

Abstract
The LodA-like proteins are a recently identified family of enzymes that rely on a cysteine tryptophylquinone (CTQ) cofactor for catalysis. They differ from other tryptophylquinone enzymes in that they are oxidases rather than dehydrogenases. GoxA is a member of this family that catalyzes the oxidative deamination of glycine. Our previous work with GoxA from Pseudoalteromonas luteoviolaceae demonstrated that this protein forms a stable intermediate upon anaerobic incubation with glycine. The spectroscopic properties of this species were unique among those identified for tryptophylquinone enzymes characterized to date. Here we use X-ray crystallography and resonance Raman spectroscopy to identify the GoxA catalytic intermediate as a product Schiff-base. Structural work additionally highlights features of the active site pocket that confer substrate specificity, intermediate stabilization and catalytic activity. The unusual properties of GoxA are discussed within the context of the other tryptophylquinone enzymes.
Keywords: Oxidase, oxidase, crystal structure, enzyme catalysis, enzyme mechanism
Graphical Abstract
INTRODUCTION
Tryptophylquinone enzymes rely on post-translational modifications of Trp residues to generate an endogenous protein-derived cofactor. The tryptophylquinone is crosslinked to the side-chain of either another Trp or a Cys residue to generate tryptophan tryptophylquinone (TTQ) 1 or cysteine tryptophylquinone (CTQ) 2, 3, respectively. Numerous structural and mechanistic studies have been conducted on the tryptophylquinone-dependent dehydrogenases: methylamine dehydrogenase (MADH, TTQ cofactor), aromatic amine dehydrogenase (AADH, TTQ cofactor) and quinohemoprotein dehydrogenase (QHNDH, CTQ cofactor). These have been reviewed in detail elsewhere 4, 5.
Several common themes have emerged from these studies. These proteins are multi-subunit dehydrogenases, shuttling electrons from substrate to type-1 copper proteins 6, 7 or, in the case of QHNDH, to c-type hemes anchored to a subunit separate from that of the catalytic site 2, 8. In the reductive half reaction (Fig. 1), a covalent imine or substrate Schiff-base adduct is formed between the amine substrate and quinone oxygen. Deprotonation of C1 of the substrate requires an active site base and generates a reduced quinol linked to a product Schiff-base. An anomalously large primary deuterium kinetic isotope effect (KIE) was observed for this reaction step, indicating that the proton abstraction from the substrate Schiff-base occurs by quantum mechanical tunneling and is at least partially rate-limiting 7–10. Reduction of the cofactor by substrate in the absence of electron acceptors yields a stable, colorless aminoquinol species 11, 12, although spectral interference from the hemes makes this difficult to assess in QHNDH. Hydrolysis of the product Schiff-base occurs rapidly and yields the aldehyde product of the reaction and a reduced aminoquinol. The catalytic mechanism of these enzymes appears to be highly conserved with one another 8, 9, 11. Another class of quinoproteins, which possess tyrosine-derived topaquinone (TPQ) cofactors, are amine oxidases that exhibit these same mechanistic features in their reductive half-reactions 13 (Fig. 1). However, the nitrogen atom of both the substrate and product Schiff-base intermediates are predicted to be protonated for the TPQ enzymes 13–15 while the protonation states of these species are unknown for TTQ and CTQ enzymes. In each of these quinoproteins, the oxidative half-reaction requires electron transfer from the reduced aminoquinol and hydrolysis of ammonia to regenerate the oxidized TTQ/CTQ cofactor, or in the case of the amine oxidases, electron transfer to O2 to regenerate the TPQ cofactor.
Figure 1.

The reductive half-reaction of tryptophylquinone and topaquinone enzymes. Schiff-base adducts are shown as neutral species as the protonation states are unknown for tryptophylquinone enzymes.
A recently identified group of CTQ-bearing enzymes called LodA-like proteins 16 are the first tryptophylquinone enzymes that function as oxidases rather than dehydrogenases and have been shown to be widespread among several classes of bacteria and fungi 17. Those characterized to date catalyze the oxidative deamination of the ε-amino group of lysine (LodA) or the amine group of glycine (GoxA). Some features of the reductive half-reaction are similar to those of MADH, AADH and QHNDH, but some are different 18, 19. The LodA-like proteins are oxidases, using molecular oxygen as an electron acceptor 18. In fact, LodA functions as an antimicrobial protein as a consequence of the hydrogen peroxide it produces 20–22. Furthermore, kinetic studies with GoxA indicate positive cooperativity and a negligible KIE for the reduction of the cofactor by substrate 19, 23, 17. Finally, crystal structures of LodA from Marinomonas mediterranea 24 and GoxA from Pseudoalteromonas luteoviolacea 17 (UniProt KB A0A161XU12) are homotetramers.
