 94 was synthesized in a three-step sequence, whereby 2-amino-5- fluorobenzoic acid 91 was converted into the thiourea derivative 92 by using ethoxycarbonyl isothiocyanate under refluxing conditions, activation of the acid with acetic anhydride enabled nucleophilic attack and closure of the thiopyrimidinone ring to give 93, and removal of the ethyl carbamate under basic conditions resulted in the isolation of 94 in good yield.
Scheme 7.
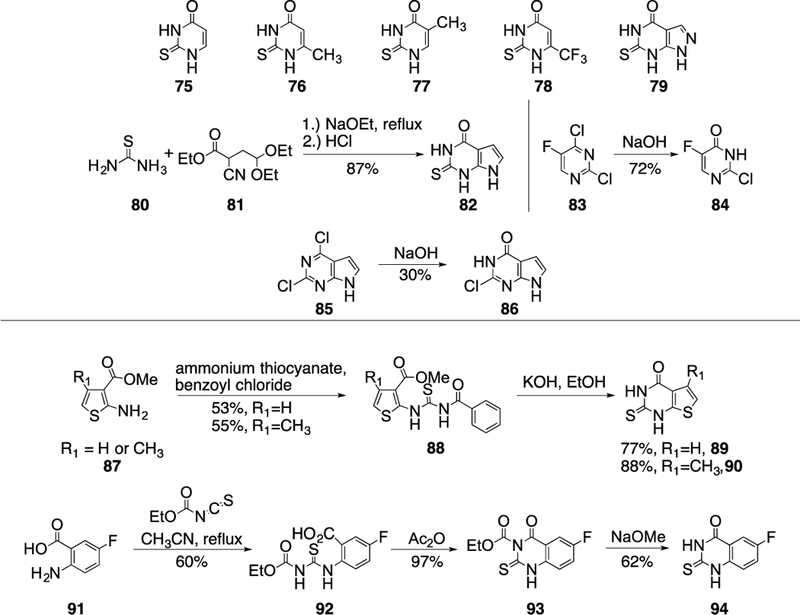
Structures and synthesis of thiopyrimidinone building blocks.
The coupling with the thio- and chloro-pyrimidinone fragments 96 and 97 proceeded via treatment with triethylamine and Huenig’s base, respectively, to afford 98. Subsequent ester hydrolysis followed by an amino acid coupling reaction provided compounds 99-110 (Scheme 8 and Table 6).
Scheme 8.
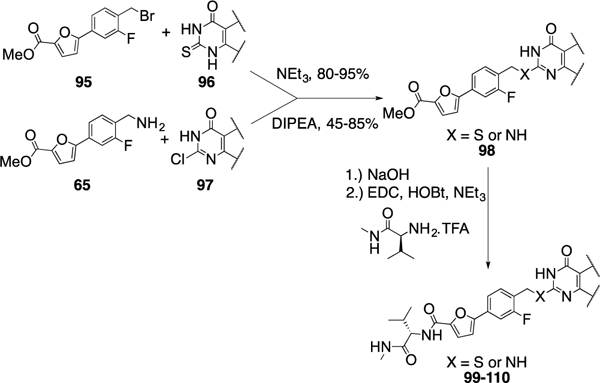
Synthesis of compounds 99–110. Compound 95 was previously described.13
Table 6.
In-vitro biological evaluation of compounds derived from Scheme 8.
