

Abstract
Background
Lacerta viridis and Lacerta bilineata are sister species of European green lizards (eastern and western clades, respectively) that, until recently, were grouped together as the L. viridis complex. Genetic incompatibilities were observed between lacertid populations through crossing experiments, which led to the delineation of two separate species within the L. viridis complex. The population history of these sister species and processes driving divergence are unknown. We constructed the first high-quality de novo genome assemblies for both L. viridis and L. bilineata through Illumina and PacBio sequencing, with annotation support provided from transcriptome sequencing of several tissues. To estimate gene flow between the two species and identify factors involved in reproductive isolation, we studied their evolutionary history, identified genomic rearrangements, detected signatures of selection on non-coding RNA, and on protein-coding genes.
Findings
Here we show that gene flow was primarily unidirectional from L. bilineata to L. viridis after their split at least 1.15 million years ago. We detected positive selection of the non-coding repertoire; mutations in transcription factors; accumulation of divergence through inversions; selection on genes involved in neural development, reproduction, and behavior, as well as in ultraviolet-response, possibly driven by sexual selection, whose contribution to reproductive isolation between these lacertid species needs to be further evaluated.
Conclusion
The combination of short and long sequence reads resulted in one of the most complete lizard genome assemblies. The characterization of a diverse array of genomic features provided valuable insights into the demographic history of divergence among European green lizards, as well as key species differences, some of which are candidates that could have played a role in speciation. In addition, our study generated valuable genomic resources that can be used to address conservation-related issues in lacertids.
Keywords: sister species, PacBio and Illumina, de novo hybrid assembly, transcripts, noncoding RNA, zinc fingers, positive selection, UV response, inversions, gene flow
Introduction
Understanding what species are and the processes driving their emergence have been two central issues in biology [1]. During the last century, genes involved in reproductive isolation were mainly identified in model organisms, such as Drosophila [2]. These studies aiming at the so-called speciation genes revealed at least three general patterns: (i) many genes involved in post-zygotic incompatibilities show accelerated evolution [2]; (ii) incompatibilities often involve a disproportionate number of genes located on sex chromosomes [3, 4]; and (iii) mis-expression is often observed in hybrids, suggesting that gene regulation is an important component of speciation [5–7]. However, the identification of incompatibilities using laborious lab crosses was only possible for model organisms, and thus the identification of loci involved in reproductive isolation [8] in natural populations remained largely unknown.
The advent of high-throughput sequencing together with the development of novel approaches for whole genome analyses opened new research avenues to study the origin of species, including non-model organisms [9]. It has been shown that genes involved in adaptation and speciation are often found in regions of low recombination, such as genomic rearrangements, suggesting that they play an important role in species diversification [10]. Several in silico tools have been developed to detect structural variation with high precision using genomic data [11–13], thus enabling us to test evolutionary hypotheses such as the role of genomic rearrangements in speciation over a wider taxonomic range [14].
The assessment of divergence in regulatory elements and transcription factors between species further adds to a more complete understanding of the link between genotypes and phenotypes. In this respect, transcriptome sequencing offers an unprecedented resolution to investigate the general importance of divergence in gene regulation in speciation. In particular, zinc-finger genes, especially Krüppel-type zinc fingers (KZNFs), a family of transcription factors, were pinpointed as strong candidates to play a role in the speciation of other vertebrates [6]. In addition, various epigenetic mechanisms between species mediated by non-coding RNA (ncRNA) can also contribute to speciation [15–18].
Nevertheless, our understanding of how speciation unfolds, as well as the mechanisms involved, will remain limited without the knowledge of the demographic history between diverging taxa [8]. Model-based methods are now available to infer the demographic history of recently diverged taxa based on genome data from a few individuals of each species [19]. Thus, patterns of gene flow and population size changes during divergence can now be inferred without extensive sampling [20].
In summary, the identification of differences in genomic features between closely related species and their demographic history can now be assessed in a cost-effective manner. The resulting information is likely to provide insights about the main candidates playing a role in diversification, upon which more specific hypotheses concerning the mechanisms of divergence can be tested.
Lizards provide an excellent model for studying speciation due to the existing knowledge of their long-term demographics and adaptive morphologies, in addition to the ease of sample collection and experimental manipulations [21]. Lizards of the genus Anolis, in particular, have been studied in detail, as their distribution on islands coupled with repeated adaptive radiations offer a perfect framework for evolutionary ecology studies [22]. Not surprisingly, the first sequenced squamate genome was an anole lizard [23]. Comparative genomic analysis of Anolis carolinensis (anole lizard) with the genomes of birds and mammals was pivotal in identifying accelerated evolution of egg proteins associated with amniote evolution [23]. Further sauropsid genomes (birds and reptiles) were sequenced in recent years, now covering a broader taxonomic range of Squamata, Archosauria, and Chelonia [24–31]. For instance, the study of Gekko japonicus (gecko lizard) contributed to the understanding of evolution and adaptation of tail regeneration, clinging, nocturnal vision, and diversification of the olfactory system [26]. In addition, the genomes of Pogona vitticeps (bearded dragon lizard) and Shinisaurus crocodilurus (Chinese crocodile lizard) have recently been characterized [24, 27]. However, comparative genome analyses of closely related lizard species pairs have been limited to anoles, where adaptive evolution of genes related to brain development and behavior was recently reported [32].
The family Lacertidae (Sauropsida, Squamata) has been well covered in terms of phylogeographic studies, providing important information about the likely timing and geographic context of speciation [21]. Within this family, the Lacerta viridis complex shows an intricate evolutionary history with secondary contact zones [33, 34]. Here, we focus on the divergence between the western clade formally described as Lacerta bilineata (NCBI:txid95620) and the eastern clade of L. viridis (NCBI:txid65476) (corresponding to lineage B and lineage V, respectively, of Marzahn et al.) that currently occupy disjointed regions in Europe [34].
Adult individuals from the two taxa are very similar: throat coloration of hatchlings and early juveniles is the only described diagnostic trait so far [35]. Gene flow between these two species was previously hypothesized in studies of allozyme variation [36, 37]. However, recent analyses based on mtDNA and one nuclear marker (fibint7) have cast doubt on the taxonomic classification of the individuals analyzed in those studies and did not provide conclusive evidence either for or against gene flow between L. viridis and L. bilineata [34].
Hybrids between different main lineages within the L. viridis complex (northern Italy and Hungary) exhibit reduced fitness under laboratory conditions [38]. This suggests that at least partial reproductive isolation between L. viridis and L. bilineata can exist in the wild due to potential genomic Bateson-Dobzhansky-Muller incompatibilities (BDMIs). Previous models have suggested that after a secondary contact, BDMIs can be maintained and further accumulate within genome rearrangements [39, 40], thus avoiding species fusion [41]. High karyotypic variability has been observed in reptiles [42], also within the L. viridis complex [43], raising the prospect that genomic rearrangements could also be involved in their diversification [44]. Finally, lizard-specific KZNF genes have recently been predicted [45], making our focal pair of taxa an excellent case study of evolution in this class of genes and their role in speciation via changes in gene regulatory networks. Overall, the L. viridis complex comprises a very interesting system where different genomic components can be studied to elucidate the demographic history and possible processes involved in speciation.
Here, we combine short Illumina and long Pacific Biosciences (PacBio) read sequencing approaches to construct high-quality de novo genomes for both L. bilineata and L. viridis, with annotation support from transcriptomic data. We investigated the demographic history of divergence between the two lacertid taxa and performed a broad comparison of key genomic features providing important insights about their divergence that can be tested in future studies aiming to identify the mechanisms ultimately leading to speciation between this closely related species pair.
Results
Genomes of L. viridis and L. bilineata
We employed a hybrid strategy of combining Illumina and PacBio sequencing data to produce separate genome assemblies for the two lacertid species. Genome sequencing coverages of 34x Illumina and 14x PacBio for L. viridis and 37x Illumina and 11x PacBio for L. bilineata aided in the construction of high-quality genome assemblies (Supplement SI-1; Supplementary Figs. S2, S3). The genome assembly sizes were 1.44 Gbp and 1.42 Gbp for L. viridis and L. bilineata, respectively. The assembled lacertid genomes achieved better contiguity than the high-coverage illumina-only contigs of G. japonicus but lower than the chromosome-level assembly of A. carolinensis (368 kbp and 663 kbp for L. bilineata and L. viridis, respectively, vs 150 Mbp in A. carolinensis) (Supplementary Table S1). The Benchmarking Universal Single-Copy Orthologs completeness in terms of single-copy ortholog (SCO) genes with vertebrate core gene set were 96% and 94%, respectively, higher than in the available lizard genomes. Since the genome of L. viridis had better contiguity than L. bilineata (higher N50 and fewer contigs), L. viridis was used as the reference to predict genomic variants, structural variants (SVs) and single-nucleotide polymorphisms between the two taxa.
The L. viridis genome consisted of a higher number of large segmental duplications (>5 kbp) compared to L. bilineata (Supplementary Fig. S4). However, no significant differences were observed in segmental duplications (>1 kbp) between the two lacertid genomes (F-test: P = 0.35 and Wilcoxon test: P = 0.55). Hence, the occurrence of fewer large segmental duplications in L. bilineata could be the result of its more fragmented genome assembly. Synteny information was used to create unordered contig clusters (minimum size of 1 Mbp covering one-third of the L. viridis genome), which roughly represent positioning on the same chromosome (Supplementary File S2). The median synonymous substitution rate (Ks) and non-synonymous substitution rate (Ka) based on 7,030 SCOs between the two lacertid species were 0.021 and 0.016, respectively. A mutation rate of 1 × 10−9 substitutions per site per generation was estimated from the four-fold degenerate sites. This mutation rate observed in the ancestral lacertid lineage is similar to the ancestral bird lineage (1.15–1.23 × 10−9 per site per generation) [46, 47].
