

ABSTRACT
Recent discoveries indicate a genetic basis for high-altitude adaptation among human groups who have resided at high altitude for millennia, including Andeans, Tibetans, and Ethiopians. Yet, genetics alone does not explain the extent of variation in altitude-adaptive phenotypes. Current and past environments may also play a role, and one way to determine the effect of the environment is through the epigenome. To characterize if Andean adaptive responses to high altitude have an epigenetic component, we analyzed DNA methylation of the promoter region of EPAS1 and LINE-1 repetitive element among 572 Quechua individuals from high- (4,388 m) and low-altitude (0 m) in Peru. Participants recruited at high altitude had lower EPAS1 DNA methylation and higher LINE-1 methylation. Altitude of birth was associated with higher LINE-1 methylation, not with EPAS1 methylation. The number of years lived at high altitude was negatively associated with EPAS1 methylation and positively associated with LINE-1 methylation. We found four one-carbon metabolism SNPs (MTHFD1 rs2236225, TYMS rs502396, FOLH1 rs202676, GLDC rs10975681) that cumulatively explained 11.29% of the variation in average LINE-1 methylation. And identified an association between LINE-1 methylation and genome-wide SNP principal component 1 that distinguishes European from Indigenous American ancestry suggesting that European admixture decreases LINE-1 methylation. Our results indicate that both current and lifetime exposure to high-altitude hypoxia have an effect on EPAS1 and LINE-1 methylation among Andean Quechua, suggesting that epigenetic modifications may play a role in high-altitude adaptation.
KEYWORDS: Epigenetics, adaptation, hypoxia, DNA methylation, Andes
Introduction
There are three main geographic regions where humans have adapted to life at high altitude including the Tibetan Plateau, the Ethiopian Plateau, and the Andean Altiplano [1]. Human populations from each of these zones exhibit unique circulatory, respiratory, and hematological adaptations with respect to low-altitude populations and with respect to each other. These adaptations include differences in resting ventilation, hypoxic ventilatory response, arterial oxygen saturation (SaO2), and hemoglobin concentration among others [2]. Genome-wide single-nucleotide polymorphism (SNP) and sequencing studies support a genetic contribution to many of these adaptations in all three high-altitude populations [3–5]. However, genetics alone does not fully explain the extent of variation observed for high-altitude adaptive phenotypes. Growth and development at high altitude also have been shown to contribute to high-altitude adaptive traits.
This is particularly the case among Andeans, for whom developmental adaptation has been shown to contribute to altitude-adaptive traits [6–8]. For example, exposure to hypoxia during growth and development is crucial for attaining the enlarged residual lung volume characteristic of high-altitude Andeans, and is related to both genetic and developmental factors [8,9]. Likewise, SaO2 exhibits both a genetic and a developmental component among Andean Quechua [10,11]. The angiotensin-converting enzyme (ACE) gene insertion/deletion (I/D) polymorphism explains ~4% of the total variance in resting and exercise SaO2 measured in Quechua individuals born and raised at high altitude in Cerro de Pasco, Peru (4,388 meters [m]) and Quechua individuals born and raised at low altitude in Lima, Peru (0 m), but the group effect of being born at high versus low altitude accounts for most of the variance in SaO2 [11]. The developmental component to high-altitude adaptation may proceed through epigenetic modification. With the rise of epigenetic methods, we now have a direct way to examine mitotically heritable changes in gene expression that occur during growth and development.
Epigenetic regulation works hand-in-hand with genetic processes to ‘prepare’ the organism for the external environment where it will live and reproduce providing a potential mechanism by which it can adapt to ever-changing environmental conditions [12,13]. Epigenetic modifications include DNA methylation, posttranslational histone tail modifications, and non-coding RNAs. DNA methylation is the most widely studied and best understood epigenetic modification [12,14]. DNA methylation occurs when a methyl group is added to the 5th carbon position of a cytosine. This modification most commonly occurs when cytosine is followed by guanine in an arrangement called a cytosine-phosphate-guanine (CpG) site [15]. DNA methylation is carried out by a group of enzymes called DNA methyltransferases (DNMT), which selectively target specific DNA sequences to be either de novo or maintenance methylated [16].
We determined DNA methylation levels at the LINE-1 repetitive DNA element and a single candidate locus, the gene EPAS1, in a cohort of indigenous Quechua Andeans. The Long Interspersed Nuclear Element-1 (LINE-1) is a repetitive DNA element that constitutes about 21% of the human genome and is often used as a marker of global DNA methylation [17]. The LINE-1 retrotransposon is highly methylated in non-diseased states [18]. LINE-1 methylation can affect the expression of nearby genes [19], and decreased methylation of LINE-1 has been shown to be associated with genomic instability, risk of cancer, diet, and exposures to toxicants [20–22]. Its high genomic frequency and its association with environmental exposures and disease state make LINE-1 a suitable genetic marker to study the effects of high-altitude hypoxia on the epigenomes of indigenous Peruvians.
Endothelial PAS domain protein 1 (EPAS1), also known as HIF-2a, is a hypoxia-responsive transcription factor that has been shown to be under selection in high-altitude Tibetans and Andeans [3,23,24]. EPAS1 is a hypoxia-activated gene. Its expression is higher in hypoxic conditions, and it was previously hypothesized to be involved in the high-altitude response [25,26]. Furthermore, EPAS1 methylation levels previously have been shown to associate with colorectal cancer, with increased methylation leading to decreased expression of the EPAS1 protein [27]. Together, these findings make EPAS1 a natural a priori candidate for studying epigenetics of high-altitude adaptation.
We used quantitative bisulfite pyrosequencing to determine DNA methylation levels at four LINE-1 CpG sites in 572 Peruvian Quechua study participants of similar genetic ancestry with three different developmental exposures to high altitude. To explore gene-specific methylation outcomes of hypoxia exposure, we used quantitative bisulfite pyrosequencing to characterize methylation at EPAS1 in this same cohort (n = 539). We identified significant DNA methylation differences among the three groups at LINE-1. Furthermore, we found that EPAS1 methylation is decreased among Peruvian Quechua living at high altitude compared to those living at low altitude.
Results
Study design and participant characteristics
We employed a migrant study design wherein we recruited a cross-sectional cohort of 603, healthy Quechua participants aged 18–35 from two locations in Peru differing by altitude: Lima (0 meters [m] above sea level) and Cerro de Pasco (4,338 m) (Figure 1(a)). From these two locations, we recruited three high-altitude exposure groups. 1) high-altitude Quechua (HAQ) (n = 300) were multigenerational high-altitude Quechua individuals who were born and raised in Cerro de Pasco. None of the HAQ study participants were mine workers even though Cerro de Pasco is a mining town. 2) migrant Quechua (MQ) (n = 152) were Quechua individuals who were born in highland Peru (> 2,500 m) and moved to low-altitude Lima (0 m) during their lifetime (Figure 1(b)). They had resided in Lima, Peru for at least two months at the time of enrollment. Thus, this study group was composed of first generation down migrants who have had full developmental exposure to high altitude. 3) low-altitude Quechua (LAQ) (n = 150) were Quechua individuals who were born, raised, and reside in in Lima (150 m). Thus, LAQ is composed of second-generation down-migrants who have had no developmental exposure to high altitude over the course of their lifetime, but their parents and both sets of grandparents were born at an altitude > 3,000 m (Figure 1(b)). It is important to note the Quechua population is a highland group. The Quechua population has a history of life at high altitude, and all individuals in this study are genetically adapted to high-altitude conditions (Bigham et al., unpublished).
Figure 1.
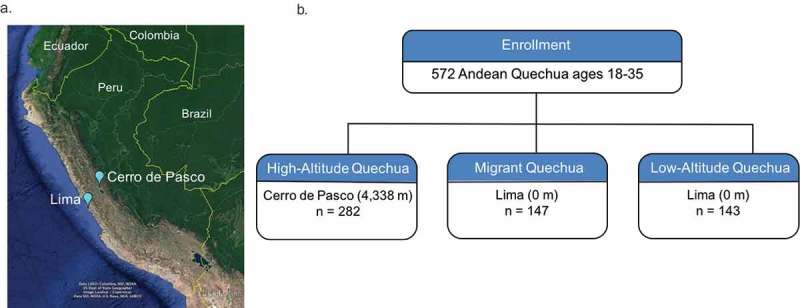
Study Design. (A) Map of Peru depicting participant recruitment locations at high-altitude, Cerro de Pasco (4,338m), and low-altitude, Lima (0m); (B) LINE-1 DNA methylation was determined in 572 Peruvian Quechua with different exposures to high-altitude hypoxia over their lifetime. High-altitude Quechua (HAQ) were born, raised, and reside in or near Cerro de Pasco, Peru. Migrant Quechua (MQ) were born and raised at high altitude (>2,500 m), but moved to low-altitude Lima (0m) during the lifetime. Low-altitude Quechua (LAQ) were born, raised, and reside at sea-level in Lima.
