

Abstract
Objective
Bioinformatics publications typically include complex software workflows that are difficult to describe in a manuscript. We describe and demonstrate the use of interactive software notebooks to document and distribute bioinformatics research. We provide a user-friendly tool, BiocImageBuilder, that allows users to easily distribute their bioinformatics protocols through interactive notebooks uploaded to either a GitHub repository or a private server.
Materials and methods
We present four different interactive Jupyter notebooks using R and Bioconductor workflows to infer differential gene expression, analyze cross-platform datasets, process RNA-seq data and KinomeScan data. These interactive notebooks are available on GitHub. The analytical results can be viewed in a browser. Most importantly, the software contents can be executed and modified. This is accomplished using Binder, which runs the notebook inside software containers, thus avoiding the need to install any software and ensuring reproducibility. All the notebooks were produced using custom files generated by BiocImageBuilder.
Results
BiocImageBuilder facilitates the publication of workflows with a point-and-click user interface. We demonstrate that interactive notebooks can be used to disseminate a wide range of bioinformatics analyses. The use of software containers to mirror the original software environment ensures reproducibility of results. Parameters and code can be dynamically modified, allowing for robust verification of published results and encouraging rapid adoption of new methods.
Conclusion
Given the increasing complexity of bioinformatics workflows, we anticipate that these interactive software notebooks will become as necessary for documenting software methods as traditional laboratory notebooks have been for documenting bench protocols, and as ubiquitous.
Keywords: bioconductor workflows, containers, reproducibility, automated, data science
BACKGROUND AND SIGNIFICANCE
Bioinformatics is an interdisciplinary research area focused on developing and applying computational methods derived from mathematics, computer science, and statistics to analyze biological data. Workflows typically involve executing a series of computational tasks. Documenting workflows has become increasingly difficult, given the growing complexity of workflows and rapidly evolving methodologies. Traditional “Materials and Methods” sections are not well suited for describing methodologies that are strongly dependent on code and parameters. Recently, public software repositories such as GitHub have made it relatively straightforward to distribute and update code. Data science notebooks such as Jupyter are the software analogs of laboratory notebooks and document the parameters and code along with the results. Formal descriptors of workflows, such as Common Workflow Language,1 that describe how different software components interact are also gaining acceptance. While these steps go a long way toward documenting computational workflows, it is estimated that >25% of computational workflows cannot be reproduced.2
The problem is that even using the correct version of each code component and executing them in the correct order and with the correct parameters on identical data is still not sufficient to ensure that the same results are obtained.3 Running the same software in a different hardware and software environment can affect the outcome. Even if this were not the case, from a practical viewpoint, these dependencies can make installing the exact version of the same components on a different system problematic. Using software suites such as Bioconductor4 has become a popular way to manage multiple packages and ensure proper installation and interoperability. However, the rapid development of new tools has made it increasingly difficult to define a base setup that is compatible with the growing number of components that are potentially written in different programming languages.
A solution to this problem is to package software inside software containers that provide all the requisite software dependencies. Docker is a very popular software container technology that has been used for large-scale deployment and re-analysis of biomedical big data.5 JupyterHub6 allows users to run Jupyter notebooks inside software containers deployed locally or through a server. Binder7 takes this a step further and generates and deploys Docker images of notebooks from GitHub on its public servers. This allows users to view and interact with notebooks in GitHub repositories without having to compile code or install software. However, using Binder or Jupyter requires writing custom Dockerfiles to specify the elements present in the Docker container.
In this paper, we demonstrate the utility of live notebooks for documenting and distributing bioinformatics workflows by presenting 4 notebooks on GitHub that use Bioconductor. In addition, we present a framework and a graphical user interface (GUI) designed to automatically generate a Dockerfile for a custom Bioconductor installation. This allows users without any technical knowledge of Docker to generate and publish live Bioconductor notebooks.
Reproducibility of bioinformatics workflows using Bioconductor
Reproducibility is essential for verification and advancement of scientific research. This is true for computational analyses, which are not reproducible in >25% of publications.2 Reproducibility in bioinformatics research refers to the ability to re-compute the data analytics results given a dataset and knowledge of the data analysis workflow.8 For this to happen, 3 elements must be available: (1) datasets, (2) code and scripts used to perform the computational analyses, and (3) metadata, details about how to obtain and process datasets, including descriptions of software and hardware environment setups.9,10 Gentleman and Lang11 proposed a compendium software framework for the distribution of dynamic documents containing text, code, data, and any auxiliary content to recreate the computations. Their framework forms the foundation of the Bioconductor project,4 which provides an online repository for software packages, data, metadata, workflows, papers, and training materials, as well as a large and well-established user community. Bioconductor also works with the broader R repository, the Comprehensive R Archive Network (CRAN),12 which also contains useful bioinformatics packages that are not included with Bioconductor.
Using software containers to enhance reproducibility of bioinformatics workflows
Unfortunately, re-running published code and data to reproduce published results is nontrivial even when using Bioconductor. Bioconductor is not static: new packages are constantly being added and other packages downgraded. Correct versioning is thus essential for reproducibility. Although Bioconductor is cross-platform, it achieves this by cross-compilation, which does not completely insulate the computation from differences in local hardware and software. The solution is to run the software in a virtual environment that is the same regardless of the underlying hardware or host operating system.
