

Abstract
The accurate inheritance of genetic material is a basic necessity in all domains of life and an unexpectedly large number of RNA processing factors are required for mitotic progression and genome stability. NRDE2 (nuclear RNAi defective-2) is an evolutionarily conserved protein originally discovered for its role in nuclear RNA interference (RNAi) and heritable gene silencing in Caenorhabditis elegans (C. elegans). The function of the human NRDE2 gene remains poorly understood. Here we show that human NRDE2 is an essential protein required for suppressing intron retention in a subset of pre-mRNAs containing short, GC-rich introns with relatively weak 5′ and 3′ splice sites. NRDE2 preferentially interacts with components of the U5 small nuclear ribonucleoprotein (snRNP), the exon junction complex, and the RNA exosome. Interestingly, NRDE2-depleted cells exhibit greatly increased levels of genomic instability and DNA damage, as well as defects in centrosome maturation and mitotic progression. We identify the essential centriolar satellite protein, CEP131, as a direct NRDE2-regulated target. NRDE2 specifically binds to and promotes the efficient splicing of CEP131 pre-mRNA, and depleting NRDE2 dramatically reduces CEP131 protein expression, contributing to impaired recruitment of critical centrosomal proteins (e.g., γ-tubulin and Aurora Kinase A) to the spindle poles during mitosis. Our work establishes a conserved role for human NRDE2 in RNA splicing, characterizes the severe genomic instability phenotypes observed upon loss of NRDE2, and highlights the direct regulation of CEP131 splicing as one of multiple mechanisms through which such phenotypes might be explained.
Keywords: splicing, nuclear RNAi, intron retention, centrosome, mitosis, genome stability
INTRODUCTION
The NRDE2 gene was initially discovered in C. elegans for its role in nuclear RNAi, i.e., the small RNA-directed silencing of nuclear transcripts (Guang et al. 2010; Burton et al. 2011). Consistent with a co-transcriptional mechanism of nuclear RNAi (Guang et al. 2010), recent studies have implicated intriguing physical and functional links between nuclear RNAi factors and components of the splicing machinery (Dumesic et al. 2013; Aronica et al. 2015; Akay et al. 2017). Indeed, the NRDE2 homolog in S. pombe, Nrl1, was found to associate with RNA splicing and decay factors to regulate cryptic intronic sequences (Lee et al. 2013; Aronica et al. 2015; Zhou et al. 2015). Nrl1 has also been linked to genomic instability, possibly through the regulation of R-loop accumulation (Aronica et al. 2015). The function of human NRDE2 remains poorly understood.
RESULTS AND DISCUSSION
Here we report the functional and biochemical characterization of the human NRDE2 (C14ORF102) gene. Unlike its homologs in nematodes and fission yeast (Guang et al. 2010; Aronica et al. 2015), we demonstrate that NRDE2 is an essential gene in human cells. Depletion of NRDE2 resulted in a complete arrest in cell growth and proliferation in all cell lines tested (Fig. 1A). si-NRDE2 specificity was confirmed by the efficient knockdown of NRDE2 mRNA and protein, and by the rescue of proliferation in cells carrying a NRDE2 overexpression construct (Supplemental Fig. 1A–C). Following NRDE2 depletion, FACS analysis revealed an accumulation of cells with 4N DNA content, indicative of an increase in the number of cells in G2 or mitosis (Fig. 1B). Cyclin B1 and phosphorylated histone H3(Ser10)—markers up-regulated in late G2 and early mitosis—were also increased (Fig. 1C), further indicating defective cell cycle progression. To investigate the nature and extent of the mitotic delay in individual cells, we performed live cell imaging using RPE-1 (retinal pigment epithelial) cells expressing H2B-GFP (for chromatin visualization) and α-tubulin-mCherry (for mitotic spindle visualization); while 50/51 NRDE2-depleted cells examined did complete mitosis, we observed a greater than threefold increase in mitotic time, with the majority of the delay occurring during prometaphase (Fig. 1D; Supplemental Movies).
FIGURE 1.

NRDE2 is an essential gene required for mitotic progression and genome stability. (A) MTT assays for cell proliferation following transfection with control or NRDE2-targeting siRNAs. Twenty nanomolar siRNAs were used in all experiments. Error bars = SD (n = 3). (B,C) Representative FACS analyses of DNA content (B) and western blots (C) of MDA-MB-231 cells collected 5 d post-transfection (n = 3). (D) Mitotic duration was quantified by time-lapse microscopy at 10 min resolution using RPE-1 cells expressing H2B-GFP (for chromatin visualization) and α-tubulin-mCherry (for mitotic spindle visualization) at 5–6 d post-transfection (n = 50 cells per condition). See also Supplemental Movies S1–S3. (E) Representative time course analysis of γH2AX levels by western blot (n = 3). (F) Categories and quantification of aberrant nuclear morphologies visualized by DAPI 4 d post si-NRDE2 transfection in MDA-MB-231 cells. “Multiple” refers to nuclear abnormalities falling under multiple categories, the most common being polylobed + micronuclei. (White) DNA. (Green) α-tubulin. More than 300 cells were scored per condition per replicate (n = 3). Bar = 10 µm. (*) P < 0.05, two-tailed t-test.
Prometaphase delays are often associated with chromosome missegregation, which can result in micronuclei formation. Micronuclei are clinically relevant markers of genomic instability, and have also been implicated as major sources of DNA damage (Zhang et al. 2015). Indeed, NRDE2-depleted HEK293T cells displayed an increased mitotic index, followed by an increase of cells containing micronuclei and γH2AX foci (a marker of double-stranded DNA breaks), as well as activation of p53 (Supplemental Fig. 1D–F). In MDA-MB-231 breast cancer cells, which are mutant for p53, NRDE2 depletion resulted in a similar, gradual accumulation of DNA damage along with a broad range of aberrant nuclear morphologies characteristic of mitotic defects (Fig. 1E,F). Taken together with recent reports identifying NRDE2 as one of ∼1600 core fitness genes in the human genome (Blomen et al. 2015; Hart et al. 2015), we conclude that NRDE2 plays an essential role in ensuring genomic stability and mitotic progression in most, if not all, human cells.