There are a number of kinetic and structural differences between the LodA-like proteins and the tryptophylquinone dehydrogenases. Perhaps most striking is that anaerobic reduction of GoxA with glycine results in the formation of a stable intermediate with a remarkably red-shifted absorption spectrum (Fig. 2) 17. The spectrum only returns to that of the resting state after exposure to air. This blue chromophore has not been observed with any other tryptophylquinone enzyme studied to date. Here we use X-ray crystallography and resonance Raman (RR) spectroscopy to characterize this unusual catalytic intermediate. The results demonstrate how the LodA-like proteins contribute to the structural and mechanistic diversity employed by tryptophylquinone enzymes.
Figure 2.

Absorption spectra of oxidized GoxA (black) and of the stable intermediate formed after the addition of 5 mM glycine in absence of O2 (Gly-GoxA, red). Samples were in 1-mm Raman capillaries at a concentration of 550 M GoxA in 50 mM potassium phosphate buffer pH 7.5.
EXPERIMENTAL PROCEDURES
Expression and Purification of GoxA —
GoxA from P. luteoviolaceae (UniProt KB A0A161XU12) was expressed in E. coli and purified as described previously17.
Resonance Raman Spectroscopy —
Resonance Raman (RR) spectra were collected on samples with enzyme concentrations ranging from ~ 400 to 600 µM in 50 mM phosphate pH 7.5 buffer. Full conversion of air-oxidized PlGoxA to the glycine adduct (Gly-GoxA) was performed inside a glovebox containing <1 ppm O2 (Omniplex System, Vacuum Atmospheres Co.) with 5 mM glycine (final concentration) and confirmed by collecting UV-vis spectra from samples inside the RR capillaries with a Cary 50 spectrometer (Varian). Isotopically labeled glycines were purchased from Cambridge Isotopes Inc. Samples in D2O and H218O (99.9% D-atom and 97% 18O atom, Sigma-Aldrich) were prepared with two concentration/re-dilution cycles of oxidized GoxA to reach >75 % solvent exchange.
Room-temperature RR spectra were obtained using a 90° geometry and a custom McPherson 2061/207 spectrograph with variable gratings and a liquid-N2 cooled CCD camera (LN-1100PB, Princeton Instruments). The Raman laser excitations at 458 (10 mW) and 647 nm (50 mW) were provided by Ar and Kr lasers (Innova I90C and I-302, Coherent). Long-pass filters (RazorEdge, Semrock) were used to attenuate the Rayleigh scattering. Rapid data acquisitions with a range of laser powers provided no evidence of photosensitivity for any of the samples and UV-vis spectra obtained before and after collection of RR spectra were unchanged. Frequencies were calibrated relative to indene, and are accurate to ±1 cm−1. Successive collections of RR spectra with different isotope labeling allow detection of ≥ 0.4 cm−1 shifts.
Electron Paramagnetic Resonance Spectroscopy—
The diamagnetic character of GoxA and Gly-GoxA was confirmed by electron paramagnetic resonance (EPR) spectroscopy with a Bruker E500 X-band spectrometer equipped with a superX microwave bridge. EPR spectra of oxidized GoxA and Gly-GoxA were measured between −180 and −20 °C with non-saturating microwave power. Quantitative analysis was performed using Cu(II)-EDTA standard solutions.
Crystallization and Structure Determination –
GoxA crystals were grown as previously described 17. Crystal trays were moved into a glovebag filled with nitrogen gas where all subsequent manipulations were performed. Crystals were transferred to nitrogen-purged mother liquor containing 10 mM glycine and 10% v/v PEG 400 as a cryoprotectant for approximately 1 min prior to cryocooling in liquid nitrogen.
Diffraction data were collected at 100 K on beamline 5.0.2 at the Advanced Light Source at Berkeley National Laboratory, indexed and integrated with XDS 25, 26 and scaled using Aimless 27. The structure was solved by molecular replacement using using Phaser-MR 28 using the native PlGoxA structure (PDB ID: 6BYW) as the search model. Manual model building was done in Coot 29 with further rounds of refinement performed using the Phenix suite 30. Figures were prepared using Pymol (http://www.pymol.org), which was also used for pairwise structural alignments.