Docker is a well-established container technology to increase the reproducibility of bioinformatics workflows.3,10,13 The Docker platform allows for virtualized application deployments within a lightweight, Linux-based wrapper or container.14 Essentially, containers are virtual environments that encapsulate only the minimum necessary dependencies, which can be quickly deployed on most major platforms in a reproducible fashion.14 Docker uses a Dockerfile that contains all the instructions to build a Docker image starting from scratch or from another Docker image. Docker images can be downloaded from repositories like Docker Hub (https://hub.docker.com/). See Supplementary File S1 for a Docker tutorial.
While Docker provides an easy, modular method to build, distribute, and replicate complex pipelines and workflows across multiple platforms, widespread adoption in the biomedical field has been hampered by the level of technical knowledge presently required to use the technology. Docker was developed for computer professionals with programming and systems administration experience who are able to write Dockerfiles to script the installation of the software environment for containers. Our group has worked on enabling graphical interfaces to interact with containers to make Docker more accessible to less technical users.10,15,16
Data science notebooks
All laboratories use notebooks to document experimental procedures and protocols. The software counterparts are data science notebooks that combine text, code, data, mathematical equations, visualizations, and rich media into a single document that can be accessed through a web browser. These software notebooks first gained popularity in mathematics research, and the Jupyter open source project has expanded the scope and audience to include many heavily computational research areas such as bioinformatics, neuroscience, and genomics.17 Project Jupyter was a spinoff project from iPython that supports the Python programming language and now maintains multiple kernels for >50 programming languages, including Ruby, Javascript, C++, and Perl.18,19 Each Jupyter notebook document is divided into individual cells that can be run independently.18 This format records every step of a computational analysis along with the scientific narrative, which makes it easier to understand, share, reproduce, and extend a published computational workflow.
To facilitate notebook sharing and reusability, Jupyter project supports nbconvert, a tool to convert notebooks to various formats such as Hypertext Markup Language (HTML), Portable Document Format (PDF), and LaTex.20 It also supports nbviewer, a similar web service, to view and download any Jupyter notebook publicly published on the web.21 In 2015, GitHub (https://github.com/), a web-based version control code repository, started supporting the Jupyter Notebook format by making it possible to render Jupyter notebooks written in any programming language on GitHub,22 which brings GitHub’s features of sharing, version control, and collaboration into the Jupyter platform. Today, there are >1 089 265 Jupyter notebooks rendered on GitHub.23 R Notebook is a similar tool that comes as part of the RStudio development environment and supports literate programming.24 It can be created and edited within RStudio IDE and can be saved in several formats, including HTML and PDF. The advantage of Jupyter Notebooks over R Notebooks is that they can be turned into executable live notebooks using JupyterHub or Binder without any extra modification needed (see detailed explanation in the next section). R Notebooks, on the other hand, can be made interactive by converting the code inside a code cell to a web application using Shiny. This process requires writing additional code to define the interactions.
Sharing live notebooks using JupyterHub and Binder
While static Jupyter notebooks can be shared and viewed using a browser without any setup via nbviewer and GitHub, sharing interactive dynamic Jupyter notebooks in which the user can execute and modify the analyses requires downloading the notebooks and installing Jupyter and a kernel to run R in the code cells. JupyterHub6 addresses this limitation and allows multiple readers access to one or more notebooks dynamically in a browser at the cost of a setting up a web server to serve the notebooks. The Binder open source project (http://mybinder.org/) goes further and provides a public server to run Jupyter notebooks hosted on GitHub. The Binder environment allows scientists to share live interactive Jupyter notebooks that are reproducible and verifiable using a web browser, with no data download or software installation requirements.17 To manage computational environments, Binder’s underlying architecture takes advantage of 2 open source projects: Docker, which builds the environments from a project’s dependencies, and Kubernetes, which schedules resources for these environments on a Google Compute Engine cluster.25 To build and launch an executable binder, a Jupyter notebook must be uploaded to a public GitHub repository, along with an environment specification such as a Dockerfile.7 See Supplementary File S2 for a tutorial to create interactive R Jupyter notebooks using Binder.
Scientists have used Binder as a publishing medium to share reproducible computational workflows.26 However, most of the use cases of Binder have been limited to the iPython kernel. An example is the Laser Interferometer Gravitational-Wave Observatory, which used iPython Jupyter notebooks and Binder to demonstrate the computational workflow corresponding to the first direct detection of gravitational waves that Einstein predicted decades ago.27,28
Although many bioinformatics workflows use R and Bioconductor, the use of interactive notebooks has been mostly restricted to Python-based workflows due to the difficulties in setup. The software dependencies required for Binder must be reconciled with the strict dependencies required for the base installation of R and Bioconductor. Any customization steps must also be included in the setup files. To enable more widespread adoption of interactive notebooks in bioinformatics, we need a more accessible method to prepare an interactive notebook as provided by BiocImageBuilder.