NRDE2 features a conserved stretch of ∼350 amino acids defined as the NRDE-2 domain, a nuclear localization sequence, and five HAT (half-a-TPR) repeats, short helical motifs found in a variety of RNA processing factors (Supplemental Fig. 2A; Hammani et al. 2012). While multiple studies have found that RNA processing factors are the most enriched functional category of genes required for mitosis and genome stability (Goshima et al. 2007; Paulsen et al. 2009; Neumann et al. 2010), to our knowledge NRDE2 has eluded the “hits” list of all such screens, possibly because of the relatively lengthy time to phenotype seen here (Supplemental Fig. 1E). To begin characterizing the molecular function of NRDE2, we examined the NRDE2 protein interactome by immunoprecipitation-mass spectrometry (IP-MS) in HEK293T cells stably expressing NRDE2-GFP. Interestingly, NRDE2 interacted almost exclusively with other RNA processing factors (Fig. 2A; Supplemental Fig. 2B,C; Supplemental Table 1). Proteins copurifying with NRDE2 included known components of the RNA exosome (e.g., EXOSC10 and SKIV2L2) (Lubas et al. 2011), the U5 small nuclear ribonucleoprotein (snRNP) (e.g., EFTUD2) (Fabrizio et al. 1997), and the exon junction complex (EJC) (e.g., eIF4A3) (Fig. 2A; Singh et al. 2012). SKIV2L2, the most abundant NRDE2-interacting protein we detected, is a conserved DExH/D box RNA helicase component of the human nuclear exosome targeting (NEXT) complex (Lubas et al. 2011); notably, previous proteomic analyses of human SKIV2L2, and its homolog in S. pombe, have both revealed specific associations with NRDE2 (Lee et al. 2013; Ogami et al. 2017).
FIGURE 2.

NRDE2 associates with the RNA exosome, the EJC, and the U5 snRNP. (A) Selected proteins copurifying with NRDE2-GFP. Known components of the RNA exosome, the EJC, and the U5 snRNP detected with >10% coverage in at least one IP replicate are shown; see also Supplemental Figure 2. The full list of proteins is shown in Supplemental Table 1. (B) Extracts prepared from HEK293T cells and HEK293T cells stably expressing NRDE2-GFP were subjected to immunoprecipitation with anti-GFP antibodies; samples were analyzed by western blotting with the indicated antibodies. (n = 2, representative blots are shown) (C) HEK293T cells stably expressing NRDE2-GFP were fixed in 3% paraformaldehyde, immunostained with the indicated antibodies, and counter-stained with DAPI (blue). Apotome-enabled images were taken on a Zeiss Axio Observer.Z1 and processed using ZEN software. Approximately 100 cells were analyzed, and representative images are shown. Bar = 2 µm. (D) RNA immunoprecipitation with anti-GFP antibodies from HEK293T cells stably expressing NRDE2-GFP followed by qPCR. Data are normalized to control immunoprecipitations with the same anti-GFP antibody in untagged HEK293T cells. Error bars = SD (n = 3).
NRDE2 interactions with SKIV2L2, EXOSC10, EFTUD2 and EIF4A3 were confirmed by co-immunoprecipitation (Fig. 2B), and further supported by indirect immunofluorescence (Fig. 2C). Like many splicing factors (Spector and Lamond 2011), the subcellular localization of NRDE2 was nucleoplasmic, reminiscent of nuclear speckles, largely excluded from nucleoli, and diffuse throughout mitotic cells (Fig. 2C). Endogenously tagged zsGreen-NRDE2 exhibited a similar expression pattern (Supplemental Fig. 3A), and endogenously tagged 3XFLAG-NRDE2 also coprecipitated with SKIV2L2 (Supplemental Fig. 3B). RNA immunoprecipitation (RIP) of NRDE2 followed by qPCR revealed an enrichment of NRDE2-bound spliceosomal RNAs, particularly the U5 small nuclear RNA (snRNA), with no detectable binding of abundant small nucleolar RNAs (snoRNAs) (Fig. 2D). To our knowledge, NRDE2 has not been previously detected in human spliceosome purifications. Our data demonstrating the association of NRDE2 with components of the RNA exosome and the U5 snRNP indicate that NRDE2 may play conserved roles in regulating RNA splicing and/or stability in human cells.
A number of reports have suggested that the mechanisms linking various splicing factor knockdowns to mitotic defects converged on the inefficient splicing of a single intron in the CDCA5 gene, which regulates sister chromatid cohesion (Oka et al. 2014; van der Lelij et al. 2014; Watrin et al. 2014). We thus performed RNA sequencing (RNA-seq) in NRDE2-depleted cells 48 h post-transfection, a time point prior to the appearance of mitotic defects and DNA damage. We examined differential gene expression as well as intron retention (IR) events; we identified 84 genes deregulated by greater than twofold in NRDE2-depleted cells (Supplemental Table 2). This set was not enriched for any biological process or molecular function by GO term analysis. A panel of these genes were validated by qPCR using an additional NRDE2 siRNA, at a different time point (96 h), and in a different (RPE-1) cell line (Supplemental Fig. 4).
To investigate intron retention (IR) events, we used the recently refined IRFinder algorithm (Middleton et al. 2017) to assess IR levels in 234,396 introns in the human genome. Our analysis identified 2172 IR events in MDA-MB-231 cells, of which 178 (8%) were differentially regulated upon NRDE2-depletion. 163 (92%) of these introns showed increased retention (Fig. 3A; Supplemental Fig. 5A; Supplemental Table 3), indicating that NRDE2 is generally required to suppress IR, i.e., promote splicing. We next surveyed the sequence properties of these retained introns. The 2172 retained introns in MDA-MB-231 cells were significantly shorter, more GC-rich, and contained weaker 5′ and 3′ splice sites (ss) compared to global intron averages, properties consistent with previous reports examining IR-prone introns (Fig. 3B; Braunschweig et al. 2014). Intriguingly, these properties were even more pronounced in NRDE2-regulated introns: NRDE2-suppressed IR events (purple boxes) occurred in introns that were shorter, more GC-rich, and contained weaker 5′ and 3′ ss even when compared to IR-prone introns (Fig. 3B).
FIGURE 3.

NRDE2 suppresses IR in a subset of short, GC-rich introns with weak 5′ and 3′ splice sites. (A) Summary of IR analysis results using IRFinder (version 1.2.3) (Middleton et al. 2017) based on gene annotations for the hg19 reference genome; see Materials and Methods for details. (B) Box plots of intron lengths, GC content, 5′ and 3′ splice site strengths for all introns (n = 234,396), retained introns (n = 2172), NRDE2-induced IR sequences (n = 15), and NRDE2-suppressed IR sequences (n = 163). Splice site strengths were calculated based on the maximum entropy model (Yeo and Burge 2004). Gray numbers indicate P-values evaluated with the two-sided Mann–Whitney U-test. (C) Bar graphs depicting the log2 expression change (evaluated by DESeq2) of genes exhibiting IR events either induced (teal) or suppressed (purple) by NRDE2. A subset of gene names are shown; all genes are listed in Supplemental Table 3.