RESULTS
Structure of the Gly-GoxA Adduct —
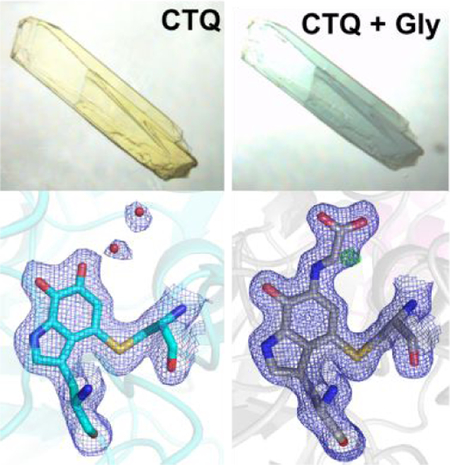
Soaking WT GoxA crystals in mother liquor containing glycine results in a dramatic color change from yellow to blue, consistent with the color change observed in solution 17. Transfer of glycine-soaked crystals to aerobic mother liquor lacking glycine resulted in the return of the yellow color over the course of several minutes (Fig. 3). Thus, it appears that the blue species formed in GoxA crystals is on the reaction pathway with properties consistent with those observed in solution.
Figure 3.

A GoxA crystal in mother liquor containing 10 mM glycine (A) was subsequently transferred to glycine-free mother liquor and imaged over approximately 5 minutes (B-F)
In order to minimize inhomogeneity at the active site due to turnover, GoxA crystals were soaked in nitrogen-purged mother liquor containing 10 mM glycine for 1–2 min prior to cryocooling and data collection. Glycine-soaked crystals were isomorphous with oxidized GoxA 17 (Table 1) and exhibited the same overall fold and tetrameric structure. Refinement of the data with an oxidized CTQ model results in clear, continuous positive difference density from the O6 quinone oxygen consistent with the formation of a covalent CTQ adduct (Fig. 4A). This adduct was modeled as a product Schiff-base resulting in a good fit to the electron density (Fig 4B). The glycine adduct is stabilized by numerous hydrogen bond interactions from His583 and Ser681within the CTQ pocket as well as Tyr766’ and His767’, which reside on a loop from a neighboring subunit (Fig. 4C). This structure is nearly identical to that of oxidized GoxA, with rmsd = 0.24 – 0.25 Å across alpha carbons comprising each dimeric unit. However, the side chains of His767’ and Ser681 reorient to interact with the carboxylate group of glycine, and the tryptophylquinone moiety of CTQ moves slightly toward these residues. Hydrogen bond partners to the O7 quinone oxygen and indole nitrogen of CTQ (i.e. the backbone atoms of His583 and Gly581, respectively) are also observed in the oxidized structure and are not disrupted by this small movement. Finally, the Schiff-base nitrogen atom is at a favorable distance and geometry to engage in a hydrogen bond with the backbone oxygen atom of His583, suggesting that this nitrogen may be protonated.
TABLE 1.
Data collection, processing and refinement statistics. Numbers in parentheses are for the high resolution shell.
| Gly-GoxA | |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.00000 |
| Space group | P21 |
| Unit cell parameters | |
| a, b, c (Å) | 110.2, 93.5, 187.9 |
| α, β, γ (°) | 90.0, 95.0, 90.0 |
| Resolution range (Å) | 48.3 – 1.82 |
| Number of reflections (measured/unique) | 1,234,529/338,105 |
| Rmerge | 0.08 (0.45) |
| I/σI | 10.0 (2.1) |
| Completeness (%) | 99.5 (99.9) |
| Redundancy | 3.7 (3.4) |
| Refinement Statistics | |
| Resolution (Å) | 1.82 |
| Rwork/Rfree | 0.176/0.213 |
| Number of atoms | |
| Protein | 24,902 |
| Mg | 4 |
| Na | 4 |
| Water | 2,952 |
| Other | 51 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.885 |
| Ramachandran | |
| Statistics | |
| Allowed | 99.8% |
| Outliers | 0.2% |
| Average B-factor (Å2) | 20.6 |
Figure 4.

The CTQ site of glycine-soaked GoxA crystals modeled as (A) oxidized CTQ and (B) a product Schiff-base. 2Fo-Fc and Fo-Fc electron density are shown as blue and green mesh contoured to 1.5σ and 5.0σ, respectively. (C) Comparison of the CTQ environment in the product Schiff-base (gray) and oxidized (PDB ID: 6BYW, green) GoxA. Probable hydrogen bonds in the glycine adduct structure are shown as dotted lines with indicated interatomic distances.