The identical structures of the HOX-cluster between the lacertid species and A. carolinensis confirmed the high genomic assembly quality since the HOX-clusters are highly conserved (Supplement SI-1). The number of chromosomes and the sex-determination system are different between A. carolinensis (2n = 36, 12 macro- and 24 microchromosomes; XY) and lacertid lizards (2n = 38, 36 macro- and 2 microchromosomes; ZW) [43, 48]. However, genomic contigs of both lacertid species were syntenic without breaks or inter-chromosomal transpositions to the macro-chromosomes of A. carolinensis (Supplementary Fig. S2), even though the lacertids and anoles split more than 150 million years ago (Mya) [49]. The only exception to this was a L. viridis contig that splits into two macro-chromosomes of the A. carolinensis genome. This particular contig of L. viridis was syntenic to five separate contigs in the L. bilineata assembly, confirming a higher fragmentation in genome assembly of the latter.
The assembled transcripts were crucial for gene annotations since the ab initio methods predicted more fragmented proteins and coding sequences (CDS) (38,000–55,000) when compared to the final gene models (22,100–22,500) (Supplementary Table S2). A majority of the longest de novo assembled transcript isoforms were from the ovarian tissue followed by the brain. Since the sequencing throughput was highest for the liver tissue in both species, this finding was not likely the result of sequencing artifacts. We identified 22,156 genes in L. viridis and 22,491 genes in L. bilineata supported by de novo assembled transcripts (Supplement SI-2; Supplementary Table S2). The larger number of genes in L. bilineata was due to the fragmentation of a few genes onto multiple contigs, which can be resolved in the future with scaffolding information. Compared to A. carolinensis, we observed an over-representation of genes involved in transfer RNA (tRNA) aminoacylation (Panther release 20170413, fold-enrichment = 2.13–2.25, P< 0.03) and tRNA metabolic process (Panther release 20170413, fold-enrichment = 1.84–1.89, P< 0.003) in both lacertids, indicating an expansion of tRNA-processing genes before their split. Putative Z-chromosome linked contigs consisted of few non-coding elements (7–11 microRNAs, 1 snoRNA, 2–3 snRNAs, and 46–53 functional tRNAs) (Supplement SI-3). The total length of the contigs assigned to the Z-chromosome in lacertids was larger (13.5–15.6 Mbp) than the Z-chromosomes of P. vitticeps (8 Mbp), but the number of identified genes were similar (205–221 and 219, respectively) [50].
The number of predicted members of the different non-coding RNA classes was similar in L. viridis and L. bilineata (Supplementary Table S3). Compared to other selected tetrapod species, there was an increase in the number of tRNAs (both functional and pseudo tRNAs) in the two lacertid species (Supplementary Figs. S5, S6). However, the numbers of tRNAs and pseudo tRNAs are known to vary significantly in eukaryotes [51]. We found an over-representation of tRNA-processing genes supported by the expansion of tRNA elements in both lacertid species maintained through deletion-duplication events. The microRNAs and snoRNAs in the lacertids exhibited losses compared to A. carolinensis (Supplementary Figs. S7, S8). Even though the numbers of snoRNAs and miRNAs were almost identical, the members in each ncRNA class diversified between the two sister species. Repeat content also differed between L. viridis and L. bilineata, with the latter exhibiting a gain of long-terminal repeat elements (Supplementary Tables S4, S5).
Demographic history of divergence
Across all sites, mean heterozygosity was slightly lower in L. bilineata than in L. viridis (π = 0.0022 and 0.0029, respectively). dxy was around 0.0123, pairwise Fst between L. viridis and L. bilineata was 0.688.
We inferred past divergence and gene flow between the two lacertid species using a composite likelihood (CL) method based on the site frequency spectrum of short sequence blocks, i.e., blockwise site frequency spectrum (bSFS) [20, 52]. Since the likelihood calculation assumes no recombination within blocks and an infinite sites mutation model, we partitioned the genome into short (i.e., 200 bases) blocks. Our dataset consisted of 5,654,020 blocks, of which 46,825 were filtered out (0.83%) since they contained both fixed differences and shared heterozygous sites, thus violating the 4-gametes criterion under the assumption of no recombination within blocks.
In total, we have 2,785 distinct mutational configurations, i.e., the counts of the four entries of the folded joint site frequency spectrum (heterozygous sites unique to L. viridis and L. bilineata; heterozygous sites shared by both lacertids; fixed differences) in each block. A total of 1,965 bSFS configurations appeared more than once in the data (Supplementary Information 3).
We compared the Akaike information criterion (AIC) scores of 13 different demographic scenarios (Fig. 1; Table 1) given the pattern of bSFS between the two lacertid species (Supplementary file 3). However, this composite likelihood computation does not account for the correlation between adjacent blocks due to the physical linkage. To correct for this, we assumed that every 1,000th block is effectively unlinked (Supplementary Information 3, Section LD), i.e., statistically independent, and corrected lnCL scores by a factor of 1/1000.
Figure 1:

Thirteen different demographic scenarios were fitted. The models M1.1, M2.1, and M3.1 are strict divergence without gene flow; M1.2, M2.2, and M3.2 allow for post-divergence gene flow from L. viridis to L. bilineata; M1.3, M2.3, and M3.3 assume gene flow in the reverse direction, i.e., from L. bilineata to L. viridis. The models M4.2 and M5.2 allow for discrete admixture from L. viridis to L. bilineata and models M4.3 and M5.3 assume the admixture in the reverse direction (from L. bilineata to L. viridis). The effective population size is either assumed to be identical between both species and their ancestor (class M1.*) or one of the species has a different effective population size compared to the other species and ancestor (classes M2.*-5.*).
Table 1:
The ΔAIC of the best model (i.e., IM 2 B(x)−>V) compared to the other scenarios .
| Model ID | Model type | ΔAIC |
|---|---|---|
| M5.3 | ADM 2B(x)−>V | −47.2 |
| M3.2 | ADM 2V (x)−>B | −141 |
| M4.3 | ADM 2B−>V (x) | −47.2 |
| M3.2 | ADM 2V −>B(x) | −141 |
| M3.3 | IM 2B(x)−>V | 0 |
| M3.2 | IM 2V(x)−>B | −77.9 |
| M3.1 | DIV 2 b | −1,380 |
| M2.3 | IM 2B−>V (x) | −32.9 |
| M2.2 | IM 2V −>B(x) | −86.5 |
| M2.1 | DIV 2 | −1,140 |
| M1.3 | IM 1B−>V | −487 |
| M1.2 | IM 1V −>B | −128 |
| M1.1 | DIV 1 | −1,380 |
To account for the effect of physical linkage between blocks, we adjusted the AIC values of each model by only sampling every 1,000 blocks. The best model is highlighted in bold. ADM: isolation with discreet admixture; IM: isolation with migration; DIV: strict divergence without gene flow.
The best of the 13 models (M3.3) supports isolation between the two lacertid species with unidirectional gene flow from L. bilineata to L. viridis and fits significantly better than simpler scenarios such as divergence without gene flow (or just a single Ne parameter) or admixture (Table 1). This model (M3.3) also suggests a smaller effective population size of L. bilineata (Ne = 37,890) compared to its ancestor and L. viridis (Ne = 95,400) (Supplement SI-4; Supplementary Table S6) and a migration rate of M = 0.288 migrants per generation from L. bilineata to L. viridis (Supplementary Table S7).
Assuming a generation time of 3.5 years and a mutation rate of 1.77 × 10−8 (based on cyt-b gene) or 1 × 10−9 (based on the four-fold degenerate sites), our estimate of the split between L. viridis and L. bilineata corresponds to 1.15 Mya and 20.37 Mya, respectively (Supplementary Table S8).
Detection of genomic rearrangements
We detected 20,160 genomic rearrangements or SVs longer than 50 bp between the two lacertids (Fig. 2; Supplementary Table S9) covering 39.4 Mbp of the L. viridis genome (2.7% of the genome). Compared to L. bilineata, 10.8 Mbp (0.7%) of the L. viridis genome was covered with large rearrangements affecting genes (covering the entire length of more than one gene). These regions were enriched for RNA-directed DNA polymerase activity (22.46 fold-enrichment, P = 5.11e-03).
Figure 2:
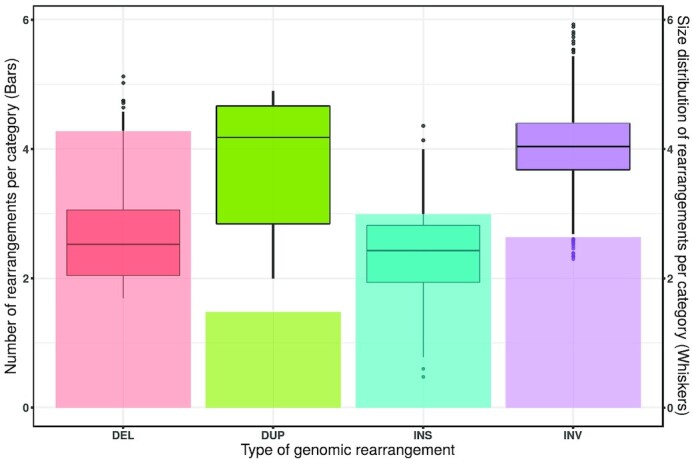
Total counts and length ranges (in bp) of genomic rearrangements of SVs between L. viridis and L. bilineata. The counts are represented by bars and length ranges by whiskers (y-axis is log10-scaled). The rearrangements plotted are categorized into deletions (DEL), duplications (DUP), insertions (INS), and inversions (INV).