DNA was extracted from whole blood, and quantitative bisulfite pyrosequencing was performed. We excluded samples that repeatedly failed pyrosequencing or produced unreliable DNA methylation data. Unreliable data were defined as samples the repeatedly failed pyrosequencing or generated DNA methylation values outside 2 standard deviations of the entire cohort. After quality control (QC), statistical analysis was performed on 572 samples for LINE1 and 539 samples for EPAS1). Please see ‘Materials and Methods’ for a more detailed description of our study cohort.
Participant characteristics are provided in Table 1. The three study groups were different by height, weight, and BMI (p < 0.05). MQ study participants were slightly but significantly older than both HAQ and LAQ study participants (p < 0.05). The study groups did not differ significantly by sex. Average age of down-migration for MQ was 15.21 ± 6.37 years of age. Principal component analysis (PCA) was performed on Quechua DNA samples and three 1000 genomes populations (90 East Asians, 60 Centre de Polymorphism Humain [CEPH] Europeans, and 60 Yorubans). We used existing SNP microarray genotype data for the Peruvians in this study from the Affymetrix (Santa Clara, CA) Axiom Biobanking array that includes ~600,000 single nucleotide polymorphisms (SNPs). PCA was performed on 328,260 autosomal SNPs with genotyping rates > 95%, MAF > 0.0001, that passed Affy QC, and that were not in strong linkage disequilibrium (R2 < 0.8). Together, all three, altitude-exposure groups formed a distinct cluster (Supp. Figure 1A-C).
Table 1.
Participant Characteristics.
| Variable | All (n = 572) | Low-altitude Quechua (n = 143) | Migrant Quechua (n = 147) | High-altitude Quechua (n = 282) |
|---|---|---|---|---|
| Average LINE-1 methylation | 69.99 ± 1.45 | 69.74 ± 1.18 | 69.82 ± 1.28 | 70.20 ± 1.63 |
| Average EPAS1 methylation | 14.42 ± 3.08 | 14.65 ± 3.06 | 15.06 ± 2.89 | 14.00 ± 3.12 |
| % Female | 49.65 (45.55, 53.75) | 48.95 (40.76, 57.14) | 53.06 (44.99, 61.13) | 48.23 (42.40,54.06) |
| Age, yr*† | 24.67 ± 5.03 | 24.38 ± 4.55 | 25.52 ± 5.49 | 24.37 ± 4.98 |
| Age migration, yr | NA | NA | 15.21 ± 6.37 | NA |
| Height (cm)*† | 158.91 ± 8.35 | 161.40 ± 8.72 | 158.01 ± 7.84 | 158.11 ± 8.19 |
| Weight (kg)*† | 61.95 ± 9.83 | 66.36 ± 11.69 | 61.76 ± 9.57 | 59.82 ± 8.07 |
| BMI** | 24.52 ± 3.28 | 25.43 ± 3.79 | 24.69 ± 3.12 | 23.96 ± 2.96 |
Data are means ± SD, 95% confidence interval (CI) for proportions in brackets; NA, not applicable
* p ≤ 0.05 for born low vs. high-to-low based on t-test
† p ≤ 0.05 for born high vs high-to-low based on t-test
** p ≤ 0.05 based on ANOVA
Sex and age effects on DNA methylation
We performed a linear mixed effects regression to determine the effects of sex and age on LINE-1 and EPAS1 methylation. Linear mixed models allow for multiple technical pyrosequencing replicates per individual. We considered average LINE-1 and EPAS1 methylation across their four respective sites in addition to DNA methylation at each of the four sites independently. Males showed higher LINE-1 and EPAS1 methylation than females on average (LINE-1: β = 0.67, p-value = 4.37e-08; EPAS1: β = 1.13, p-value = 2.47e-05) and at each CpG site when adjusted for age (S1 Table) consistent with previous research showing DNA methylation associations with sex [28]. At the first LINE-1 CpG site (CpG1), older individuals displayed significantly lower levels of methylation (β = −0.037, p-value<0.05, adjusted for sex and altitude). All other LINE-1 CpG positions and the average LINE-1 methylation were not associated with age. EPAS1 methylation levels were positively associated with age (β = 0.09, p-value<0.05, adjusted for sex and city of recruitment).
Genetic ancestry contributes to LINE-1 methylation
DNA methylation signatures differ by human population and are known to contribute to variation between populations [29,30]. Moreover, a previous study has shown higher LINE-1 methylation in non-Hispanic whites compared to non-Hispanic blacks in the United States [31]. Peruvians today exhibit varying levels of European admixture as a result of the Spanish colonization of South America [32]. We implemented a linear mixed model analysis to test whether European admixture proportion contributed to the level of LINE-1 DNA methylation. We used the data from the first principal component (gPC1) (adjusted for the altitude of birth, age, sex, and BMI) that separated Quechua from Europeans to test for associations with LINE-1 methylation. We saw a significant positive association between LINE-1 DNA methylation and Andean ancestry (β = −45.05, p-value = 0.0004) (Figure 2) suggesting that individuals with less European admixture have higher LINE-1 methylation. The association between EPAS1 and gPC1 was not significant, but approaching significance (β = 51.27, p-value = 0.052).
Figure 2.
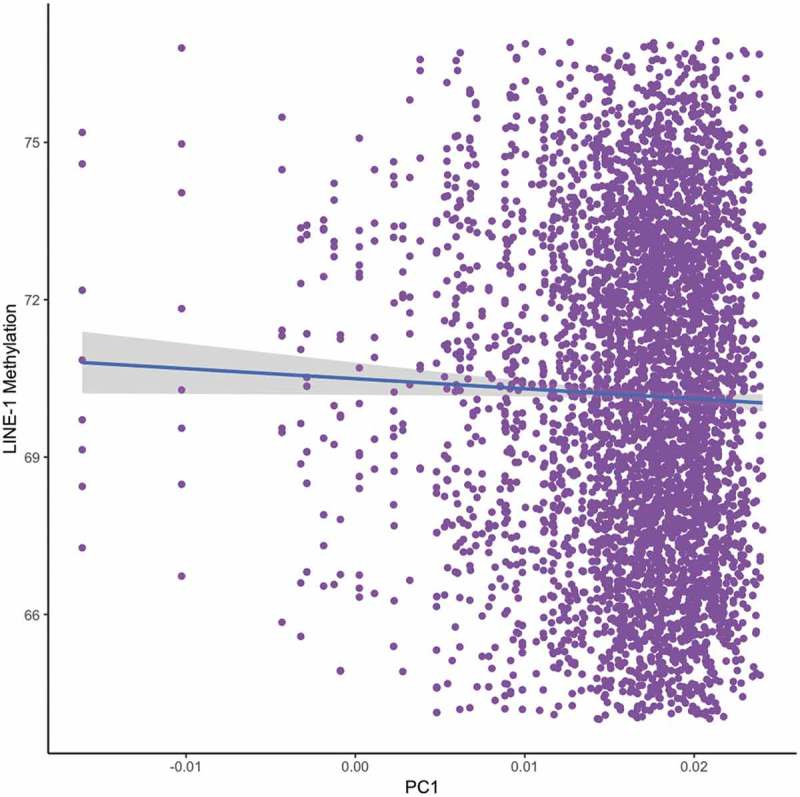
Association between LINE-1 methylation and the first principle component (PC1) from the genome-wide PCA analysis (n = 572). PCA was performed on genome-wide SNP data generated using the Affymetrix Axiom Biobanking Array. All study participants are shown as filled circles. The blue line represents X. X is shown by the grey shading. Lower levels of European admixture are associated with higher levels of LINE-1 methylation (p-value = 0.0004). Individuals with lower levels of European admixture have higher values for PC1.
