

Abstract
Protein synthesis in all organisms proceeds by stepwise translocation of the ribosome along messenger RNAs (mRNAs), during which the helicase activity of the ribosome unwinds encountered structures in the mRNA. This activity is known to occur near the mRNA tunnel entrance, which is lined by ribosomal proteins uS3, uS4, and uS5. However, the mechanism(s) of mRNA unwinding by the ribosome and the possible role of these proteins in the helicase activity are not well understood. Here, we present a crystal structure of the Escherichia coli ribosome in which single-stranded mRNA is observed beyond the tunnel entrance, interacting in an extended conformation with a positively charged patch on ribosomal protein uS3 immediately outside the entrance. This apparent binding specificity for single-stranded mRNA ahead of the tunnel entrance suggests that product stabilization may play a role in the unwinding of structured mRNA by the ribosomal helicase.
Keywords: ribosome, mRNA, helicase, structure, uS3
INTRODUCTION
The ribosome carries out templated protein synthesis in all cells by reading the genetic information in messenger RNAs (mRNAs) (decoding), catalyzing the addition of amino acids to nascent polypeptide chains (peptidyl transfer), and moving mRNA and tRNA molecules in concert through its internal binding sites (translocation). Concomitant with these major activities are a host of coordinated ribosome functions at different stages of translation. Key among them is the intrinsic ability of the ribosome to actively unwind structured mRNAs prior to the decoding step (Takyar et al. 2005; Qu et al. 2011). This is to ensure unhindered translation on mRNA, a molecule capable of forming secondary and tertiary structures, in its single-stranded state. Challenging the helicase, highly stable structures in the mRNA can stall the ribosome, and in the right sequence context, shift the translational reading frame (Baranov et al. 2002; Giedroc and Cornish 2009; Tholstrup et al. 2012). This frameshifting can occur endogenously, or it can be induced by viruses, some of which are potent human and animal pathogens (Brierley 1995; Namy et al. 2004).
The path of mRNA on the ribosome has been observed by crystallography to wrap around the neck of the small ribosomal subunit (Yusupova et al. 2001). The mRNA enters the ribosome through a tunnel between the head and shoulder of the subunit, the entrance of which is lined by ribosomal proteins uS3, uS4, and uS5. The electron density for the ∼5-nt mRNA segment inside the tunnel (between the entrance and the A-site codon) is typically weak in crystal structures of the ribosome and is rarely seen to continue outside of the tunnel (Yusupova et al. 2006; Jenner et al. 2010b). Similarly, in cryo-electron microscopy (cryo-EM), structures of classical-state ribosome–mRNA complexes (Fischer et al. 2016; Zhang et al. 2018), the local resolution for the mRNA in the vicinity of the tunnel entrance is typically low. Notably, the crystal structure of the chimeric hybrid-state ribosome shows a longer stretch of mRNA, ∼8 nt, inside an elongated tunnel formed as a result of small subunit head rotation (Zhou et al. 2014).
It is not clear how a translating ribosome interacts with an encountered mRNA structure such as a hairpin. One possibility (Yusupova et al. 2001) is that the head domain of the small subunit (which contains protein uS3) interacts with one strand of the duplex, while the body domain (containing proteins uS4 and uS5) interacts with the other strand. During translocation, the head domain moves relative to the body (Ratje et al. 2010; Guo and Noller 2012), and this could physically pry apart the two strands as a result. In this scheme, the geometry of the tunnel entrance in the classical state would complement that of encountered mRNA structures such as duplexes. However, for a helicase that encounters base-paired segments only sporadically, such a strategy might be counterproductive because binding to a duplex would favor its formation and disfavor its opening. Indeed, the opposite strategy is used by RNA helicases of the DEAD-box family, which have a higher binding affinity for the helicase product (i.e., single-stranded RNA) than for the substrate (e.g., a duplex) if they bind the latter at all (Jarmoskaite and Russell 2011; Linder and Jankowsky 2011). If product stabilization is also used by the ribosomal helicase, it seems more likely that the geometry of the tunnel entrance in the classical state evolved not to fit common mRNA structures such as duplexes, but to preferentially bind single-stranded mRNA.
We have determined the crystal structure of a classical-state E. coli ribosome–mRNA complex in a novel packing arrangement, in which single-stranded mRNA is seen to interact, via its backbone and in an extended conformation, with positively charged residues in protein uS3 immediately outside the tunnel. This strongly favors the involvement of product stabilization in the mechanism of unwinding structured mRNA by the ribosome, as described above. In vitro assays with reconstituted ribosomes indicate that mRNA binding to uS3 near the tunnel entrance is important for mRNA-dependent tRNA binding on the ribosome.
RESULTS
Obtaining crystals with a novel crystal packing arrangement
The structure presented here was obtained using wild-type E. coli 70S ribosomes in complex with a hairpin-containing mRNA. The pH03H9 mRNA contained a Shine–Dalgarno (SD) sequence, a start (AUG) codon, three consecutive valine (GUA) codons, a spacer sequence, and a 4-bp hairpin capped by a UUCG tetraloop (Fig. 1A; Supplemental Table S1). The ribosome was first bound on the mRNA at the start codon in the presence of charged initiator tRNA. This was followed by in vitro translation in the presence of valyl-tRNAVal·EF-Tu·GTP ternary complex, EF-G, and GTP. Only partial translocation appears to have taken place at this stage (see below). The resulting elongation complex was then used for crystallization, and X-ray diffraction data were collected to solve the structure by molecular replacement at a resolution of 3.9 Å (Table 1).
FIGURE 1.

Crystal packing rearrangement in the presence of hairpin-containing mRNA. (A) Sequence of the hairpin-containing mRNA used to obtain the new crystal form. Nucleotide positions are numbered according to the most likely mRNA register, in which the first valine codon occupies the ribosomal P-site in the crystal structure following in vitro translation (see Supplemental Fig. S5B for alternative possible registers). The segment from −15 to +15 (highlighted in yellow) is ordered in the crystal structure. (B) The crystal packing of ribosomes in the hairpin complex (right; this study) is compared to that in the classical/hybrid-state complex with single-stranded mRNA (left; PDB 4V9D) (Dunkle et al. 2011), when aligned on 16S rRNA of ribosome B. Note that more space has become available in front of protein uS3 (red) in the expected location of the mRNA hairpin, indicated by the thick arrow. (C) Contact between adjacent ribosomes in the crystal includes the docking of the protruding protein bL9 from one ribosome (A or B, in red and orange, respectively) on the 16S rRNA of the other. (D) In complexes formed with no mRNA or with single-stranded mRNA (e.g., PDB 4V9D) (Dunkle et al. 2011), the bL9 contacts are very similar for ribosomes A and B (pink and light orange, respectively), when aligned on the 16S rRNA of the two ribosomes. The conserved interaction between A55 of 16S rRNA and Phe91 of bL9 is indicated. (E) In the hairpin complex, both bL9 contacts are significantly rearranged compared to panel D, as indicated by the arrows, and are markedly different for ribosomes A and B (red and orange, respectively).