The small positive difference density near the glycine alpha carbon in the product Schiff-base model prompted us to model several other possible intermediate structures at this position (Fig. 5). Included are hydroxylated intermediates (carbinolamines) shown to be formed during hydrolysis of the product Schiff-base in AADH 31. For every intermediate model, there is residual difference density at > |4.0 σ| except for the hydroxylated intermediate with S-stereochemistry about the alpha carbon. Although this model represents the best fit to the density, the hydroxyl group occupies a similar position occupied by a water molecule in the oxidized CTQ structure (Fig. 5E). Thus, the residual difference density in the Schiff-base models could be due to a proportion of oxidized GoxA and its associated water molecule in these crystals. Refinement of a model with the oxidized structure waters at occupancies of 0.25–0.5 (depending on chain) and corresponding partial occupancies on the product Schiff-base also eliminated significant residual difference density. Thus, we are unable to differentiate between a S-hydroxylated intermediate and a mixture of product Schiff-base and oxidized CTQ at the current resolution.
Figure 5.

The glycine adduct modeled as (A) substrate Schiff-base, (B) product Shiff-base, (C) R-hydroxylated intermediate and (D) S-hydroxylated intermediate. (E) The oxidized CTQ and associated water molecule are overlaid on B. Positive and negative Fo-Fc electron density are shown as green and red mesh, respectively, contoured to 4.0 . Square brackets in chemical drawings of (A) and (B) reflect uncertainty of the protonation state of the nitrogen atom.
Despite these ambiguities, the S-hydroxylated structure is a likely catalytic intermediate based on its geometry. Hydroxylation from the side of the cofactor that would result in the R-configuration is unlikely as this side is tightly packed against Trp696 (Fig. 6). In contrast, the other side of CTQ faces a relatively open pocket containing Asp678 and a number of water molecules engaged in a hydrogen bonding network. Weak electron density around the carboxylate indicates conformational flexibility of the side chain, which was also observed in the oxidized structure. As modeled, the closest approach of an Asp carboxylate oxygen to the glycine nitrogen and alpha carbon is 4.3 and 5.1 Å, respectively. The positioning of this residue suggests a likely function in activation of water for proton abstraction and/or hydroxylation.
Figure 6.

Water structure around the glycine adduct. (A and B) The adduct modeled as the product Schiff-base with relevant residues are shown as sticks and hydrogen bonds as dotted lines with indicated interatomic distances. 2Fo-Fc electron density is shown as blue mesh contoured to 1.0 .
Resonance Raman Characterization of GoxA CTQ —
RR spectra of air-oxidized GoxA obtained with 458-nm excitation exhibit intense v(C=C) indole ring vibrations at 1554 and 1517 cm−1 and keto v(C=O) modes at 1633 and 1653 cm−1 reminiscent of other tryptophan-based quinoprotein cofactors (Fig. 7) 32–35. Attempts were made to confirm these keto carbonyl stretching mode assignments through overnight exposure of GoxA to H218O water, but no significant changes were observed in the RR spectra.
Figure 7.

RR spectra of oxidized GoxA (black) and Gly-GoxA (red) with 458 nm excitation at room temperature; the final protein concentration was ~550 μM in 50 mM KPi pH 7.5.
Redox-cycling of GoxA through aerobic exposure to 5 equivalents of glycine in H218O contributes to an increase in non-specific fluorescent background in the RR spectra obtained with 458-nm excitation. However, RR spectra obtained with a 407-nm excitation provided further resonance enhancement of the Raman spectra of the oxidized CTQ cofactor and confirm the assignment of the 1633 cm−1 as a v(C=O) stretch since it shifts ~ 30-cm−1 after multiple turnovers in 18OH2 (Fig. S1). This v(C=O) frequency is 5 to 20 cm−1 higher than those measured earlier in MADH homologues, supporting a lack of strong hydrogen-bond donors to the keto carbonyls in GoxA and is best described as an in-phase stretching vibration of the two keto carbonyls 32.
Anaerobic exposure of GoxA to glycine results in a major reorganization of the RR spectra obtained with 458-nm excitation and a loss of the v(C=O) modes without the appearance of new signals above 1600 cm−1 (Fig. 7). As expected, changing the laser excitation from 458 to 647 nm produces significantly different RR spectra for Gly-GoxA, but all modes are observed with both resonance enhancement conditions (Fig. S2), and the absence of signal above 1600 cm−1 remains true with the red excitation. These results support the formation of a glycine adduct while ruling out an iminoquinone structure, which would be anticipated to show enhancement v(C=O) and v(C=N) vibrations. EPR spectra of the glycine adduct revealed only a very weak organic radical signal centered around g ~ 2.00 (Figure S3). This signal corresponds to < 0.2 equivalents of CTQ and rules out a stable semiquinone radical structure for the intermediate.