| IC50(nM)† | Microsome stability (min) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd. | R1 | MMP-13 | MMP-2 | MMP-8 | logD7.4a | logPb | rat | mouse | human |
| 2 | 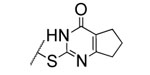 |
2.7±0.6 | >5000 | >5000 | 4.2 | 3.0 | 9 | 20 | 12 |
| 99 | 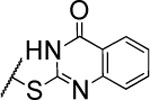 |
9.4±1.7* | >5000 | 400±38.0 | 4.6 | 3.6 | nd | nd | nd |
| 100 | 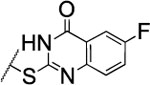 |
2.5±0.5* | >5000 | 270±38.0 | 4.8 | 3.8 | 4 | 28 | 8 |
| 101 | 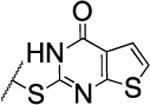 |
8.4±1.4 | nd | nd | 4.4 | 3.4 | 6 | 29 | 14 |
| 102 | 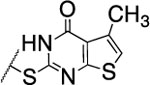 |
13±2.7 | nd | nd | 4.9 | 3.9 | 4 | 28 | 13 |
| 103 | 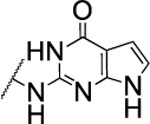 |
>5000 | nd | nd | 3.7 | 2.9 | nd | nd | nd |
| 104 | 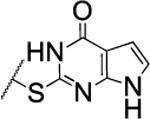 |
2000±850 | nd | nd | 3.7 | 2.9 | nd | nd | nd |
| 105 | 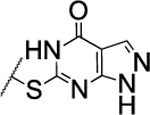 |
274±29.0 | nd | nd | 3.1 | 2.4 | nd | nd | nd |
| 106 | 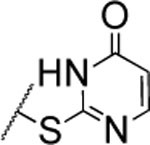 |
153±13.0 | nd | nd | 3.1 | 2.0 | 33 | 57 | 160 |
| 107 | 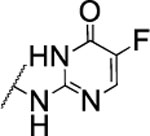 |
2600±480 | nd | nd | 1.7 | 1.9 | nd | nd | nd |
| 108 | 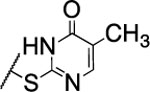 |
1300±110 | nd | nd | 3.5 | 2.5 | nd | nd | nd |
| 109 | 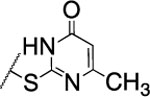 |
88±5.6 | nd | nd | 3.7 | 2.5 | nd | nd | nd |
| 110 | 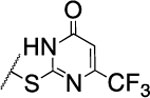 |
1200±200 | nd | nd | 4.3 | 3.0 | 41 | 43 | >120 |
IC50 values for MMP-1, MMP-9, and MT1-MMP > 5 μM.
Some selectivity profiles among MMP-1, MMP-2, MMP-8, MMP-9 and MT1-MMP were not determined due to either low activity or poor microsomal stability of the compound. nd = not determined.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Calculated with ChemDraw.
The substitution of the cyclopentene ring with a benzene (99) or a fluoro-benzene ring (100) gave two highly potent MMP-13 inhibitors (IC50 = 9.4 and 6.6 nM, respectively). Both compounds showed a good selectivity profile among the collagenases (MMP-1, MMP-2, MMP-8, MMP-9 and MT1-MMP) with only moderate activity for MMP-8 (IC50 = 400 and 270 nM, respectively). Additionally, the stability of 100 in human and rat liver microsomes dropped to 8 and 4 min, respectively, presumably due to increased lipophilicity (clogD7.4 = 4.8 and clogP = 3.8) of the compound compared to 2. The incorporation of a thiophene ring resulted 101 and 102, which were active MMP-13 inhibitors but only moderately inhibited collagen cleavage at an inhibitor concentration of 20 μM (77% and 51%, respectively). The potency of the pyrrole containing compounds 103 and 104 decreased significantly; compound 103 was not active at the highest concentration tested (5 μM) and 104 exhibited an IC50 value for MMP-13 of 2 μM. The pyrazole derivative of 2 (105) also lost potency against MMP-13 (IC50 = 274 nM). Removing the cyclopentene ring completely (106) or substituting the thiopyrimidinone ring with a fluorine atom (107) or a trifluoromethyl group (110) also resulted in a decrease of activity towards MMP-13 (IC50 = 153 nM, 2.6 μM, and 1.2 μM, respectively), although 106 and 110 showed excellent stability especially in human liver microsomes (>120 min for both compounds). These data supported our hypothesis that the saturated carbon atoms in the thiopyrimidone part of 2 are metabolically labile sites and easily accessible for hepatic oxidation.
After the comparison and analysis of the in vitro properties of the compounds described herein, seven were selected for further evaluation of their permeability and solubility properties (Table 7). Inhibitors 21, 22, and 23 were part of the first compound set, which was focused on the variation of the terminal methylamine group in 2. The three compounds have an excellent selectivity profile within the collagenases (IC50 values for MMP-1, MMP-2, MMP-8, MMP-9, and MT1-MMP >5 μM) and all three have improved half-lives in rat liver microsomes (72, 29, and 25 min, respectively) compared with 2. A special focus has been placed on the in vitro hepatic stability in rats as further in vivo PK is planned to be perfomed in rats. Compounds 31 and 28 possessed unnatural amino acids, which could protect these compounds from proteolytic cleavage in vivo. Compound 52 does not exhibit improved microsomal half-life but is lacking the furan ring from 2 which has been attributed to toxicity for other compounds in preclinical studies.24, 27 Finally, compound 72, in which the metabolically labile sulfur was replaced by a nitrogen atom, exhibited good microsomal stability in rat liver microsomes (22 min) and had significantly reduced clogD7.4 and clogP values (3.0 and 2.4, respectively), which contribute to improved solubility properties.
Table 7.