TABLE 1.
Crystallographic statistics for the E. coli 70S–hairpin complex
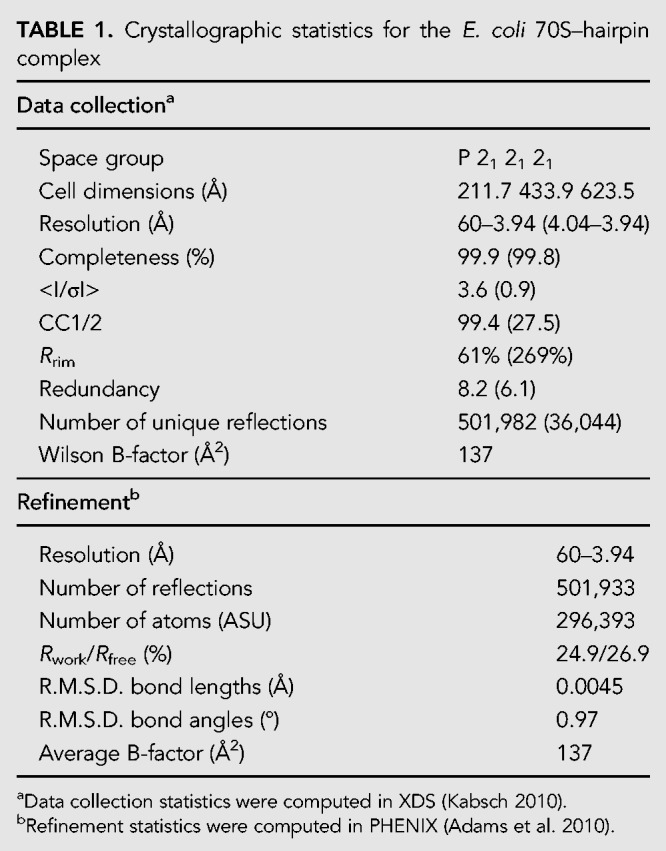
The complex crystallized in the P 21 21 21 space group, with two ribosomes (A and B) in the asymmetric unit, both in the classical state. This structure represents a new crystal form, distinct from previously published structures of 70S ribosomes in this space group that contained unstructured mRNA or no mRNA (Schuwirth et al. 2006; Selmer et al. 2006; Jenner et al. 2010b; Dunkle et al. 2011). Compared to those structures, we observe a large-scale rigid-body rearrangement of the ribosomes that opens up cavities on the solvent side of protein uS3, near the 3′ end of the mRNA where the hairpin is positioned (Fig. 1B; Supplemental Fig. S1), suggesting that the rearrangement may have occurred to accommodate the hairpin. Notably, crystal contacts involving protein uS3, which are seen in other crystal structures of 70S ribosomes in this space group, are absent in both ribosomes in this structure.
Another major rearrangement of crystal contacts is observed between the large subunit protein bL9 in one ribosome and 16S rRNA in the neighboring ribosome (Fig. 1C–E; Supplemental Fig. S2). In ribosome A in the present structure, the C-terminal domain of bL9 forms new crystal contacts by moving ∼30 Å toward the spur helix (h6) in the neighboring 16S rRNA, while in ribosome B, it moves by ∼15 Å toward the body of the neighboring 16S rRNA (Fig. 1E). These results suggest that the presence of the mRNA hairpin may influence ribosome–ribosome packing, resulting in novel inter-ribosomal contacts not seen in complexes with single-stranded mRNA.
Single-stranded mRNA interacts with the ribosome inside and outside of the downstream tunnel
With no mRNA in the initial structure model, unbiased simulated-annealing mFo–DFc maps showed traceable electron density for the mRNA from position −15 to +15 (where position +1 is defined as the first nucleotide of the P-site codon) (Fig. 2A). After placing the mRNA model (Fig. 2B; Supplemental Fig. S3A), the A- and P-site codons (positions +1 to +6) show the strongest 2mFo–DFc electron density compared to the rest of the mRNA. Upstream of the E-site, the mRNA forms an imperfect SD helix in an orientation similar to the previously observed post-initiation state (Supplemental Fig. S3B; Yusupova et al. 2006). The mRNA tunnel downstream from the A-site houses five nucleotides from position +7 to +11 (Fig. 2B), beyond which the mRNA continues as a single strand up to position +15, interacting mainly with protein uS3 (Fig. 2C).
FIGURE 2.

mRNA structure in the hairpin complex. (A) Difference electron density peaks (red) for mRNAs in simulated annealing mFo–DFc mRNA omit maps for ribosomes A and B, contoured at 2.5σ and blurred by 400 Å2. The inset shows the map overlaid on the mRNA models. (B) The 2mFo–DFc electron density map for mRNA and its associated anti-SD sequence in ribosome B, contoured at 1σ (blue). The mRNA and the A- and P-site tRNAs are also shown. The inset compares the path of mRNA in ribosomes A and B (blue and red, respectively) after alignment on the 16S rRNA. (C) mRNA path beyond the tunnel entrance between the head and the body (shoulder) of the small ribosomal subunit in ribosome B. The inset shows a close-up view of the entrance.
Inside the downstream tunnel, mRNA bases at positions +6 and +7 are stacked, and nucleotide C1397 of 16S rRNA is tucked in away from the +7 base (Fig. 3), similar to what was seen in elongation complexes previously (Jenner et al. 2010b). After position +9 or +10, the mRNA orientation appears to flip such that its backbone is facing uS3 when mRNA emerges at the tunnel entrance at position +12 (Fig. 3). The conserved positively charged residues Lys44 and Arg46 in protein uS4, and Arg131 in protein uS3, interact with the mRNA backbone near the entrance (Fig. 3).
FIGURE 3.