Figure 8 compares RR spectra of adducts prepared with Gly, 15N-Gly, 13C2-Gly, 13C213C1-Gly, and D2C2-Gly, in both H2O and D2O. The 15N-subsitution results in small but unambiguous shifts in all v(C=C) modes, i.e., 1587 (−1), 1556 (−2), 1511 (+2), and 1499 (−2) cm−1, as well as in the fingerprint v(C-C) bands, i.e., 1371 (−4), 1330 (−5), 1300 (−2) cm−1. The 13C2-Gly, 13C213C1-Gly substitutions minimally affect the v(C=C) modes, but they have a cumulative effect on the intense v(C-C) mode at 1371 (−5 and −11) cm−1 which supports a dominant single-bond character to the Gly C2-C1 bond in the adduct. The sensitivity of this 1371-cm−1 mode (−6) and nearby 1330 cm−1 band to the D2C2-Gly substitution highlights mixing of C-CH stretching and rocking vibrations. For all glycine isotopic substitutions, the size of the shifts are small and the number of RR bands affected is large, which support the formation of a Schiff-base adduct that extends the indole ring aromaticity to the entire glycyl moiety. H2O/D2O solvent exchange results in larger downshifts for nearly all the high- and mid-frequency RR bands and suggests the presence of multiple exchangeable protons in the chromophoric species. In addition to the indole N-H of CTQ, exchangeable protons in the glycine adduct may include quinol O-H, protonated Schiff-base, and C1-carboxylic acid O-H group.
Figure 8.

High-frequency RR spectra of isotopically labeled Gly-GoxA in H2O (black) and D2O (red) with 647 nm excitation at room temperature.
Intense low-frequency modes at 513 and 458 cm−1 correspond to bending/deformation modes. The 458-cm−1 band is unchanged by the 13C-isotope substitutions and only minimally by the 15N-Gly substitution, but this band exhibits a −14 cm−1 shift with D2C2-Gly and an additional −6 cm−1 shift with H2O/D2O exchange (Fig. 9). The 513-cm−1 shows a similar behavior. These H/D shifts are indicative of coupled hydrogen bending motions across the glycine C2 and the rest of the chromophore. It would be tempting to suggest that these cumulative H/D shifts are indicative of a protonated Schiff-base structure consistent with the other results obtained, but vibrational coupling across the indole adduct could just as easily lead to mixed glycine C2-H and indole N-H motions.
Figure 9.

Low-frequency RR spectra of isotopically labeled Gly-GoxA in H2O (black) and D2O (red) with 647 nm excitation at room temperature.
H/D exchange also affects bands best enhanced with the 458-nm excitation (Fig. 10). Specifically, bands at 1376, 1153 and 913 cm−1 shift minimally with 15N- and 13C-glycines but virtually disappear in favor of a strong band at 988 cm−1 upon H/D exchange. These H/D effects are consistent with couplings of C-C stretches and N-H and/or O-H rocks and are reminiscent of prior RR work on bacteriorhodopsin. Specifically, inferring contributions from in-plane N-H rocking and C-C stretching, Mathies and coworkers have associated a loss of a 1348 cm−1 band to a gain at 977 cm−1 upon H/D-exchange for the protonated Schiff-base of bacteriorhodopsin 36. This supports the suggestion from the results in Fig. 9 that the Schiff-base in the glycine-reduced GoxA is protonated.
Figure 10.

Combined low- and high-frequency RR spectra of isotopically labeled Gly-GoxA spectra obtained with 458 nm excitation at room temperature.
DISCUSSION
Several pieces of evidence identify the blue intermediate in GoxA catalysis as a product Schiff-base. First, among the possible intermediates only the substrate and product Schiff-base have the potential to exhibit the increased conjugation relative to the resting CTQ needed to account for the red shifts of its absorption features (Fig. 5). Secondly, RR shows the loss of keto v(C=O) modes at 1633 and 1653 cm−1 in the intermediate, indicating reduction of CTQ that would be observed in the product but not the substrate Schiff-base. Moreover, 15N-Gly results in small but unambiguous shifts in all v(C=C) modes between 1600–1500 cm−1 and fingerprint v(C-C) bands between 1400–1300 cm−1 without the appearance of strong vibrations above 1600 cm−1, supporting reduction of CTQ to the product Schiff-base rather than substrate Schiff-base, which would show enhanced v(C=O) and v(C=N) vibrations. 13C2-Gly and 13C2,13C1-Gly substitutions minimally affect the v(C=C) modes, but they show additive effect from these two C-atoms on the intense v(C-C) mode at 1371 (−5 with 13C2-Gly and −11 with 13C2,13C1-Gly) cm−1 which support a dominant single bond character to the C2-C1 bond in the adduct. H/D shifts further demonstrated extensive vibrational coupling between CTQ and the glycine adduct. Although not diagnostic, this may indicate that the product Schiff-base is protonated. Finally, the crystal structure of the adduct is consistent with a Schiff-base intermediate. Although a hydroxylated intermediate model best accommodated residual difference density, the presence of a proportion of oxidized CTQ with its associated water molecule could also explain this density. Based on the cumulative evidence, the final structure deposited to the Protein Data Bank is that of the product Schiff-base.