OUR CONTRIBUTIONS
We present a novel software tool, BiocImageBuilder, that automates the technical step of creating Dockerfiles for live Jupyter notebooks using Bioconductor. A web-based graphical interface allows the user to choose the Bioconductor packages that need to be installed and whether the Dockerfile is to be used with Binder on GitHub or in a local installation. Additional R packages from CRAN can also be included. The tool then builds the appropriate Dockerfile for the user to upload with his or her notebook. While it is possible to use a generic image and embed install commands within the code cells to specify the R/Bioconductor environment, building a customized image or Dockerfile avoids wasteful package downloads at runtime that would occur each time the code is run by the reader. The generated Dockerfile can be verified in 3 ways depending on how it is used. If a JupyterHub server is being used to author notebooks, the Dockerfile can be used to construct the author’s code cells, so that by default, any shared notebook with the same Dockerfile will be identical. If a local installation of Jupyter is being used for authoring, the Dockerfile can be tested by serving the notebook using a local JupyterHub installation and verifying that the code cells produce the expected output. Alternatively, one can create a GitHub/Binder repository and create and click on a binder badge to launch and test the served notebook. This is described in Supplementary File S2.
We illustrate the feasibility of our tool in 4 case studies: differential expression analysis for ectopic pregnancy, pattern discovery of gene expression data across human cell lines, a published RNA sequencing workflow, and drug signature prediction using KINOMEscan data. All of our case studies use the R programming language and software packages from Bioconductor/CRAN. The live Jupyter notebooks for these case studies represent new work. Figure 1 shows an overview of our approach.
Figure 1.

Overview of our approach. The author of the Bioconductor workflow uses BiocImageBuilder to generate a Dockerfile that describes the Bioconductor and CRAN packages installed. The Dockerfile and the notebook files are uploaded to a server or GitHub repository. A custom container is then built with the default Linux base image for Bioconductor, dependencies for Jupyter, JuptyerHub, and/or Binder, and the Bioconductor packages. For GitHub installations, the Binder server builds the container and provides a link to run the container on its public cluster. JupyterHub provides the same functionality locally or on a private server. Using the container, the end user is able to view the notebook and execute, modify, and save the code on his or her local machine regardless of whether it uses Linux, MacOS, or Windows. In the case where the container is run remotely, no additional installation of software is required on the part of the end user.
Table 1 summarizes differences between options for installing and using data science notebooks. Installing Jupyter and R/Bioconductor does not allow readers to run R/Bioconductor in Jupyter notebooks – downloading and installing a kernel to support R is required. The functionality added by JupyterHub is to allow multiple readers to run notebooks interactively within their browser at the cost of setting up a server (or using a Docker container to run the server locally). Binder provides the server, but the user needs to have the files available on GitHub. In both cases, the author needs to provide a Dockerfile specifying his/her environment. Where BiocImageBuilder comes in is to help the author provide a customized Dockerfile. Specifically, BiocImageBuilder moves the R/Bioconductor package installation instructions into the Dockerfile. Since our default option is to specify the versions of R/Bioconductor packages in the Dockerfile, reproducibility of results is ensured even between R/Bioconductor releases.
Table 1.
Comparison of different options to install and use data science notebooks.
| Platform | Author requirements | Reader requirements | R/Bioconductor | Multi-user share |
|---|---|---|---|---|
| Jupyter | Python, Jupyter | Python, Jupyter | No | No |
| Jupyter/IRkernel | Python, Jupyter, IRkernel | Python, Jupyter, IRkernel | Yes | No |
| R Notebook | Rstudio | Rstudio | Yes | No |
| JupyterHub | Docker server, Dockerfile | Browser | Yes | Yes |
| Binder | GitHub repository, Dockerfile | Browser | Yes | Yes |
AUTOMATIC GENERATION OF DOCKERFILES FOR BIOCONDUCTOR WORKFLOWS
Bioconductor compiles and tests each component of its suite on a set of stock Windows, MacOS, and Linux machines. This ensures that all components in Bioconductor are compatible and should install on most hardware configurations. In addition, the Bioconductor core team provides Docker containers for the release and development versions of the complete suite.29 No facility exists, however, for building custom images with a smaller, specified set of Bioconductor components.
We have developed a GUI-based tool, BiocImageBuilder, for this purpose. BiocImageBuilder starts with a base image that is based on the stock Linux machine used by Bioconductor to test packages. The base image is modified to include components for JupyterHub or Binder compatibility and the kernels necessary to run R. For images to be run by Binder, Dockerfile commands using the Linux Conda utility are used to install the Bioconductor and CRAN packages. Otherwise, commands using Bioconductor’s biocLite utility are used to install the components. The notebook’s author simply starts up the container with BiocImageBuilder and points his or her browser to a local URL. The author will then see a form for choosing the desired starting image, the desired components, and the option of specifying a custom startup script to be run as part of the container build process (see Figure 2 and Supplementary File S3). BiocImageBuilder will then produce a Dockerfile. This can be uploaded to GitHub, along with a Jupyter notebook file, to create a repository that distributes an interactive notebook that can be viewed using Binder. Alternatively, a Dockerfile can be produced that is suitable for private deployment using JupyterHub. Users can also use the Dockerfile themselves to directly build an actual image of their notebook to use, store, or distribute on DockerHub and other repositories. Currently, we support R 3.4 and Bioconductor 3.5. We intend to add support for other versions in the future so that downgraded packages can be run. Containerizing Jupyter notebooks ensures that they will always be viewable, insulating the user from future changes to Bioconductor or R.