Insertions-deletions (indels) are the most frequent genomic rearrangements mainly affecting introns, repeat elements, and pseudo-tRNAs (Supplement SI-5; Supplementary Table S10). This is similar to the observations made with respect to SVs in humans and pigs [12, 53]. Most SVs overlapping exons cover entire exons and do not result in frame shift mutations, with the exception of EXD2 and HERC2, suggesting that their functions can be complemented by other genes (Supplement SI-6).
Structural selection of ncRNAs
MicroRNAs (miRNA) were the most structurally conserved family of ncRNAs followed by small nucleolar RNAs (snoRNA) (Supplementary Fig. S9). The four types of ncRNAs and the number of groups in each category are shown in Supplementary Table S11 (Supplement SI-7). High levels of diversity were observed in tRNAs, especially in pseudo-tRNA, which was further supported by high copy numbers of tRNAs with a low conservation among tRNA orthologs between the two lacertid species.
All ncRNAs with low structural diversity across orthologs were computationally tested for sites with positive selection in either species. The positively selected snoRNA families belong to the H/ACA box class, which can introduce changes in post-translational mechanisms and pseudouridylation between the two species [54]. SNORD61 (small nucleolar RNA, C/D Box 61) (Supplementary Fig. S10a) was inferred to have evolved under positive selection in L. bilineata. The human ortholog of SNORD61 occurs in the intron of a RBMX (RNA binding motif protein, X-linked gene), known to be involved in the dosage compensation and cohesion regulation of sister chromatids [55]. Two microRNAs showed signs of positive selection in L. viridis: MIR6516 (mir-6516-3p) (Supplementary Fig. S10b), associated with urea synthesis in pigs [56], and MIR27 (mir-27a and mir27-d) (Supplementary Fig. S10c), known to play a role in regeneration and osteoblast differentiation in mice [57, 58]. However, mir-27d was absent in L. bilineata, so the structural divergence in the mir-27 family between the two lacertid species can be due to the presence of an additional MIR27 sequence in L. viridis. Two long intergenic non-coding (lincRNA) orthologs (LiNC66 and LiNC29) overlapping genomic regions conserved across tetrapods were structurally divergent between the two species, as indicated by high selection scores and stable secondary structures (Supplementary Fig. S10d-e).
Varying selection pressures in protein-coding genes
The visual opsins are pivotal for adaptation to diurnal habitats in Squamata [24, 59]. For instance, the nocturnal G. japonicus lost two of the five functional opsin paralogs compared to diurnal anoles [26]. All five paralogs of visual opsins in A. carolinensis (22 transcripts from ENSEMBL) were also present in L. viridis and L. bilineata (20 transcript sequences), indicating conservation of genes for diurnal vision. We observed high conservation of SWS1 (opsins related to UV vision), described to be involved in sexual selection [60, 61], and of the pigmentation protein MC1R, previously associated with adaptive coloration in sand lizards [62] (Supplement SI-8).
Genes involved in neuronal activity, behavior, auditory perception, and female reproductive system development were conserved in the lacertid ancestor, i.e., before the split between the two species (compared to five other vertebrates in the background). Genes with different selective constraints between the two species (i.e., differently influenced by purifying selection after their split) were related to brain and neural development, embryo and cartilage development, along with behavioral responses (Supplementary Table S12).
The test for positive selection in either of the two species was performed with the branch-site model of codeml (model M2) using a subset of other lizards as background branches. The number of genes with positively selected sites (PSS) in different foreground branches (L. viridis, L. bilineata, or the ancestor of L. viridis and L. bilineata) are shown in Supplementary Table S13 (Supplement SI-9). The predicted ontologies of genes with PSS in either of the two species indicate variation in growth and developmental processes, behavioral responses (temperature and pH), and transcriptional regulation (Supplementary Table S14). One of the genes with PSS in L. bilineata (STAR7) is located on the Z-chromosome. We identified two transcription factor genes, UBIP1 and RPA2, involved in gene silencing and reproductive functions [63, 64], with adaptive differences between the two species. Three genes with PSS overlapped inverted regions; GPR155 gene with PSS in L. bilineata, both TDRD3 and UGPA with PSS in L. viridis. GPR155 is involved in cognitive functions and expressed in mice forebrain [65], while TDRD3 is directly associated with oocyte formation and X-linked developmental disorders [66, 67]. Three genes, NASP, PDL11, and RTKN, were positively selected in the ancestor of the lacertid branch compared to background branches that include more distant classes such as mammals and birds (Supplement SI-9, Supplementary Table S15).
The prostacyclin synthase (PTGIS) involved in regeneration through prostaglandin synthesis is positively selected in A. carolinensis and G. japonicus [26]. This gene evolved under positive selection in the lacertid ancestor with A. carolinensis and G. japonicus as the background, hinting at evolutionary changes in regenerative mechanisms among lacertid lizards.
Diversification of UV-responsive genes
We identified three paralogs of the hyaluronidases (HYAL1, HYAL2, and HYAL4) in both the lacertid genomes. Two genes (STIK1 and HYAL2) coding for proteins in the extra-cellular matrix of the skin reacting to UV-B light (GO:0071493) [68] were positively selected in the ancestral branch of the two species, while the HYAL1 paralog was positively selected in the L. viridis branch (Supplement SI-8). Arylsulfatase gene (ARSB), which is involved in the chondroitin sulfate biosynthesis pathway along with HYAL, was also positively selected in L. viridis. Significant pathway enrichment of chondroitin sulfate biosynthesis was observed for PSGs in L. viridis (P = 2.6e-06, q = 1.3e-05).
Divergence of Kruppel-type zinc-finger proteins
To investigate the role of Krüppel-associated box (KRAB)-ZNFs in reproductive isolation of the two lacertid species, we compared the DNA-binding domains of KZNF orthologs. From the 53 KZNF orthologs, 6 C2H2 zinc-finger proteins showed binding-specific differences between the two lacertid species (Supplement SI-10). While the longest transcripts of these six KZNFs were assembled from ovarian tissues (Supplementary Table S16), they were also expressed in all the other tissues analyzed (brain, heart, liver, and kidneys).
Impact of rearrangements on sequence evolution
Deletions are the most frequent type of SVs in the genome and occurred on both positively selected genes and those with no signs of positive selection. Duplications and insertions only occurred in genes evolving without signs of positive selection, while deletions and inversions occurred in genes irrespective of their selective regime. The ratio between number of regions with rearrangements or SVs to those with no detected rearrangements was not significantly different between genes under positive selection and those with no signs of positive selection (Boschloo's exact test, two-sided; difference in proportion = 0.125, P = 0.06, q = 0.1). Since this can be due to abundant indels obscuring the association in other categories of SVs, we tested the association between each SV category with PSGs separately, applying independent Boschloo exact-tests (Supplementary Table S17). An association of PSGs within inversions when compared to other SV categories was observed, but this did not remain significant after multiple testing (P = 0.028, q = 0.06). We also observed a significant association of PSGs over genes with no signs of positive selection within inversions compared to both non-rearranged regions (P = 0.009, q = 0.03) and collinear regions (P = 0.006, q = 0.03). The inversions overlapping PSGs seem to reflect independent events, since the inversions are located on different contigs in the genome with size ranges between 70 kbp and 700 kbp.
Discussion
We provide high quality assembled genomes of two closely related lacertid species, L. viridis and L. bilineata, investigate their history of divergence, and analyze the patterns of genomic variation between these species.
The assembly contiguity was highest with partial error correction of PacBio reads (without splitting at chimeric junctions) followed by hybrid assembly through DBG2OLC implementing removal of chimeric joins. This hybrid assembly strategy aided in generating high-quality contig-level genomes with moderate genome coverages (∼35X Illumina and ∼15X PacBio). Our lacertid genome assemblies showed higher completeness than the available lizard genomes (Supplementary Table S1).
The time of population divergence between L. viridis and L. bilineata was estimated as at least 1.15 Mya (per generation), whereas the previously estimated mitochondrial divergence time was 2.6–3.4 Mya [33, 69]. Lacerta viridis and L. bilineata show a high level of genome-wide differentiation (FST = 0.688). The best demographic model (M3.3) supported unidirectional gene flow from L. bilineata to L. viridis and higher effective population size for L. viridis than L. bilineata, consistent with the difference in genetic diversity between the two lacertid species.
Species-specific diversity within various ncRNA classes and adaptive differences in ncRNA orthologs capable of altering their secondary structures are two important factors contributing to evolutionary divergence, since varying ncRNA structures imply functional changes [17]. Copy number variation and differences in the content of miRNA families hint at variability in gene regulatory networks between the lacertid sister species. Species-specific splicing mechanisms can be attributed to the losses of snoRNA families (SNORA17 and SNORA20) in L. bilineata and structural changes in SNORD61, which is involved in dosage compensation in humans [70].