Path of the mRNA in ribosomes A and B. The mRNA (red) runs from right to left in the 5′–3′ direction. It has few interactions with the components of the tunnel. Outside of the tunnel, the mRNA interacts with the C-α3 helix in protein uS3.
Outside of the tunnel, the mRNA interacts with the base of the C-terminal α helix 3 (C-α3 helix) in uS3 (Fig. 3; Supplemental Figs. S3D, S4). In ribosome A, nucleotides +13 to +14 interact via their backbone with the conserved positively charged residues Arg132 and Lys135 in the C-α3 helix, while in ribosome B, the backbone of nucleotides +14 and +15 interacts with Arg132 and Arg136 in the same α helix (Fig. 3). The path of mRNA differs slightly in ribosomes A and B (Fig. 2B, inset; Fig. 3, right panels), apparently due to the close approach of crystal contacts in front of the tunnel entrance in ribosome A but not ribosome B (Supplemental Fig. S3C). However, in both cases, the mRNA segment outside the tunnel adopts an extended and straightened conformation that deviates significantly from the curvature of A-form geometry (Figs. 2B, 3). This conformation has a mean phosphate–phosphate distance of ∼6.5 Å, which implies C2′-endo sugar puckers for at least some nucleotides.
Despite ample space for a hairpin inside the cavities between ribosomes in this crystal form (Supplemental Figs. S1, S5A), no clear electron density corresponding to the 3′-mRNA hairpin is observed, suggesting that the hairpin is unwound or otherwise disordered.
The data resolution is insufficient to distinguish individual bases and to directly establish the mRNA register relative to the ribosome. However, only three registers are probably based on the structure (Supplemental Fig. S5B), none of which correspond to full ribosome translocation after the in vitro translation step. Of these, the most likely register corresponds to translocation by one codon, placing the beginning of the hairpin at position +15 (Supplemental Fig. S5B, possibility 2), although the other two possibilities cannot be formally excluded.
uS3 mutations reduce tRNA-binding affinity in an mRNA-dependent manner
It has been shown previously that mutation of conserved uS3 residues Arg131, Arg132, and Lys135 to alanine affects the helicase activity of the ribosome (Takyar et al. 2005). In the present structure, we observe these positively charged residues in the binding site for single-stranded mRNA, suggesting their involvement not only in the helicase activity of the ribosome, but also in its basic mRNA binding activity. To test the functional significance of uS3–mRNA binding, we tested ribosomes reconstituted with the same uS3 triple mutant for their ability to bind tRNA in an mRNA-dependent manner, using the filter binding assay. This indirect measure of mRNA binding was used in order to exclude the appreciable nonspecific mRNA binding that we observe when using a direct measure of total bound mRNA (data not shown). 35S-fMet-tRNAfMet binding to ribosomes at 10 mM Mg2+ is indeed mRNA-dependent: Estimated tRNA-binding affinity for wild-type reconstituted ribosomes in the presence of the single-stranded fMVVV mRNA (at twofold molar excess; Supplemental Table S1) is about 150-fold higher than in the absence of mRNA (Table 2, rows 1 and 4).
TABLE 2.
Binding of fMet-tRNAfMet to reconstituted 70S ribosomes

Binding affinity for 35S-fMet-tRNAfMet in the presence of mRNA is lower by about 50-fold in ribosomes reconstituted with the triple-mutant uS3 protein compared to those with wild-type uS3 (Fig. 4A; Table 2, rows 1 and 2). Despite lower affinity, the extent of tRNA-binding in the triple-mutant approaches that of wild-type at saturating tRNA concentrations, suggesting that the mutant ribosome population is not simply partially inactive. Reconstituted ribosomes lacking uS3 altogether show a more severe tRNA binding defect of about 100-fold lower affinity and less than half the extent of wild-type (Table 2, row 3; Fig. 4A). tRNA binding affinities of the wild-type and triple-mutant ribosomes are similar to each other in the absence of mRNA (Table 2, rows 4 and 5), or under mRNA-independent binding conditions (at 20 mM Mg2+) (Table 2, rows 6–9), suggesting that the mutation mainly affects specific mRNA-dependent tRNA binding. In the absence of mRNA or at 20 mM Mg2+, the inferred binding stoichiometry is higher (Table 2, right column), consistent with nonspecific binding at multiple tRNA sites under those conditions. Finally, in toeprinting assays (Hartz et al. 1988) using the same mRNA, mutant ribosomes show weaker toeprints at the start codon compared to wild-type ribosomes (Fig. 4B, lanes P), but are capable of translocation once bound at the start codon (Fig. 4B, lanes T). Together, these results indicate that the triple-mutant ribosomes are defective in mRNA-dependent P-site tRNA binding, and suggest that efficient tRNA binding requires uS3–mRNA interactions.
FIGURE 4.
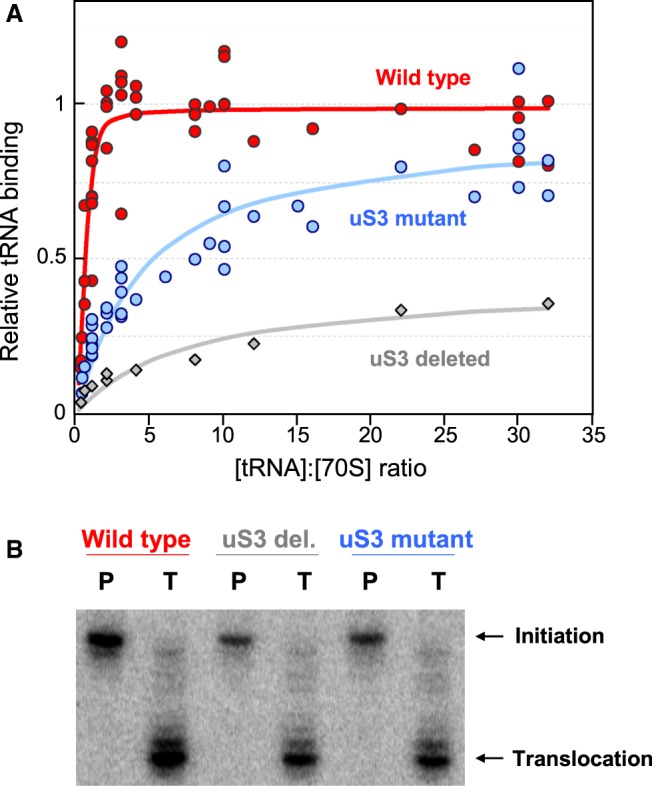
Mutational analysis of uS3 residues near the mRNA tunnel entrance. (A) Binding of 35S-labeled fMet-tRNAfMet to reconstituted E. coli ribosomes containing wild-type uS3 (red), triple-mutant uS3 (blue), or no uS3 (gray), in the presence of fMVVV mRNA, as measured by filter binding. Binding is normalized to that of the wild type at saturation. (B) P-site binding (P) and translocation (T) toeprints of reconstituted ribosomes containing wild-type uS3, no uS3, or mutant uS3, programmed with the fMVVV mRNA, showing weaker toeprints when using mutant or no uS3.