Permeability and solubility of compounds selected by their in vitro microsomal stability.
| Compd. | Permeability [nm/s] |
Retention | Solubility [μM] |
|---|---|---|---|
| 2 | 23.8 | 47% | 15.5 |
| 21 | 3.4 | 61% | nd |
| 22 | 13.2 | 34% | nd |
| 23 | 0.3 | 30% | 58.7 |
| 28 | 72.9 | 17% | 16.0 |
| 31 | 21.1 | 24% | 47.5 |
| 52 | 34.3 | 40% | nd |
| 72 | 1.98 | 45% | 36.7 |
The artificial membrane permeability assay (PAMPA assay)15 is a rapid, low-cost, fast, and high-throughput in vitro method to assess the gastrointestinal permeability properties of a compound. The passive diffusion of a potential drug through a phospholipid bilayer can be monitored to obtain in vivo permeability insights. The permeability in the PAMPA assay for the compounds 21 (3.4 nm/s) and 23 (0.3 nm/s) was extremely low. Compound 21, which exhibits clogP and clogD74 values of 4.1 and 5.1, respectively, shows 61% retention and is too lipophilic to pass the lipid bilayer. On the other hand the retention of compound 23 is lower (30%) but presumably the secondary amine within the azetidine ring is protonated under the weakly acidic conditions used in the PAMPA assay mimicing the pH in the intestine, which restricts the compound from diffusing through the lipid bilayer.
The two best compounds which exhibited low retention (<30%), good permeability (ca. 20 nm/s), and good kinetic solubility (> 15 μM) were 28 and 31 (17%, 72.9 nm/s, and 16.0 μM for 28 and 24%, 19.1 nm/s, and 47.5 μM for 31, respectively).
In vivo PK properties.
For a first assessment of the in vivo stability of the synthesized MMP-13 inhibitors, compounds 2, 28 and 30-32 were subjected to PK analysis after IV administration (1 mg/kg dose; Table 8) in rats. Lead compound 2 provided a reference, but as our future goal is to develop an orally bioavailable MMP-13 inhibitor, we focused on compounds 28 and 30-32 as the substitution of L-valine in 2 by unnatural amino acids should minimize possible cleavage by peptidases and proteases in the gastrointestinal tract. Compounds 28, 30 and 32 did not show the best selectivity profiles within the different MMPs tested but compound 28 exhibited excellent permeability (72.9 nm/s) in the PAMPA assay as well as reasonable solubility (16 μM). Compounds 30 and 32 were selected for further evaluation in in vivo PK studies due to their increased stabilities in human liver microsomes (29 and 59 min, respectively). There was not much difference in the rat in vivo PK parameters after IV dosing for the five compounds but all of them exhibited excellent plasma half-lives (> 2.4 h), reached maximum concentrations higher than 50 μM, and had very low clearance rates with less than 0.3 mL/min/kg. Furthermore, the concentration of 2 in the synovial fluid of the rat knee after IP injection was evaluated to determine if the inhibitor was reaching its needed place of action. At the 4 h time point the concentration of 2 in the synovial fluid was 380 nM, which is ca. 140 times higher than its IC50 for MMP-13.
Table 8.
Rat in vivo PK parameters of selected MMP-13 inhibitors.
| Compd. | T1/2 (h) | cmax(μM) | Cl (mL/min/kg) |
|---|---|---|---|
| 2 | 2.9 | 47.6 | 0.18 |
| 28 | 2.6 | 59.4 | 0.25 |
| 30 | 2.8 | 75.6 | 0.16 |
| 31 | 3.0 | 56.8 | 0.23 |
| 32 | 2.4 | 56.9 | 0.22 |
Dose: IV, 1 mg/kg; formulation = 1 mg/mL in 10/10/80 DMSO/Tween 80/water.
CYP inhibition.
The inhibition of cytochrome P450 isoforms, especially CYP 3A4, can be a major drawback in drug development, potentially indicating toxicity. CYP 3A4 inhibition has also been responsible for the termination of the development of MMP-13 inhibitors.28 Therefore, we further assessed the in vitro inhibition of CYP 1A2, 2C9, 2D6, and 3A4 for the compounds 2, 28 and 30-32 (Table 9) at a concnetration of 10 μM. The lead compound 2 inhibited CYP 2C9 and 3A4 at levels of 60 and 16%, respectively. Interestingly, compound 28 inhibited CYP 2C9 and 3A4 even more strongly, 75 and 43%, respectively. Compounds 30, 31, and 32 exhibited lower inhibition of CYP 2C9 (35, 29, and 25%, respectively) and did not inhibit the activity of CYP 3A4.