Figure 2.

Screenshot of BiocImageBuilder. The user selects from a menu the Bioconductor and CRAN packages required for his or her notebook. BiocImageBuilder then generates the Dockerfile describing a minimal Linux container that contains these packages. The Dockerfile can be uploaded to GitHub, where it can be viewed interactively using Binder.
BiocImageBuilder is written in Python3 using PyQt5 (https://wiki.python.org/moin/PyQt), which is a Python binding for the Quicktime engine that renders the graphical interface. Although PyQt5 is meant to be cross-compatible over different platforms, the installation of many dependencies can be quite complicated for some user environments. To avoid these problems, BiocImageBuilder is packaged using our GUIdock-noVNC container.15 This container creates a mini–web server that serves the rendered graphics through a local port and can be run on any Docker-compatible platform (Windows, MacOS, Linux). Most modern browsers that support HTML5 (eg, Chrome, Firefox, Safari, Opera) can be used to access the BiocImageBuilder.
Note that BiocImageBuilder is designed for those who wish to author an interactive Bioconductor notebook – it is not required for end users who wish to interact with a published notebook. The source code of BiocImageBuilder is publicly available at https://github.com/Bioconductor-notebooks/BiocImageBuilder and its Docker image is publicly available at https://hub.docker.com/r/biodepot/bioc-builder/.
CASE STUDIES
In this section, we present 4 case studies in which we illustrate the use of R and Bioconductor packages within Jupyter notebooks. Static snapshots of these notebooks are included as Supplementary Files S4–7. The corresponding fully interactive notebooks are available from https://github.com/Bioconductor-notebooks. In case study 1, we extended the published differential expression analyses of ectopic pregnancy. In case study 2, we created our own workflows for cross-platform omics data. In case study 3, we replicated a published RNA sequencing workflow in our proposed framework. In case study 4, we demonstrated the utility of these live interactive notebooks when applied to KINOMEscan data. None of these case studies overlap with any case studies in our previously published work. These case studies span different applications and illustrate general analytical techniques, such as clustering and data visualization, that are generally applicable to high-throughput data.
All the case studies are available on GitHub as static notebooks. However, to increase the reproducibility and turn them into live executable notebooks using Binder, environmental specifications need to be uploaded in the form of a Dockerfile for each case study. To achieve that, BiocImageBuilder was used to create Binder-compatible Dockerfiles, and for each case study the list of specific required packages was chosen from the checklist menus. These Dockerfiles are saved and uploaded to GitHub, along with Jupyter notebooks and data. Binder uses these Dockerfiles to create temporary executable environments for each case study that can be rerun using only a browser, without any further installations.
Case study 1: Identifying differentially expressed genes for ectopic pregnancy
Motivation and overview
When a woman's pregnancy test result is positive, initial testing of the uterus is visualized on a transvaginal ultrasound scan (TVS). As shown in Figure 3, the possible outcomes of the TVS are: (1) intrauterine pregnancy (IUP), which is normal pregnancy with fertilized egg implanted inside the uterus; (2) ectopic pregnancy (EP), where the fertilized egg can be seen in the TVS scan, but it is implanted outside the uterus; or (3) pregnancy of unknown location (PUL), when the pregnancy test is positive, but no evidence of pregnancy is seen on TVS.30
Figure 3.
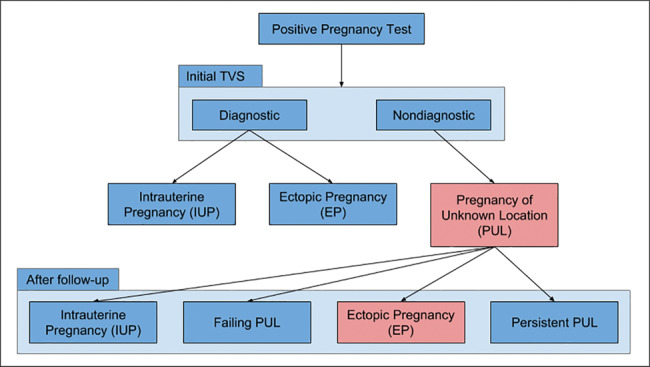
Outcome of initial TVS scan. PUL = pregnancy of unknown location; TVS = transvaginal ultrasound scan; EP = ectopic pregnancy.
Cases of PUL can subsequently lead to one of the following outcomes: (1) failing PUL (miscarriage): majority of cases (50%–70%); (2) normal IUP: it is too early to visualize the fertilized egg on TVS; (3) EP: 7%–20% of PUL cases; the EP was not seen on the initial TVS examination.30,31
In the case of PUL, close surveillance is required, consisting of serial office visits, ultrasounds, and blood draws over a period as long as a 6 weeks.32 During this surveillance period, no medical or surgical intervention is taken until a conclusive diagnosis of EP is reached and the nonviability of the embryo is concluded.32 Thus, the clinicians’ objectives are to: (1) diagnose EP as early as possible to avoid health risks and (2) ensure that this early diagnosis is correct, to avoid ending a viable pregnancy erroneously.33 Delayed diagnosis of EP is the most common life-threatening emergency in early pregnancy.32 Despite the high frequency of this serious condition, early diagnosis of EP can be challenging. In practice, there are several methods used to detect EP in the case of PUL, and they largely depend on biochemical markers such as serum progesterone34 and serum human chorionic gonadotrophin levels.35 However, the biochemical markers used are not consistent,36 and the International Society of Ultrasound in Obstetrics and Gynecology encourages the use of mathematical models to expedite EP detection.37
In this case study, we aimed to identify differentially expressed genes among patients with EP by analyzing gene expression data. Differentially expressed genes are the subset of genes that exhibit expression patterns associated with EP.