A-site tRNA bending is accompanied by A-site finger interactions
In initial maps calculated before placement of mRNA and tRNAs in our structure, strong difference electron density for tRNA was observed in both the A-site and the P-site of each ribosome (and the E-site of ribosome A only) (Fig. 5A). The A-site and P-site tRNAs in each ribosome are base-paired with the mRNA codons and are present at approximately full occupancy (Supplemental Fig. S6A). Flipping of 16S rRNA bases G530, A1492, and A1493 in the decoding center of the small subunit, and closure of the shoulder of the small subunit in the presence of cognate A-site tRNA are observed, consistent with previous reports (Supplemental Fig. S6B–D; Ogle et al. 2002). The A-site tRNA is seen in a bent conformation due to an ∼10° hinge motion near its 26–44 purine–purine pair. This moves the tRNA acceptor arm by ∼5 Å to one side (Fig. 5B), and widens the gap between the A- and P-site tRNAs. The elbow of the A-site tRNA interacts with helix H38 (A-site finger) of 23S rRNA, which has twisted and moved toward the tRNA elbow compared to E. coli complexes with a vacant A-site (Fig. 5C; Dunkle et al. 2011; Noeske et al. 2015). Notably, the conserved bulged A896 in H38 is stacked on the G19:C56 tertiary base pair in the elbow of the A-site tRNA (Fig. 5C; Supplemental Fig. S6E).
FIGURE 5.

tRNA structures in the hairpin complex. (A) Electron density peaks (orange) for tRNAs in mFo–DFc tRNA + mRNA omit maps for ribosomes A and B, contoured at 2σ and blurred by 200 Å2. The insets show the map overlaid on the tRNA models. (B) Cartoon representation of the A-site tRNA (red) viewed from the subunit interface (left) or from the large subunit (right). The A-site tRNA in the E. coli ribosome (red, this study) is bent compared to unbound tRNA (light and dark blue, PDB 1EHZ and 4TRA, respectively) (Westhof et al. 1988; Shi and Moore 2000). The nonbent A-site tRNA in the T. thermophilus ribosome (green, PDB 4V5D) (Voorhees et al. 2009) is also shown for comparison. Position of the 26:44 base pair is indicated by a black dot. All tRNA structures are aligned by their anticodon stem–loop. (C) In the presence of A-site tRNA (yellow), the tip of helix H38 of 23S rRNA (red) interacts with the tRNA elbow, moving by up to 5 Å toward it compared to when the A-site is vacant (faint green, PDB 4YBB) (Noeske et al. 2015). The stacking of A896 base of H38 on the G19:C56 tertiary base pair of the tRNA is shown. Note that the terminal loop of H38 was not modeled in the vacant complex.
DISCUSSION
The crystal structure of the E. coli ribosome presented here reveals interaction of single-stranded mRNA (the helicase product) with protein uS3 outside the mRNA tunnel (Fig. 3). The straightened path of the bound mRNA is incompatible with curvature of the backbone in an A-form RNA duplex, suggesting a binding specificity for the single-stranded form of mRNA. This observation favors the possibility that product stabilization contributes to the mechanism of unwinding structured mRNAs by the ribosome.
The crystal form presented here has cavities between adjacent ribosomes that provide sufficient space for the mRNA hairpin (Fig. 1B; Supplemental Figs. S1, S5A). It also allows us to observe binding of single-stranded mRNA to uS3, as it lacks the usual uS3 crystal contacts (seen in previous crystals of wild-type ribosomes) that could otherwise interfere with mRNA binding to uS3.
In a recent cryo-EM study of the T. thermophilus ribosome in complex with an mRNA containing a hairpin at position +12 (Zhang et al. 2018), a low-resolution envelope for the hairpin was observed starting near Gln136 in the C-α3 helix of uS3 outside of the tunnel. This corresponded to position ∼+15 on the mRNA rather than +12 where the hairpin started, indicating that the segment from +12 to +15 may have become single-stranded. Indeed, SHAPE probing of the ribosome-bound mRNA (Zhang et al. 2018) revealed high reactivity for the strand complementary to the +12–15 segment, as expected for single-stranded RNA. However, the +12–15 segment itself showed little SHAPE reactivity, suggesting that despite being unpaired, its backbone may have been immobilized by binding to the ribosome, in agreement with the observations in the present study. Taken together, the current evidence points to the preferential binding of single-stranded mRNA to protein uS3 outside the tunnel, i.e. the involvement of product stabilization in mRNA unwinding by the ribosome.
Further insight into the mechanism of action of the ribosomal helicase during translocation comes from comparing the current classical-state ribosome structure with the crystal structure of a translocation intermediate, the chimeric hybrid-state ribosome in complex with EF-G, in which the head domain of the small ribosomal subunit is rotated relative to the body domain (Fig. 6; Zhou et al. 2014). In the classical state, mRNA is held at the tunnel entrance via interaction with conserved positively charged residues in proteins uS3 (head) and uS4 (body) that lie directly across from each other (Figs. 3, 6A). Mutation of these residues confers defects in the helicase activity of the ribosome (Takyar et al. 2005). During translocation, the 30S head domain rotates by 21°, pulling uS3 away from uS4 (Fig. 6B); mRNA interaction with uS3 is maintained, but the uS4–mRNA interaction shifts from approximately+12 to +15, effectively lengthening the mRNA tunnel by one codon (Fig. 6B). The tunnel is too narrow to accommodate a helix, so any bulky structure in the intervening mRNA segment (+12 to +14) would have to be unwound prior to or concomitant with head rotation. Our observation of interaction between uS3 and non-A-form single-stranded mRNA up to position +15 in the classical state (i.e., product stabilization) would predispose the mRNA segment +12 to +14 to becoming single-stranded even before head rotation takes place, providing a significant kinetic enhancement of mRNA unwinding (H Amiri, in prep.).