Table 9.
CYP isoform inhibition of selected MMP-13 inhibitors.
| % Inhibition of human CYPs at 10 μM | ||||
|---|---|---|---|---|
| Compd. | 1A2 | 2C9 | 2D6 | 3A4 |
| 2 | 8 | 60 | −33 | 16 |
| 28 | −2 | 75 | −35 | 43 |
| 30 | −5 | 35 | −31 | −6 |
| 31 | −12 | 29 | −45 | −17 |
| 32 | −3 | 25 | −19 | −3 |
Molecular Modeling.
Docking studies were performed to rationalize potency trends within the SAR study of our MMP-13 inhibitors. In particular, we investigated the lack of discrimination of inhibition potency between the enantiomeric pairs 9/10, 11/12, 13/14, and 15/16 (Table 2), in contrast to 2/25, 26/27, 28/29 (Table 3). We have already demonstrated that a combination of hydrophobic interactions of the isopropyl group and hydrogen bonding interactions between the amide group in compound 2 with the backbone amide groups of Gly183 and Tyr244 in MMP-13 is responsible for a 40-fold higher inhibition potency compared to its enantiomeric counterpart.13 A similar trend is observed for the enantiomeric pairs 26/27 and 28/29, respectively, which possess hydrophobic groups in their R positions (Table 3). However, in 9/10, 11/12, 13/14, and 15/16, where the terminal methyl group of 2 is substituted by hydrophilic moieties (R1-groups, Table 2), which are able to form additional hydrogen bonding interactions with Tyr214 and/or Ser182 as shown in our docking models (Figure 3A-D), an analogous drop in inhibition potency was not observed. We surmised from these observations that the hydrophobic contact between the isopropyl unit and Ile243 is less significant than the additional hydrogen bonding interactions by the polar R1-groups. Alternatively, the (R)-enantiomers could bind to MMP-13 in the same binding pose as (R)-2 in the X-ray co-crystal structure (PDB code: 5UWM).13 In this binding mode (Figures 3E, 3F), the polar R1-groups can form hydrogen bonding interactions with Glu223 or backbone amides of MMP-13, and the loss of hydrogen bonds of the amide units of the (R)-enantiomers can be compensated by the polar interactions of the corresponding R1- group. Consequently, the enantiomeric pairs 9/10, 11/12, 13/14, and 15/16 exhibit high inhibition potency regardless of their stereoconfiguration.
Figure 3.
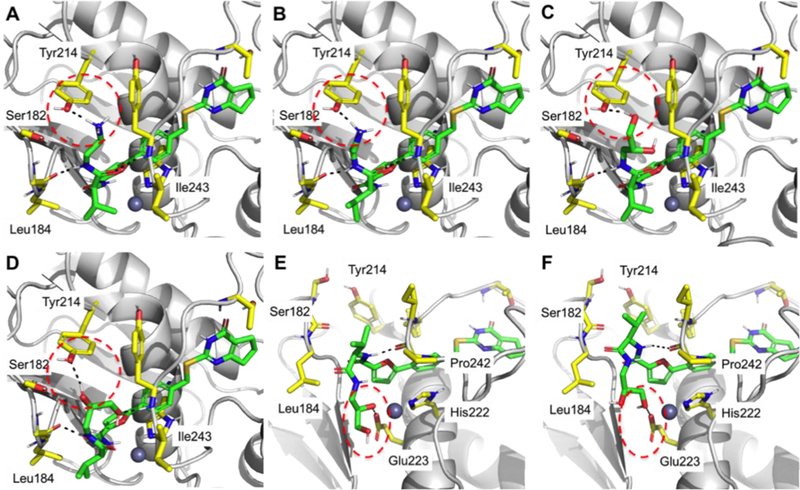
Predicted binding poses of compounds 9 (A), 10 (B), 11 (C), 12 (D,E), and 14 (F) in the active site of MMP-13. (E, F) Alternative binding poses of 12, 14, and other (R)-enantiomers. Ligand-protein interactions are shown in black dashed lines. Hydrogen bond interactions are marked with a red dashed circle. Inhibitors are presented in green sticks, and amino acids near inhibitors are in yellow sticks. Pymol was used to generate figures.