We do note that our ability to detect IR events from the RNA-seq data is not saturated. For instance, an alternative IR analysis in SeqMonk identified 47 affected IR events following NRDE2-depletion, many of which were not detected by IRFinder (Supplemental Table 4; Supplemental Fig. 5A). While part of this discrepancy is due to different intron annotations in different programs, it is almost certain that increasing RNA-seq read depths will reveal additional NRDE2-dependent IR events not detected in this study. Nonetheless, properties of NRDE2-regulated introns remained consistent: 43 of 47 (91%) introns exhibited increased retention following NRDE2 depletion, again indicating that NRDE2 generally suppresses IR. Moreover, these introns were also significantly more GC-rich and contained weaker 5′ ss compared to global intron averages (Supplemental Fig. 5B). Global intron levels were not measurably affected by NRDE2 depletion (Supplemental Fig. 5C). Thus, unlike many core splicing factors, together these data suggest that NRDE2 is only required for the efficient splicing of a small subset of particularly weak introns.
Intron-retaining transcripts are susceptible to degradation via nonsense-mediated decay (NMD) (Ge and Porse 2014), representing a potential mechanism of post-transcriptional gene regulation. Strikingly, in NRDE2-depleted cells, we found that genes identified by IRFinder exhibiting increased IR were predominantly down-regulated, whereas genes exhibiting decreased IR were predominantly up-regulated (Fig. 3C). Furthermore, GO term analyses revealed that the 163 IR events suppressed by NRDE2 (which occurred in 138 unique genes) were enriched in chromosome components (Bonferroni-corrected P = 0.02), and included the histone acetyltransferase KAT2A, and the condensin II complex subunit NCAPG2, whose mRNA expression based on RNA-seq analysis were reduced by ∼1.6- and ∼1.7-fold, respectively (Supplemental Tables 2, 3). These data not only suggest that dysregulated IR contributes to the gene expression changes in NRDE2-depleted cells, but also that these changes may cumulatively be an important source of genomic instability.
We used qPCR to confirm IR events in a number of genes previously implicated in directly promoting accurate chromosome segregation and mitotic progression, including CEP131 (Staples et al. 2012), TUBGCP2 (Neben et al. 2004), and KIF2C (Fig. 4A; Andrews et al. 2004). We sought to further investigate the role of NRDE2 in regulating CEP131 (Centrosomal Protein 131) expression, as both of our IR analyses identified CEP131 as a major NRDE2-regulated transcript. CEP131 was also one of only four genes affected by an IR event identified by IRFinder that increased greater than twofold and resulted in an IR ratio >0.5 in NRDE2-depleted cells (Fig. 4B). The IR ratio is defined as the number of intronic reads divided by those of flanking, spliced exonic reads. Moreover, CEP131 had previously been identified as an essential centriolar satellite factor and regulator of genome stability (Staples et al. 2012). The IR event suppressed by NRDE2 occurs within the 16th intron of CEP131, where loss of NRDE2 resulted in intronic RNA levels to nearly those of flanking exons (Fig. 4C). Critically, RIP-qPCR revealed that NRDE2 preferentially bound to unspliced CEP131 transcripts (Fig. 4D). As the retained intron in CEP131 contained multiple in-frame stop codons (Supplemental Fig. 6), we expected NRDE2 depletion to reduce CEP131 protein levels. Indeed, a striking loss of CEP131 protein was observed in NRDE2-depleted HEK293T and MDA-MB-231 cells (Fig. 4E; Supplemental Fig. 7A,B). The expression of CEP68, another centrosome factor, was unaffected (Supplemental Fig. 7B). Taken together, we conclude that NRDE2 is a major regulator of CEP131 expression, likely through its role in suppressing IR in CEP131 pre-mRNA.
FIGURE 4.

NRDE2 is required for CEP131 pre-mRNA splicing and protein expression. (A) qPCR analysis of IR in CEP131, TUBGCP2, and KIF2C. Primers detecting spliced transcripts are located on exons flanking the retained intron. Primers detecting unspliced transcripts flanked either the 5′ splice junctions or 3′ splice junctions. Error bars = SD (n = 3). (*) P < 0.05, two-tailed t-test. (B) Genes displaying greater than twofold increased IR following NRDE2 depletion are shown where the IR ratio in NRDE2-depleted cells is >0.5. The IR ratio measures the fraction of transcripts retaining a given intron (Middleton et al. 2017). (C) Integrated Genomics Viewer (IGV) visualization of RNA-seq data from si-Control and si-NRDE2-2 samples at the CEP131 locus. The retained intron is highlighted in pink. (D) RNA immunoprecipitation with anti-GFP antibodies from HEK293T cells stably expressing NRDE2-GFP followed by qPCR. Data are normalized to control immunoprecipitations with the same anti-GFP antibody in untagged HEK293T cells. Error bars = SD (n = 3). (E) Representative fluorescent images and quantification of CEP131-immunostained HEK293T cells 6 d post-si-transfection. More than 70 cells were scored per condition. (*) P < 0.05, two-tailed t-test.
Interestingly, unlike most other genes exhibiting increased IR, our RNA-seq data indicated that CEP131 transcripts were up-regulated ∼1.5-fold in NRDE2-depleted cells (Supplemental Table 2). We hypothesized that, given the interactions between NRDE2 and nuclear RNA exosome components, NRDE2 may couple the regulation of RNA splicing and decay of certain transcripts. We thus tested the effect of NRDE2 on CEP131 pre-mRNA stability. Consistent with a role for NRDE2 in promoting pre-mRNA decay, unspliced CEP131 transcripts were significantly stabilized relative to actin mRNA in NRDE2-depleted cells (Supplemental Fig. 7C). These data suggest that the increase in unspliced CEP131 transcripts may be due to contributions from both reduced splicing and a reduced rate of RNA decay. Nonetheless, by 4 d post-NRDE2 depletion, overall CEP131 RNA was reduced to ∼60% that of control-treated cells (Supplemental Fig. 7D), consistent with the eventual decay of aberrantly spliced transcripts and the concomitant reduction in CEP131 protein.
Centriolar satellites are thought to regulate protein trafficking to the centrosomes (Tollenaere et al. 2015). Centrosomes are large proteinaceous complexes critical for spindle microtubule organization, whose activity and duplication must be tightly regulated to maintain genome stability (Nigg and Stearns 2011). We tested whether the reduction of CEP131 protein following NRDE2 depletion was sufficient to cause defects in mitosis. Indeed, siRNA-mediated depletion of CEP131, which reduced CEP131 protein to levels comparable to those following si-NRDE2 treatment (Supplemental Fig. 8A), significantly impaired the centrosomal recruitment of Aurora Kinase A (AURKA), a major mitotic kinase required for centrosome maturation (Supplemental Fig. 8B; Hochegger et al. 2013). CEP131 depletion also resulted in the loss of cell proliferation (data not shown) and a range of aberrant nuclear morphologies remarkably similar to those induced by NRDE2 depletion (Supplemental Fig. 8C). These results support previous findings that CEP131 is essential for centrosome maturation and genome stability (Staples et al. 2012; Li et al. 2017), and indicate that impaired CEP131 expression was sufficient to partially explain the genomic instability seen upon NRDE2 depletion. However, ectopic CEP131 cDNA expression failed to rescue a range of NRDE2 knockdown phenotypes (Supplemental Fig. 9), suggesting that dysregulation of additional genes, at least in part through IR-dependent mechanisms, cumulatively contribute to the genomic instability following loss of NRDE2.