High-altitude exposure at birth and the time of recruitment influence global DNA methylation
Average LINE-1 methylation was significantly higher in HAQ compared to both LAQ (p-value = 0.0010) and MQ (p-value = 0.0038). LAQ and MQ did not differ in their levels of methylation (p-value = 0.69). To isolate the effect of growth and development at altitude on global methylation patterns, we combined HAQ and MQ into a single group wherein all individuals were born at high altitude (HAQ + MQ; n = 429). We used linear mixed effects models to account for multiple technical pyrosequencing replicates per individual and to avoid averaging the LINE-1 CpG sites. Based on linear mixed models adjusted for sex, age, group, PC1, and BMI, individuals born at high altitude (HAQ+MQ) showed significantly higher levels of LINE-1 methylation compared to LAQ who were born and raised at low altitude (β = 0.58, p-value = 0.00027, marginal R2 = 0.014, conditional R2 = 0.726) (Table 2, Figure 3(a)). Furthermore, when considering only individuals who had lived at high altitude for some or all of their lifetime (HAQ+MQ), LINE-1 methylation significantly increased with the number of years an individual lived at high altitude (β = 0.033, p-value = 0.0047, marginal R2 = 0.012, conditional R2 = 0.737) and when considering all participants (HAQ, MQ, and LAQ) regardless of altitude of birth (β = 0.024, p-value = 4.73e-05, marginal R2 = 0.015, conditional R2 = 0.726) (Table 2, Supp. Figure 2). The relationship between the altitude of birth and DNA methylation was not significant for EPAS1 (Table 2).
Table 2.
Significant linear mixed effects models.
| Beta | p-value | R2 marginal | R2 conditional | |
|---|---|---|---|---|
| LINE1 Entire cohort (n = 572) | ||||
| Altitude of Birth* | 0.598 (High) | 0.0002 | 0.014 | 0.729 |
| City of Recruitment** | 0.474 (Cerro) | 0.0002 | 0.014 | 0.729 |
| Years at high altitude** | 0.025 (years HA) | 3.51E-05 | 0.015 | 0.729 |
| LINE1 Individuals born at high altitude (n = 429) | ||||
| City of Recruitment** | 0.387 (Cerro) | 0.0170 | 0.011 | 0.740 |
| Years at high altitude** | 0.033 (years HA) | 0.0047 | 0.012 | 0.740 |
| LINE1 Quechua Low-Altitude Migrants MQ (n = 146) | ||||
| Years at high altitude** | 0.029 (years HA) | 0.071 | 0.012 | 0.729 |
| EPAS1 Entire cohort (n = 539) | ||||
| Altitude of Birth *** | −0.392 (High) | 0.2119 | 0.036 | 0.887 |
| City of Recruitment *** | −0.828 (Cerro) | 0.0025 | 0.044 | 0.887 |
| Years at high altitude *** | −0.029 (years HA) | 0.0193 | 0.040 | 0.887 |
| EPAS1 Individuals born at high altitude (n = 380) | ||||
| City of Recruitment*** | −0.912 (Cerro) | 0.0054 | 0.049 | 0.879 |
| Years at high altitude*** | −0.059 (years HA) | 0.0092 | 0.047 | 0.879 |
| EPAS1 Quechua Low-Altitude Migrants MQ (n = 123) | ||||
| Years at high altitude*** | −0.049 (years HA) | 0.1895 | 0.021 | 0.862 |
Altitude refers to the altitude of birth (High vs. low)
City refers to the city of recruitment (Cerro de Pasco vs Lima)
Years HA refers to the number of years one lived at high altitude
LA, Low altitude
HA, high altitude
REML, restricted maximum likelihood
Marginal R2 represents the variance explained by the fixed factors, and conditional R2 represents the variance explained by both fixed and random factors
* Model was adjusted for sex, age, group, BMI, and PC1
** Models were adjusted for sex, age, BMI, and PC1
*** Models were adjusted for sex, age, and PC1
Figure 3.
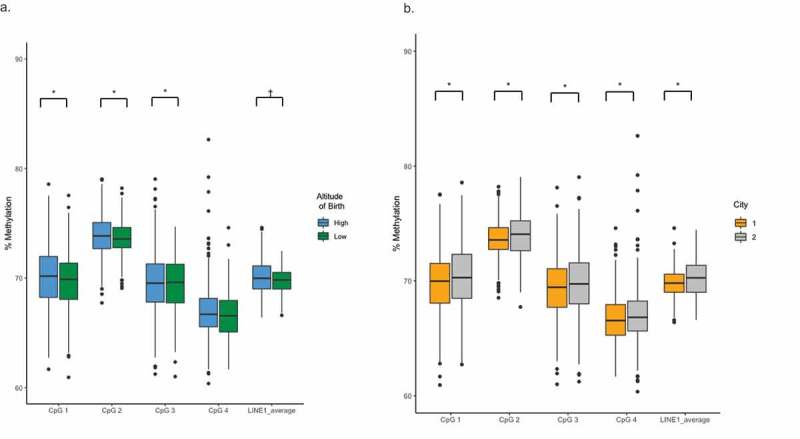
(A) LINE-1 methylation level comparison by CpG position between individuals born at high altitude (HAQ+MQ) and individuals born at low altitude (LAQ). LINE-1 methylation is significantly higher at p < 0.05 among born at high-altitude participants for CpG positions 1 and 2 and the average CpG, and at p < 0.10 for position 4. DNA methylation is not significantly different at position 3. Linear mixed models were corrected for sex, age, recruitment group (i.e. HAQ, MQ, or LAQ), PC1, and BMI. * significant at p < 0.05, † significant at p < 0.10 (B) LINE-1 methylation level comparison between individuals recruited in Lima, Peru (LAQ+MQ) and individuals recruited in Cerro de Pasco, Peru (HAQ). LINE-1 methylation is significantly higher in participants recruited in Cerro de Pasco for all 4 LINE-1 CpG positions and the average CpG. Linear mixed models were corrected for sex, age, recruitment group (i.e. HAQ, MQ, or LAQ), PC1, and BMI. * Significant at p < 0.05.Average CpG was calculated over 4 LINE-1 CpG sites including the technical replicates.
We tested whether the city of recruitment in this study influenced LINE-1 and EPAS1 methylation using linear mixed models adjusted for sex, age, BMI, and gPC1. LINE-1 methylation was higher in individuals recruited in Cerro de Pasco compared to Lima (β = −0.47, p-value = 0.00026, marginal R2 = 0.014, conditional R2 = 0.727). Conversely, EPAS1 methylation was significantly lower in individuals recruited in Cerro de Pasco (HAQ) compared to individuals recruited in Lima (LAQ+MQ) (β = −0.83, p-value = 0.0025, marginal R2 = 0.044, conditional R2 = 0.887, Figure 4). When only considering individuals born at high altitude (HAQ+MQ), LINE-1 methylation was higher among HAQ (β = 0.38, p-value = 0.017, marginal R2 = 0.011, conditional R2 = 0.737) (Table 2, Figure 3(b)), but EPAS1 methylation was lower (β = −0.91, p-value = 0.005, marginal R2 = 0.049, conditional R2 = 0.879, Figure 4). These data suggest that current exposure to hypoxia could influence the epigenome. However, factors other than hypoxia might be at play in determining the degree of methylation in our cohort. One potential environmental difference between the recruitment sites is inorganic lead exposure.
Figure 4.
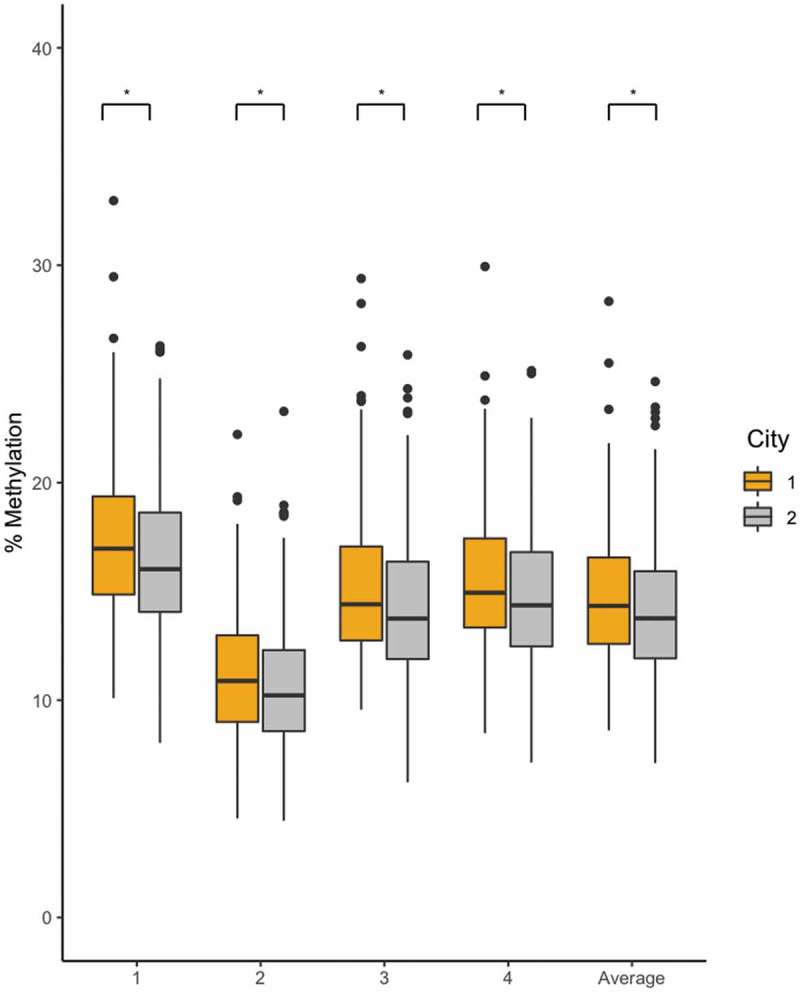
EPAS1 methylation level comparison between individuals recruited in Lima, Peru (LAQ+MQ) and individuals recruited in Cerro de Pasco, Peru (HAQ). EPAS1 methylation is significantly higher in participants recruited in Lima for all 4 EPAS1 CpG positions and the average methylation. * Significant at p < 0.05.