Positive selection of sites in NASP and PDLIM1 in the lacertid ancestral branch compared to distant background branches, including mammals and birds, may indicate disparate evolutionary changes in the ancestor of L. viridis and L. bilineata with regard to reproductive processes, i.e., spermatogenesis, fertilization, and embryo implantation [71–74]. In contrast, positive selection acting on coding sites in just one lacertid species after their split suggests adaptive differences that could play a role in the speciation process [75–77].
UV-reflectance of plumages in birds is an important trait involved in the sexual selection of morphologically similar sibling species of Passeriformes [78]. Sexual selection in L. viridis has also been linked to UV-response. Males with more UV-reflective patches on the skin are preferably selected by the females [79, 80]. We show that hyaluronidases, known to be differentially expressed on exposure to UV-B in the skin of mice [68, 81, 82], evolved rapidly in L. viridis. We speculate that differential cutaneous response to UV through changes in the chondroitin sulfate biosynthesis pathway could be driven by mating preferences, which could ultimately contribute to speciation. Further studies are needed to test this hypothesis.
KRAB-ZNFs or KZNFs are transcriptional regulators confined to tetrapod vertebrates [83] and are known to play a role in species-specific changes in gene regulatory network through binding domain differences between humans and chimps [6, 84–87]. The divergence of transcription factors, especially differences in DNA-binding regions of KZNFs as observed here, could eventually have contributed to some degree of reproductive isolation between the two species, which should be further tested. This receives further support from adaptive differences in the transcription factors (UBIP1 and RPA2) crucial for spermatogenesis [63, 64]. Varying levels of purifying selection in genes influencing forebrain development and behavior suggest different selective constraints between L. viridis and L. bilineata. The behavioral differences can be related to varying ecological habitats and environmental conditions [35] after the split of L. viridis and L. bilineata. Selective differences in genes related to behavior and brain development have been reported to be involved in the diversification of anoles [32].
Genomic regions harboring inversions are known to suppress recombination in heterokaryotypes facilitating speciation in the presence of gene flow [88] and in maintaining favorable combinations of locally adapted alleles at different loci [89]. Genomic inversions between the two lacertid species are significantly associated with positively selected genes (PSGs). Two of the three PSGs occurring within inversions play a role in cognitive and reproductive functions (GPR155 and TDRD3), suggesting that they could be involved in speciation. However, it is currently unknown if these inversions represent fixed differences between the two species, and the lengths of these inversions is at a lower scale (less than 1 Mbp) than those known to play a role in adaptation and speciation [10]. Future studies should try to address these issues, as well as the role of these genes in reproductive isolation between L. viridis and L. bilineata.
Conclusions
We assembled the first high-quality genomes of two closely related species of European green lizards with a cost-effective strategy. Genes related to transcriptional regulation, behavior, and neural and reproductive development have diversified the most between the two lacertid species. Species-specific diversity of ncRNAs, adaptive evolution in regulatory elements, and transcription factors (including binding domain differences in KZNFs) indicate variation in gene regulatory networks between the two species. Adaptive evolution of genes responsible for differential cutaneous response to UV exposure, in particular, could be driven by mate choice and ultimately contribute to reproductive isolation. Altogether, we provide the first comprehensive study of the evolutionary history and genic, structural, and regulatory differences between the genomes of two closely related lacertid species. This comprises an important baseline for understanding the genomic regions and mechanisms involved in the speciation of European green lizards. In addition to a detailed analysis of the demographic history and evolutionary scenario of European green lizards, our study provides valuable resources that will help establish conservation guidelines for lacertids experiencing population declines due to habitat loss [90].
Materials and Methods
Sampling
Two adult females were sampled for this study, a L. viridis from Tokaj, northeastern Hungary (21.39775oE, 48.11363oN) (September 2013) and a L. bilineata from Mâlain, France (4o48’2.01”E, 47o21’16.27”N) (July 2014). There is no known morphological variation between the individuals of the two species (Supplementary Fig. S1). These represent two of the four main clades within the L. viridis complex [33, 34, 38, 91]. Animals were captured with permits of the issuing authorities (please refer to the Acknowledgements) and handled according to the guidelines of the Herpetological Animal Care and Use Committee of the American Society of Ichthyologists and Herpetologists. Tissues from the brain, heart, liver, kidney, and ovaries were dissected for tissue-specific transcriptome sequencing, and the remaining tissues were stored separately at –80°C.
Whole-genome and transcriptome sequencing
Tail tissue from each sample was digested with proteinase K, and genomic DNA was extracted using a chloroform-based method [92]. The whole genome was sequenced using both short (Illumina) and long read (PacBio) sequencing techniques. Short-read libraries with insert sizes of 380 bp and 450 bp were prepared for each individual separately. The Illumina paired-end sequences were double-indexed using a multiplexing sequencing protocol [93, 94] on a HiSeq2500. SMRTbell template library was prepared according to the instructions from PacBio (Menlo Park, CA), following the Procedure and Checklist—Greater Than 10 kb Template Preparation. Briefly, for preparation of 15 kb libraries 10 µg (L. bilineata) and 20 µg (L. viridis) genomic DNA was damage-repaired twice, end-repaired, and ligated overnight to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 from PacBio. Reactions were carried out according to the manufacturer's instructions. BluePippin Size-Selection to greater than 15 kb was performed according to the manufacturer's instructions (Sage Science, Beverly, MA). Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbell template were assessed with the Calculator in RS Remote, PacBio. Long-read sequencing was carried out for both genomes with 20 SMRT Cells applying P6-C4 chemistry on a PacBio RS-II sequencer. Average PacBio read lengths of 14 kb and 12 kb were retrieved for L. viridis and L. bilineata, respectively.
RNA from each tissue was extracted using Trizol Reagent (Life Technologies, Carlsbad, CA) and purified with the RNeasy Mini Kit (Qiagen, Hilden, Germany). The mRNA was purified using the Dynabeads mRNA Purification Kit (Life Technologies). The purity and concentration of RNA and cDNA were checked using Nanodrop and Bioanalyzer 2100 (Agilent Technologies, CA), and fragments of length 200–250 bp were obtained using Ambion RNA fragmentation reagent. The first and second strands of cDNA were synthesized using random hexamer primers with SuperScript II reverse transcriptase (Life Technologies) and DNA Polymerase I with RNase H treatment (Life Technologies) respectively.
Lacerta viridis was sequenced on a single lane for a more accurate estimate of genome size and repeat content. In order to avoid lane and run biases, sequencing was distributed over three lanes with all genomes and transcriptomes.
Non-coding RNA annotation and repeat analysis
Small ncRNAs were annotated on the genomic contigs by performing an infernal cmscan v1.1.1 (Infernal, RRID:SCR_011809) using the RFAM covariance models as input, and homologous ncRNA genes were filtered with a cutoff of 1e-06 [95, 96].
Additionally, ncRNA class-specific annotation methods were used for tRNAs, snoRNAs, and miRNAs. tRNAs were annotated using tRNAscan-SE v1.3.1 software (tRNAscan-SE, RRID:SCR_010835) with default parameters [97]. The Basic Local Alignment Search Tool (BLAST)-based snoStrip pipeline [98] was used to annotate snoRNAs. A comprehensive set of snoRNAs from vertebrates and aves were used as query set [99]. To detect miRNAs, the avian set of miRNAs were used as query sequences for a BLAST search in the lizard genomes. All resulting blast hits were filtered for the conservation of the seed region. The annotated snoRNAs and miRNAs in lacertids were validated by blast searches against this reference database, and mature miRNA sequence homologies were used. In the case of overlapping miRNA and snoRNA annotations, both were retained as it is known that snoRNAs can be processed into small derived RNAs from miRNA-like RNAs [100]. Putative lincRNAs were predicted based on the transcripts with no coding potential as assessed by Transdecoder of the Trinity v2.6.5 suite (Trinity, RRID:SCR_013048) [101] and mapping on their respective genome without chimeric paths. Furthermore, only the conserved lincRNAs with one-to-one orthologs between lacertids were retained.
For comparison, ncRNA families (except lincRNA) were also annotated in other selected sauropsid genomes. A reference database was created using sequenced and annotated genomes from reptiles, aves, and other vertebrates. The program ePoPe [102] was used to understand the evolution of snoRNAs and miRNAs in the lacertids through the construction of phylogenetic trees based on the gains and losses of ncRNA families.
The RepeatModeler v1.0.4 pipeline (RepeatModeler, RRID:SCR_015027) [103] was used to predict repeats in the genomes of lacertids. The predicted repeat families were used as initial libraries for de novo annotation of repeats using RepeatMasker v4.0.5 (RepeatMasker, RRID:SCR_012954) [104]. The evolution of these repeats was investigated using the repeat library available for tetrapod species (Database: 20140131).
Population histories, gene flow, and coalescence
To assess the demographic history between L. viridis and L. bilineata, we used the blockwise composite likelihood approach. We analytically computed the probabilities of mutational configurations in blocks of fixed length using the bSFS framework [20].
We mapped the illumina reads from L. viridis and L. bilineata to the L. viridis reference genome with BWA MEM v0.7.12-r1039 software [105]. The homozygosity/heterozygosity of each site in both lacertids was predicted based on the reference genome with FreeBayes v9.9.13 (FreeBayes, RRID:SCR_010761) [106] with a minimum read support of five and minimum allele frequency of 0.2. The intergenic regions of the genome were chopped into blocks of length 200 bp; this resulted in 5,654,020 blocks in total. The number of four mutation types defined by the joint SFS (Fig. 3) were counted using Heffalump query ([107] commit 7773784). In total, 2,785 distinct mutational configurations were obtained, of which 1,965 appeared more than once. We then summarized the frequency of each polymorphism pattern across all blocks [108, 109]. This data summary is referred to as distribution of bSFS.