Nonetheless, six additional lines of evidence suggest that the impaired function of centrosomes, which organize and nucleate spindle microtubules to ensure accurate chromosome segregation, underlie the requirement for NRDE2 in proper mitotic progression. First, we confirmed that the centrosomal localization of CEP131 was also reduced in NRDE2-depleted mitotic cells (Fig. 5A). Second, we confirmed that the centrosomal localization of both active AURKA (Fig. 5B) and total AURKA (Supplemental Fig. 10A) was similarly reduced. This was a specific centrosomal recruitment defect, as overall AURKA expression was in fact increased (Supplemental Fig. 10B,C), consistent with AURKA levels peaking during mitosis (Hochegger et al. 2013) and an increased fraction of cells in mitosis following NRDE2 depletion. Third, we observed that centrosomal γ-tubulin, which directly mediates microtubule nucleation and is recruited to centrosomes by AURKA (Hochegger et al. 2013; Oakley et al. 2015), was also significantly reduced—most notably in mitotic cells (Fig. 5C)—further indicating impaired centrosome maturation. A moderate reduction in total γ-tubulin expression was also observed (Supplemental Fig. 10D), possibly exacerbating centrosomal recruitment defects.
FIGURE 5.
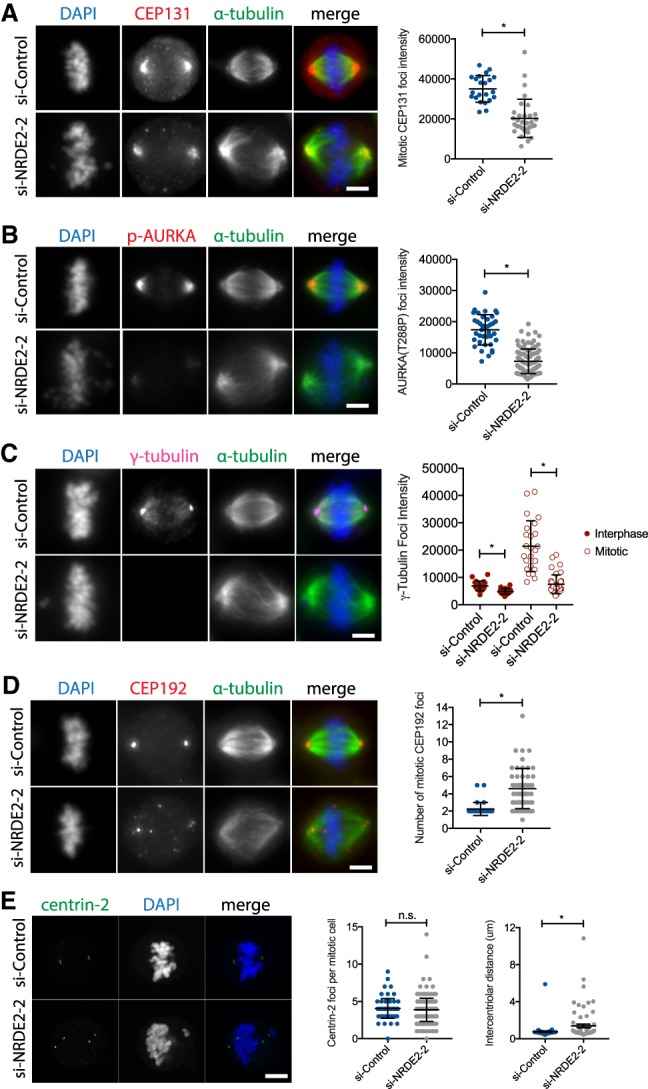
NRDE2 is required for centrosome maturation, integrity, and function. (A–D) MDA-MB-231 cells were fixed and stained with the indicated antibodies 4 d post-transfection. Maximum intensity z-projections of representative image stacks are shown. Mean foci intensities were measured in a 1–2 µm2 circle (depending on the marker) drawn around the brightest point of each marker. More than 20 foci were quantified per condition per replicate (n ≥ 3). (E, left) Maximum intensity z-projections of confocal image stacks of mitotic MDA-MB-231 cells fixed and stained with the centriole marker centrin-2 4 d post-transfection. (Right) Quantification of centriole number and the distance between centriole pairs; at least 70 centriole pairs were analyzed per condition. All intensity and distance measurements were performed using the Zen Blue software (Zeiss). Bar = 5 µm. n.s, not significant. (*) P < 0.05, two-tailed t-test.
Fourth, we found that CEP192 (Centrosomal protein 192), which is required for both γ-tubulin and AURKA recruitment, and therefore central to centrosome function (Gomez-Ferreria et al. 2007; Joukov et al. 2014), was not only reduced at spindle poles but also formed additional, nonmicrotubule-nucleating foci (Fig. 5D), suggesting impaired centrosome integrity and mislocalization/aggregation of certain centrosome proteins. Interestingly, through mechanisms that remain unclear, total CEP192 protein was also considerably reduced, without a major effect on CEP192 mRNA levels (Supplemental Fig. 10E,F). Reduced CEP192 expression is known to cause a complete loss of centrosome function (Gomez-Ferreria et al. 2007), and thus likely plays a key role (in addition to the loss of CEP131) in contributing to mitotic defects in NRDE2-depleted cells. Fifth, analysis of the centriole marker centrin-2 revealed that while centriole duplication was largely unaffected by NRDE2 depletion, a significantly increased number of mitotic cells displayed prematurely disengaged centrioles (Fig. 5E), providing further evidence for impaired centrosome integrity. Finally, consistent with multiple centrosome defects, frequencies of aberrant mitotic spindles were significantly increased following NRDE2 depletion (Supplemental Fig. 10G). We conclude that NRDE2 is required for the function of centrosomes during mitosis.
Lastly, we tested a possible nonmitotic mechanism, the impaired processing of R-loops, that might also contribute to NRDE2-dependent genome maintenance. R-loops are RNA–DNA hybrids known to threaten genomic stability, and have been shown to be regulated by the NRDE2 homolog in fission yeast (Lee et al. 2013; Aronica et al. 2015). However, we did not observe any significant increases in R-loop accumulation upon NRDE2 depletion in human cells (Supplemental Fig. 11). These data are in agreement with a recent study (Richard et al. 2018), which also concluded that the role of human NRDE2 in the DNA damage response was likely independent of R-loop processing.