We also performed docking studies to examine the preference of the sulfur linkage (2) vs. nitrogen (72), carbon (73), or oxygen (74) linkages (Table 5) in terms of inhibition potency. We assume that due to the smaller dihedral angle for Csp2-S-Csp3 (101.5°, 2) compared to those for Csp2-N-Csp3 (119.3°, 72), Csp2- C-Csp3 (114.4°, 73), and Csp2-O-Csp3 (117.3°, 74), respectively, the cyclopentyl unit in compound 2 is aligned in an optimal position for forming hydrophobic contacts with the protein surface composed of Phe252, Pro255, and Leu239 (Supporting Fig S1 A). Furthermore, the increased dihedral angles in 72, 73, and 74 lead to a slightly deviated alignment of the carbonyl oxygen within the pyrimidinone part of the structures, which results in a less favorable orientation for the formation of hydrogen bonding interactions with Thr247 and explains the decreased inhibition of MMP-13. Interestingly, compound 72 has the largest dihedral angle, which should lead to the least favorable orientation for the formation of hydrogen bonding interaction as well as hydrophobic contacts, but an additional hydrogen bond by the hydrogen atom of the amine linkage with Thr245 might be the reason which compensates the drop in inhibition potency to only twofold compared to compound 2. (Supporting Fig S1B).
The significance of the hydrophobic contact between the cyclopentyl unit in 2 and the hydrophobic surface of MMP-13 is also reflected within the SAR of the compounds shown in Table 6. The compounds 106, 107, 108, 109, and 110, which are lacking the cyclopentyl moiety and can not form hydrophobic contacts, exhibit decreased inhibition potency by 30 ~ 1,000 fold compared to 2, while 99, 100, 101, and 102 have similar IC50 values as compound 2.
We also investigated the replacement of the furan ring in compound 2 with other heterocycles (Table 4) by docking studies. The central part of the structures 2, 52, 53, 54, 55, and 56, comprising the phenyl ring connected to the corresponding 5- membered heterocycles, occupies the same area within the binding site of MMP-13 with high alignment to each other. However, the methyl group attached to the imidazole ring in 53 causes a non-planar conformation of the amide unit connected to the imidazole ring with a dihedral angle of 34.5°. This non-planar conformation results in the loss of hydrogen bond interaction between the N-methylamide unit of 53 and the amide backbone of Tyr244, and might be responsible for a decreased inhibition of MMP-13 (Supporting Fig s2a, B).
Conclusions.
Starting from compound 2 a comprehensive SAR/SPR study was performed by systematically varying all five subunits (Figure 2). As a goal of this study, we were interested in finding a set of selective and potent MMP-13 inhibitors that can be used in in vivo efficacy studies in a rat OA model. Analogs of 2 were designed and synthesized to investigate their SAR/SPR in terms of inhibition potency, specificity, microsomal stability, solubility, and permeability. As hepatic metabolization is a significant challenge in drug discovery, we focused on the optimization of the in vitro microsomal stability at the beginning of the lead optimization process. We found that substituting the terminal methyl group of 2 with cyclohexanol (compound 21) gave a highly selective MMP-13 inhibitor with increased in vitro microsomal stabilities in rats and mice but the hepatic stability in humans was not improved compared to 2. The addition of small saturated heterocycles such as an oxetane (22) or azetidine ring (23) gave potent and selective MMP-13 inhibitors with increased microsomal stabilities in human liver microsomes. The variation of the amino acid part of 2 provided three compounds (30-32) with very promising human microsomal stabilities (t1/2 = 29, 74, and 59 min, respectively). Out of these three compounds only 31 showed a good selectivity profile within the collagenases; inhibition of MMP-8 resulted in an IC50 value of 832 nM, which was a 100-fold worse compared with MMP-13.
Displacement of the furan-phenyl ring system with other heterocyclic rings gave highly active compounds with increased human microsomal stabilites (Table 4). However, only 52 was able to inhibit collagen cleavage at a concentration of 20 μM, while all other compounds inhibited the cleavage of the natural substrate by less than 30%. Substitution of the sulfur atom within the thiopyrimidone scaffold of 2 with a nitrogen (72) or carbon (73) atom improved the human microsomal half-life (17 and 26 min, respectively). This result suggested that the sulfur atom could be oxidized by human hepatic metabolizing enzymes. Finally, the removal of the saturated carbon atoms within the S1 ‘subsite binding thiopyrimidone scaffold enhanced metabolic stability, as compound 106 showed greatly improved microsomal stabilities (rat/mouse/human 33/57/160 min) compared to 2. Unfortunately, removal of the CH2-groups resulted in a loss of activity (106-110) and/or selectivity (99 and 100).