Cerro de Pasco is a major mining center of Peru. High levels of inorganic lead in the soil have been reported as well as high whole-blood lead levels (WBLL) in women and children living there [33,34]. We determined WBLL in a random subset of the individuals (n = 294) (148 from Lima [LAQ (69) and MQ (79)] and 157 from Cerro de Pasco [HAQ]) using atomic absorption spectroscopy. Individuals recruited in Cerro de Pasco [HAQ] had significantly higher WBLLs (4.75 ± 1.53 μg/dL) compared to the individuals recruited in Lima [LAQ (2.05 ± 0.65 μg/dL) and MQ (2.01 ± 0.60 μg/dL)] (p-value<2.2e-16) (Figure 5). WBLLs were not associated with LINE-1 methylation (β = 0.024, p-value = 0.63) (Supp. Figure 3). The association between WBLLs and EPAS1 methylation was approaching significance when adjusted for sex, age, BMI, and gPC1 (β = −0.20, p-value = 0.08, Sup. Figure 4). When WBLLs were tested in participants recruited in Lima and Cerro de Pasco separately, the relationship was not significant (Lima: β = −0.56, p-value = 0.23; Cerro: β = −0.18, p-value = 0.35).
Figure 5.
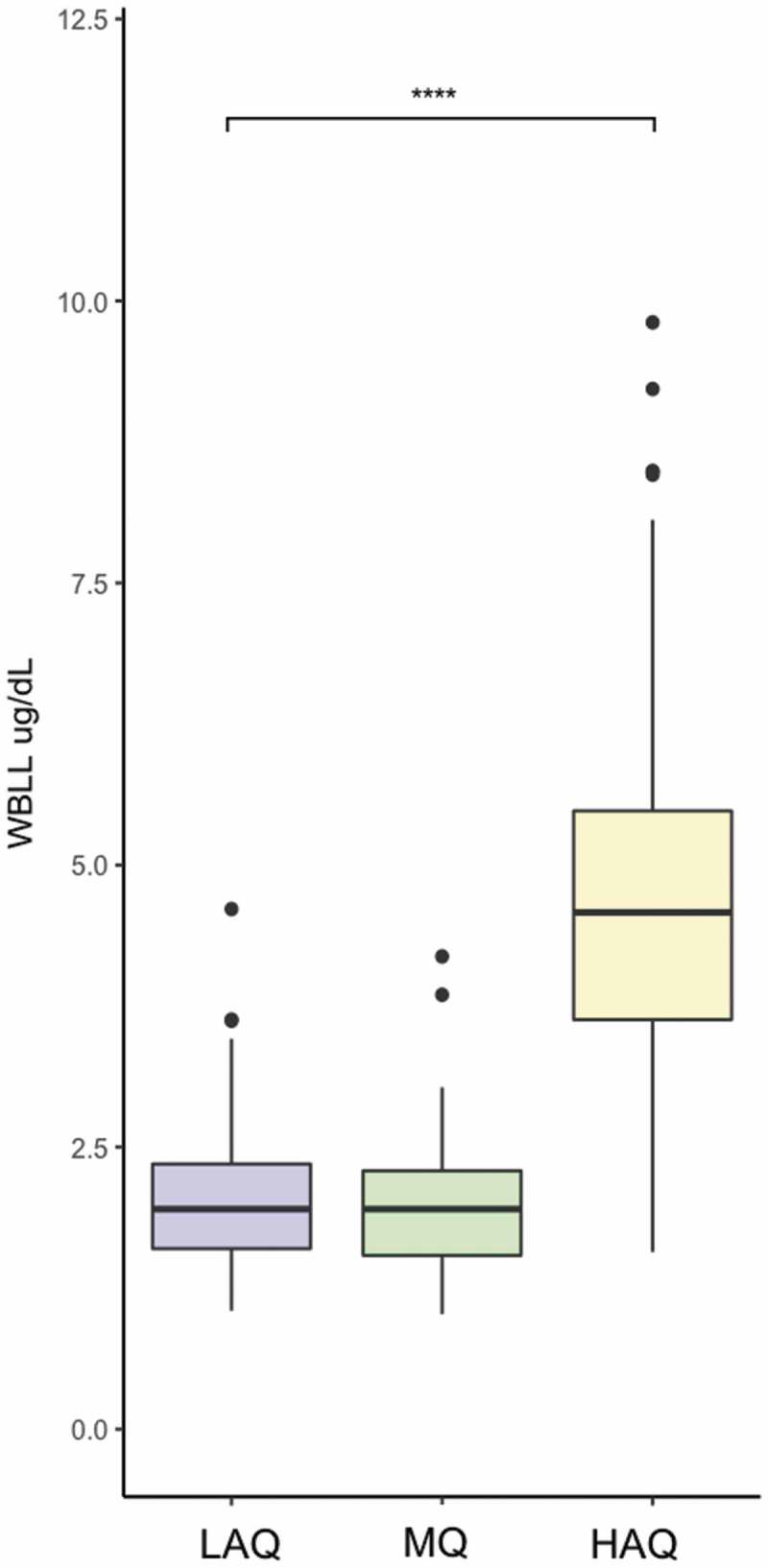
Whole-blood lead level WBLL (μg/dL) comparison between the participants from the three different groups: LAQ, MQ, and HAQ. WBLLs are significantly lower in LAQ (p-value = 3.56E-45) and MQ (p-value = 3.33E-48) compared to HAQ. WBLLs do not differ significantly between LAQ and MQ (p-value = 0.76).
One-carbon metabolism associations
The folate-mediated one-carbon (1C) metabolism pathway is involved in the biosynthesis of amino acids, metabolism of nucleotides, maintenance of epigenetic modifications, and mediation of redox mechanisms [35]. S-adenosylmethionine (SAM) is the primary methyl donor for DNA methylation. It is generated from methionine with the help of DNA methyltransferases in the 1C metabolism pathway [22,36]. Previous studies have shown that polymorphisms in the 1C metabolism pathway can affect LINE-1 methylation [37,38]. We identified 44 SNPs in the 1C metabolism pathway that have been found to associate with changes in LINE-1 methylation and for which we had genotype data from the Affymetrix (Santa Clara, CA) Axoim Biobanking array [36–42]. We first tested each SNP for an association with average LINE-1 methylation using an additive model of inheritance in R (R Core Group, 2018) (Supplementary Table 2). Models were adjusted for the altitude of birth (high vs. low), sex, BMI, and admixture measured as the first PC of the genome-wide SNP data. In the filtering step, out of the 44 SNPs tested four (MTHFD1 rs2236225, TYMS rs502396, FOLH1 rs202676, GLDC rs10975681), were significant at p-value<0.05 (Table 3). These genes are involved in the 1C pathway generating methyl groups for DNA methylation, which is why the we hypothesized that they would act in a cumulative fashion. We included the four SNPs in the same model. This four-SNP-model explained 11.29% variation in the LINE-1 methylation, while models with each SNP alone explained 9% or less of the variation in the LINE-1 methylation. EPAS1 methylation was not associated with the one-carbon metabolism SNPs.
Table 3.