Taken together, our work identifies NRDE2 as a novel human U5 snRNP-associated splicing factor required for centrosome function and mitotic progression. Further, NRDE2 is directly required for the efficient splicing of a single intron in an essential centrosome gene (CEP131). However, there is no reason to believe that NRDE2 is a specific regulator of mitosis. Our data simply positions NRDE2 as a peripheral splicing factor required for suppressing IR in a subset of pre-mRNAs, particularly those with short, weak, GC-rich introns. The fact that NRDE2-regulated IR events happen to occur in mitotic genes including CEP131, TUBGCP2, and KIF2C should nonetheless provide a partial mechanistic explanation for the mitotic defects observed following reduced NRDE2 expression (Supplemental Fig. 12). How might NRDE2 selectively regulate specific introns? It is interesting that our IP-MS data uncovered ACIN1 as a NRDE2-interacting protein (Fig. 2A). ACIN1 is a peripheral EJC component that has been shown to preferentially bind short, GC-rich introns with weak splice signals (Rodor et al. 2016)—precisely the types of introns retained following NRDE2 depletion. It will be interesting to determine whether the intron specificity of NRDE2 is intrinsic or mediated by its protein partners. Moreover, it was recently reported that the preferential killing of spliceosome-mutant cancer cells could be mediated by a small molecule that induced the retention of short, GC-rich introns (Seiler et al. 2018). Whether certain populations of low NRDE2-expressing cells show specific sensitivities to spliceosome and/or mitotic inhibitors may also be an interesting area of future investigation.
MATERIALS AND METHODS
Cell culture and siRNA transfections
HEK293T, MDA-MB-231, H23, Hela, and RPE-1 cells were obtained from the American Type Culture Collection (ATCC), grown at 37°C in a 5% CO2 atmosphere, and passaged according to standard procedures, in 10% FBS and 1% Penicillin/Streptavidin. All siRNA transfections were performed at 20 nM concentration, with Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher) according to the manufacturer's protocol. siRNAs used were: si-Control (Ambion AM4611), si-NRDE2-1 (Ambion Silencer Select s30063, 5′ GGUGUUGUUUGAUGAUAUUTT 3′), si-NRDE2-2 (Ambion Silencer Select s30064, 5′ GUUUAGUACCUUUUCGAUATT 3′), si-CEP131 (AZI1) (Ambion Silencer AM16708 5′ GCUGAUUGCAAGGCACAAGTT 3′).
Cell proliferation
Cell proliferation was determined by MTT assays according to the manufacturer's protocol (Thermo Fisher). Transfected cells were seeded in 96-well plates 48 h (or 24 h for MDA-MB-231) post-transfection at ∼1000 cells/well. The following day was considered Day 1 of the MTT assay, to which all A560 readings were normalized.
RNA extraction, reverse transcription, and quantitative PCR
Cells were collected in TRIzol (Invitrogen) and total RNA was isolated with Direct-zol RNA Miniprep Plus spin columns (Zymo Research) according to the manufacturer's protocol, including the on-column DNase I treatment. Total cDNA synthesis was performed on 50 ng–1 µg total RNA using Superscript IV Reverse Transcriptase (Thermo Fisher Scientific), with random hexamers. cDNA was diluted a minimum of 1:2 before expression analysis by qPCR with SYBR Green I Master Mix (Roche) in a Roche LightCycler 480 II (qPCR program: 95°C for 5 min, followed by 45 cycles of [95°C for 10 sec, 54°C for 10 sec, 72°C for 10 sec], followed by a melting curve analysis). Fold changes were calculated using the 2−ΔΔCT method.
For small nuclear/nucleolar RNAs: Total RNA was collected as above. Polyadenylation-coupled reverse transcription was performed in a standard 20 µL Superscript IV reverse transcription reactions, but in 0.5× Superscript IV buffer and with the following additions: 1 µL poly(A) buffer (NEB #B0276S), 1 µL poly(A) polymerase (NEB # M0276S), 1 mM ATP, and 2.5 µM anchored oligod(T) primer (5′-GCTGTCAACGATACGCTACGTAACGGCATGACAGTGTTTTTTTTTTTTTTTTTVN-3′). Samples were incubated at 37°C for 1 h, then 85°C for 8 min before placing on ice prior to qPCR. qPCRs were run using gene-specific forward primers and a universal reverse primer. All primers were used at a final concentration of 5 µM each, and their sequences are listed in Supplemental Table 4.
RNA-seq
Triplicate RNA samples from si-Control and si-NRDE2-2-treated MDA-MB-231 cells were collected 48 h post-transfection using the mirVana RNA isolation kit (Thermo Fisher). Libraries were synthesized using Illumina TruSeq Stranded mRNA sample preparation kits from 500 ng of purified total RNA according to the manufacturer's protocol. The final dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR using the Kapa Biosystems library quantification kit according to manufacturer's protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75 bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. The total read depths for the three control-treated replicates were 61.4, 63.9, and 56.1 M. The total read depths for the three si-NRDE2-treated replicates were 57.8, 58.9, and 46.4 M. Sequenced reads were aligned to the hg19 reference genome assembly; >90% of reads were mappable in all samples. Gene counts were quantified using STAR (v2.5.1b) and differential expression testing was performed by DESeq2 (v1.10.1) as part of the VIPER analysis pipeline (https://bitbucket.org/cfce/viper/).
Intron analysis (IRFinder)
Intron retention analysis was performed using the IRFinder tool (version 1.2.3) (Middleton et al. 2017) based on the gene annotations for the hg19 reference genome, using the RNA-seq data obtained from triplicate si-Control and si-NRDE2-2-treated cells. All potential introns (n = 234,396) were tested, and introns were defined as any region between two exon features in any transcript. The exon coordinates were derived from reference gene annotation files. The Audic and Claverie test for small numbers of replicates was applied to the quantified IR events to determine differential expression between control and NRDE2 knockdown conditions. Significant IR events were defined as previously reported (Middleton et al. 2017), according to these criteria: Benjamini–Hochberg-adjusted P < 0.05; IRratio in either control or NRDE2 knockdown >0.1 (i.e., at least 10% of transcripts exhibit retention of the intron for at least one of the conditions), and retained introns exhibit a coverage (i.e., intron depth) of at least three reads after excluding nonmeasurable intronic regions. Additional details are described in Supplemental Table 3.