After testing the solubility and permeability of a selected compound set (Table 7) we identified compound 31 which exhibited improved microsomal half-life times (human/rat/mouse 74/13/31 min). Although the permeability coefficient for 31 (21.1 nm/s) is in the same range as for compound 2, its retention within the artificial phospholipid bilayer in the PAMPA assay is lower (24% for 31 versus 47% for 2) and the kinetic solubility is improved by three-fold. Furthermore, compound 31 shows no inhibition of CYP 3A4 and exhibited excellent in vivo PK properties after IV dosing. As 31 is structurally closely related to 2 which could be detected in the synovial fluid at a concentration of 380 nM, we expect 31 to show a similar distribution into the rat knee. Therefore, considering the improved pharmacokinetic properites, compounds 31 appears to be an appropriate candidate for a first round of in vivo efficacy studies in a rat OA model (Table 10). Additionally, combining the best results observed here for each subunit modification, i.e. (22 or 23) and (28 or 31) and (72 or 73), may provide improved compounds for in vivo use.
Table 10.
Comparison of PK properties of compounds 2 and 31.
| Property | Compound 2 | Compound 31 |
|---|---|---|
| Kinetic solubility | 15.7 μM | 47.5 μM |
| Permeability / Retention | 23.8 nm/s / 47% | 21.1 nm/s / 24% |
| t1/2 (human/rat/mouse) | 12/9/20 min | 74/13/31 min |
| CYP 3A4 inhibition at 10μM | 16% | No inhibition |
Recent studies have produced a variety of selective MMP-13 inhibitors.29 However, distinct drawbacks to these inhibitors have been reported. For inhibitors presented as organic anions, binding to human organic anion transporter 3 resulted in nephrotoxicity.30 Inhibitors possessing carboxylic acids may generate reactive metabolites through protein conjugation of the resulting acyl glucuronide.30,31 Pyrimidine-2-carboxamide-4-one-based inhibitors have exhibited poor bioavailability, low volume of distribution, poor metabolic stability, and/or P450 3A4 inhibition.28 Obtaining appropriate kinetic solubilities for MMP- 13 inhibitors has proved challenging.12, 32 Due to design considerations based on activity profiles and prior data, several of the compounds described herein avoid many of these pitfalls, particularly poor solubility and metabolic stability as well as the potential for nephrotoxicity and generation of reactive metabolites.
Supplementary Material
Acknowledgments
This research was supported by NIH R01 grant AR063795 (GBF and WRR). RF was supported by a Schroedinger Postdoctoral Fellowship, financed by the Austrian Science Fund (FWF).
Footnotes
Supplementary Material
All procedures for the synthesis of the compounds presented in the manuscript as well as methods and experimental details can be found in the Supplementary data associated with this article at https://
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen A, Gupte C, Akhtar K, Smith P, Cobb J. The global economic cost of osteoarthritis: How the UK compares. Arthritis. 2012; D01: 10.1155/2012/698709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee AS, Ellman MB, Yan D, Kroin JS, Cole BJ, Van Wijnen AJ, Im HJ. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene. 2013;527:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bijlsma JWJ, Berenbaum F, Lafeber PJG. Osteoarthritis: an update with relevance for clinical practice. The Lancet 2011;377:2115–2126. [DOI] [PubMed] [Google Scholar]
- 4.(a) Flood J The role of acetaminophen in the treatment of osteoarthritis. Am. J. Manag. Care 2010;16:48–54; [PubMed] [Google Scholar]; (b) Chen YF, Jobanputra P, Barton P, Bryan S, Fry-Smith A, Harris G, Taylor RS Cycloxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol. Asses. 2008;12:1–278. [DOI] [PubMed] [Google Scholar]
- 5.Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: Pathobiology—targets and ways for therapeutic intervention. Adv. Drug Deliver. Rev. 2006;58:128–149. [DOI] [PubMed] [Google Scholar]
- 6.Mengshol JA, Mix KS, Brinckerhoff CE, Matrix metalloproteinases as therapeutic targets in arthritic diseases: Bull’s-eye or missing the mark? Arthritis Rheum. 2002;46:13–20. [DOI] [PubMed] [Google Scholar]
