

Abstract
Clarifying the relationships between neuropsychiatric symptoms and Alzheimer’s disease (AD)-related pathology may open avenues for effective treatments. Here, we investigate the odds of developing neuropsychiatric symptoms across increasing burdens of neurofibrillary tangle and amyloid-β pathology. Participants who passed away between 2004 and 2014 underwent comprehensive neuropathologic evaluation at the Biobank for Aging Studies from the Faculty of Medicine at the University of São Paulo. Postmortem interviews with reliable informants were used to collect information regarding neuropsychiatric and cognitive status. Of 1,092 cases collected, those with any non-Alzheimer pathology were excluded, bringing the cohort to 455 cases. Braak staging was used to evaluate neurofibrillary tangle burden, and the CERAD neuropathology score was used to evaluate amyloid-β burden. The 12-item neuropsychiatric inventory was used to evaluate neuropsychiatric symptoms and CDR-SOB score was used to evaluate dementia status. In Braak I/II, significantly increased odds were detected for agitation, anxiety, appetite changes, depression, and sleep disturbances, compared to controls. Increased odds of agitation continue into Braak III/IV. Braak V/VI is associated with higher odds for delusions. No increased odds for neuropsychiatric symptoms were found to correlate with amyloid-β pathology. Increased odds of neuropsychiatric symptoms are associated with early neurofibrillary tangle pathology, suggesting that subcortical neurofibrillary tangle accumulation with minimal cortical pathology is sufficient to impact quality of life and that neuropsychiatric symptoms are a manifestation of AD biological processes.
Keywords: Alzheimer’s disease, amyloid plaques, anxiety, appetite behavior, depression, neurofibrillary tangles, neuropathology, neuropsychiatry, sleep, tau protein
INTRODUCTION
Alzheimer’s disease (AD) features a long prodrome during which pathology accumulates without causing neurocognitive symptoms [1]. AD is characterized by the accumulation of neurofibrillary tangles (NFT) and amyloid-β plaques, as well as neuron loss [2]. Neuron loss is the best predictor of cognitive decline, followed by NFT burden [3, 4]. The Braak staging system is a highly reproducible scheme capturing NFT spread in cortical regions [5]. PET-tau imaging studies corroborate the validity of Braak staging in vivo [6]. In Braak stages I/II, cortical NFT are confined to the entorhinal cortex and parts of hippocampus. NFT spread to paralimbic cortices in Braak stages III/IV and finally reach the higher-order association and primary neocortex in Braak stages V/VI [5]. On average, the first signs of cognitive decline occur during Braak stage III, meanwhile most cases at Braak stages I/II remain cognitively normal [5, 7].
Longitudinal studies suggest that a higher prevalence of neuropsychiatric symptoms (NPS) exist years preceding cognitive decline in individuals who later develop AD-type dementia, compared to age-matched controls [8–22]. Some suggest that NPS such as depression and anxiety are risk factors for AD [23–27]. However, a growing number of investigations using biomarkers suggest that NPS are driven by AD-related neuropathological changes [28–36]. Nevertheless, the question of whether NPS are part of the AD spectrum or exist independently or as epiphenomena is still open, especially in precognitive stages.
In addition to the cortex, NFT are abundant in subcortical nuclei. In fact, NFT pathology onset in key brainstem and hypothalamic nuclei consistently precedes cortical NFT pathology, correlates with neuronal death, and increases in severity along Braak stages [12, 37–45]. In 2011, Braak and colleagues revised their staging system to include the onset of NFT pathology in subcortical nuclei as a precortical stage [42, 46]. Among the subcortical nuclei accumulating NFT at Braak stage 0 are the locus coeruleus, dorsal raphe nucleus, and perifornical nucleus of the hypothalamus, which modulate anxiety, depression, and sleep disturbances, respectively. This early subcortical involvement corroborates claims that NPS may be part of the constellation of early clinical symptoms, corresponding to stage 2 of the NIA-AA neurocognitive staging system, and that effective treatment of these symptoms will require targeting AD pathology [47]. However, direct validation of this hypothesis is missing. Determining if AD pathology underlie specific NPS at pre-cognitive stages has important implications, including the possibility of using specific NPS as outcomes for clinical trials targeting prodromal AD phases, or designing effective treatment for NPS associated with AD.