Significant one-carbon metabolism SNPs.
| Gene | SNP | Model | MAF | Minor allele | Function | β (95% CI) | p-value |
|---|---|---|---|---|---|---|---|
| FOLH1 | rs202676 | Additive | 0.39 | G | Loss of stop | 0.21 (0.04, 0.38) | 0.018 |
| GLDC | rs10975681 | Additive | 0.30 | C | Intronic | −0.17 (−0.33, −0.01) | 0.042 |
| MTHFD1 | rs2236225 | Additive | 0.34 | A | Missense | 0.22 (0.05, 0.40) | 0.014 |
| TYMS | rs502396 | Additive | 0.40 | T | Intronic | 0.23 (0.06, 0.40) | 0.008 |
All models were adjusted for the altitude of birth, sex, age, BMI, and PC1
Discussion
Genetic and epigenetic factors have been hypothesized to contribute to high-altitude adaptive phenotypes in Andeans [43]. However, it is not understood whether epigenetic processes play a role in their establishment. We hypothesized that epigenetic modification may underlie Andean developmental adaptation to high-altitude hypoxia. Our results suggest that high-altitude hypoxia increases global levels of DNA methylation in Peruvian Quechua measured at the LINE-1 repetitive element. Additionally, they show that high-altitude residence decreases gene-specific EPAS1 methylation. Our study marks the first time that epigenetic modifications have been explored in the context of differential high-altitude exposure among Andean Quechua. Furthermore, our findings support the role of epigenetic processes in contributing to high-altitude adaptive phenotypes in the Andes.
We chose to assess global levels of DNA methylation using the LINE-1 element as its methylation state may reflect the overall state of methylation in the epigenome [44–46]. We found that hypoxia exposure was associated with increased global methylation. This was supported by three observations: 1) HAQ have significantly higher levels of DNA methylation compared to MQ and LAQ 2) Individuals born at high altitude (HAQ+MQ) have significantly higher levels of DNA methylation compared to individuals born at low altitude (LAQ), and 3) the number of years an individual has lived at high altitude significantly increases DNA methylation. The differences in LINE-1 methylation we observed between altitude exposure groups were small in magnitude. However, LINE-1 elements are present at a high copy number in the genome (~17%), and thus even a small difference in DNA methylation may reflect significant global epigenomic differences [47].
DNA methylation signatures differ between populations [29,30]. We observed a significant association between LINE-1 methylation and the amount of European admixture (β = 0.29, p-value = 0.0011) reflected by the first PC from a PCA performed using genome-wide SNPs indicating that underlying genetic variation may significantly impact global LINE-1 methylation levels, and thus must be considered in DNA methylation analyses.
We identified lower EPAS1 promoter region methylation associated with current altitude of residence in Andean Quechua. EPAS1 is a transcription factor involved in the body’s response to oxygen levels that is active in hypoxic condition [48,49]. Mutations in EPAS1 are associated with erythrocytosis [50], and EPAS1 SNPs are associated with high-altitude adaptation in Tibetans [3]. Lower methylation in the EPAS1 promoter region has been shown to correlate with higher expression of EPAS1 [27]. Therefore, in our cohort, this suggests that high-altitude residence is potentially associated with increased EPAS1 expression [27]. An observation consistent with EPAS1 being activated in hypoxic conditions [25].
A previous study conducted in Cerro de Pasco demonstrated an increase in oxidative stress markers like plasma glutathione and lipid peroxidation products [51], and increased levels of ROS have been found to associate with lower DNA methylation levels [52]. In our cohort, individuals from Cerro de Pasco have higher levels of LINE-1 methylation compared to the individuals from Lima.
DNA methylation and oxidative stress come together in the 1C metabolism pathway, as it stimulates synthesis of glutathione and reduces homocysteine, thus interfering with methylation [53]. We found associations between average LINE-1 methylation and four one-carbon metabolism SNPs (MTHFD1 rs2236225, TYMS rs502396, FOLH1 rs202676, GLDC rs10975681). These genes are involved in various aspects of the 1C pathway, including the conversion of tetrahydrofolate (THF) into various methylenetetrahydrofolate derivatives (MTHFD1), conversion of deoxyuridylate to thymidylate (TYMS), the ability to absorb dietary folate (FOLH1), and the degradation of glycine (GLDC) [54–57].
The effects of high-altitude adaptation on DNA methylation have been explored in other studies of Andeans and in high-altitude Ethiopians. Previous studies of DNA methylation in high-altitude adaptation have shown that increased methylation of a different high-altitude adaptive gene, EGLN1, was associated with excessive erythrocytosis in Andean men suggesting that it plays a role in the development of chronic mountain sickness characterized by increased levels of erythrocytes, which is commonly seen among Andean highlanders [58]. Differences in DNA methylation have also been explored between Ethiopian highlanders and lowlanders, however, the study did not find significant methylation differences in hypoxia-associated genes [59].
Although epigenetic processes involved in human adaptation to high altitude remain largely unknown, epigenetic aspects of hypoxia have been well studied in relation to cancer, as hypoxia is a hallmark of most tumors [60]. Tumor cells grow and proliferate at a rate that outpaces their blood supply. Therefore, they must adapt to an environment with low oxygen concentrations [61,62], similar to humans living in the hypobaric environment of high altitude. In cancerous cells, hypoxia inducible factors (HIFs) have been shown to work in concert with epigenetic mechanisms to respond to hypoxia [63]. For example, in renal carcinomas and multiple myelomas the HIF pathway gene von-hippel lindau (VHL) is silenced via hypermethylation of its 5ʹ CpG island [64,65]. Additionally, transcription of a known altitude-adaptive candidate gene, Endothelial PAS Domain Protein 1 (EPAS1), is mediated by DNA methylation in colorectal cancer [27]. These findings highlight the ability of DNA methylation to play a role in mediating the cellular response to hypoxia. Furthermore, they support the notion that human populations may adapt to high-altitude hypoxia through epigenetic mechanisms.
In our cohort, the city of recruitment (Lima vs. Cerro de Pasco) had a significant effect on DNA methylation. We hypothesized that this could be a result of environmental exposure to lead given that Cerro de Pasco is a mining town and lead exposure has been shown to decrease LINE-1 methylation in previous studies [66–68]. We did not find strong associations between DNA methylation and lead exposure in our study. However, WBLLs are representative of recent lead exposure, and it is unknown what the lifetime exposure levels were for all participants. Another potential difference between the two recruitment sites is diet, which is known to affect DNA methylation [69]. Lima is an urban center and Cerro de Pasco is a remote, Andean town. Any potential differences in diet between participants recruited in Lima and Cerro de Pasco may contribute to the differences in DNA methylation that we observed.
DNA methylation signatures differ by tissue and cell type [70]. We used DNA extracted from whole blood, which is comprised of a mixture of DNA-containing cells. It is possible that these DNA-containing blood cell types are different between high- and low-altitude individuals. For example, high-altitude Andeans have elevated hemoglobin levels [71] compared to low-altitude individuals suggesting a higher number of red blood cells associated with altitude. We tested the association between hemoglobin levels and LINE-1 and EPAS1, which was not significant (data not shown), suggesting that differences in blood cell types are potentially not affecting our results.
The environment affects DNA methylation signatures, and exposures to toxicants, pharmaceutical agents, exercise, stress, and other factors have been shown to associate with changes in epigenetic marks, such as DNA methylation [72–75]. Our findings suggest that being born at high altitude leaves a persistent mark on the epigenome through adulthood by leaving lasting effects on global levels of DNA methylation measured at LINE-1. We also show that altitude of residence affects EPAS1 potentially activating it. Based on our findings, genome-wide DNA methylation studies of high-altitude adaptation are a natural next step in determining the individual genes and pathways that may undergo changes in DNA methylation in association with adaptation to high altitude.
Materials and methods
Study population
Study participants consisted of Peruvian Quechua individuals recruited based on three high-altitude/developmental exposure patterns (Table 1): 1) high-altitude Quechua (HAQ) (n = 300): Quechua individuals born and raised at high altitude who were recruited from the city of Cerro de Pasco, Peru located at an elevation of 4,338 m; 2) migrant Quechua (MQ) (n = 150): Quechua individuals who were born in various high-altitude locations (>2,500 m) and moved to low-altitude Lima at some point in their lifetime; 3) low-altitude Quechua (LAQ) (n = 152): Quechua individuals born and raised at low altitude who were recruited in Lima, Peru at an elevation of 0 m. Male and female participants were unrelated, healthy, non-pregnant/lactating, non-smokers, between 18 and 35 years old (Table 1). Relatedness was determined during the recruitment process using a pre-screening questionnaire. Genetic relatedness was estimated with the program King [76] using genome-wide SNP data from the Affymetrix Axiom Biobanking array. All individuals were in good health at the time of enrollment with no history of chronic disease or recent illness. All study participants were screened for anemia using altitude specific cut-offs. None of the study participants recruited in Cerro de Pasco were mine workers even though Cerro de Pasco is a mining town.