- 7.(a) Takaishi H, Kimura T, Dalal S, Okada Y, D’Armiento J. Joint diseases and matrix metalloproteinases: A role for MMP-13. Curr. Pharm. Biotechnol. 2008;9:47–54; [DOI] [PubMed] [Google Scholar]; (b) Neuhold LA, Killar L, Zhao WG, Sung MLA, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, DeGennaro LJ. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001;107:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: Role in arthritis. Front. Biosci. 2006;11:529–543; [DOI] [PubMed] [Google Scholar]; (b) Fingleton B. MMPs as therapeutic targets—Still a viable option? Semin. Cell Dev. Biol. 2008;19:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a)Chen JM, Nelson FC, Levin JI, Mobilio D, Moy FJ, Nilakantan R, Zask A, Powers R. Structure-based design of a novel, potent, and selective inhibitor for MMP-13 utilizing NMR spectroscopy and computer-aided molecular design. J. Am. Chem. Soc. 2000;122:9648–9654; [Google Scholar]; (b)Engel CK, Pirard B, Schimanski S, Kirsch R, Habermann J, Klingler O, Schlotte V, Weithmann KU, Wendt KU. Structural basis for the highly selective inhibition of MMP-13. Chemistry & Biology 2005;12:181–189; [DOI] [PubMed] [Google Scholar]; (c)Reiter LA, Freeman-Cook KD, Jones CS, Martinelli GJ, Antipas AS, Berliner MA, Datta K, Downs JT, Eskra JD, Forman MD, Greer EM, Guzman R, Hardink JR, Janat F, Keene NF, Laird ER, Liras JL, Lopresti- Morrow LL, Mitchell PG, Pandit J, Robertson D, Sperger D, Vaughn-Bowser ML, Waller DM, Yocum SA. Potent, selective pyrimidinetrione-based inhibitors of MMP-13. Bioorg. Med. Chem. Lett. 2006;16:5822–5826; [DOI] [PubMed] [Google Scholar]; (d)Johnson AR, Pavlovsky AG, Ortwine DF, Prior F, Man CF, Bornemeier DA, Banotai CA, Mueller WT, Mcconnell P, Yan C, Baragi V, Lesch C, Roark WH, Wilson M, Datta K, Guzman R, Han HK, Dyer RD. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 2007;282:27781–27791; [DOI] [PubMed] [Google Scholar]; (e)Gooljarsingh LT, Lakdawala A, Coppo F, Luo L, Fields GB. Tummino PJ, Gontarek RR. Characterization of an exosite binding inhibitor of matrix metalloproteinase 13. Protein Sci. 2008;17:66–71; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f)Li JJ, Nahra J, Johnson AR, Bunker A, O’Brien P, Yue WS, Ortwine DF, Man CF, Baragi V, Kilgore K, Dyer RD, Han HK. Quinazolinones and pyrido[3,4-d]pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2008:51:835–841; [DOI] [PubMed] [Google Scholar]; (g)Heim-Riether A, Taylor SJ, Liang S, Gao DA, Xiong Z, Michael August E, Collins BK, Farmer Ii BT, Haverty K, Hill-Drzewi M, Junker HD, Mariana Margarit S, Moss N, Neumann T, Proudfoot JR, Keenan LS, Sekul R, Zhang Q, Li J, Farrow NA. Improving potency and selectivity of a new class of non- Zn-chelating MMP-13 inhibitors. Bioorg. Med. Chem. Lett. 2009;19:5321– 5324. [DOI] [PubMed] [Google Scholar]
- 10.(a) Nara H, Kaieda A, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. Discovery of novel, highly potent, and selective matrix metalloproteinase (MMP)-13 inhibitors with a 1,2,4-triazol-3-yl moiety as a zinc binding group using a structure-based design approach. J. Med. Chem. 2017;60:608–626; [DOI] [PubMed] [Google Scholar]; (b) Gege C, Bao B, Bluhm H, Boer J, Gallagher BM, Korniski B, Powers TS, Steeneck C, Taveras AG, Baragi VM. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular Treatment of osteoarthritis. J. Med. Chem. 2011;55: 709–716. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lauer-Fields JL, Minond D, Chase PS, Baillargeon PE, Saldanha SA, Stawikowska R, Hodder P, Fields GB. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triplehelical FRET substrate. Bioorg. Med. Chem. 2009;17:990–1005; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roth J, Minond D, Darout E, Liu Q, Lauer J, Hodder P, Fields GB, Roush WR. Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg. Med. Chem. Lett. 2011;21:7180–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spicer TP, Jiang JW, Taylor AB, Choi JY, Hart PJ, Roush WR, Fields GB, Hodder PS, Minong D. Characterization of selective exosite- binding inhibitors of matrix metalloproteinase 13 that prevent articular cartilage degradation in vitro. J. Med. Chem. 2014;57:9598–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi JY, Fuerst R, Knapinska AM, Taylor A, Smith L, Cao X, Hart PJ, Fields GB, Roush WR. Structure-based design and synthesis of potent and selective matrix metalloproteinase 13 inhibitors. J. Med. Chem. 2017;60:3814–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Fields GB. Interstitial collagen catabolism. J. Biol. Chem. 2013;288:8785–8793; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Amar S, Smith L, Fields GB. Matrix metalloproteinase collagenolysis in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:1940– 1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998;41:1007–1010. [DOI] [PubMed] [Google Scholar]