Previous investigations have used postmortem evaluation to examine the relationships between AD pathology and several NPS [12, 48–53], but they either were limited by a relatively small sample [48, 49], were enriched for individuals at moderate or severe disease stages with cognitive decline, rarely excluded cases on the basis of co-existing pathologies which are confounders, or focused on one or few NPS.
Studies focused on determining the neurobiological basis of prodromal NPS in AD are lacking, and the relationships between pathology and NPS remain unclear. Despite recent improvements, neuroimaging and fluid-biomarker studies remain insensitive to subcortical AD pathology, likely critical loci for studies of this nature. To examine our hypothesis that AD pathology constitutes a neurobiological basis of NPS, even at prodromal stages, we analyze a large, population-based clinicopathological series to examine the relationships between the burdens of protein hallmarks of AD—NFT and amyloid-β—and domains from the neuropsychiatric inventory (NPI) [54].
MATERIALS AND METHODS
Participants
The local ethical committees approved this study, and all informants signed an informed consent. Cases were sourced from the Biobank for Aging Studies from the Faculty of Medicine at the University of São Paulo (BBAS-USP), formerly known as the Brain Bank of the Brazilian Aging Brain Study Group [7, 55]. Cases were collected between 2004 and 2014 by a city autopsy service. The inclusion criteria of the BBAS-USP includes age at death of 50 years or older and availability of a knowledgeable informant with at least weekly contact with the deceased in the six months before death. Cases with inconsistent data or brain tissue incompatible for neuropathological analyses were excluded [55]. Out of the 1,092 available cases, we excluded all cases with any level of non-AD pathology, including Lewy bodies, TDP-43 inclusions, primary tauopathies, and cerebrovascular changes, bringing the sample to 455 cases with a broad gradient of AD-type pathology across a range of clinical statuses.
Evaluation of symptoms
Scores from the 12-item NPI were collected in semi-structured postmortem informant interviews and reflect the participant’s status three months before death to avoid influence of peri-agonal events [54]. The NPI evaluates 12 domains: agitation, apathy, anxiety, appetite, delusions, depression, disinhibition, elation, hallucinations, irritability, motor, and sleep. Scores are typically calculated by multiplying frequency (1–4) and severity (1–3) for domains with any disturbance. As the median domain scores of our participants are zero, this was set as the cutoff for a negative diagnosis, with any score above zero receiving a positive diagnosis. Functional cognitive status was collected using the informant version of the Clinical Dementia Rating Sum of Boxes score (CDR-SOB) [56, 57]. The informant version of the CDR-SOB has been validated in the Brazilian population [58].
Neuropathological assessment
The BBAS-USP uses a 14-region immunohistochemistry panel to detect neurodegeneration and universally-accepted criteria to stage and diagnose cases [7, 55]. NFT pathology was assessed in formalin-fixed paraffin-embedded sections by immunostaining for phospho-Ser396/Ser404 tau (PHF-1, 1:2000; gift from Peter Davies, New York), scored by Braak stage, encoded into four groups following the conventional categorization [5, 59]: Braak stages 0, I/II, III/IV, and V/VI. Amyloid-β pathology was scored using CERAD-NP for the density of neuritic plaques, scored as none, sparse, moderate, or frequent [60].
Statistics
One-way ANOVA and Chi-squared tests were used to compare demographic and clinical metrics across groups. NPS were assessed across groups using logistic regression. The independent variables were the four Braak groups and CERAD-NP, using “Braak stage 0” and “CERAD-NP None” as the reference groups. The dependent variables were the NPI score and each of the domains (binary: 0/>0). Logistic regression models assessed the odds of having a NPS for given pathologic groups compared to reference groups. Models were adjusted for CDR-SOB. We further adjusted models for age, sex, years of education, and the other hallmark (e.g., adjusting for CERAD-NP in the Braak stage model). Multicollinearity was examined using variation inflation factors, with factors less than ten accepted. Statistics were conducted using RStudio. The α-level was set at 0.05 for two-tailed tests.