Schiff-base intermediates are invoked in the mechanisms for all tryptophylquinones and have been trapped and structurally characterized in the TTQ enzyme AADH 31 and the CTQ enzyme LodA 24. In fact, structures for an entire series of catalytic intermediates have been observed for AADH including substrate and product Schiff-bases as well as a hydroxylated intermediate 31. The adduct formed upon soaking of LodA crystals with lysine was modeled as a product Schiff-base 24. What these structures share in common is an Asp residue in close proximity to the substrate adduct where it can act as a general acid/base. In fact, to our knowledge all tryptophylquinone amine dehydrogenase structures show the presence of a structurally-conserved Asp residue near the quinone moiety of the cofactor 2, 3, 31, 37–40. In AADH, Asp128β is proposed to facilitate formation of the substrate Schiff-base, its deprotonation to form the product Schiff-base, as well as activation of a water for hydrolysis 31. It is positioned at a similar distance to the substrate adduct as what is observed in the GoxA structure, suggesting that GoxA follows a similar mechanism with Asp678 acting in the same capacity as Asp128β in AADH. Mutation of the corresponding active site Asp residue in MADH 41 and LodA 42 prevented maturation of the TTQ and CTQ cofactors, respectively, making their role in catalysis difficult to assess directly. However, in the TPQ enzyme copper amine oxidase, mutation of the invariant Asp298 to Ala resulted in a six order of magnitude drop in catalytic efficiency 43, highlighting the essential nature of this residue to catalysis.
A significant difference in the mechanism of GoxA catalysis from that described for the other quinoproteins, is that substrate hydrolysis depends on O2. Indeed, the Schiff-base intermediate is remarkably stable under anaerobic conditions. While the large number of hydrogen bonds to the bound substrate adduct may account for significant stabilization of this intermediate species, it cannot explain this puzzling oxygen dependence. Further study will be required to determine how hydrolysis is coupled to CTQ oxidation by molecular oxygen.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the staff at the Berkeley Center for Structural Biology at Lawrence Berkeley National Laboratory. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231. The atomic coordinates and structure factors for glycine-soaked GoxA (6EER) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
FUNDING SOURCES
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R37GM41574 (VLD).
Footnotes
SUPPORTING INFORMATION
Additional resonance Raman spectra and the EPR spectrum of Gly-GoxA.
REFERENCES
- [1].McIntire WS, Wemmer DE, Chistoserdov A, and Lidstrom ME (1991) A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase, Science 252, 817–824. [DOI] [PubMed] [Google Scholar]
- [2].Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, and Mathews FS (2001) Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking, Proc. Natl. Acad. Sci. U. S. A 98, 14268–14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Satoh A, Kim JK, Miyahara I, Devreese B, Vandenberghe I, Hacisalihoglu A, Okajima T, Kuroda S, Adachi O, Duine JA, Van Beeumen J, Tanizawa K, and Hirotsu K (2002) Crystal structure of quinohemoprotein amine dehydrogenase from Pseudomonas putida. Identification of a novel quinone cofactor encaged by multiple thioether cross-bridges, J. Biol. Chem 277, 2830–2834. [DOI] [PubMed] [Google Scholar]
- [4].Davidson VL (2005) Structure and mechanism of tryptophylquinone enzymes, Bioorg. Chem 33, 159–170. [DOI] [PubMed] [Google Scholar]
- [5].Yukl ET, and Davidson VL (2018) Diversity of structures, catalytic mechanisms and processes of cofactor biosynthesis of tryptophylquinone-bearing enzymes, Arch. Biochem. Biophys 654, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Husain M, and Davidson VL (1985) An inducible periplasmic blue copper protein from Paracoccus denitrificans. Purification, properties, and physiological role, J. Biol. Chem 260, 14626–14629. [PubMed] [Google Scholar]