- 16.Kerns EH, Di Li. Drug-like Properties: Concepts, Structure, Design and Methods: From ADME to Toxicity Optimization. Oxford, UK: Elsevier; 2008. [Google Scholar]
- 17.Comer JEA. High-Throughput Measurement of logD and pKa In: Van de Waterbeemd H, Lennernäs H, Artursson P, eds. Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability. Weinheim, D: Wiley-VCH Verlag GmbH & Co. KGaA; 2004:pp21–45. [Google Scholar]
- 18.Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. Potent, selective and cell penetrant Inhibitors of SF-1 by functional ultra-high-throughput screening. Mol. Pharmacol. 2008;73:1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 20.Van de Waterbeemd H, Smith DA, Beaumont K, Walker DK. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001;44:1313–1333. [DOI] [PubMed] [Google Scholar]
- 21.Nassar AEF, Kamel AM, Clarimont C. Improving the decision making process in the structural modification of drug candidates: Enhancing metabolic stability. Drug Discovery Today 2004;9:1020–1028. [DOI] [PubMed] [Google Scholar]
- 22.Lauer-Fields JL, Fields GB. Triple-helical peptide analysis of collagenolytic protease activity. Biol. Chem. 2002;383:1095–1105. [DOI] [PubMed] [Google Scholar]
- 23.Hamman JH, Enslin GM, Kotze AF. Oral delivery of peptide drugs. BioDrugs 2005;19:165–177. [DOI] [PubMed] [Google Scholar]
- 24.Nelson SD. Metabolic activation and drug toxicity. J. Med. Chem. 1982;25:753–765. [DOI] [PubMed] [Google Scholar]
- 25.Zhou S, Chan SY, Goh BC, Chan E, Duan W, Huang M, McLeod HL. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005;44:279–304. [DOI] [PubMed] [Google Scholar]
- 26.Song YH, Son HY. Synthesis of New 1- Phenylthieno[1,2,4]triazolo[4,3-a]pyrimidin-5(4H)-one derivatives. J. Heterocycl. Chem. 2011;48:597–603. [Google Scholar]
- 27.Peterson LA. Electrophilic intermediates produced by bioactivation of furan. Drug Metab. Rev. 2006:38:615–626. [DOI] [PubMed] [Google Scholar]
- 28.Nara H, Sato K, Kaieda A, Oki H, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. Design, synthesis, and biological activity of novel, potent, and highly selective fused pyrimidine-2-carboxamide-4-one- based matrix metalloproteinase (MMP)-13 zinc-binding inhibitors. Bioorg. Med. Chem. 2016;24:6149–6165. [DOI] [PubMed] [Google Scholar]
- 29.Xie XW, Wan RZ, Liu ZP. Recent research advances in selective matrix metalloproteinase-13 inhibitors as anti-osteoarthritis agents. ChemMedChem 2017;12:1157–1168. [DOI] [PubMed] [Google Scholar]
- 30.Ruminski PG, Massa M, Strohbach J, Hanau CE, Schmidt M, Scholten JA, Fletcher TR, Hamper BC, Carroll JN, Shieh HS, Caspers N, Collins B, Grapperhaus M, Palmquist KE, Collins J, Baldus JE, Hitchcock J, Kleine HP, Rogers MD, McDonald J, Munie GE, Messing DM, Portolan S, Whiteley LO, Sunyer T, Schnute ME. Discovery of N-(4-fluoro-3- methoxybenzyl)-6-(2-(((2S,5R)-5-(hydroxymethyl)-1,4-dioxan-2-yl)methyl)- 2H-tetrazol-5-yl)-2-methylpyrimidine-4-carboxamide. A highly selective and orally bioavailable matrix metalloproteinase-13 inhibitor for the potential treatment of osteoarthritis. J. Med. Chem. 2016;59:313–327. [DOI] [PubMed] [Google Scholar]
- 31.Sallusti BC, Sabordo L, Evans AM, Nation RL. Hepatic disposition of electrophilic acyl glucuronide conjugates. Curr. Drug Metab. 2000;1,:163–180. [DOI] [PubMed] [Google Scholar]
- 32.Nara H, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. Thieno[2,3 d]pyrimidine-2-carboxamides bearing a carboxybenzene group at 5-position: Highly potent, selective, and orally available MMP-13 inhibitors interacting with the S1 “ binding site. Bioorg. Med. Chem. 2014;22:5487–5505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.