A secondary analysis using conditional logistic regression in age-matched pairs of Braak stage 0 and Braak stage I-II cases (n = 198) was done to examine these relationships free of possibly confounding age-related effects. This model was corrected for sex, years of education, CDR-SOB, and CERAD-NP.
RESULTS
For the 455 cases in the study, the mean age was 70.5 (SD = 12.1) years with a mean educational level of 4.7 (SD = 4.0) years. 52% of the sample was male and the median CDR-SOB was 1.83 (IQR = 4.83). Demographics for this sample, stratified by pathologic groups, are depicted in Tables 1 and 2.
Table 1.
Demographic and clinical statistics for the study population by Braak group (n = 455)
| Characteristic | Braak 0 (n = 129) |
Braak I-II (n = 200) |
Braak III-IV (n = 78) |
Braak V-VI (n = 48) |
p valuea |
|---|---|---|---|---|---|
| Age, Mean (SD), y | 61.9 (9.7) | 70.2 (11.0) | 79.1 (8.8) | 80.9 (9.2) | <0.001b |
| Education, Mean (SD), y | 5.7 (4.2) | 5.0 (4.1) | 3.1 (3.0) | 3.3 (3.1) | <0.001b |
| CDR-SOB, Median (IQR) | 0 (0) | 0 (0) | 0 (3) | 15.5 (18) | <0.001b |
| CERAD-NP score, Median (IQR)c | 0 (0) | 0 (0) | 2 (1) | 3 (0) | <0.001b |
| Male sex, No. (%) | 83 (64) | 107 (54) | 34 (44) | 12 (25) | <0.001 |
| NPI >0, No. (%) | 49 (3) | 112 (56) | 54 (69) | 36 (75) | <0.001 |
| NPI Agitation >0, No. (%)d | 7 (5) | 33 (17) | 10 (13) | 23 (48) | <0.001 |
| NPI Apathy >0, No. (%) | 15 (12) | 22 (11) | 13 (17) | 13 (27) | 0.024 |
| NPI Anxiety >0, No. (%) | 18 (14) | 49 (25) | 23 (30) | 13 (27) | 0.035 |
| NPI Appetite >0, No. (%) | 18 (14) | 51 (26) | 22 (28) | 24 (50) | <0.001 |
| NPI Delusions >0, No. (%)d | 2 (2) | 6 (3) | 8 (10) | 13 (27) | <0.001 |
| NPI Depression >0, No. (%)d | 13 (10) | 32 (16) | 14 (18) | 12 (25) | 0.087 |
| NPI Disinhibition >0, No. (%) | 2 (2) | 2 (1) | 6 (8) | 9 (19) | <0.001 |
| NPI Elation >0, No. (%) | 2 (2) | 7 (3) | 2 (3) | 1 (2) | 0.75 |
| NPI Hallucinations >0, No. (%) | 4 (3 | 6 (3) | 7 (9) | 17 (35) | <0.001 |
| NPI Irritability >0, No. (%) | 12 (9) | 29 (15) | 15 (19) | 11 (23) | 0.077 |
| NPI Motor >0, No. (%) | 3 (2) | 4 (2) | 2 (3) | 12 (25) | <0.001 |
| NPI Sleep >0, No. (%) | 15 (12) | 41 (21) | 18 (23) | 16 (33) | 0.009 |
p value determined using chi-square test unless otherwise stated
p value determined using one-way ANOVA
CERAD-NP scores were assigned as 0 for none, 1 for sparse, 2 for moderate, and 3 for frequent
Agitation, delusions, and depression have one missing case each
Table 2.