Data
Duncan et al.38 collected gestation-matched endometrial tissue from women with EP (n = 11) and IUP (n = 13), and samples were profiled using the Affymetrix Human Genome U133 Plus 2.0 platform. The resulting CEL files were normalized using Robust Multi-array Average38 and are publicly available from ArrayExpress (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-680.
Analysis
We filtered the Robust Multi-array Average normalized gene expression data to keep the probe sets that are common with prospective validation samples profiled using Affymetrix genechip Human Gene 2.0 ST. AnnotationDbi39 and Stringr40 Bioconductor packages were used to access, map, and process gene identifiers in specific chip annotation databases.41,42 Duncan et al.38 identified genes differentially expressed in EP vs IUP using the t test, with multiple comparison correction using the Benjamini-Hochberg false discovery detection method with a corrected P < .05. In our analysis, we started by performing a standard t test without corrections, with a range of threshold values. We also performed other multiple test correction methods, including the Bonferroni correction, SAM,43 and LIMMA.44 Our resulting lists of differentially expressed genes showed considerable overlap with the results from Duncan et al. In particular, Duncan's top upregulated gene, CSH1, resulted from most of our differential expression analyses, and Duncan's top downregulated gene, CRISP3, resulted from SAM analysis and the Benjamini-Hochberg detection method. We also generated heatmaps to visualize these differentially expressed genes. We observed that EP and IUP samples were mostly assigned to distinct clusters, with the exception of 2 IUP samples that clustered with EP samples, which Duncan et al. referred to as the effect of decidualization degree. Furthermore, we performed Gene Set Enrichment Analysis45 to identify pathways and functional categories among the differentially expressed genes. The details of the analyses are provided in Supplementary File S4.
Case study 2: Cross-platform analysis of human cell line genomics data
Motivation and overview
The Library of Integrated Network based Cellular Signatures (LINCS) program, funded by the National Institutes of Health, generates different types of data, including gene expression, proteomic, and cell imaging data, in response to drug and genetic perturbations (http://lincsproject.org/).46 One of the main objectives of the LINCS program is to study gene signatures resulting from perturbations applied to human cell lines. In particular, the LINCS L1000 gene expression data measure the expression level of approximately 1000 landmark genes in response to drug and genetic perturbation experiments across multiple human cell lines. We aimed to study the similarity of patterns in the L1000 data across different cell lines. The LINCS L1000 gene expression data are publicly available from the Gene Expression Omnibus (GEO) database under accession number GSE70138.
Our goal was to study the consistency of cell line similarities across the LINCS L1000 data and other data sources. In particular, we used the LINCS L1000 gene expression data to explore similarities between different cell lines using different analysis methods, including clustering and dimension reduction techniques. Our work was inspired by Zhang et al.,47 in which multiple datasets, including Cancer Cell Line Encyclopedia (CCLE) data48 and Cancer Genome Project data,49 were used to explore the similarities of cell lines and drugs. The results of this study suggested that similar cell lines would be expected to have similar drug responses, and similar drugs would be expected to have similar effects on a cell line.
Data
We used L1000 data processed by the L1K++ pipeline, an alternative data-processing pipeline for the L1000 gene expression data that we developed at the University of Washington Tacoma. L1K++ is implemented in C++ using linear algorithms to make it more than 1000× faster than the available pipelines.50 We substantiated our results from L1K++ processed data using published cell line gene expression data generated using microarray and RNA sequencing (RNA-seq) technology. The CCLE gene expression data used Affymetrix microarrays to profile the genome-wide transcription activities across approximately 1000 human cancer cell lines.48 The CCLE data are publicly available from the GEO database under accession number GSE36133. Similarly, Klijn et al.51 used RNA-seq technology to profile the expression across 675 untreated human cancer cell lines. These data are publicly available from the ArrayExpress database under accession number E-MTAB-2706 (www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2706/).
Analysis
In the Jupyter notebook (see Supplementary File S5), we read the 3 datasets (L1K++, CCLE, RNA-seq), including all cell lines and genes. Then we standardized each of the 3 datasets separately by computing the z-score for gene expression across all cell lines. In order to compare the results from the L1K++ data to those from the other 2 datasets, we first computed the intersection of genes and cell lines in common between L1K++ and CCLE, which resulted in 55 cell lines and the landmark genes. For L1K++ and RNA-seq, we found 41 cell lines in common. Subsequently, we calculated the pairwise distances (including Euclidean distances and squared Mahalanobis distances) and correlation coefficients (including Pearson’s correlation and rank-based Kendall’s correlation) between each pair of cell lines based on their gene expression profiles. We then applied hierarchical clustering and model-based clustering52 to cluster L1K++ vs CCLE and L1K++ vs RNA-seq cell lines.