FIGURE 6.

Comparison with the head-rotated state. The contact region between mRNA and the mRNA tunnel becomes extended during translocation. (A) In the classical state (this work), three mRNA nucleotides (red) are held at the tunnel entrance formed by proteins uS3, uS4, and uS5 (pink) via interactions with six conserved positively charged residues in uS3 and uS4 (blue). (B) In the chimeric hybrid state (PDB 4W29) (Zhou et al. 2014), rotation of the 30S head domain during translocation results in elongation of the tunnel, thus allowing accommodation of an additional three nucleotides, or one codon (orange), within the tunnel.
A role for protein uS3 in ribosome–mRNA interaction is supported by biochemical studies. Mutation of uS3 residues Arg131, Arg132, and Lys135 to alanines was previously shown to significantly reduce the mRNA helicase activity of the ribosome (Takyar et al. 2005). Our results show that mRNA-dependent tRNA binding activity is also significantly compromised in the uS3 mutant (Fig. 4), suggesting that these uS3 residues may help bind the mRNA, thus favoring stable ribosome–tRNA–mRNA interactions. Mutation of homologous residues in yeast uS3 has also been shown to affect translation initiation in an mRNA-dependent fashion in vivo (Dong et al. 2017).
The ribosome is commonly found to protect mRNA residues up to position +15 or beyond (Steitz 1969; Beyer et al. 1994; Hüttenhofer and Noller 1994; Mohammad et al. 2016). Given that mRNA emerges at the tunnel entrance around position +12 (Yusupova et al. 2006; Jenner et al. 2010b), this implies that mRNA–ribosome interactions are maintained outside of the tunnel up to at least position +15, as observed in the present structure. The electrostatic interaction observed here between the mRNA and protein uS3 appears to extend even to the binding of an mRNA mimic, the Stm1 protein, to yeast 80S ribosomes. This protein occupies the path of the mRNA inside yeast ribosomes harvested under nutrient starvation. Remarkably, a negatively charged α helix in Stm1 runs along the C-α3 helix of protein uS3 outside of the tunnel entrance (Ben-Shem et al. 2011), following a path similar to that reported here for the mRNA (Fig. 7).
FIGURE 7.
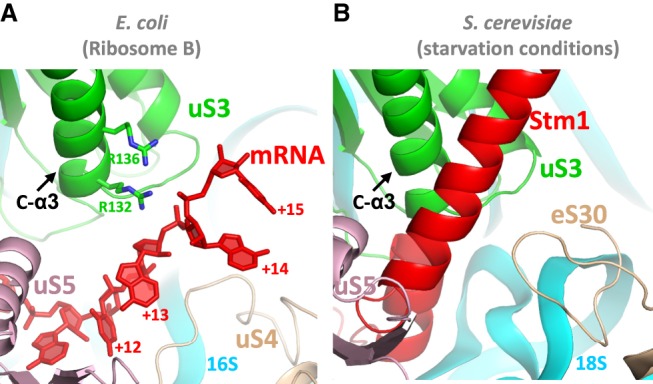
Comparison with the path of Stm1 in yeast 80S ribosomes. (A) The straightened path of mRNA (red) along protein uS3 (green) in ribosome B in this study resembles (B) the path of protein Stm1 in the nutrient-starved yeast 80S ribosome (PDB 4V88) (Ben-Shem et al. 2011).
Lastly, the A-site tRNA is seen here to bend near the 26–44 purine–purine base pair, a common hinge point for tRNA flexing during translation (Korostelev et al. 2006; Zhou et al. 2013). At the same time, the H38 helix of 23S rRNA has moved to allow stacking of its conserved bulged A896 on the G19:C56 tertiary base pair in the tRNA elbow (Fig. 5C). This is analogous to stacking of the G2112:A2169 tertiary base pair of 23S rRNA (in the L1 stalk) on the G19:C56 pair of the P/E and E/E tRNAs (Mohan and Noller 2017), suggesting a role for the universally conserved G19:C56 pair as a stacking platform in tRNA interactions with rRNA. A-site tRNA bending and H38 movement can also be found in cryo-EM structures of classical-state E. coli ribosomes (Fischer et al. 2016; Huter et al. 2017; Loveland and Korostelev 2018). In contrast, in T. thermophilus ribosomes, H38 movement to the extent seen here has not been detected (Jenner et al. 2010a), and the bent A-site tRNA conformation has been observed only in the presence of viomycin and capreomycin, antibiotics thought to inhibit translocation by stabilizing A-site tRNA binding (Fig. 3B; Supplemental Fig. S3F; Stanley et al. 2010). The functional significance of A-site tRNA bending and the H38–tRNA interaction thus remains to be understood.
MATERIALS AND METHODS
Ribosome preparation
Tight-couple ribosome preparation was performed essentially as previously described (Moazed and Noller 1989). E. coli cells (strain MRE600) were grown to mid-log phase at 37°C, and then cooled on ice for 1 h. Clarified French-press lysates were layered on a 38% sucrose cushion in 20 mM Tris-HCl (pH 7.0), 500 mM NH4Cl, 15 mM MgCl2, 6 mM βME and centrifuged in a Beckman Ti60 rotor at 40,000 rpm for 20 h at 4°C to pellet the ribosomes. The pellets were salt-washed twice by resuspending in the same buffer containing 500 mM NH4Cl and centrifuging in a Ti60 rotor at 55,000 rpm for 2 h at 4°C. Resuspended ribosomes were then layered on a 10%–35% sucrose gradient in 20 mM Tris-HCl (pH 7.0), 100 mM NH4Cl, 6 mM MgCl2, 6 mM βME and centrifuged in a SW28 rotor at 19,000 rpm for 16 h at 4°C. The 70S fraction was collected manually and pelleted by centrifugation in a Ti45 rotor at 36,000 rpm for 22 h at 4°C, resuspended in the same buffer (no sucrose) containing 10 mM MgCl2 to a final ribosome concentration of about 25 mg/mL, and flash frozen. Analytical sucrose gradient centrifugations were performed to confirm the purity (>95%) of the 70S particles.
mRNA, tRNA, and elongation factor preparations
The mRNAs (Supplemental Table S1) were made in vitro by T7 RNA polymerase transcription using DNA templates obtained from plasmids pH03H9 (by linearization) or pH03_fMVVV (by PCR). About 100 µg of the DNA template was incubated in a 1 mL reaction containing 80 mM K+-HEPES (pH 7.5), 50 mM NaCl, 2 mM spermidine, 30 mM DTT, 20 mM MgCl2, and 12 mM of total NTPs (4.8 mM ATP, 3.6 mM GTP, 2.4 mM UTP, 1.2 mM CTP, adjusted for the base composition of the pH03H9 mRNA) in the presence of 100 µg of purified T7 RNA polymerase and 2.5 units of inorganic pyrophosphatase (Fermentas) for 3 h at 37°C. The RNA was then purified on an 8% denaturing polyacrylamide gel.