Figure 3:

The folded blockwise site frequency spectrum (bSFS). The variation in alleles represented by different colors (the ancestral state showed in red). Given a single genealogy (a diploid genome from two populations can form six possible genealogies), each block contains four mutation types: (i) unique heterozygous sites in L. bilineata, (ii) unique heterozygous sites in L. viridis, (iii) shared heterozygous sites between L. viridis and L. bilineata, or (iv) homozygous sites that are different between L. viridis and L. bilineata, i.e., homozygous fixed differences. The bSFS (spectrum of SFS) has been calculated by counting the number of occurrences of each SFS.
Blocks containing both fixed differences and shared heterozygous sites violate the four-gametes criterion, and 46,825 blocks (0.83%) were removed under the assumption of no recombination within blocks. To account for physical linkage between adjacent blocks, we assumed that every 1000th block is effectively unlinked, i.e., statistically independent, and corrected lnCL scores by a factor of 1/1000. We fitted 13 different demographic scenarios (Fig. 2) accounting for the presence or absence of gene flow, direction of gene flow, continuous or discrete migration, and changes in effective population sizes. Models were compared using the AIC of their composite log-likelihoods.
We estimated the generation length based on the mean age of the mothers of all offspring [110] given the age structure data by Elbing [111] and Saint Girons et al. [112] for three German populations of L. viridis and two French populations of L. bilineata, respectively. In captivity, females that breed for the first time lay on average 8.5 eggs, whereas older females lay 11.1 eggs [113]. Given this data, we estimated a mean generation length of 3.6 and 2.9 years for L. viridis and L. bilineata, respectively. We therefore assumed a generation time of about 3–4 years for both species. To scale our result in real time, we used a mutation rate of 1 × 10−9 per site per generation based on the four-fold degenerate sites of the single-copy gene orthologs between L. viridis and L. bilineata (Supplementary Methods SM-4). The lower limit of the mutation rate was assumed as 1.77 × 10−8 per site per generation (3.5 years as the generation time) based on the pairwise distance of 6.19% in the cytochrome b gene between L. viridis and L. bilineata [34]. This assumption is similar to the mutation rate of NADH-2 in A. carolinensis (1.3% mutations per million years) [114].
Detection of genomic rearrangements from read-based pipelines and syntenic blocks
Genomic rearrangements between the lacertids were detected based on both read-based methods and syntenic blocks information. Lacerta viridis was used as the reference genome since the assembly was more contiguous for this species. Genomic reads from L. bilineata were used as the query, and the reads of L. viridis mapped against the reference were used as control.
Read-based pipelines
Genomic rearrangements were detected between lacertids using read mapping-based methods for Illumina paired-end reads and for PacBio-reads separately, followed by SV callers specifically developed to deal with short and long read sequences, respectively. In both approaches, reads of L. bilineata (query) and of L. viridis (control) were separately mapped against the same reference (L. viridis).
The alignment of Illumina reads was carried out with BWA MEM v0.7.12 [105], and rearrangements were detected with MetaSV v0.5.2 pipeline [115], which uses BREAKDANCER v1.1.2 (BREAKDANCER, RRID:SCR_001799) [116] to infer SVs using paired-end read information, CNVnator v0.3.1 (CNVnator, RRID:SCR_010821) [117] to predict copy-number variants from abnormal read-coverages and Pindel v0.2.4 (Pindel, RRID:SCR_000560) [118] to detect large SV-related breakpoint events. The insert size was estimated as 400±50 from 1 million observations based on the alignment of paired-end Illumina reads. A minimum support of five reads and mapping quality of 30 was set as the threshold to support SVs from BREAKDANCER. A bin size of 500 was used to run CNVnator, and only precise SV events were called. While for Pindel, only variants with minimum read support of five paired-reads were used. MetaSV pipeline was used to merge the SVs from these three different SV callers, and local de novo assemblies were constructed using the ABYSS assembler for insertions. In order to maintain a high level of sensitivity and specificity (>90%) in the detection of SVs, only the rearrangements called with a minimum support of eight uniquely mapped paired-end reads were used for further analyses [119].
The PacBio reads were aligned to the reference with NGMLR v0.2.1, and the alignment was fed to Sniffles v1.0.3 SV-caller [11] to call variants with a minimum support of seven reads (at least half of the PacBio genome coverage of 14X).
Syntenic blocks approach
In addition to read-based methods, rearrangements were also detected from the blocks of synteny obtained through the UCSC pipeline [120]. The alignments were converted to single-coverage genomes using single_cov2 of the MultiZ pipeline [121] to avoid spurious assignments. Strand changes within syntenic blocks were clustered as inversions (I) based on the orientation of the successive (I+1) and preceding (I-1) blocks. Regions with missing bases in the query alone were predicted to be deletions, while gaps in the reference genome alone were considered as insertions. Additionally, hierarchical alignment (HAL) format [122] of the single-coverage genomes was used to predict rearrangements with the halBranchMutations tool. This tool generates annotations for the location of rearrangements based on the branch of interest in the HAL file (between L. viridis and L. bilineata in our case). The events detected in both directions, i.e., L. viridis reference with L. bilineata as query and L. bilineata reference and L. viridis as query were retained. The length threshold was set to 50 bp, and the predicted rearrangements were filtered based on quality to reduce false positives (Supplement SM-7).
Segmental duplications in the two lacertid species were detected by self-aligning the two genomes separately with chained LASTZ [123] (step = 9, H = 3000, K = 5000). High identity matches (90% identity) within each genome of 1 kb or more were defined as segmental duplications.
Structural selection in non-coding RNAs
The predicted ncRNAs (miRNA, snoRNA, tRNA, and lincRNA) in lacertids were tested for structural selection (selection of sites acting on secondary structure in either of the lacertids) with Gekko japonicus as outgroup. We used the Selection on the Secondary Structure (SSS) test [124], a statistical test that assigns selection scores for each given sequence based on the comparison between the structure of the given sequence and the structure of group consensus. It also provides a diversity value for the family that indicates its structural conservation. The diversity value (d-score) is the family's median base-pair distance to its consensus. The miRNAs, snoRNAs, and tRNAs were divided into sub-groups based on their families or their anti-codon sequences, and only those sub-groups with at least three sequences were tested. The groups that exhibited high structural diversity (median base pair distance to the consensus, d ≥10.0) were excluded from further analyses.
A ncRNA structural test to detect positively selected structures is only appropriate for structurally conserved groups. Low d-score values (d <10.0) were used to distinguish conservation chosen based on structural uniformity of the groups. This cutoff was based on the visual inspection of the secondary structures of families with d-scores of 1 to 20. Secondary structures of ncRNA sequences were predicted using RNAfold [125]. In a similar fashion, structures with selection scores of 0 to 30 were visually compared to the structure of their group consensus. High selection scores (s ≥10.0) were used to predict the positively selected sequences of small ncRNAs. Secondary structures with high selection scores were manually inspected to remove false positives. Specifically, the candidates with structures of low stability or those fundamentally dissimilar to the family consensus indicating loss of function were excluded.
The selection test was adapted for lincRNAs and performed only on the two lacertid species without any outgroup since lincRNA annotations of other closely related species were unavailable. Since the positive selection of secondary structure cannot be determined without outgroups, we instead detected divergence of lincRNA structure within the lacertids. Local conserved structure blocks were predicted for the orthologous lincRNA families, and these blocks were subjected to an adaptation of SSS test based on local structures. The structural selection for lincRNAs was assessed locally, since most base-pairings occur between nucleotides within a short distance [124, 126]. Local blocks of high structural diversity were excluded from further analysis. Since outgroups were not used for lincRNAs, a lower selection score threshold (s ≥4.0) was applied to detect divergent candidates that were visually inspected later to exclude false positives.
Ortholog prediction and selection tests
In order to investigate the selection pressure in the lacertid branch (ancestor of L. viridis and L. bilineata) compared to other vertebrates, the CDS of five species, namely, anole lizard (Anolis carolinensis), chicken (Gallus gallus), frog (Xenopus tropicalis), spotted garfish (Lepisosteus oculatus), and human (Homo sapiens) were downloaded from the Ensembl database version 83 (Ensembl, RRID:SCR_002344) [127]. To keep the data consistent and avoid re-annotations, the CDS annotations were also extracted from the Ensembl database. The orthologs between the protein-coding sequences of the species were identified with ProteinOrtho V5 using the synteny option to reduce false orthologs assignments. The output was converted to run the POTION pipeline [128], which tests for selection acting on protein coding genes. Only the single-copy orthologs in each species were retained for each orthologous group.
The protein identity filtering in POTION was set to 30% in each orthologous group and sequence size limits to more than 10 times or less than 0.2 of the median size in the group. Only groups with at least four species were retained. The sequences in each orthologous group (after filtering paralogs) were aligned and gap trimmed; phylogenetic trees were constructed, and groups with recombinants were excluded from the selection tests. The intermediates files from the POTION pipeline were used to generate unrooted trees with lacertids (L. viridis and L. bilineata) in the foreground branches. The remaining species were used as the background to test for positive selection using the branch-site model of codeml within the PAML v4.8 package (PAML, RRID:SCR_014932) [129]. A likelihood ratio test based on χ2 distribution was used to detect genes with significant positive selection followed by multiple testing through the Benjamini–Hochberg procedure. Genes with P < 0.05 and q <0.05 were retained and referred to as being positively selected in the lacertid branch.