Height and weight were collected at the time of enrollment, and BMI was calculated using the kg/m2 equation. Participants provided a blood sample drawn from an antecubital vein into a vacutainer collection tube containing EDTA for DNA extraction. Whole blood was field stabilized in lysis buffer and transported to the University of Michigan. Genomic DNA was extracted using the Puregene DNA purification system (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
DNA methylation
Quantitative pyrosequencing was performed to assess DNA methylation levels at four CpG sites of the LINE-1 repetitive element and EPAS1. One µg of DNA from each sample was bisulfite converted using the EZ-96 DNA Methylation™ Kit (Zymo Research, Irvine, CA). Bisulfite converted DNA was amplified in duplicate using EPAS1 and LINE-1 primers and HotstarTaq plus Master Mix (Qiagen, Valencia, CA). LINE-1 forward primer sequence: 5ʹ-TTGAGTTAGGTGTGGGATATAGTT-3ʹ, LINE-1 reverse primer sequence: 5ʹ-[biotin]-CAAAAAATCAAAAAATTCCCTTTCC-3ʹ, LINE-1 sequencing primer: 5ʹ-AGGTGTGGATATAGT-3ʹ. The following EPAS1 region was targeted chr2:46,526,762–46,527,105 (hg19). EPAS1 forward primer sequence: 5ʹ-TGGGAGTAGGGGAAAAATGAT-3ʹ, EPAS1 reverse primer sequence: 5ʹ-[biotin]- ACAATCCCCACAATAATTCTTAA-3ʹ, EPAS1 sequencing primer: 5ʹ-AGTAGGGGAAAAATGATT-3ʹ.
DNA methylation levels at each CpG site for EPAS1 and LINE-1 were assessed using the Pyromark Q96 pyrosequencer (Qiagen, Valencia, CA), for more detail see [77]. A bisulfite conversion check ensured full bisulfite conversion of the DNA. The assay was validated in duplicate using a DNA methylation scale of DNA with known values of methylation (0%, 20%, 40%, 60%, and >80%). The DNA methylation scale was created from whole genome amplified DNA (representing 0% DNA methylation), and DNA treated with CpG methyltransferase M.SssI (New England Biolabs, Ipswich, MA) (representing >80% DNA methylation). PCR amplification followed by pyrosequencing was performed on each bisulfite treated sample in duplicate for LINE-1. For EPAS1, 10% of the samples were duplicated. The data were checked via built in quality control measures, and samples with coefficients of variance (CV), which was calculated as standard deviation for all samples over the average for all samples multiplied by 100, exceeding 10 were excluded from the further analyses. Of the 603 samples, 64 either failed pyrosequencing or had high CV for EPAS1 and 31 for LINE-1 and were excluded from the statistical analyses. Statistical analysis was performed on 572 samples (282 HAQ, 147 MQ, and 143 LAQ).
Microarray genotyping
Microarray genotype data were generated for all Andean Quechua DNA samples using the Affymetrix (Santa Clara, CA) Axoim Biobanking array. The Axiom array features approximately 610,000 markers including haplotype tagging SNPs as well as exonic and eQTL variants. We performed a PCA on autosomal variants with call rates > 95%, major allele frequency > 0.0001 that passed the QC of Affymetrix (n = 373,260).
To identify candidate genes from the one-carbon metabolism pathway we performed a literature search for publications reporting one-carbon metabolism SNPs in association with DNA methylation, cancer, and diet among others [36–42,78–80]. Based on the previously published one-carbon metabolism SNP associations, we selected 81 SNPs that were present on the Affymetrix (Santa Clara, CA) Axoim Biobanking array. After removing SNPs that were monomorphic in our samples, were in high LD, and had MAF less than 0.05, we ended up with 44 SNPs used in the analysis. The following model was used to determine the combined effect of 1C SNPs on LINE-1 methylation. Yi = B00 + B01(rs2236225) + B02(rs202676) + B03(rs502396) + B04(rs10975681) + Altitude + Sex + Age + PC1 + BMI + E. where Yi = average LINE-1 methylation.
Statistical analyses
All statistical analyses were conducted using R version 3.4.0 (R Core Team, 2018). Packages lme4 [81], lmerTest [82], ggplot2 [83], and MuMIn (Barton, 2009) were employed. We used both linear mixed effects models and simple linear models to be able to show results while accounting for multiple technical pyrosequencing replicates per individual and to also test the effects on average LINE-1 or EPAS1 methylation. The following model was tested for linear mixed models:
Where X = altitude, city of recruitment, years lived at high altitude, One-carbon metabolism SNPs, Pos = Individual CpG site position, and ID = sample ID. Position and ID were coded as random effects.
Funding Statement
This work was supported by the National Science Foundation (grant number BCS-1132310 to T.D.B, F.L., and A.W.B.); The University of Michigan (to A.W.B and A.C.); and the Michigan Lifestage Environmental Exposures and Disease (M-LEEaD) National Institute of Environmental Health Sciences Core Center (P30 ES017885 to D.C.D.). A.C. was supported by a Baldwin Fellowship from the L.S.B. Leakey Foundation.
Acknowledgments
First and foremost, we would like to thank the study participants of Lima and Cerro de Pasco, Peru. We are also thankful to Dr. Sudipta Ghosh (NEHU University, Shillong, India), Obed Garcia (University of Michigan), Nate Bartman, Jason Howard, Jacqueline Imse, and Kevin Heffernan (Syracuse University), Mark Olfert (University of West Virginia), and Francisco Villafuerte (Universidad Peruana Cayetano Heredia). We also are grateful for the help of several participant recruiters and physicians, including Cesar De Albertis, Laura Mori, Josseline Honorio, and Alejandro Zamudio.