Case study 3: Alignment and differential analyses of RNA-seq analysis workflows
Motivation and overview
With the rapidly decreasing costs of sequencing technology, RNA-seq has become a well-established technology to measure gene expression. Here, we demonstrate the feasibility and merits of using an interactive Jupyter notebook to document a published RNA-seq data analysis workflow in Bioconductor53 (www.bioconductor.org/help/workflows/rnaseqGene/).
Data
We used the RNA-seq data from the Bioconductor “airway” package, in which airway smooth-muscle cells were treated with dexamethasone, a synthetic glucocorticoid steroid with anti-inflammatory effects.54 Glucocorticoids are used, for example, by patients with asthma to reduce inflammation of the airways. In the experiment, 4 primary human airway smooth-muscle cell lines were treated with 1 µM dexamethasone for 18 h. For each of the 4 cell lines, we have a treated and an untreated sample. The data are also publicly available in the GEO database under accession number GSE52778.
Analysis
We followed the steps of analysis published by Love et al.53 In particular, we started with the BAM files that provide the alignment data in a binary format. After normalizing the table of read counts, we performed differential expression analysis using DESeq,55 visualization using heatmaps for sample distances, and a mean average plot for the estimated model coefficients.
Case study 4: Drug signature prediction using KINOMEscan data
Motivation and overview
Protein kinases are key regulators of cell function, and thus play critical roles in many biological processes. A protein kinase is an enzyme that transfers phosphate groups from molecules to specific substrates. Kinases are important targets for therapeutic drugs.56 Many drugs have been developed to target kinases in the treatment of cancer.57 However, the underlying biology of many of these kinases is not yet fully elucidated. Therefore, drug-target interactions profiling the binding of small-molecule drugs to kinases are of great interest.
Data
The Harvard Medical School LINCS project measured kinase biochemical profiles using a competition binding assay called DiscoverRx KINOMEscan.58 Specifically, the researchers profiled a panel of 478 kinases against 78 small molecule compounds.56 The KINOMEscan data are publicly available from http://lincs.hms.harvard.edu/kinomescan/.
Analysis
We aimed to identify protein targets of drug compounds using the KINOMEscan data. We downloaded and parsed the KINOMEscan data. We found 87 profiles of small-molecule drugs at 10 μM drug concentration across 440 kinases. These data represent the percentage of kinase bound by ligand in the presence of a given drug concentration compared to the DMSO control reaction. The Harvard Medical School web site graphed the results using percentage thresholds of 35%, 5%, and 1%. In our Jupyter notebook (see Supplementary File S7), we established a threshold for the KINOMEscan data at 35% and presented a heatmap of those data showing which compounds are considered active among which kinases. Since there is no established threshold for this “percent of control” – for example, Vidovic et al.59 defined compounds as active if they inhibit a kinase >90% – this case study illustrates the added value of these interactive live notebooks by allowing the user to easily change the threshold and update the drug-target interaction visualization results with a few simple clicks.
DISCUSSION
We present a web-based framework and a GUI designed to automatically generate a Dockerfile to create and publish live R and Bioconductor notebooks for bioinformatics workflows without any technical knowledge of Docker containers. These web-based notebooks can be published and viewed with modifiable and executable code in a browser without having to install any software. We demonstrate the applications of these interactive notebooks using 4 case studies, in which we show the revolutionary aspects of dynamic live notebooks compared to traditional static reports and visualizations for data analysis. Notebooks generated in our framework ensure reproducibility of analysis through the use of software containers. Our interactive notebooks enable clinicians and biomedical scientists to visually interact with the analyses while exploring the results through different types of interactive visualizations (eg, Plotly60 in case studies 2 and 4). In addition, parameters can be modified easily. Our approach and BiocImageBuilder are not limited to bioinformatics applications that use Bioconductor software packages, but can be used for any applications that use the R programming language and software packages from CRAN.
A limitation of Jupyter notebooks is that each notebook is limited to one kernel supporting a single programming language. All of our 4 case studies used IRkernel, which assumes an R programming environment. However, modern bioinformatics workflows consist of modules that are potentially written in different programming languages. Existing options to use both R and Python in the same notebook include rpy2, which is an R interface embedded in a Python process61; Beaker, with individual cells supporting different languages62; and the creation of custom hybrid kernels. Another possibility is to use the Docker Python application programming interface to launch containers within a custom cell, which would allow running workflows with components written in different languages and with different software specifications. For future work, we would like to explore these options to allow for a modular structure consisting of different computing environments for bioinformatics applications.
FUNDING
LHH and KYY are supported by National Institutes of Health grant U54-HL127624. RA and AA gratefully acknowledge a full sponsorship from the Saudi Arabian Cultural Mission scholarship program 2015–2017.
AUTHOR CONTRIBUTIONS
RA drafted the manuscript and was primarily responsible for figuring out how Binder works. JH implemented and wrote documentation for BiocImageBuilder. LHH added the noVNC container to BiocImageBuilder and refined the container. KYY designed and coordinated the study. AA created the Jupyter notebooks for case studies 2 and 4. RA and YT created the Jupyter notebooks for case studies 1 and 3, respectively. RA and NO performed the analysis of the EP data. JH, LHH, and RA created the figures. JH created the movie uploaded as Supplementary File S3. All authors tested BiocImageBuilder and edited the manuscript.