To detect adaptive evolution through positive selection within either lacertids, additional tests (PAML branch-site models) were performed with less distant outgroups using a set of five lizard species, namely, L. viridis, L. bilineata, Anolis carolinensis, Gekko japonicus, and Pogona vitticeps. The single-copy orthologs were identified with ProteinOrtho with a minimum protein identity of 70%, e-value of 1e-06, and minimum similarity of 0.99 for additional hits. The orthologous coding sequences from the five lizard species were aligned with MACSE while accounting for frame-shifts, and the stop codon at the end of the sequence was removed. Unrooted trees were generated with three different foreground branches: (i) lacertids (L. viridis and L. bilineata), (ii) L. viridis alone, and (iii) L. bilineata alone. The rest of the workflow for detection of recombinants, removal of gaps, and codeml tests was similar to the POTION pipeline followed by filtering for significant candidate genes (P < 0.05, q < 0.05). In order to avoid false predictions of PSS at the beginning or toward the end of alignments, where mismatches were allowed, the candidate genes predicted to contain PSS in either species were visually inspected.
Availability of supporting data
The assembled genomes, their annotations, transcript data, variant calls (variant call formats), snapshots of the code, and other supporting datasets are available in Zenodo [130] and in the GigaScience GigaDB [131] repositories.
Additional files
Additional file 1: This supplement contains methods SM1-SM11, information SI1-SI11, Figures S1–S10, Tables S1–S17 and References.
Additional file 2: The figure for the contig clusters in lacertids generated from synteny information between L. viridis and L. bilineata.
Additional file 3: Mathematica notebook containing the code used and other supporting information from the demography analysis of L. viridis and L. bilineata.
Abbreviations
AIC: Akaike information criterion; BDMI: Bateson-Dobzhansky-Muller incompatibility; BLAST: Basic Local Alignment Search Tool; bSFS: blockwise site frequency spectrum; CDS: coding sequence; CL: composite likelihood; HAL: hierarchical alignment; HYAL: hyaluronidase; indels: insertions and deletions; KRAB: Krüppel-associated box; KZNF: Krüppel-type zinc finger; lincRNA: long intergenic non-coding RNA; miRNA: microRNA; Mya: Million years ago; NCBI: National Center for Biotechnology information; ncRNA: non-coding RNA; PacBio: Pacific Biosciences; PSG: positively selected gene; PSS: positively selected site; RBMX: RNA binding motif protein, X-linked gene; rRNA: ribosomal RNA; SCO: single-copy ortholog; SNORD61: small nucleolar RNA, C/D Box 61; SFS: site frequency spectrum; snoRNA: small nucleolar RNA; SSS: Selection on the Secondary Structure; SV: structural variants; tRNA: transfer RNA.
Competing interests
The authors declare that they have no competing interests. The authors alone are responsible for the content and writing of the paper.
Funding
This project was funded by the Deutsche Forschungsgemeinschaft (FZT 118, SFB 1052 and SPP 1738).
Author contributions
K.H. and A.G. collected the samples; A.W. Illumina sequenced the genome and transcriptome; C.S., B.B., and J.O. were involved in the PacBio genome sequencing; S.R.R.K. performed the assembly and annotation; S.K. and S.R.R.K. annotated the non-coding RNA; M.B.W.C. analyzed selection in non-coding RNA; S.R.R.K. and H.I. performed the positive selection analysis; S.R.R.K. and M.C. analyzed structural variants; A.S. and K.L. analyzed the population histories; S.R.R.K., R.F., K.N., P.F.S., and M.S. wrote the initial draft of the manuscript; H.I., M.C., A.S., K.L., S.K., M.B. W.C., J.O., B.B., C.B., and K.H. edited the manuscript; R.F., K.N., K.H., P.F.S., and M.S. conceived the study.
Supplementary Material
6/20/2018 Reviewed
6/22/2018 Reviewed
10/28/2018 Reviewed
ACKNOWLEDGEMENTS
The authors thank the reviewers, curators, and the journal editors for their constructive criticisms on our manuscript. We gratefully acknowledge the support of the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig. We also thank Conrad Helm, Detlef Bernhard, Michael Gerth, Ronny Wolf, and Stefan Schaffer for their help in the laboratory; Steve Hoffmann and Thomas Hackl for valuable discussions; and Stephane Peyrégne for helpful suggestions and proofreading. The specimen from Hungary was kindly collected and provided by the team of Szabolcs Lengyel, Centre for Ecological Research, Hungarian Academy of Science. Capture permit (no. 13778-7/2013) was issued by the North Hungarian Environmental Protection, Nature Conservation and Water Management Inspectorate. Capture permit (no. 36) for the French specimen was issued by the Préfet de la Côte d'Or. P.F.S and S.K. thank the DFG for funding under the project SFB 1052 "Obesity Mechanisms" (project number: 209933828). P.F.S, K.N and M.B.W.C thank the DFG for funding under the grant SPP 1738 (project numbers: STA 850/19-1 and STA 920/6-1). K.N. and R.F. thank the Volkswagen Foundation for funding within the framework “Support for Europe.” R.F. was financed by FCT (grant SFRH/BPD/89313/2012) and is currently funded by the European Union's Horizon 2020 Research and Innovation Programme, under the Marie Sklodowska-Curie grant agreement 706376. KL is supported by an Independent Research fellowship from the Natural Environmental Research Council (NERC) UK (NE/L011522/1).
References
- 1. Coyne JA, Orr HA. Speciation, Sunderland, Massachussets, US, Sinauer Associates, 2004. [Google Scholar]
- 2. Presgraves DC. The molecular evolutionary basis of species formation. Nat Rev Genet. 2010;11(3):175–80. [DOI] [PubMed] [Google Scholar]
- 3. Charlesworth B, Coyne JA, Barton NH. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 1987;130(1):113–46. [Google Scholar]
- 4. Masly JP, Presgraves DC. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 2007;5(9):e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mack KL, Campbell P, Nachman MW. Gene regulation and speciation in house mice. Genome Res. 2016;26(4):451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nowick K, Carneiro M, Faria R. A prominent role of KRAB-ZNF transcription factors in mammalian speciation?. Trends Genet. 2013;29(3):130–9. [DOI] [PubMed] [Google Scholar]
- 7. Ortiz-Barrientos D, Kane NC. Meeting review: American Genetics Association Symposium on the genetics of speciation. Mol Ecol. 2007;16(14):2852–4. [DOI] [PubMed] [Google Scholar]
- 8. Ravinet M, Faria R, Butlin RKet al. . Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J Evol Biol. 2017;30(8):1450–77. [DOI] [PubMed] [Google Scholar]
- 9. Seehausen O, Butlin RK, Keller I, et al. . Genomics and the origin of species. Nat Rev Genet. 2014;15(3):176–92. [DOI] [PubMed] [Google Scholar]
- 10. Wellenreuther M, Bernatchez L. Eco-evolutionary genomics of chromosomal inversions. Trends in Ecology & Evolution. 2018;33(6):427–40. [DOI] [PubMed] [Google Scholar]
- 11. Sedlazeck FJ, Rescheneder P, Smolka Met al. . Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sudmant PH, Rausch T, Gardner EJ, et al. . An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davey JW, Barker SL, Rastas PMet al. . No evidence for maintenance of a sympatric Heliconius species barrier by chromosomal inversions. Evolution Letters. 2017;1(3):138–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walter Costa MB, Honer Zu Siederdissen C, Tulpan D, et al. . Temporal ordering of substitutions in RNA evolution: uncovering the structural evolution of the human accelerated region 1. J Theor Biol. 2017;438:143–50. [DOI] [PubMed] [Google Scholar]
- 16. Frías-Lasserre D, Villagra CA. The importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Frontiers in Microbiology. 2017;8:2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhartiya D, Scaria V. Genomic variations in non-coding RNAs: structure, function and regulation. Genomics. 2016;107(2-3):59–68. [DOI] [PubMed] [Google Scholar]
- 18. Gardner PP, Fasold M, Burge SWet al. . Conservation and losses of non-coding RNAs in avian genomes. PLoS One. 2015;10(3):e0121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sousa V, Hey J. Understanding the O\origin of species with genome-scale data: the role of gene flow. Nat Rev Genet. 2013;14(6):404–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lohse K, Chmelik M, Martin SH, et al. . Efficient strategies for calculating blockwise likelihoods under the coalescent. Genetics. 2016;202(2):775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Camargo A, Sinervo B, Sites JW Jr. Lizards as model organisms for linking phylogeographic and speciation studies. Mol Ecol. 2010;19(16):3250–70. [DOI] [PubMed] [Google Scholar]