Intron analysis (SeqMonk)
Differential expression analysis of 315,405 intronic probes predefined in SeqMonk (Babraham Bioinformatics) was performed with DESeq2 on si-Control and si-NRDE2-treated triplicate RNA-seq samples. Introns that passed all three filters (P < 0.01, an average read count difference of >100, and a minimum threefold change), were considered significantly deregulated. Removal of seven probes due to overlap with alternative exons resulted in the identification of the 47 deregulated introns within 45 different genes shown in Supplemental Table 4. Average read counts from flanking exons, also analyzed by DESeq2 in SeqMonk, were used to calculate intron levels relative to flanking exon expression.
Flow cytometry
For cell cycle analysis, cells were harvested 5 d post-transfection, washed once in PBS, and fixed in 75% ice cold ethanol for >30 min. Cells were then washed once in PBS and stained in 10 µg/mL Hoechst 33342 (Thermo Fisher) in PBS. DNA content was analyzed by flow cytometry (BD Biosciences FACSCalibur). At least 10,000 events were collected per sample.
Lentiviral production and infection
Generation two lentiviral constructs (0.75 µg of pMD2.G [Addgene plasmid 12259] and 0.75 µg psPAX2) (Addgene plasmid 12260) and 1.5 µg of either NRDE2-mGFP expression plasmid (Origene RC224847L2) or a dox-inducible 3xFLAG-CEP131 construct (a kind gift from L. Shi, Tianjin Medical University), were transfected into HEK293T cells in 6-well plates with Lipofectamine 3000. Media was changed after 6 h and viral supernatant was collected 36—48 h later, 0.2 µm filter sterilized and frozen at −80°C. For NRDE2 expression, HEK293T and MDA-MB-231 cells were infected with either control GFP (Origene TR30021V) or NRDE2-GFP lentiviral supernatant. GFP-positive cells were sorted by flow cytometry ∼1 wk post-infection. For generating stable dox-inducible 3XFLAG-CEP131 expressing lines, infected MDA-MB-231 cells were selected and maintained in 1.5 µg/mL of Puromycin.
Immunofluorescence and confocal microscopy
Antibodies used: γH2AX, Millipore 05-636, 1:500; phospho-H3(Ser10), Cell Signaling Technology #9701, 1:500; α-tubulin, Cell Signaling Technology #3873, 1:2000; 1:500; γ-tubulin, Abcam ab11317, 1:500; Aurora A, Cell Signaling Technology #14475, 1:200; phospho-Aurora A/B/C, Cell Signaling Technology #2914, 1:300; CEP-131, Proteintech 25735-1-AP, 1:300; SKIV2L2, Proteintech 12719-2-AP, 1:600; EXOSC10, Proteintech 11178-1-AP, 1:300; EFTUD2, Proteintech 10208-1-AP, 1:300; eIF4A3, Proteintech 17504-1-AP, 1:300; CEP192, Proteintech 18832-1-AP, 1:300; Centrin-2, Millipore 04-1624, 1:300; S9.6, Kerafast ENH001, 1:100.
Cells were grown on coverslips coated with 0.1% gelatin (Sigma G-9382) in 6-well plates, fixed in chilled 100% methanol for 7 min at −20°C (unless otherwise indicated), and washed 3× in PBS. Fixed cells were blocked in 5% BSA (Sigma) in TBST for 1–2 h at room temperature and incubated in primary antibodies in 2.5% BSA in TBST overnight at 4°C. The following day, cells were washed 3× in PBS, incubated in Alexa Fluor 488/Alexa Fluor 555/Alexa Fluor 647 (Molecular Probes) conjugated secondary antibodies (1:1000 in 2% BSA in TBST) for 1 h at room temperature in the dark, washed 3× in PBS, and mounted onto microscope slides with ProLong Diamond Antifade with DAPI (Life Technologies). For fluorescence microscopy, slides were visualized using a Zeiss Axio Observer.Z1 under a Plan-Apochromat 63×/1.40 Oil DIC objective. Fluorescent images were captured using a Hamamatsu Camera, and processed using the ZEN 2 Blue Edition Software (2011). For analysis of MDA-MB-231 nuclear morphologies and quantification of HEK293T H3S10P-positive mitotic cells, slides were imaged with a monochrome camera on an EVOS FL Imaging System under a 20× fixed objective. For confocal analyses, cells were examined under a LSM 880 Laser Scanning Microscope.
Live cell imaging
RPE-1 cells stably expressing H2B-GFP and α-tubulin-mCherry were seeded in black 24-well plates (ibidi #82406) 48 h following siRNA transfections. Two days later, beginning at 96 h post-transfection, plates were mounted on a Nikon Ti-E inverted microscope equipped with the Nikon Perfect Focus system, and cells were imaged through a 20× 0.75 NA Plan Apo objective using a Zyla 4.2 sCMOS camera (Andor). Imaging was performed in an environmentally controlled chamber maintained at 37°C with humidified 5% CO2. Z-stacks (8-µm volume at 2-µm spacing) were collected at 10-min intervals over a total of 48 h. Mitotic duration was visually scored from the recorded videos using MetaMorph (Molecular Devices), beginning with the first sign of chromosome condensation and ending with decondensed chromosomes in daughter nuclei.
Western blotting
Antibodies used: Monoclonal ANTI-FLAG, M2 (Sigma, F1804); β-actin (Santa Cruz, sc-47778); Cyclin B1 (Cell Signaling Technology #12231); phospho-p53(Ser15) (Cell Signaling Technology #9286); GFP (Abcam, ab290); CEP68 (Proteintech, 15147-1-AP); Antibodies for phospho-H2AX(Ser139), phospho-H3(Ser10), γ-tubulin, CEP-131, CEP192, Aurora A, SKIV2L2, EFTUD2, eIF4A3, and EXOSC10 are the same ones used for immunofluorescence (above).
Cell pellets were resuspended in RIPA buffer, rotated at 4°C for 30 min, and centrifuged at 13,000 rpm for 20 min. Laemmli buffer was added to the supernatant and protein samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were blocked in 5% milk or BSA (for phospho-proteins), and incubated in primary antibodies overnight at 4°C at 1:2000 dilutions, except for β-actin (1:7500) and DNAPKcs(S2056P) (1:1000). Blots were then washed 3× in TBST, incubated in HRP-conjugated secondary antibodies (Santa Cruz) (∼1:10000) for 1 h at room temperature, washed 3× in TBST, and detected by enhanced chemiluminescence (BioRad).