Demographic and clinical statistics for the study population by CERAD-NP (n = 455)
| Characteristic | None (n = 293) |
Sparse (n = 50) |
Moderate (n = 57) |
Frequent (n = 55) |
p valuea |
|---|---|---|---|---|---|
| Age, Mean (SD), y | 66.1 (11.3) | 75.8 (10.0) | 79.8 (7.8) | 79.4 (9.7) | <0.001b |
| Education, Mean (SD), y | 5.3 (4.2) | 4.5 (4.0) | 3.1 (2.7) | 3.2 (3.1) | <0.001b |
| CDR-SOB, Median (IQR) | 0 (0) | 0 (0) | 0 (5) | 12 (18) | <0.001b |
| Braak stage, Median (IQR) | 1 (2) | 2 (2) | 3 (1) | 5 (1.5) | <0.001b |
| Male sex, No. (%) | 166 (57) | 28 (56) | 26 (46) | 16 (29) | 0.002 |
| NPI >0, No. (%) | 141 (48) | 28 (56) | 40 (70) | 42 (76) | <0.001 |
| NPI Agitation >0, No. (%)c | 35 (12) | 8 (16) | 7 (12) | 23 (42) | <0.001 |
| NPI Apathy >0, No. (%) | 37 (13) | 5 (10) | 10 (18) | 11 (20) | 0.34 |
| NPI Anxiety >0, No. (%) | 57 (19) | 14 (28) | 17 (30) | 15 (27) | 0.18 |
| NPI Appetite >0, No. (%) | 62 (21) | 14 (28) | 13 (23) | 26 (47) | <0.001 |
| NPI Delusions >0, No. (%)c | 7 (2) | 1 (2) | 9 (16) | 12 (22) | <0.001 |
| NPI Depression >0, No. (%)c | 41 (14) | 6 (12) | 11 (19) | 13 (24) | 0.23 |
| NPI Disinhibition >0, No. (%) | 4 (1) | 1 (2) | 3 (5) | 11 (20) | <0.001 |
| NPI Elation >0, No. (%) | 9 (3) | 0 (0) | 2 (3) | 1 (2) | 0.60 |
| NPI Hallucinations >0, No. (%) | 10 (3) | 2 (4) | 6 (11) | 16 (29) | <0.001 |
| NPI Irritability >0, No. (%) | 34 (12) | 5 (10 | 13 (23) | 15 (27) | 0.005 |
| NPI Motor >0, No. (%) | 7 (2) | 1 (2) | 2 (3) | 11 (20) | <0.001 |
| NPI Sleep >0, No. (%) | 47 (16) | 10 (20) | 14 (25) | 19 (35) | 0.012 |
p value determined using chi-square test unless otherwise stated
p value determined using one-way ANOVA
Agitation, delusions, and depression have one missing case each.
Models comparing Braak stages I/II to Braak stage 0, adjusted for CDR-SOB, age, sex, education level, and CERAD-NP, showed significantly higher odds of agitation, anxiety, appetite changes, depression, sleep disturbances, and overall NPI. Increased odds for agitation continued in Braak stages III/IV. Braak stages V/VI had higher odds of delusions compared to Braak stage 0 (Table 3).
Table 3.
Odds ratios and 95% confidence intervals for neuropsychiatric symptoms by Braak stage group (n = 455)
| Symptom | Braak stage group | OR, [95% CI]a | p value |
|---|---|---|---|
| Agitationb | Braak I-II | 6.08, [2.44, 17.52] | <0.001 |
| Braak III-IV | 9.16, [1.35, 58.95] | 0.019 | |
| Braak V-VI | 10.67, [0.19, 411.83] | 0.209 | |
| Apathy | Braak I-II | 0.88, [0.39, 2.02] | 0.758 |
| Braak III-IV | 2.4, [0.59, 9.04] | 0.201 | |
| Braak V-VI | 31.76, [0.59, 1729.08] | 0.073 | |
| Anxiety | Braak I-II | 2.14, [1.13, 4.19] | 0.022 |
| Braak III-IV | 2.86, [0.82, 9.67] | 0.092 | |
| Braak V-VI | 3.86, [0.09, 106.65] | 0.44 | |
| Appetite | Braak I-II | 2.57, [1.37, 5.01] | 0.004 |
| Braak III-IV | 2.75, [0.81, 9.14] | 0.10 | |
| Braak V-VI | 17.34, [0.67, 558.88] | 0.082 | |