- [7].Hyun YL, and Davidson VL (1995) Electron transfer reactions between aromatic amine dehydrogenase and azurin, Biochemistry 34, 12249–12254. [DOI] [PubMed] [Google Scholar]
- [8].Sun D, Ono K, Okajima T, Tanizawa K, Uchida M, Yamamoto Y, Mathews FS, and Davidson VL (2003) Chemical and kinetic reaction mechanisms of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans, Biochemistry 42, 10896–10903. [DOI] [PubMed] [Google Scholar]
- [9].Brooks HB, Jones LH, and Davidson VL (1993) Deuterium kinetic isotope effect and stopped-flow kinetic studies of the quinoprotein methylamine dehydrogenase, Biochemistry 32, 2725–2729. [DOI] [PubMed] [Google Scholar]
- [10].Basran J, Patel S, Sutcliffe MJ, and Scrutton NS (2001) Importance of barrier shape in enzyme-catalyzed reactions. Vibrationally assisted hydrogen tunneling in tryptophan tryptophylquinone-dependent amine dehydrogenases, J. Biol. Chem 276, 6234–6242. [DOI] [PubMed] [Google Scholar]
- [11].Bishop GR, Valente EJ, Whitehead TL, Brown KL, Hicks RP, and Davidson VL (1996) Direct Detection by 15N NMR of the Tryptophan Tryptophylquinone Aminoquinol Reaction Intermediate of Methylamine Dehydrogenase, J. Am. Chem. Soc 118, 12868–12869. [Google Scholar]
- [12].Bishop GR, Zhu Z, Whitehead TL, Hicks RP, and Davidson VL (1998) Identification of reaction products and intermediates of aromatic-amine dehydrogenase by 15N and 13C NMR, Biochem. J 330 (Pt 3), 1159–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mure M, Mills SA, and Klinman JP (2002) Catalytic mechanism of the topa quinone containing copper amine oxidases, Biochemistry 41, 9269–9278. [DOI] [PubMed] [Google Scholar]
- [14].Hartmann C, and Klinman JP (1987) Reductive trapping of substrate to bovine plasma amine oxidase, J. Biol. Chem 262, 962–965. [PubMed] [Google Scholar]
- [15].Mure M, and Klinman JP (1995) Model studies of topa quinone: synthesis and characterization of topa quinone derivatives, Methods Enzymol 258, 39–52. [DOI] [PubMed] [Google Scholar]
- [16].Campillo-Brocal JC, Chacón-Verdú MD, Lucas-Elío P, and Sánchez-Amat A (2015) Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity, BMC Genomics 16, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Andreo-Vidal A, Mamounis KJ, Sehanobish E, Avalos D, Campillo-Brocal JC, Sanchez-Amat A, Yukl ET, and Davidson VL (2018) Structure and Enzymatic Properties of an Unusual Cysteine Tryptophylquinone-Dependent Glycine Oxidase from Pseudoalteromonas luteoviolacea, Biochemistry 57, 1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sehanobish E, Shin S, Sanchez-Amat A, and Davidson VL (2014) Steady-state kinetic mechanism of LodA, a novel cysteine tryptophylquinone-dependent oxidase, FEBS Lett 588, 752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sehanobish E, Williamson HR, and Davidson VL (2016) Roles of Conserved Residues of the Glycine Oxidase GoxA in Controlling Activity, Cooperativity, Subunit Composition, and Cysteine Tryptophylquinone Biosynthesis, J. Biol. Chem 291, 23199–23207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lucas-Elio P, Hernandez P, Sanchez-Amat A, and Solano F (2005) Purification and partial characterization of marinocine, a new broad-spectrum antibacterial protein produced by Marinomonas mediterranea, Biochim. Biophys. Acta 1721, 193–203. [DOI] [PubMed] [Google Scholar]
- [21].Lucas-Elío P, Gómez D, Solano F, and Sanchez-Amat A (2006) The antimicrobial activity of marinocine, synthesized by Marinomonas mediterranea, is due to hydrogen peroxide generated by its lysine oxidase activity, J. Bacteriol 188, 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mai-Prochnow A, Lucas-Elio P, Egan S, Thomas T, Webb JS, Sanchez-Amat A, and Kjelleberg S (2008) Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several gram-negative bacteria, J. Bacteriol 190, 5493–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campillo-Brocal JC, Lucas-Elio P, and Sanchez-Amat A (2013) Identification in Marinomonas mediterranea of a novel quinoprotein with glycine oxidase activity, Microbiologyopen 2, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Okazaki S, Nakano S, Matsui D, Akaji S, Inagaki K, and Asano Y (2013) X-ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in L-lysine epsilon-oxidase from Marinomonas mediterranea, J. Biochem 154, 233–236. [DOI] [PubMed] [Google Scholar]