For charging of fMet and Val tRNAs, 10 nmol of each purified tRNA (Subriden) was incubated in a 330 µL reaction containing 50 mM HEPES (pH 7.5), 50 mM KCl, 10 mM MgCl2, 5 mM DTT, 4 mM ATP, 80 nmol of the appropriate amino acid, and 60 µL of DEAE-purified E. coli S-100 fraction (Moazed and Noller 1989) for 20 min at 37°C. For fMet, 300 nM of neutral N10-formyltetrahydrofolate was also present in the mixture. The tRNAs were phenol–chloroform extracted, ethanol precipitated, and passed through a 1 mL Sephadex G25 spin column. Successful charging was confirmed by acid PAGE analysis (Walker and Fredrick 2008).
For expression and purification of elongation factors, BL21(DE3) cells expressing 6xHis-tagged EF-G or EF-Tu (Takyar et al. 2005) were grown to mid-log and then induced with IPTG at 1 mM for 3 h. Purification was performed at 4°C in buffers containing 50 mM Tris-HCl (pH 7.5), 60 mM NH4Cl, 7 mM MgCl2, 6 mM βME, and 15% glycerol. All buffers for EF-Tu also contained 10 µM GDP. Affinity purification of clarified French-pressed lysates was performed with Ni-NTA resin (Qiagen) and the proteins were eluted in buffer containing 250 mM imidazole. Dialyzed fractions were loaded on a Resource Q FPLC column (GE), eluted in the same buffer plus 150 mM KCl (for EF-G) or 130 mM KCl (for EF-Tu), and dialyzed to remove KCl prior to storage.
Complex formation and crystallization
To form the ribosome complex for crystallization, 400 pmol of E. coli 70S ribosome (at 10 µM) were prewarmed at 37°C for 10 min. Separately, 800 pmol of the hairpin-containing mRNA pH03H9 (at 100 µM) in water were heated at 90°C for 3 min, snap-cooled in an ice bath for 10 min, brought to 37°C, added to the ribosome along with 800 pmol of charged fMet tRNA in a total volume of 60 µL in 20 mM Tris-HCl (pH 7.0), 100 mM NH4Cl, 10 mM MgCl2, 6 mM βME, and incubated at 37°C for 20 min for P-site tRNA binding. In parallel, 1800 pmol of the E. coli EF-Tu protein (at 120 µM) was incubated in its storage buffer plus 2 mM GTP and 2 mM MgCl2 at 37°C for 10 min. To this EF-Tu mixture, 1600 pmol of charged Val-tRNA was added, and incubation was continued for another 5 min for ternary complex formation in the same buffer as in P-site binding. The ternary complex was then added, plus 1200 pmol of EF-G and GTP to a final concentration of 1 mM, to the P-site binding reaction, and incubated in a total volume of 100 µL at 37°C for 15 min for ribosome translocation. The complex was then brought to room temperature and used for crystallization.
For sitting-drop crystallization with vapor diffusion, 0.5 µL of the complex was mixed with 0.5 µL of the reservoir mix containing 100 mM Tris acetate (pH 7.0), 100 mM KSCN, 4.2% PEG 20,000 (Hampton), and 10%–12% pentaerythritol propoxylate 629 (17/8 PO/OH) (Hampton) and equilibrated against 50 µL of the reservoir mixture in a 96-well plate at 16°C for up to 2 wk. Large rod-shaped crystals (100 µm in the smallest dimension) were then cryoprotected by stepwise addition of the reservoir mixture containing 12%, 15%, and 20% pentaerythritol propoxylate 629 (17/8 PO/OH), respectively, and equilibration against the same mixture for about 24 h at each step. The crystals were then harvested from the drop through a layer of Paratone-N oil (Hampton), flash frozen in liquid nitrogen, and screened for X-ray diffraction. The deposited structure was solved using data collected from a single 10-d-old crystal.
Data collection and refinement
Diffraction data were collected at Beamline 12.3.1 at the Advanced Light Source (LBNL) and processed to obtain structure factor amplitudes using the XDS package (Kabsch 2010), with 2% of reflections designated as the free set. A previously published structural model of the E. coli ribosome (Dunkle et al. 2011) (without tRNAs) was directly used for multiple rounds of rigid-body refinement in PHENIX (Adams et al. 2010) with progressively smaller rigid bodies. Coordinates for proteins uL10, bL31, and uS2 were obtained from PDB 4YBB, 5NWY, and 4TOI, respectively (Byrgazov et al. 2015; Noeske et al. 2015; Su et al. 2017). tRNA models were placed into initial difference electron density peaks at this stage. Further rounds of editing and refitting in COOT (Emsley et al. 2010), restrained refinement of atomic coordinates, isotropic B-factors, and TLS and group-occupancy parameters in PHENIX and Refmac (Murshudov et al. 1997; Winn et al. 2011), as well as multistart simulated annealing refinement (Korostelev et al. 2009) and real-space coordinate refinement in PHENIX, improved the electron density maps and the R-factors. The mRNA was then built into the difference density peaks using KiNG (Chen et al. 2009) and RCrane (Keating and Pyle 2012) with further rounds of refinement using tight geometry restraints. Structural alignments and image preparations were performed in PyMOL.
Mutant ribosome preparation
A triple mutant (R131A, R132A, K135A) version of the E. coli uS3 gene was generated using the Kunkel method (Kunkel et al. 1987). Mutant proteins were expressed, purified, and used for in vitro 30S reconstitution and 70S preparation as previously described (Culver and Noller 2000), except that the S protein mixture (containing 3.5-fold molar excess of each of the proteins uS2, uS4–uS21, and either wild-type, triple-mutant, or no uS3) was added to 16S rRNA in a single step rather than sequentially. Reconstituted 70S ribosomes were concentrated to about 1 µM in buffer containing 20 mM Tris-HCl (pH 7.0), 100 mM NH4Cl, 20 mM MgCl2, and 6 mM βME. Successful assembly of triple-mutant uS3 was confirmed by separating 8 pmol of reconstituted 70S on a 15% SDS-PAGE gel, noting that the mutant protein shows a higher electrophoretic mobility than wild-type.