Author Contributions
A.W.B., A.C., T.D.B., F.L.V., M.R., M.K., D.C.D., and J.M.G., conceived and designed the research; A.C. and T.R.J performed experiments; A.W.B., A.C., J.M.G. analyzed data; A.W.B., A.C., and wrote the manuscript with contributions from all authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Moore LG. Human genetic adaptation to high altitude. High Alt Med Biol. 2001. Summer;2(2):257–279. PubMed PMID: 11443005. [DOI] [PubMed] [Google Scholar]
- [2].Bigham AW, Julian CG, Wilson MJ, et al. Maternal PRKAA1 and EDNRA genotypes are associated with birth weight, and PRKAA1 with uterine artery diameter and metabolic homeostasis at high altitude. Physiol Genomics. 2014. September 15;46(18):687–697. PubMed PMID: 25225183; PubMed Central PMCID: PMC4166715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beall CM, Cavalleri GL, Deng L, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 2010. June 22;107(25):11459–11464. PubMed PMID: 20534544; PubMed Central PMCID: PMCPMC2895075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010. September;6(9):e1001116 PubMed PMID: 20838600; PubMed Central PMCID: PMC2936536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Simonson TS, Yang Y, Huff CD, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010. July 2;329(5987):72–75. PubMed PMID: 20466884. [DOI] [PubMed] [Google Scholar]
- [6].Brutsaert TD. Genetic and environmental adaptation in high altitude natives. Conceptual, methodological, and statistical concerns. Adv Exp Med Biol. 2001;502:133–151. PubMed PMID: 11950135. [DOI] [PubMed] [Google Scholar]
- [7].Frisancho AR, Martinez C, Velasquez T, et al. Influence of developmental adaptation on aerobic capacity at high altitude. J Appl Physiol. 1973. February;34(2):176–180. PubMed PMID: 4686351. [DOI] [PubMed] [Google Scholar]
- [8].Frisancho AR, Frisancho HG, Albalak R, et al. Developmental, genetic, and environmental components of lung volumes at high altitude. Am J Hum Biol. 1997;9(2):191–203. PubMed PMID: 28561519. [DOI] [PubMed] [Google Scholar]
- [9].Brutsaert TD, Araoz M, Soria R, et al. Higher arterial oxygen saturation during submaximal exercise in Bolivian Aymara compared to European sojourners and Europeans born and raised at high altitude. Am J Phys Anthropol. 2000. October;113(2):169–181. PubMed PMID: 11002203. [DOI] [PubMed] [Google Scholar]
- [10].Kiyamu M, Leon-Velarde F, Rivera-Chira M, et al. Developmental Effects Determine Submaximal Arterial Oxygen Saturation in Peruvian Quechua. High Alt Med Biol. 2015. June;16(2):138–146. PubMed PMID: 25977978; PubMed Central PMCID: PMCPMC4490701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bigham AW, Kiyamu M, Leon-Velarde F, et al. Angiotensin-converting enzyme genotype and arterial oxygen saturation at high altitude in Peruvian Quechua. High Alt Med Biol. 2008. Summer;9(2):167–178. PubMed PMID: 18578648; PubMed Central PMCID: PMCPMC3140306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lam LL, Emberly E, Fraser HB, et al. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A. 2012. October 16;109(Suppl 2):17253–17260. PubMed PMID: 23045638; PubMed Central PMCID: PMCPMC3477380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lv J, Xin Y, Zhou W, et al. The epigenetic switches for neural development and psychiatric disorders. J Genet Genomics. 2013. July 20;40(7):339–346. PubMed PMID: 23876774. [DOI] [PubMed] [Google Scholar]
- [14].Mohn F, Schubeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet. 2009. March;25(3):129–136. PubMed PMID: 19185382. [DOI] [PubMed] [Google Scholar]
- [15].Tollefsbol TO. Advances in epigenetic technology. Methods Mol Biol. 2011;791:1–10. PubMed PMID: 21913067; PubMed Central PMCID: PMCPMC3227536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fraga MF, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques. 2002. September;33(3):632–649. PubMed PMID: 12238773. [DOI] [PubMed] [Google Scholar]
- [17].Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001. February 15;409(6822):860–921. PubMed PMID: 11237011. [DOI] [PubMed] [Google Scholar]
- [18].Nelson HH, Marsit CJ, Kelsey KT. Global methylation in exposure biology and translational medical science. Environ Health Perspect. 2011. November;119(11):1528–1533. PubMed PMID: 21669556; PubMed Central PMCID: PMCPMC3226501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Belancio VP, Deininger PL, Roy-Engel AM. LINE dancing in the human genome: transposable elements and disease. Genome Med. 2009. October 27;1(10):97 PubMed PMID: 19863772; PubMed Central PMCID: PMCPMC2784310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Woo HD, Kim J, Christensen BC. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One. 2012;7(4):e34615 PubMed PMID: 22509334; PubMed Central PMCID: PMCPMC3324531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ogino S, Nosho K, Kirkner GJ, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008. December 3;100(23):1734–1738. PubMed PMID: 19033568; PubMed Central PMCID: PMCPMC2639290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem. 2012. August;23(8):853–859. PubMed PMID: 22749138; PubMed Central PMCID: PMCPMC3405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Foll M, Gaggiotti OE, Daub JT, et al. Widespread signals of convergent adaptation to high altitude in Asia and america. Am J Hum Genet. 2014. October 2;95(4):394–407. PubMed PMID: 25262650; PubMed Central PMCID: PMCPMC4185124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Eichstaedt CA, Pagani L, Antao T, et al. Evidence of early-stage selection on EPAS1 and GPR126 genes in Andean high altitude populations. Sci Rep. 2017. October 12;7(1):13042 PubMed PMID: 29026132; PubMed Central PMCID: PMCPMC5638799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Semenza GL. Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatr Res. 2001. May;49(5):614–617. PubMed PMID: 11328942. [DOI] [PubMed] [Google Scholar]
- [26].Nanduri J, Semenza GL, Prabhakar NR. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2017. December 1;313(6):L1096–L1100. PubMed PMID: 28839104; PubMed Central PMCID: PMCPMC5814703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rawluszko-Wieczorek AA, Horbacka K, Krokowicz P, et al. Prognostic potential of DNA methylation and transcript levels of HIF1A and EPAS1 in colorectal cancer. Mol Cancer Res. 2014. August;12(8):1112–1127. MCR-14-0054. PubMed PMID: 24825851. [DOI] [PubMed] [Google Scholar]
- [28].El-Maarri O, Becker T, Junen J, et al. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet. 2007. December;122(5):505–514. PubMed PMID: 17851693. [DOI] [PubMed] [Google Scholar]
- [29].Heyn H, Moran S, Hernando-Herraez I, et al. DNA methylation contributes to natural human variation. Genome Res. 2013. September;23(9):1363–1372. PubMed PMID: 23908385; PubMed Central PMCID: PMCPMC3759714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fraser HB, Lam LL, Neumann SM, et al. Population-specificity of human DNA methylation. Genome Biol. 2012. February 9;13(2):R8 PubMed PMID: 22322129; PubMed Central PMCID: PMCPMC3334571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang FF, Cardarelli R, Carroll J, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011. May;6(5):623–629. PubMed PMID: 21739720; PubMed Central PMCID: PMCPMC3230547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sandoval JR, Salazar-Granara A, Acosta O, et al. Tracing the genomic ancestry of Peruvians reveals a major legacy of pre-Columbian ancestors. J Hum Genet. 2013. September;58(9):627–634. PubMed PMID: 23863748. [DOI] [PubMed] [Google Scholar]
- [33].Conklin L, Sánchez CA, Neri A et al. Exposiciones a metales pesados en niños y mujeres en edad fértil en tres comunidades mineras Cerro de Pasco. Perú: CDC; 2008. [Google Scholar]
- [34].van Geen A, Bravo C, Gil V, et al. Lead exposure from soil in Peruvian mining towns: a national assessment supported by two contrasting examples. Bull World Health Organ. 2012. December 1;90(12):878–886. PubMed PMID: 23284193; PubMed Central PMCID: PMCPMC3524960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017. January 10;25(1):27–42. PubMed PMID: 27641100; PubMed Central PMCID: PMCPMC5353360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bleich S, Semmler A, Frieling H, et al. Genetic variants of methionine metabolism and DNA methylation. Epigenomics. 2014;6(6):585–591. PubMed PMID: 25531253. [DOI] [PubMed] [Google Scholar]
- [37].Tajuddin SM, Amaral AF, Fernandez AF, et al. Genetic and non-genetic predictors of LINE-1 methylation in leukocyte DNA. Environ Health Perspect. 2013. June;121(6):650–656. PubMed PMID: 23552396; PubMed Central PMCID: PMCPMC3672919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wernimont SM, Clark AG, Stover PJ, et al. Folate network genetic variation, plasma homocysteine, and global genomic methylation content: a genetic association study. BMC Med Genet. 2011. November;21(12):150 PubMed PMID: 22103680; PubMed Central PMCID: PMCPMC3266217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hazra A, Fuchs CS, Kawasaki T, et al. Germline polymorphisms in the one-carbon metabolism pathway and DNA methylation in colorectal cancer. Cancer Causes Control. 2010. March;21(3):331–345. PubMed PMID: 19936946; PubMed Central PMCID: PMCPMC3570978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gibson TM, Brennan P, Han S, et al. Comprehensive evaluation of one-carbon metabolism pathway gene variants and renal cell cancer risk. PLoS One. 2011;6(10):e26165 PubMed PMID: 22039442; PubMed Central PMCID: PMCPMC3198392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chang SC, Chang PY, Butler B, et al. Single nucleotide polymorphisms of one-carbon metabolism and cancers of the esophagus, stomach, and liver in a Chinese population. PLoS One. 2014;9(10):e109235 PubMed PMID: 25337902; PubMed Central PMCID: PMCPMC4206280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Llanos AA, Chandwani S, Bandera EV, et al. Associations between sociodemographic and clinicopathological factors and breast cancer subtypes in a population-based study. Cancer Causes Control. 2015. December;26(12):1737–1750. PubMed PMID: 26376894; PubMed Central PMCID: PMCPMC4628597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Frisancho AR. Developmental functional adaptation to high altitude: review. Am J Hum Biol. 2013. Mar-Apr;25(2):151–168. PubMed PMID: 24065360. [DOI] [PubMed] [Google Scholar]