CONFLICTS OF INTEREST
The authors have no competing conflicts of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Mr Yuvi Panda and Mr Chris Holdgraf from the Berkeley Institute of Data Science for providing access to the latest version of Binder.
REFERENCES
- 1. Peter A, Michael RC, Nebojša T, et al. Common Workflow Language, v 1.0. 2016. [Google Scholar]
- 2. Freedman LP, Cockburn IM, Simcoe TS. The economics of reproducibility in preclinical research. PLoS Biol. 2015;136:e1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meiss T, Hung L-H, Xiong Y, Sobie E, Yeung KY. Software solutions for reproducible RNA-seq workflows. bioRxiv. 2017:099028. [Google Scholar]
- 4. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;510:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vivian J, Rao A, Nothaft FA, et al. Rapid and efficient analysis of 20,000 RNA-seq samples with Toil. bioRxiv. 2016:062497. [Google Scholar]
- 6. Ragan-Kelley M, Kelley K, Kluyver T. JupyterHub: deploying Jupyter notebooks for students and researchers. 2016. https://github.com/minrk/jupyterhub-pydata-2016. Accessed April 27, 2017. [Google Scholar]
- 7. Binder. 2017. http://docs.mybinder.org/. Accessed April 29, 2017.
- 8. Leek JT, Peng RD. Opinion: Reproducible research can still be wrong: adopting a prevention approach. Proc Natl Acad Sci USA. 2015;1126:1645–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buffalo V. Bioinformatics Data Skills: Reproducible and Robust Research with Open Source Tools. Sebastopol, CA: O'Reilly Media; 2015. [Google Scholar]
- 10. Hung LH, Kristiyanto D, Lee SB, Yeung KY. GUIdock: using docker containers with a common graphics user interface to address the reproducibility of research. PLoS One. 2016;114:e0152686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gentleman RC, Lang DT. Statistical analyses and reproducible research. J Comput Graphical Stats. 2007;161:1–23 [Google Scholar]
- 12. The Comprehensive R Archive Network (CRAN). https://cran.r-project.org/. Accessed January 4, 2017.
- 13. Boettiger C. An introduction to Docker for reproducible research. ACM SIGOPS Operating Systems Review, Special Issue on Repeatability and Sharing of Experimental Artifacts. 2015;491:71–79. [Google Scholar]
- 14. Schulz WL, Durant TJ, Siddon AJ, Torres R. Use of application containers and workflows for genomic data analysis. J Pathol Inform. 2016;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mittal V, Hung LH, Keswani J, Kristiyanto D, Lee SB, Yeung KY. GUIdock-VNC: Using a graphical desktop sharing system to provide a browser-based interface for containerized software. Gigascience. 2017;64:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hung L-H, Meiss T, Keswani J, Xiong Y, Sobie E, Yeung KY. Building containerized workflows for RNA-seq data using the BioDepot-workflow-Builder (BwB). bioRxiv. 2017:099010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kluyver T, Ragan-Kelley B, Pérez F, et al. Jupyter Notebooks – a publishing format for reproducible computational workflows. In: Loizides F, Schmidt B, ed. Positioning and Power in Academic Publishing: Players, Agents and Agendas. Clifton, VA: IOS Press; 2016:87–90. [Google Scholar]
- 18. Perez F, Granger BE. IPython: A System for Interactive Scientific Computing. Computing Sci Eng. 2007;93:21–29. [Google Scholar]
- 19. Jupyter kernels. 2017. https://github.com/jupyter/jupyter/wiki/Jupyter-kernels. Accessed March 20, 2017.
- 20. Jupyter Notebook Conversion. 2016. https://github.com/jupyter/nbconvert. Accessed March 9, 2017.
- 21. nbviewer: A simple way to share Jupyter Notebooks. https://nbviewer.jupyter.org/. Accessed March 17, 2017.
- 22. Rendering Notebooks on GitHub. 2015. https://blog.jupyter.org/rendering-notebooks-on-github-f7ac8736d686. Accessed April 12, 2017.
- 23. Search results on GitHub. 2015. https://github.com/search?l=&q=nbformat+extension%3Aipynb&ref=advsearch&type=Code&utf8=%E2%9C %93. Accessed May 3, 2017.
- 24. R Notebook. 2016. http://rmarkdown.rstudio.com/r_notebooks.html. Accessed May 12, 2017.
- 25. Toward publishing reproducible computation with Binder. 2016. https://elifesciences.org/elife-news/toward-publishing-reproducible-computation-binder. Accessed April 18, 2017.
- 26. Sofroniew NJ, Vlasov YA, Hires SA, Freeman J, Svoboda K. Neural coding in barrel cortex during whisker-guided locomotion. eLife. 2015;4:e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collaboration LS, Virgo C, Abbott BP, et al. GW151226: observation of gravitational waves from a 22-solar-mass binary black hole coalescence. Phys Rev Lett. 2016;11624:241103. [DOI] [PubMed] [Google Scholar]
- 28. PyCBC: Python Software for Astrophysical Analysis of Gravitational Waves from Compact Object Coalescence. 2016. https://github.com/ligo-cbc/. Accessed April 21, 2017.