Protein/RNA immunoprecipitation
HEK293T, HEK293T(3xFLAG-NRDE2), and HEK293T(NRDE2-GFP) cells were grown to confluency in 10 cm dishes, washed once in cold PBS, and lysed on ice for 5 min in 1× nondenaturing lysis buffer (Cell Signaling Technology #9803) with freshly added HALT protease inhibitor (Thermo Fisher #78430). Lysates were centrifuged at 14,000g for 10 min and precleared with Protein G Dynabeads (Thermo Fisher #10003D) for 1 h at 4°C. Five microliters of anti-FLAG M2 (Sigma F1804) or 1 µL of anti-GFP (Abcam ab290) was added per IP (equivalent of one 10 cm dish) and rotated overnight at 4°C. The next day, protein G Dynabeads were added and rotated for 2 h at 4°C. Beads were washed five times in lysis buffer for 5–10 min each at 4°C. Protein was eluted in Laemmli sample buffer (Bio-Rad) with 10% 2-mercaptoethanol and subjected to analysis by either western blotting (see above) or mass spectrometry. For RNA immunoprecipitation, the same procedure was used except all incubations were performed in the presence of Superase Inhibitor (Invitrogen AM2694), and RNA was eluted from beads with TRIzol reagent and isolated with Direct-zol RNA Miniprep Plus spin columns (Zymo Research).
Tagging the endogenous NRDE2 locus in HEK293T cells using CRISPR/Cas9 technology
To insert a 3xFLAG tag at the N terminus of the NRDE2 gene, we used a CRISPR/Cas9 recombination strategy as previously described (Cong et al. 2013; Ran et al. 2013). In short, a guide RNA targeting the 5′ end of NRDE2 with the sequence 5′-AGCCTCACTAAGCCCCGCAA-3′ was cloned into the pX458 vector (Addgene, plasmid #48138) following the protocol described here (http://www.genome-engineering.org/crispr/wp-content/uploads/2014/05/CRISPR-Reagent-Description-Rev20140509.pdf). The pX458 vector expresses a Cas-9-GFP fusion protein that can be used as a selection marker. The primers used for the guide RNA cloning were 5′-CACCGAGCCTCACTAAGCCCCGCAA-3′ for the forward primer and 5′-AAACTTGCGGGGCTTAGTGAGGCTC-3′ for the reverse. A repair template that included 1 kb upstream of NRDE2 ATG, the in-frame N-terminal 3xFLAG tag, and 1 kb downstream from the ATG was amplified using standard PCR methods. Then, the repair template was cloned into the HindIII and EcoRV sites of a pUC19 vector. To improve the chances of recombination, a silent mutation was introduced at the PAM sequence in our repair template (from GGG to GGA). Both vectors were transfected into HEK293T cells at 70% confluency using Lipofectamine 2000 (Thermo Fisher) following the company's protocol for transfections in 24-well plates. The total amount of DNA transfected into cells was 1 µg (500 ng each vector). After transfection, cells were incubated at 37°C, 5% CO2 for 48 h in DMEM media supplemented with 10% FBS and 100 U/mL penicillin, and 100 µg/mL streptomycin. Cells were then FACS sorted for GFP-positive cells into two 96-well plates with a single cell per well. After 2 wk, clones that grew colonies were then genotyped by PCR for the presence of the 3xFLAG sequence. Positive clones were subjected to WB to confirm proper 3xFLAG-NRDE2 fusion protein expression.
To insert the zsGreen tag at the N terminus of NRDE2, we followed a similar approach, except the gRNA was cloned into the pX330 vector (Addgene, plasmid #42230), which has no selectable marker. The repair template also had 1 kb upstream of ATG, in-frame zsGreen tag, and 1 kb downstream from the ATG with the silent mutation in the PAM sequence and cloned into pUC19 as described above. HEK293T cells at 70% confluency were transfected with 1 µg (500 ng each vector) total DNA using Lipofectamine 2000 in 24-well plates. After transfection, cells were incubated at 37°C, 5% CO2 for 48 h in DMEM media supplemented with 10% FBS and 100 U/mL penicillin, and 100 µg/mL streptomycin. Cells were then FACS sorted for zsGreen positive cells into two 96-well plates with a single cell per well. After 2 wk, clones that grew colonies were then genotyped by PCR for the presence of the zsGreen sequence. Expression of zsGreen-NRDE2 was confirmed by fluorescence microscopy. Primers and sequences of 3xFLAG and zsGreen repair templates are available upon request.
Mass spectrometry
Immunoprecipitated samples were run on a 10% polyacrylamide gel; Coomassie Blue-stained bands corresponding to ∼25 kDa (IgG light chain) and ∼50 kDa (IgG heavy chain) were excised and removed; the remaining gel slices were cut into ∼1 mm3 pieces and subjected to a modified in-gel trypsin digestion. Gel pieces were washed and dehydrated with acetonitrile for 10 min followed by removal of acetonitrile. Pieces were then completely dried in a speed-vac. Rehydration of the gel pieces was with 50 mM ammonium bicarbonate solution containing 5 ng/µL modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. After 45 min, the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution to just cover the gel pieces. Samples were then placed in a 37°C room overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac (∼1 h) and reconstituted in 5–10 µL of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 µm C18 spherical silica beads into a fused silica capillary (100 µm inner diameter × ∼30 cm length) with a flame-drawn tip. After equilibrating the column each sample was loaded via a Famos auto sampler (LC Packings) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As peptides eluted they were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher). All databases include a reversed version of all the sequences and the data was filtered to between a 1%–2% peptide false discovery rate.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Slack, Kennedy, and Pellman laboratories for insightful discussions, experimental suggestions, and critical reading of the manuscript. We also thank Zach Herbert and the Dana Farber Molecular Biology Core Facilities for RNA-seq and gene expression analysis; the Harvard Chan Bioinformatics Core for computational guidance; K. Schukken and the Foijer laboratory (University Medical Center Groningen, Netherlands) for generously sharing cell lines used for live imaging; L. Shi (Tianjin Medical University, China) for generously sharing the inducible CEP131 lentiviral construct; Ross Tomaino and the Taplin Mass Spectrometry Facility for mass spectrometry analyses. N.T.U. is a HHMI fellow of the Damon Runyon Cancer Research Foundation (DRG-2238-15). This work was supported by the National Institutes of Health (R01AG033921) to F.J.S., funding from the Ludwig Center at Harvard and a pilot grant from the Harvard Medical School Epigenetics Initiative to F.J.S. and S.K.
Author contributions: A.L.J., R.P., N.T.U., J.R.H., D.P., S.K., and F.J.S. designed the experiments. A.L.J., R.P., N.T.U., J.R.H., M.E.P., and B.D.A. performed experiments and analyzed results. A.L.J., R.P., J.R.H., S.K., and F.J.S. wrote/edited the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.069773.118.