Filter binding assay
Nitrocellulose filter binding assays were typically performed by premixing 2 pmol of wild-type or mutant reconstituted 70S ribosomes and 4 pmol of fMVVV mRNA in buffer containing 20 mM Tris-HCl (pH 7.0), 100 mM NH4Cl, 10 mM or 20 mM MgCl2, 6 mM βME, and 0.25 mg/mL BSA on ice, followed by the addition of serial dilutions (0.6–64 pmol) of 35S-labeled fMet-tRNAfMet in a total volume of 20 µL, and incubation at 37°C for 15 min for P-site tRNA binding. After chilling on ice for 5 min, 10 µL of each reaction was blotted on a presoaked nitrocellulose HA filter (Millipore), which was then washed three times with a total of 15 mL ice-cold buffer without BSA, dried, and quantified using a scintillation counter. Background filter binding in the absence of ribosomes was subtracted. tRNA binding affinities (Kd) were estimated in Origin (OriginLab) by least-squares minimization fitting of the counts (count) at each total tRNA concentration (tRNA), and total ribosome concentration (70S), to the hyperbolic formula for equilibrium binding,
where Cmax is the estimated maximum count per unit of ribosome concentration (the product of tRNA specific activity, reaction volume, and binding stoichiometry). All counts were divided by the value of Cmax estimated for wild-type binding at 10 mM Mg2+ to obtain relative tRNA binding (Fig. 4A) and calculate relative stoichiometry (Table 2).
Toeprinting assay
For toeprinting assays (Hartz et al. 1988), 10 pmol of E. coli 70S ribosome were incubated in a 10 µL reaction containing 20 pmol of the fMVVV mRNA and 20 pmol of fMet-tRNAfMet in 20 mM Tris-HCl (pH 7.0), 100 mM NH4Cl, 10 mM MgCl2, and 6 mM βME at 37°C for 15 min for P-site binding. For each translocation reaction, 40 pmol of Val-tRNAVal were incubated with 45 pmol of EF-Tu·GTP (prepared in the same way as described above for crystallization) at 37°C for 5 min, and the resulting ternary complex was added, along with 30 pmol of EF-G and 1 mM of GTP and 1 mM MgCl2 to a P-site binding reaction and incubated at 37°C for 10 min to perform translocation. Primer extension was carried out by addition of ∼40 fmol of 32P-end-labeled toeprinting oligonucleotide (100 kcpm radioactivity), 70 µM each dNTP, and 0.3 unit of AMV reverse transcriptase (Seikagaku), and incubation at 37°C for 3 min. The mixture was then ethanol precipitated, resuspended in urea loading dye, and run on an 8% polyacrylamide toeprinting gel containing 8 M urea. The gel was dried and exposed to a phosphorimager screen overnight for quantification.
DATA DEPOSITION
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data bank under accession number 6BY1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the beamline staff at the LBNL and SLAC National Laboratories for help with diffraction screening and data collection; John Paul Donohue for assistance with data processing; Laura Lancaster for contributions to the manuscript and valuable discussions; Shervin Takyar, Jianyu Zhu, Manuel Ares, William Scott, and Doug Kellogg for expert advice; and members of the Noller laboratory for their critical feedback. This work was supported by grant no. R35 GM118156 to H.F.N. from the National Institute of General Medical Sciences, National Institutes of Health.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068965.118.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov PV, Gesteland RF, Atkins JF. 2002. Recoding: translational bifurcations in gene expression. Gene 286: 187–201. 10.1016/S0378-1119(02)00423-7 [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334: 1524–1529. 10.1126/science.1212642 [DOI] [PubMed] [Google Scholar]
- Beyer D, Skripkin E, Wadzack J, Nierhaus KH. 1994. How the ribosome moves along the mRNA during protein synthesis. J Biol Chem 269: 30713–30717. [PubMed] [Google Scholar]
- Brierley I. 1995. Ribosomal frameshifting viral RNAs. J Gen Virol 76: 1885–1892. 10.1099/0022-1317-76-8-1885 [DOI] [PubMed] [Google Scholar]
- Byrgazov K, Grishkovskaya I, Arenz S, Coudevylle N, Temmel H, Wilson DN, Djinovic-Carugo K, Moll I. 2015. Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res 43: 661–673. 10.1093/nar/gku1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Davis IW, Richardson DC. 2009. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci 18: 2403–2409. 10.1002/pro.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, Noller HF. 2000. In vitro reconstitution of 30S ribosomal subunits using complete set of recombinant proteins. Methods Enzymol 318: 446–460. 10.1016/S0076-6879(00)18069-3 [DOI] [PubMed] [Google Scholar]
- Dong J, Aitken CE, Thakur A, Shin B-S, Lorsch JR, Hinnebusch AG. 2017. Rps3/uS3 promotes mRNA binding at the 40S ribosome entry channel and stabilizes preinitiation complexes at start codons. Proc Natl Acad Sci 114: E2126–E2135. 10.1073/pnas.1620569114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JH. 2011. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332: 981–984. 10.1126/science.1202692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Bock LV, Maracci C, Wang Z, Paleskava A, Konevega AL, Schröder GF, Grubmüller H, Ficner R, et al. 2016. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 540: 80–85. 10.1038/nature20560 [DOI] [PubMed] [Google Scholar]
- Giedroc DP, Cornish PV. 2009. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res 139: 193–208. 10.1016/j.virusres.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Noller HF. 2012. Rotation of the head of the 30S ribosomal subunit during mRNA translocation. Proc Natl Acad Sci 109: 20391–20394. 10.1073/pnas.1218999109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz D, McPheeters DS, Traut R, Gold L. 1988. Extension inhibition analysis of translation initiation complexes. Methods Enzymol 164: 419–425. 10.1016/S0076-6879(88)64058-4 [DOI] [PubMed] [Google Scholar]
- Huter P, Arenz S, Bock LV, Graf M, Frister JO, Heuer A, Peil L, Starosta AL, Wohlgemuth I, Peske F, et al. 2017. Structural basis for polyproline-mediated ribosome stalling and rescue by the translation elongation factor EF-P. Mol Cell 68: 515–527.e6. 10.1016/j.molcel.2017.10.014 [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A, Noller HF. 1994. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J 13: 3892–3901. 10.1002/j.1460-2075.1994.tb06700.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I, Russell R. 2011. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip Rev RNA 2: 135–152. 10.1002/wrna.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Demeshkina N, Yusupova G, Yusupov M. 2010a. Structural rearrangements of the ribosome at the tRNA proofreading step. Nat Struct Mol Biol 17: 1072–1078. 10.1038/nsmb.1880 [DOI] [PubMed] [Google Scholar]