- [44].van Bemmel D, Lenz P, Liao LM, et al. Correlation of LINE-1 methylation levels in patient-matched buffy coat, serum, buccal cell, and bladder tumor tissue DNA samples. Cancer Epidemiol Biomarkers Prev. 2012. July;21(7):1143–1148. EPI-11-1030. PubMed PMID: 22539607; PubMed Central PMCID: PMCPMC3397796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li C, Yang X, Xu M, et al. Epigenetic marker (LINE-1 promoter) methylation level was associated with occupational lead exposure. Clin Toxicol (Phila). 2013. May;51(4):225–229. PubMed PMID: 23528182. [DOI] [PubMed] [Google Scholar]
- [46].Wangsri S, Subbalekha K, Kitkumthorn N, et al. Patterns and possible roles of LINE-1 methylation changes in smoke-exposed epithelia. PLoS One. 2012;7(9):e45292 PubMed PMID: 23028911; PubMed Central PMCID: PMCPMC3445447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Breton CV, Marsit CJ, Faustman E, et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the children’s environmental health and disease prevention research center’s epigenetics working group. Environ Health Perspect. 2017. April;125(4):511–526. PubMed PMID: 28362264; PubMed Central PMCID: PMCPMC5382002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997. January 1;11(1):72–82. PubMed PMID: 9000051. [DOI] [PubMed] [Google Scholar]
- [49].Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001. August;7(8):345–350. PubMed PMID: 11516994. [DOI] [PubMed] [Google Scholar]
- [50].Percy MJ, Beer PA, Campbell G, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008. June 1;111(11):5400–5402. PubMed PMID: 18378852; PubMed Central PMCID: PMCPMC2396730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jefferson JA, Simoni J, Escudero E, et al. Increased oxidative stress following acute and chronic high altitude exposure. High Alt Med Biol. 2004. Spring;5(1):61–69. PubMed PMID: 15072717. [DOI] [PubMed] [Google Scholar]
- [52].Pogribny IP, Tryndyak VP, Woods CG, et al. Epigenetic effects of the continuous exposure to peroxisome proliferator WY-14,643 in mouse liver are dependent upon peroxisome proliferator activated receptor alpha. Mutat Res. 2007. December 1;625(1–2):62–71. PubMed PMID: 17586532; PubMed Central PMCID: PMCPMC2111058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Menezo YJ, Silvestris E, Dale B, et al. Oxidative stress and alterations in DNA methylation: two sides of the same coin in reproduction. Reprod Biomed Online. 2016. December;33(6):668–683. PubMed PMID: 27742259. [DOI] [PubMed] [Google Scholar]
- [54].Brody LC, Conley M, Cox C, et al. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet. 2002. November;71(5):1207–1215. PubMed PMID: 12384833; PubMed Central PMCID: PMCPMC385099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Trinh BN, Ong CN, Coetzee GA, et al. Thymidylate synthase: a novel genetic determinant of plasma homocysteine and folate levels. Hum Genet. 2002. September;111(3):299–302. PubMed PMID: 12215845. [DOI] [PubMed] [Google Scholar]
- [56].Ji Y, Hebbring S, Zhu H, et al. Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: pharmacometabolomics-informed pharmacogenomics. Clin Pharmacol Ther. 2011. January;89(1):97–104. PubMed PMID: 21107318; PubMed Central PMCID: PMCPMC3034442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guo J, Xie H, Wang J, et al. The maternal folate hydrolase gene polymorphism is associated with neural tube defects in a high-risk Chinese population. Genes Nutr. 2013. March;8(2):191–197. PubMed PMID: 22918695; PubMed Central PMCID: PMCPMC3575888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Julian CG. Epigenomics and human adaptation to high altitude. J Appl Physiol (1985). 2017. November 1;123(5):1362–1370. PubMed PMID: 28819001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Alkorta-Aranburu G, Beall CM, Witonsky DB, et al. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 2012;8(12):e1003110 PubMed PMID: 23236293; PubMed Central PMCID: PMCPMC3516565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996. August 9;86(3):353–364. PubMed PMID: 8756718. [DOI] [PubMed] [Google Scholar]
- [61].Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007. June;26(2):225–239. PubMed PMID: 17440684. [DOI] [PubMed] [Google Scholar]
- [62].Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011. June;11(6):393–410. PubMed PMID: 21606941. [DOI] [PubMed] [Google Scholar]
- [63].Watson JA, Watson CJ, McCann A, et al. Epigenetics, the epicenter of the hypoxic response. Epigenetics. 2010. May 16;5(4):293–296. PubMed PMID: 20418669. [DOI] [PubMed] [Google Scholar]
- [64].Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994. October 11;91(21):9700–9704. PubMed PMID: 7937876; PubMed Central PMCID: PMCPMC44884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hatzimichael E, Dranitsaris G, Dasoula A, et al. Von Hippel-Lindau methylation status in patients with multiple myeloma: a potential predictive factor for the development of bone disease. Clin Lymphoma Myeloma. 2009. June;9(3):239–242. PubMed PMID: 19525194. [DOI] [PubMed] [Google Scholar]
- [66].Li C, Yang X, Xu M, et al. Epigenetic marker (LINE-1 promoter) methylation level was associated with occupational lead exposure. Clin Toxicol (Phila). 2013;51(4):225–229. [DOI] [PubMed] [Google Scholar]
- [67].Goodrich JM, Dolinoy DC, Sánchez BN, et al. Adolescent epigenetic profiles and environmental exposures from early life through peri-adolescence. Environ Epigenet. 2016;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wright RO, Schwartz J, Wright RJ, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect. 2010;118(6):790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang FF, Morabia A, Carroll J, et al. Dietary patterns are associated with levels of global genomic DNA methylation in a cancer-free population. J Nutr. 2011. June;141(6):1165–1171. PubMed PMID: 21525250; PubMed Central PMCID: PMCPMC3095144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kitamura E, Igarashi J, Morohashi A, et al. Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics. 2007. March;89(3):326–337. PubMed PMID: 17188838; PubMed Central PMCID: PMCPMC1847344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Beall CM, Brittenham GM, Strohl KP, et al. Hemoglobin concentration of high-altitude Tibetans and Bolivian Aymara. Am J Phys Anthropol. 1998. July;106(3):385–400. PubMed PMID: 9696153. [DOI] [PubMed] [Google Scholar]
- [72].Senut MC, Cingolani P, Sen A, et al. Epigenetics of early-life lead exposure and effects on brain development. Epigenomics. 2012. December;4(6):665–674. PubMed PMID: 23244311; PubMed Central PMCID: PMCPMC3555228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Csoka AB, Szyf M. Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Med Hypotheses. 2009. November;73(5):770–780. PubMed PMID: 19501473. [DOI] [PubMed] [Google Scholar]
- [74].Non AL, Hollister BM, Humphreys KL, et al. DNA methylation at stress-related genes is associated with exposure to early life institutionalization. Am J Phys Anthropol. 2016. September;161(1):84–93. PubMed PMID: 27218411; PubMed Central PMCID: PMCPMC5278953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ling C, Ronn T. Epigenetic adaptation to regular exercise in humans. Drug Discov Today. 2014. July;19(7):1015–1018. PubMed PMID: 24632002. [DOI] [PubMed] [Google Scholar]
- [76].Manichaikul A, Mychaleckyj JC, Rich SS, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010. November 15;26(22):2867–2873. PubMed PMID: 20926424; PubMed Central PMCID: PMCPMC3025716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Virani S, Dolinoy DC, Halubai S, et al. Delivery type not associated with global methylation at birth. Clin Epigenetics. 2012. June 9;4(1):8 PubMed PMID: 22682523; PubMed Central PMCID: PMCPMC3404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lucock M, Yates Z, Martin C, et al. Methylation diet and methyl group genetics in risk for adenomatous polyp occurrence. BBA Clin. 2015. June;3:107–112. PubMed PMID: 26673393; PubMed Central PMCID: PMCPMC4661521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Aneiros-Guerrero A, Lendinez AM, Palomares AR, et al. Genetic polymorphisms in folate pathway enzymes, DRD4 and GSTM1 are related to temporomandibular disorder. BMC Med Genet. 2011. May;26(12):75 PubMed PMID: 21615938; PubMed Central PMCID: PMCPMC3129576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kim W, Woo HD, Lee J, et al. Dietary folate, one-carbon metabolism-related genes, and gastric cancer risk in Korea. Mol Nutr Food Res. 2016. February;60(2):337–345. PubMed PMID: 26833750. [DOI] [PubMed] [Google Scholar]
- [81].Bates D, Machler M, Bolker BM, et al. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015. October;67(1):1–48. PubMed PMID: WOS:000365981400001; English. [Google Scholar]
- [82].Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest package: tests in linear mixed effects models. J Stat Softw. 2017. December;82(13):1–26. PubMed PMID: WOS:000417711600001; English. [Google Scholar]
- [83].Wickham H. ggplot2: elegant graphics for data analysis. Use R. 2009;1–212. PubMed PMID: WOS:000269437100014; English DOI: 10.1007/978-0-387-98141-3 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.