REFERENCES
- Akay A, Di Domenico T, Suen KM, Nabih A, Parada GE, Larance M, Medhi R, Berkyurek AC, Zhang X, Wedeles CJ, et al. 2017. The helicase aquarius/EMB-4 is required to overcome intronic barriers to allow nuclear RNAi pathways to heritably silence transcription. Dev Cell 42: 241–255.e6. 10.1016/j.devcel.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. 2004. Aurora B regulates MCAK at the mitotic centromere. Dev Cell 6: 253–268. 10.1016/S1534-5807(04)00025-5 [DOI] [PubMed] [Google Scholar]
- Aronica L, Kasparek T, Ruchman D, Marquez Y, Cipak L, Cipakova I, Anrather D, Mikolaskova B, Radtke M, Sarkar S, et al. 2015. The spliceosome-associated protein Nrl1 suppresses homologous recombination-dependent R-loop formation in fission yeast. Nucleic Acids Res 44: 1703–1717. 10.1093/nar/gkv1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. 2015. Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096. 10.1126/science.aac7557 [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NO, Burkhart KB, Kennedy S. 2011. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc Natl Acad Sci 108: 19683–19688. 10.1073/pnas.1113310108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR III, Bartel DP, Madhani HD. 2013. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152: 957–968. 10.1016/j.cell.2013.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Laggerbauer B, Lauber J, Lane WS, Lührmann R. 1997. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J 16: 4092–4106. 10.1093/emboj/16.13.4092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Porse BT. 2014. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 36: 236–243. 10.1002/bies.201300156 [DOI] [PubMed] [Google Scholar]
- Gomez-Ferreria MA, Rath U, Buster DW, Chanda SK, Caldwell JS, Rines DR, Sharp DJ. 2007. Human Cep192 is required for mitotic centrosome and spindle assembly. Curr Biol 17: 1960–1966. 10.1016/j.cub.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, Stuurman N. 2007. Genes required for mitotic spindle assembly in Drosophila S2 cells. Science 316: 417–421. 10.1126/science.1141314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. 2010. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465: 1097–1011. 10.1038/nature09095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammani K, Cook WB, Barkan A. 2012. RNA binding and RNA remodeling activities of the half-a-tetratricopeptide (HAT) protein HCF107 underlie its effects on gene expression. Proc Natl Acad Sci 109: 5651–5656. 10.1073/pnas.1200318109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. 2015. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163: 1515–1526. 10.1016/j.cell.2015.11.015 [DOI] [PubMed] [Google Scholar]
- Hochegger H, Hégarat N, Pereira-Leal JB. 2013. Aurora at the pole and equator: overlapping functions of Aurora kinases in the mitotic spindle. Open Biol 3: 120185 10.1098/rsob.120185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V, Walter JC, De Nicolo A. 2014. The Cep192-organized aurora A-Plk1 cascade is essential for centrosome cycle and bipolar spindle assembly. Mol Cell 55: 578–591. 10.1016/j.molcel.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, et al. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. 10.1016/j.cell.2013.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Song N, Liu L, Liu X, Ding X, Song X, Yang S, Shan L, Zhou X, Su D, et al. 2017. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat Commun 8: 14866 10.1038/ncomms14866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. 2011. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43: 624–637. 10.1016/j.molcel.2011.06.028 [DOI] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJL, Bomane A, Cosson B, Eyras E, Rasko JEJ, et al. 2017. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18: 51 10.1186/s13059-017-1184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neben K, Tews B, Wrobel G, Hahn M, Kokocinski F, Giesecke C, Krause U, Ho AD, Krämer A, Lichter P. 2004. Gene expression patterns in acute myeloid leukemia correlate with centrosome aberrations and numerical chromosome changes. Oncogene 23: 2379–2384. 10.1038/sj.onc.1207401 [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Hériché J-K, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464: 721–727. 10.1038/nature08869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Stearns T. 2011. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154–1160. 10.1038/ncb2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley BR, Paolillo V, Zheng Y. 2015. γ-Tubulin complexes in microtubule nucleation and beyond. Mol Biol Cell 26: 2957–2962. 10.1091/mbc.E14-11-1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogami K, Richard P, Chen Y, Hoque M, Li W, Moresco JJ, Yates JR III, Tian B, Manley JL. 2017. An Mtr4/ZFC3H1 complex facilitates turnover of unstable nuclear RNAs to prevent their cytoplasmic transport and global translational repression. Genes Dev 31: 1257–1271. 10.1101/gad.302604.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y, Varmark H, Vitting-Seerup K, Beli P, Waage J, Hakobyan A, Mistrik M, Choudhary C, Rohde M, Bekker-Jensen S, et al. 2014. UBL5 is essential for pre-mRNA splicing and sister chromatid cohesion in human cells. EMBO Rep 15: 956–964. 10.15252/embr.201438679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. 2009. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239. 10.1016/j.molcel.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. 2013. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Ogami K, Chen Y, Feng S, Moresco JJ, Yates JR III, Manley JL. 2018. NRDE-2, the human homolog of fission yeast Nrl1, prevents DNA damage accumulation in human cells. RNA Biol 15: 868–876. 10.1080/15476286.2018.1467180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodor J, Pan Q, Blencowe BJ, Eyras E, Cáceres JF. 2016. The RNA-binding profile of Acinus, a peripheral component of the exon junction complex, reveals its role in splicing regulation. RNA 22: 1411–1426. 10.1261/rna.057158.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. 2018. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24: 497–504. 10.1038/nm.4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764. 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Lamond AI. 2011. Nuclear speckles. Cold Spring Harb Perspect Biol 3: a000646 10.1101/cshperspect.a000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples CJ, Myers KN, Beveridge RDD, Patil AA, Lee AJX, Swanton C, Howell M, Boulton SJ, Collis SJ. 2012. The centriolar satellite protein Cep131 is important for genome stability. J Cell Sci 125: 4770–4779. 10.1242/jcs.104059 [DOI] [PubMed] [Google Scholar]
- Tollenaere MAX, Mailand N, Bekker-Jensen S. 2015. Centriolar satellites: key mediators of centrosome functions. Cell Mol Life Sci 72: 11–23. 10.1007/s00018-014-1711-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P, Stocsits RR, Ladurner R, Petzold G, Kreidl E, Schmitz J, Neumann B, Ellenberg J, Peters JM. 2014. SNW 1 enables sister chromatid cohesion by mediating the splicing of sororin and APC 2 pre-mRNAs. EMBO J 33: 2643–2658. 10.15252/embj.201488202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrin E, Demidova M, Watrin T, Hu Z, Prigent C. 2014. Sororin pre-mRNA splicing is required for proper sister chromatid cohesion in human cells. EMBO Rep 15: 948–955. 10.15252/embr.201438640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. 2015. Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184. 10.1038/nature14493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu J, Schermann G, Ohle C, Bendrin K, Sugioka-Sugiyama R, Sugiyama T, Fischer T. 2015. The fission yeast MTREC complex targets CUTs and unspliced pre-mRNAs to the nuclear exosome. Nat Commun 6: 7050 10.1038/ncomms8050 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.