- [25].Kabsch W (2010) Xds, Acta Crystallogr. D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kabsch W (2010) Integration, scaling, space-group assignment and post-refinement, Acta Crystallogr. D Biol. Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [30].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, Mulholland AJ, Sutcliffe MJ, Scrutton NS, and Leys D (2006) Atomic description of an enzyme reaction dominated by proton tunneling, Science 312, 237–241. [DOI] [PubMed] [Google Scholar]
- [32].Backes G, Davidson VL, Huitema F, Duine JA, and Sanders-Loehr J (1991) Characterization of the tryptophan-derived quinone cofactor of methylamine dehydrogenase by resonance Raman spectroscopy, Biochemistry 30, 9201–9210. [DOI] [PubMed] [Google Scholar]
- [33].Gorren AC, Moënne-Loccoz P, Backes G, de Vries S, Sanders-Loehr J, and Duine JA (1995) Evidence for a methylammonium-binding site on methylamine dehydrogenase of Thiobacillus versutus, Biochemistry 34, 12926–12931. [DOI] [PubMed] [Google Scholar]
- [34].Moënne-Loccoz P, Nakamura N, Itoh S, Fukuzumi S, Gorren AC, Duine JA, and Sanders-Loehr J (1996) Electrostatic environment of the tryptophylquinone cofactor in methylamine dehydrogenase: evidence from resonance Raman spectroscopy of model compounds, Biochemistry 35, 4713–4720. [DOI] [PubMed] [Google Scholar]
- [35].McIntire WS, Bates JL, Brown DE, and Dooley DM (1991) Resonance Raman spectroscopy of methylamine dehydrogenase from bacterium W3A1, Biochemistry 30, 125–133. [DOI] [PubMed] [Google Scholar]
- [36].Smith SO, Braiman MS, Myers AB, Pardoen JA, Courtin JML, Winkel C, Lugtenburg J, and Mathies RA (1987) Vibrational analysis of the all-trans-retinal chromophore in light-adapted bacteriorhodopsin, J. Am. Chem. Soc 109, 3108–3125. [Google Scholar]
- [37].Cavalieri C, Biermann N, Vlasie MD, Einsle O, Merli A, Ferrari D, Rossi GL, and Ubbink M (2008) Structural comparison of crystal and solution states of the 138 kDa complex of methylamine dehydrogenase and amicyanin from Paracoccus versutus, Biochemistry 47, 6560–6570. [DOI] [PubMed] [Google Scholar]
- [38].Chen L, Durley R, Poliks BJ, Hamada K, Chen Z, Mathews FS, Davidson VL, Satow Y, Huizinga E, Vellieux FM, and Hol WGJ (1992) Crystal structure of an electron-transfer complex between methylamine dehydrogenase and amicyanin, Biochemistry 31, 4959–4964. [DOI] [PubMed] [Google Scholar]
- [39].Vellieux FM, Huitema F, Groendijk H, Kalk KH, Jzn JF, Jongejan JA, Duine JA, Petratos K, Drenth J, and Hol WG (1989) Structure of quinoprotein methylamine dehydrogenase at 2.25 A resolution, EMBO J 8, 2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vellieux FM, Kalk KH, Drenth J, and Hol WG (1990) Structure determination of quinoprotein methylamine dehydrogenase from Thiobacillus versutus, Acta Crystallogr. B 46 (Pt 6), 806–823. [DOI] [PubMed] [Google Scholar]
- [41].Jones LH, Pearson AR, Tang Y, Wilmot CM, and Davidson VL (2005) Active site aspartate residues are critical for tryptophan tryptophylquinone biogenesis in methylamine dehydrogenase, J. Biol. Chem 280, 17392–17396. [DOI] [PubMed] [Google Scholar]
- [42].Chacón-Verdú MD, Campillo-Brocal JC, Lucas-Elío P, Davidson VL, and Sánchez-Amat A (2015) Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-epsilon-oxidase and glycine oxidase from Marinomonas mediterranea, Biochim. Biophys. Acta 1854, 1123–1131. [DOI] [PubMed] [Google Scholar]
- [43].Chiu YC, Okajima T, Murakawa T, Uchida M, Taki M, Hirota S, Kim M, Yamaguchi H, Kawano Y, Kamiya N, Kuroda S, Hayashi H, Yamamoto Y, and Tanizawa K (2006) Kinetic and structural studies on the catalytic role of the aspartic acid residue conserved in copper amine oxidase, Biochemistry 45, 4105–4120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.