- 29. Docker containers for Bioconductor. https://http://www.bioconductor.org/help/docker/. Accessed March 10, 2017.
- 30. Kirk E, Bourne T. Predicting outcomes in pregnancies of unknown location. Women's Health. 2008;45:491–99. [DOI] [PubMed] [Google Scholar]
- 31. Banerjee S, Aslam N, Woelfer B, Lawrence A, Elson J, Jurkovic D. Expectant management of early pregnancies of unknown location: a prospective evaluation of methods to predict spontaneous resolution of pregnancy. BJOG. 2001;1082:158–63. [DOI] [PubMed] [Google Scholar]
- 32. Goldner TE, Lawson HW, Xia Z, Atrash HK. Surveillance for ectopic pregnancy: United States, 1970–1989. MMWR. CDC surveillance summaries 1993;426:73–85. [PubMed] [Google Scholar]
- 33. Boyraz G, Bozdag G. Pregnancy of unknown location. J Turkish German Gynecol Assoc. 2013;142:104–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mol BW, Lijmer JG, Ankum WM, van der Veen F, Bossuyt PM. The accuracy of single serum progesterone measurement in the diagnosis of ectopic pregnancy: a meta-analysis. Human Reproduction. 1998;1311:3220–27. [DOI] [PubMed] [Google Scholar]
- 35. Kadar N, Bohrer M, Kemmann E, Shelden R. The discriminatory human chorionic gonadotropin zone for endovaginal sonography: a prospective, randomized study. Fertility Sterility. 1994;616:1016–20. [DOI] [PubMed] [Google Scholar]
- 36. Silva C, Sammel MD, Zhou L, Gracia C, Hummel AC, Barnhart K. Human chorionic gonadotropin profile for women with ectopic pregnancy. Obstetrics Gynecol. 2006;1073:605–10. [DOI] [PubMed] [Google Scholar]
- 37. Condous G, Timmerman D, Goldstein S, Valentin L, Jurkovic D, Bourne T. Pregnancies of unknown location: consensus statement. Ultrasound Obstet Gynecol. 2006;282:121–22. [DOI] [PubMed] [Google Scholar]
- 38. Duncan WC, Shaw JL, Burgess S, McDonald SE, Critchley HO, Horne AW. Ectopic pregnancy as a model to identify endometrial genes and signaling pathways important in decidualization and regulated by local trophoblast. PLoS One. 2011;68:e23595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. AnnotationDbi: Annotation Database Interface [program]. R package version 1.36.2. version. 2017.
- 40. stringr: Simple, Consistent Wrappers for Common String Operations [program]. R package version 1.2.0 version. 2017.
- 41. hgu133plus2.db: Affymetrix Human Genome U133 Plus 2.0 Array annotation data (chip hgu133plus2). [program]. R package version 3.2.3. version. 2016.
- 42. hugene20stprobeset.db: Affymetrix hugene20 annotation data (chip hugene20stprobeset). [program]. R package version 8.5.0. version. 2016.
- 43. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;989:5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- 45. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;10243:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Musa A, Ghoraie LS, Zhang SD, et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform. pii: bbw112. doi: 10.1093/bib/bbw112 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang N, Wang H, Fang Y, Wang J, Zheng X, Liu XS. Predicting anticancer drug responses using a dual-layer integrated cell line-drug network model. PLoS Comput Biol. 2015;119:e1004498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;4837391:603–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cancer Genome Project. http://www.sanger.ac.uk/science/groups/cancer-genome-project. Accessed January 25, 2017.
- 50. Hung LH. L1K++: A Fast Pipeline that Increases the Accuracy of L1000 Gene Expression Data. YouTube video from BD2K-LINCS; 2015. [Google Scholar]
- 51. Klijn C, Durinck S, Stawiski EW, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;333:306–12. [DOI] [PubMed] [Google Scholar]
- 52. Yeung KY, Fraley C, Murua A, Raftery AE, Ruzzo WL. Model-based clustering and data transformations for gene expression data. Bioinformatics. 2001;1710:977–87. [DOI] [PubMed] [Google Scholar]
- 53. Love MI, Anders S, Kim V, Huber W. RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000Research. 2015;4:1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Himes BE, Jiang X, Wagner P, et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;96:e99625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;1110:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McAllister FE, Niepel M, Haas W, Huttlin E, Sorger PK, Gygi SP. Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Analytical Chem. 2013;859:4666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;1255:1780–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DiscoverRx KINOMEscan technology. Secondary DiscoverRx KINOMEscan technology. http://www.discoverx.com/technologies-platforms/competitive-binding-technology/kinomescan-technology-platform. Accessed February 15, 2017.
- 59. Vidovic D, Koleti A, Schurer SC. Large-scale integration of small molecule–induced genome-wide transcriptional responses, Kinome-wide binding affinities and cell-growth inhibition profiles reveal global trends characterizing systems-level drug action. Front Genet. 2014;5:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Plotly: visualize data together [program]. https://plot.ly. Accessed March 5, 2017.
- 61. rpy2. https://rpy2.bitbucket.io/. Accessed May 31, 2017.
- 62. Beaker. http://beakernotebook.com/. Accessed May 31, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.