- 22. Thorpe RS, Surget-Groba Y, Johansson H. The relative importance of ecology and geographic isolation for speciation in anoles. Philosophical Transactions of the Royal Society B: Biological Sciences. 2008;363(1506):3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alfoldi J, Di Palma F, Grabherr M, et al. . The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477(7366):587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao J, Li Q, Wang Zet al. . Sequencing, de novo assembling, and annotating the genome of the endangered Chinese crocodile lizard Shinisaurus crocodilurus. GigaScience. 2017;6(7):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rice ES, Kohno S, John JSet al. . Improved genome assembly of American alligator genome reveals conserved architecture of estrogen signaling. Genome Res. 2017;27(5):686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y, Zhou Q, Wang Yet al. . Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nature Communications. 2015;6:10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Georges A, Li Q, Lian J, et al. . High-coverage sequencing and annotated assembly of the genome of the Australian dragon lizard Pogona vitticeps. GigaScience. 2015;4(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Castoe T, Koning A, Hall K, et al. . The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A. 2013;110:20645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vonk F, Casewell N, Henkel Cet al. . The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A. 2013;110:20651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaffer H, Minx P, Warren D, et al. . The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Z, Pascual-Anaya J, Zadissa Aet al. . The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet. 2013;45:701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tollis M, Hutchins ED, Stapley J, et al. . Comparative genomics reveals accelerated evolution in conserved pathways during the diversification of anole lizards. Genome Biology and Evolution. 2018;10(2):489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Böhme MU, Fritz U, Kotenko T, et al. . Phylogeography and cryptic variation within the Lacerta viridis complex (Lacertidae, Reptilia). Zoologica Scripta. 2007;36(2):119–31. [Google Scholar]
- 34. Marzahn E, Mayer W, Joger U, et al. . Phylogeography of the Lacerta viridis complex: mitochondrial and nuclear markers provide taxonomic insights. Journal of Zoological Systematics and Evolutionary Research. 2016;54(2):85–105. [Google Scholar]
- 35. Tvrtkovi N, Lazar B, Tome Set al. . The western green lizard Lacerta (viridis) bilineata Daudin, 1804 (Sauria: Lacertidae) in Slovenia and Croatia. Natura Croatica. 1998;7(4):363–9. [Google Scholar]
- 36. Amann T, Rykena S, Joger U, et al. . Zur artlichen Trennung von Lacerta bilineata DAUDIN, 1802 und L. viridis (LAURENTI, 1768). Salamandra. 1997;33(4):255–68. [Google Scholar]
- 37. Joger U, Amann T, Veith M. Phylogeographie und genetische differenzierung im Lacerta viridis/bilineata komplex. Mertensiella. 2001;13:60–68. [Google Scholar]
- 38. Rykena S. Kreuzungsexperimente zur Prüfung der Artgrenzen im Genus Lacerta sensu stricto. Zoosystematics And Evolution. 1991;67(1):55–68. [Google Scholar]
- 39. Noor MA, Grams KL, Bertucci LA, et al. . Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci U S A. 2001;98(21):12084–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Navarro A, Barton NH. Chromosomal speciation and molecular divergence–accelerated evolution in rearranged chromosomes. Science. 2003;300(5617):321–4. [DOI] [PubMed] [Google Scholar]
- 41. Faria R, Navarro A. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends Ecol Evol. 2010;25(11):660–9. [DOI] [PubMed] [Google Scholar]
- 42. Olmo E. Rate of chromosome changes and speciation in reptiles. Genetica. 2005;125(2-3):185–203. [DOI] [PubMed] [Google Scholar]
- 43. Olmo E, Odierna G, Cobror O. C-band variability and phylogeny of Lacertidae. Genetica. 1986;71:63–74. [Google Scholar]
- 44. Kupriyanova L, Kuksin A, Odierna G. Karyotype, chromosome structure, reproductive modalities of three southern Eurasian populations of the common lacertid lizard, Zootoca vivipara (Jacquin, 1787). Acta Herpetologica. 2008;3(2):2008. [Google Scholar]
- 45. Liu H, Chang LH, Sun Y, et al. . Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol Evol. 2014;6(3):510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smeds L, Qvarnström A, Ellegren H. Direct estimate of the rate of germline mutation in a bird. Genome Res. 2016;26(9):1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nam K, Mugal C, Nabholz Bet al. . Molecular evolution of genes in avian genomes. Genome Biol. 2010;11(6):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chiarelli AB. Cytotaxonomy and Vertebrate Evolution, 1973. Capanna Eand Chiarelli AB. Academic Press, Cambridge, Massachusetts, US. [Google Scholar]
- 49. Townsend TM, Mulcahy DG, Noonan BPet al. . Phylogeny of iguanian lizards inferred from 29 nuclear loci, and a comparison of concatenated and species-tree approaches for an ancient, rapid radiation. Mol Phylogenet Evol. 2011;61(2):363–80. [DOI] [PubMed] [Google Scholar]
- 50. Deakin JE, Edwards MJ, Patel H, et al. . Anchoring genome sequence to chromosomes of the central bearded dragon (Pogona vitticeps) enables reconstruction of ancestral squamate macrochromosomes and identifies sequence content of the Z chromosome. BMC Genomics. 2016;17:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bermudez-Santana C, Attolini CS-O, Kirsten T, et al. . Genomic organization of eukaryotic tRNAs. BMC Genomics. 2010;11(1):270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bunnefeld L, Frantz LA, Lohse K. Inferring bottlenecks from genome-wide samples of short sequence blocks. Genetics. 2015;201(3):1157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao P, Li J, Kang H, et al. . Structural variant detection by large-scale sequencing reveals new evolutionary evidence on breed divergence between Chinese and European pigs. Sci Rep. 2016;6:18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kiss T, Fayet-Lebaron E, Jády BE. Box H/ACA small ribonucleoproteins. Mol Cell. 2010;37(5):597–606. [DOI] [PubMed] [Google Scholar]
- 55. Matsunaga S, Takata H, Morimoto A, et al. . RBMX: a regulator for maintenance and centromeric protection of sister chromatid cohesion. Cell Rep. 2012;1(4):299–308. [DOI] [PubMed] [Google Scholar]
- 56. Sun R-P, Xi Q-Y, Sun J-J, et al. . In low protein diets, microRNA-19b regulates urea synthesis by targeting SIRT5. Sci Rep. 2016;6:33291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang T, Xu Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem Biophys Res Commun. 2010;402(2):186–9. [DOI] [PubMed] [Google Scholar]
- 58. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346(6205):1248012. [DOI] [PubMed] [Google Scholar]
- 59. Ricklefs RE, Losos JB, Townsend TM. Evolutionary diversification of clades of squamate reptiles. J Evol Biol. 2007;20(5):1751–62. [DOI] [PubMed] [Google Scholar]
- 60. van Hazel I, Sabouhanian A, Day L, et al. . Functional characterization of spectral tuning mechanisms in the great bowerbird short-wavelength sensitive visual pigment (SWS1), and the origins of UV/violet vision in passerines and parrots. BMC Evol Biol. 2013;13:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Lanuza GPi, Font E. Ultraviolet vision in lacertid lizards: evidence from retinal structure, eye transmittance, SWS1 visual pigment genes and behaviour. J Exp Biol. 2014;217(16):2899. [DOI] [PubMed] [Google Scholar]
- 62. Laurent S, Pfeifer SP, Settles ML, et al. . The population genomics of rapid adaptation: disentangling signatures of selection and demography in white sands lizards. Mol Ecol. 2016;25(1):306–23. [DOI] [PubMed] [Google Scholar]
- 63. Kim JS, Chae JH, Cheon YP, et al. . Reciprocal localization of transcription factors YY1 and CP2c in spermatogonial stem cells and their putative roles during spermatogenesis. Acta Histochem. 2016;118(7):685–92. [DOI] [PubMed] [Google Scholar]
- 64. Elmayan T, Proux F, Vaucheret H. Arabidopsis RPA2: a genetic link among transcriptional gene silencing, DNA repair, and DNA replication. Curr Biol. 2005;15(21):1919–25. [DOI] [PubMed] [Google Scholar]
- 65. Trifonov S, Houtani T, Shimizu J-I, et al. . GPR155: gene organization, multiple mRNA splice variants and expression in mouse central nervous system. Biochem Biophys Res Commun. 2010;398(1):19–25. [DOI] [PubMed] [Google Scholar]
- 66. Laisk-Podar T, Kaart T, Peters Met al. . Genetic variants associated with female reproductive ageing – potential markers for assessing ovarian function and ovarian stimulation outcome. Reprod Biomed Online. 2015;31(2):199–209. [DOI] [PubMed] [Google Scholar]
- 67. Linder B, Plöttner O, Kroiss Met al. . Tdrd3 is a novel stress granule-associated protein interacting with the fragile-X syndrome protein FMRP. Hum Mol Genet. 2008;17(20):3236–46. [DOI] [PubMed] [Google Scholar]
- 68. Tobiishi M, Sayo T, Yoshida Het al. . Changes in epidermal hyaluronan metabolism following UVB irradiation. J Dermatol Sci. 2011;64(1):31–38. [DOI] [PubMed] [Google Scholar]
- 69. Kolora SR, Faria R, Weigert A, et al. . The complete mitochondrial genome of Lacerta bilineata and comparison with its closely related congener L. viridis. Mitochondrial DNA Part A. 2017;28(1):116–8. [DOI] [PubMed] [Google Scholar]
- 70. Sperveslage J, Hoffmeister M, Henopp T, et al. . Establishment of robust controls for the normalization of miRNA expression in neuroendocrine tumors of the ileum and pancreas. Endocrine. 2014;46(2):226–30. [DOI] [PubMed] [Google Scholar]
- 71. Finn RM, Ellard K, Eirin-Lopez JM, et al. . Vertebrate nucleoplasmin and NASP: egg histone storage proteins with multiple chaperone activities. FASEB J. 2012;26(12):4788–804. [DOI] [PubMed] [Google Scholar]
- 72. Nagatomo H, Kohri N, Akizawa H, et al. . Requirement for nuclear autoantigenic sperm protein mRNA expression in bovine preimplantation development. Anim Sci J. 2016;87(3):457–61. [DOI] [PubMed] [Google Scholar]
- 73. Shang Y, Wang H, Jia P, et al. . Autophagy regulates spermatid differentiation via degradation of PDLIM1. Autophagy. 2016;12(9):1575–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yin J, Ni B, Tian ZQ, et al. . Regulatory effects of autophagy on spermatogenesis. Biol Reprod. 2017;96(3):525–30. [DOI] [PubMed] [Google Scholar]