- Jenner LB, Demeshkina N, Yusupova G, Yusupov M. 2010b. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 17: 555–560. 10.1038/nsmb.1790 [DOI] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating KS, Pyle AM. 2012. RCrane: semi-automated RNA model building. Acta Crystallogr D Biol Crystallogr 68: 985–995. 10.1107/S0907444912018549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF. 2006. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 126: 1065–1077. 10.1016/j.cell.2006.08.032 [DOI] [PubMed] [Google Scholar]
- Korostelev A, Laurberg M, Noller HF. 2009. Multistart simulated annealing refinement of the crystal structure of the 70S ribosome. Proc Natl Acad Sci 106: 18195–18200. 10.1073/pnas.0909287106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol 154: 367–382. 10.1016/0076-6879(87)54085-X [DOI] [PubMed] [Google Scholar]
- Linder P, Jankowsky E. 2011. From unwinding to clamping—the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12: 505–516. 10.1038/nrm3154 [DOI] [PubMed] [Google Scholar]
- Loveland AB, Korostelev AA. 2018. Structural dynamics of protein S1 on the 70S ribosome visualized by ensemble cryo-EM. Methods 137: 55–66. 10.1016/j.ymeth.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Noller HF. 1989. Interaction of tRNA with 23S rRNA in the ribosomal A, P, and E sites. Cell 57: 585–597. 10.1016/0092-8674(89)90128-1 [DOI] [PubMed] [Google Scholar]
- Mohammad F, Woolstenhulme CJ, Green R, Buskirk AR. 2016. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep 14: 686–694. 10.1016/j.celrep.2015.12.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S, Noller HF. 2017. Recurring RNA structural motifs underlie the mechanics of L1 stalk movement. Nat Commun 8: 14285 10.1038/ncomms14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- Namy O, Rousset JP, Napthine S, Brierley I. 2004. Reprogrammed genetic decoding in cellular gene expression. Mol Cell 13: 157–168. 10.1016/S1097-2765(04)00031-0 [DOI] [PubMed] [Google Scholar]
- Noeske J, Wasserman MR, Terry DS, Altman RB, Blanchard SC, Cate JHD. 2015. High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol 22: 336–341. 10.1038/nsmb.2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. 10.1016/S0092-8674(02)01086-3 [DOI] [PubMed] [Google Scholar]
- Qu X, Wen J-D, Lancaster L, Noller HF, Bustamante C, Tinoco I. 2011. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature 475: 118–121. 10.1038/nature10126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratje AH, Loerke J, Mikolajka A, Brünner M, Hildebrand PW, Starosta AL, Dönhöfer A, Connell SR, Fucini P, Mielke T, et al. 2010. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature 468: 713–716. 10.1038/nature09547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuwirth BS, Day JM, Hau CW, Janssen GR, Dahlberg AE, Cate JHD, Vila-Sanjurjo A. 2006. Structural analysis of kasugamycin inhibition of translation. Nat Struct Mol Biol 13: 879–886. 10.1038/nsmb1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FV IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942. 10.1126/science.1131127 [DOI] [PubMed] [Google Scholar]
- Shi H, Moore PB. 2000. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA 6: 1091–1105. 10.1017/S1355838200000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley RE, Blaha G, Grodzicki RL, Strickler MD, Steitz TA. 2010. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat Struct Mol Biol 17: 289–293. 10.1038/nsmb.1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. 1969. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature 224: 957–964. 10.1038/224957a0 [DOI] [PubMed] [Google Scholar]
- Su T, Cheng J, Sohmen D, Hedman R, Berninghausen O, von Heijne G, Wilson DN, Beckmann R. 2017. The force-sensing peptide VemP employs extreme compaction and secondary structure formation to induce ribosomal stalling. Elife 6: e25642 10.7554/eLife.25642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takyar S, Hickerson RP, Noller HF. 2005. mRNA helicase activity of the ribosome. Cell 120: 49–58. 10.1016/j.cell.2004.11.042 [DOI] [PubMed] [Google Scholar]
- Tholstrup J, Oddershede LB, Sørensen MA. 2012. mRNA pseudoknot structures can act as ribosomal roadblocks. Nucleic Acids Res 40: 303–313. 10.1093/nar/gkr686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. 2009. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol 16: 528–533. 10.1038/nsmb.1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SE, Fredrick K. 2008. Preparation and evaluation of acylated tRNAs. Methods 44: 81–86. 10.1016/j.ymeth.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, Dumas P, Moras D. 1988. Restrained refinement of two crystalline forms of yeast aspartic acid and phenylalanine transfer RNA crystals. Acta Crystallogr A 44: 112–123. 10.1107/S010876738700446X [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242. 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupova GZ, Yusupov MM, Cate JH, Noller HF. 2001. The path of messenger RNA through the ribosome. Cell 106: 233–241. 10.1016/S0092-8674(01)00435-4 [DOI] [PubMed] [Google Scholar]
- Yusupova G, Jenner L, Rees B, Moras D, Yusupov M. 2006. Structural basis for messenger RNA movement on the ribosome. Nature 444: 391–394. 10.1038/nature05281 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hong S, Ruangprasert A, Skiniotis G, Dunham CM. 2018. Alternative mode of E-site tRNA binding in the presence of a downstream mRNA stem loop at the entrance channel. Structure 26: 437–445.e3. 10.1016/j.str.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2013. Crystal structures of EF-G–ribosome complexes trapped in intermediate states of translocation. Science 340: 1236086 10.1126/science.1236086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2014. How the ribosome hands the A-site tRNA to the P site during EF-G–catalyzed translocation. Science 345: 1188–1191. 10.1126/science.1255030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.