- 75. Chen J, Shishkin AA, Zhu X, et al. . Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wiberg RA, Halligan DL, Ness RW, et al. . Assessing recent selection and functionality at long noncoding RNA loci in the mouse genome. Genome Biol Evol. 2015;7(8):2432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhen Y, Andolfatto P. Methods to detect selection on noncoding DNA. Methods Mol Biol. 2012;856:141–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bleiweiss R. Ultraviolet plumage reflectance distinguishes sibling bird species. PNAS. 2004;101(47):16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bajer K, Molnár O, Török J, et al. . Female European green lizards (Lacerta viridis) prefer males with high ultraviolet throat reflectance. Behav Ecol Sociobiol. 2010;64(12):2007–14. [Google Scholar]
- 80. Bajer K, Molnár O, Török Jet al. . Ultraviolet nuptial colour determines fight success in male European green lizards (Lacerta viridis). Biol Lett. 2011;7(6):866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Averbeck M, Gebhardt CA, Voigt S, et al. . Differential regulation of hyaluronan metabolism in the epidermal and dermal compartments of human skin by UVB irradiation. J Invest Dermatol. 2007;127(3):687–97. [DOI] [PubMed] [Google Scholar]
- 82. Kurdykowski S, Mine S, Bardey V, et al. . Ultraviolet-B irradiation induces differential regulations of hyaluronidase expression and activity in normal human keratinocytes. Photochem Photobiol. 2011;87(5):1105–12. [DOI] [PubMed] [Google Scholar]
- 83. Urrutia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003;4:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212. [DOI] [PubMed] [Google Scholar]
- 85. Englbrecht CC, Schoof H, Böhm S. Conservation, diversification and expansion of C2H2 zinc finger proteins in the Arabidopsis thaliana genome. BMC Genomics. 2004;5(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Emerson RO, Thomas JH. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009;5(1):e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nowick K, Fields C, Gernat Tet al. . Gain, loss and divergence in primate zinc-finger genes: a rich resource for evolution of gene regulatory differences between species. PLoS One. 2011;6(6):e21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rieseberg LH. Chromosomal rearrangements and speciation. Trends in Ecology & Evolution. 2001;16(7):351–8. [DOI] [PubMed] [Google Scholar]
- 89. Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173(1):419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Böhme MU, Schneeweiß N, Fritz U, et al. . Small edge populations at risk: genetic diversity of the green lizard (Lacerta viridis viridis) in Germany and implications for conservation management. Conservation Genetics. 2007;8(3):555–63. [Google Scholar]
- 91. Joger U, Fritz U, Guicking D, et al. . Phylogeography of western Palaearctic reptiles – Spatial and temporal speciation patterns. Zoologischer Anzeiger - A Journal of Comparative Zoology. 2007;246(4):293–313. [Google Scholar]
- 92. Weigert A, Helm C, Meyer Met al. . Illuminating the base of the annelid tree using transcriptomics. Mol Biol Evol. 2014;31(6):1391–401. [DOI] [PubMed] [Google Scholar]
- 93. Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40(1):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010(6):pdb prot5448. [DOI] [PubMed] [Google Scholar]
- 95. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nawrocki EP, Burge SW, Bateman A, et al. . Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(Database issue):D130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bartschat S, Kehr S, Tafer H, et al. . snoStrip: a snoRNA annotation pipeline. Bioinformatics. 2014;30(1):115–6. [DOI] [PubMed] [Google Scholar]
- 99. Jorjani H, Kehr S, Jedlinski DJ, et al. . An updated human snoRNAome. Nucleic Acids Res. 2016;44(11):5068–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Taft RJ, Glazov EA, Lassmann T, et al. . Small RNAs derived from snoRNAs. RNA. 2009;15(7):1233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haas BJ, Papanicolaou A, Yassour M, et al. . De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. 2013;8(8):1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hertel J, Stadler PF. The expansion of animal microRNA families revisited. Life (Basel). 2015;5(1):905–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Smit A, Hubley R. RepeatModeler Open-1.0. 2008–2015. https://www.repeatmasker.org/repeatmodeler/, Accessed 17 Sep 2018.
- 104. Smit A, Hubley R, Green P. RepeatMasker Open-4.0. 2013–2015. https://www.repeatmasker.org/RMDownload.html, Accessed 17 Sep 2018.
- 105. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint. 2013, https://arxiv.org/abs/1303.3997v2, Accessed 17 Sep 2018.
- 106. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint. 2012, https://arxiv.org/abs/1207.3907, Accessed 17 Sep 2018. [Google Scholar]
- 107. Saffari A, Lohse K. reverseGame, Bitbucket. 2018. https://bitbucket.org/amin-saffari/reversegame/src/master/, Accessed 17 Sep 2018.
- 108. Lohse K, Harrison RJ, Barton NH. A general method for calculating likelihoods under the coalescent process. Genetics. 2011;189(3):977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nurnberger B, Lohse K, Fijarczyk Aet al. . Para-allopatry in hybridizing fire-bellied toads (Bombina bombina and B. variegata): inference from transcriptome-wide coalescence analyses. Evolution. 2016;70(8):1803–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Caughley G. Parameters for seasonally breeding populations. Ecology. 1967;48(5):834–9. [DOI] [PubMed] [Google Scholar]
- 111. Elbing K. Zur Wiederbesiedlung brandgeschädigter Teilhabitate durch Smaragdeidechsen (Lacerta viridis). Salamandra. 2000;36(3):175–84. [Google Scholar]
- 112. Saint Girons H, Castanet J, Bradshaw D, et al. . Démographie comparée de deux populations françaises de Lacerta viridis (Laurenti, 1768). Journal of Ecology. 1989;44(4):361–86. [Google Scholar]
- 113. Rykena S, Nettmann HK. Lacerta viridis (Laurenti, 1768) – Smaragdeidechse.; Böhme W, Handbuch der Reptilien und Amphibien Europas, Band 2/I: Echsen II (Lacerta), (Wiesbaden: Aula-Verlag, 1984), 129–180. [Google Scholar]
- 114. Tollis M, Ausubel G, Ghimire D, et al. . Multi-locus phylogeographic and population genetic analysis of Anolis carolinensis: historical demography of a genomic model species. PLoS One. 2012;7(6):e38474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mohiyuddin M, Mu JC, Li J, et al. . MetaSV: an accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics. 2015;31(16):2741–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen K, Wallis JW, McLellan MDet al. . BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Abyzov A, Urban AE, Snyder Met al. . CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ye K, Schulz MH, Long Q, et al. . Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kent WJ, Baertsch R, Hinrichs A, et al. . Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A. 2003;100(20):11484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Blanchette M, Kent WJ, Riemer C, et al. . Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14(4):708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hickey G, Paten B, Earl D, et al. . HAL: a hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics. 2013;29(10):1341–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Harris R. Improved pairwise alignment of genomic DNA. Ph.D. Thesis. The Pennsylvania State University; 2007. [Google Scholar]
- 124. Walter Costa MB, Höner zu Siederdissen C, Stadler PFet al. . SSS-test: a novel test for detecting selection on the secondary structures of non-coding RNAs. 2017. https://www.bioinf.uni-leipzig.de/~choener/pdfs/wal-hoe-2017b.pdf, Accessed 17 Sep 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lorenz R, Bernhart SH, Höner zu Siederdissen C, et al. . ViennaRNA Package 2.0. Algorithms for Molecular Biology. 2011;6(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lange SJ, Maticzka D, Mohl M, et al. . Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012;40(12):5215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yates A, Akanni W, Amode MRet al. . Ensembl 2016. Nucleic Acids Res. 2016;44(D1):D710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hongo JA, de Castro GM, Cintra LCet al. . POTION: an end-to-end pipeline for positive Darwinian selection detection in genome-scale data through phylogenetic comparison of protein-coding genes. BMC Genomics. 2015;16:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 130. Kolora R, Weigert A, Saffari Aet al. . Divergent evolution between sister species of European green lizards (Version 1) [Data set]. Zenodo. 2018. 10.5281/zenodo.1219810. [DOI] [Google Scholar]
- 131. Kolora R, Weigert A, Saffari Aet al. . Supporting data for "Divergent evolution in the genomes of closely-related lacertids, Lacerta viridis and L.bilineata and implications for speciation.". GigaScience Database. 2018. 10.5524/100533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
6/20/2018 Reviewed
6/22/2018 Reviewed
10/28/2018 Reviewed
Data Availability Statement
The assembled genomes, their annotations, transcript data, variant calls (variant call formats), snapshots of the code, and other supporting datasets are available in Zenodo [130] and in the GigaScience GigaDB [131] repositories.