| Delusionsb | Braak I-II | 3.09, [0.58, 24.75] | 0.21 |
| Braak III-IV | 0.26, [0.00, 8.89] | 0.50 | |
| Braak V-VI | 262.7, [4.35, 17965.89] | 0.006 | |
| Depressionb | Braak I-II | 2.32, [1.08, 5.22] | 0.035 |
| Braak III-IV | 1.15, [0.22, 5.04] | 0.86 | |
| Braak V-VI | 0.14, [0.00, 10.94] | 0.43 | |
| Disinhibition | Braak I-II | 2.51, [0.23, 43.50] | 0.45 |
| Braak III-IV | 12.69, [0.25, 385.58] | 0.15 | |
| Braak V-VI | 0.00, [0.00, 5.31] | 0.17 | |
| Elation | Braak I-II | 3.97, [0.72, 39.08] | 0.15 |
| Braak III-IV | 1.21, [0.01, 40.51] | 0.93 | |
| Braak V-VI | 0.04, [0.00, 1942.23] | 0.71 | |
| Hallucinations | Braak I-II | 1.93, [0.48, 8.85] | 0.36 |
| Braak III-IV | 0.73, [0.03, 8.94] | 0.82 | |
| Braak V-VI | 18.91, [0.21, 941.76] | 0.15 | |
| Irritability | Braak I-II | 1.71, [0.80, 3.84] | 0.18 |
| Braak III-IV | 0.76, [0.13, 3.58] | 0.74 | |
| Braak V-VI | 0.05, [0.00, 6.00] | 0.27 | |
| Motor | Braak I-II | 0.97, [0.19, 5.37] | 0.97 |
| Braak III-IV | 1.01, [0.02, 16.79] | 0.99 | |
| Braak V-VI | 2.88, [0.00, 382.79] | 0.72 | |
| Sleep | Braak I-II | 2.33, [1.18, 4.82] | 0.018 |
| Braak III-IV | 0.76, [0.15, 3.21] | 0.72 | |
| Braak V-VI | 4.8, [0.11, 150.12] | 0.38 | |
| Total NPI | Braak I-II | 2.54, [1.53, 4.30] | <0.001 |
| Braak III-IV | 2.33, [0.84, 6.67] | 0.11 | |
| Braak V-VI | 1.83, [0.08, 40.23] | 0.69 |
Models adjusted for age, sex, years of education, CERAD-NP, and CDR-SOB. Reference group: Braak stage = None
Agitation, delusions, and depression have one missing case each.
The models for the associations between NPS and CERAD-NP, adjusted for CDR-SOB, age, sex, years of education, and Braak stage, showed lower odds of agitation and appetite in cases with a CERAD-NP of “moderate” compared to those with a CERAD-NP of “none”. In addition, cases with a CERAD-NP of “frequent” had lower odds of apathy, depression, and elation (Table 4).
Table 4.
Odds ratios and 95% confidence intervals for neuropsychiatric symptoms by CERAD-NP score (n = 455)
| Symptom | CERAD-NP Score | OR, [95% CI]a | p value |
|---|---|---|---|
| Agitationb | Sparse | 1.02, [0.35, 2.69] | 0.97 |
| Moderate | 0.42, [0.17, 0.87] | 0.032 | |
| Frequent | 0.65, [0.35, 1.11] | 0.14 | |
| Apathy | Sparse | 0.39, [0.10, 1.17] | 0.12 |
| Moderate | 0.56, [0.26, 1.07] | 0.01 | |
| Frequent | 0.25, [0.08, 0.59] | 0.006 | |
| Anxiety | Sparse | 1.28, [0.58, 2.71] | 0.53 |
| Moderate | 1.03, [0.65, 1.60] | 0.90 | |
| Frequent | 0.70, [0.43, 1.10] | 0.14 | |
| Appetite | Sparse | 1.09, [0.49, 2.30] | 0.83 |
| Moderate | 0.57, [0.33, 0.93] | 0.03 | |
| Frequent | 0.70, [0.44, 1.07] | 0.11 | |
| Delusionsb | Sparse | 0.53, [0.02, 5.36] | 0.63 |
| Moderate | 2.23, [0.66, 7.47] | 0.19 | |
| Frequent | 1.23, [0.49, 2.82] | 0.64 | |
| Depressionb | Sparse | 0.64, [0.19, 1.81] | 0.43 |
| Moderate | 0.80, [0.44, 1.40] | 0.46 | |
| Frequent | 0.54, [0.28, 0.95] | 0.049 | |
| Disinhibition | Sparse | 1.48, [0.06, 13.70] | 0.75 |
| Moderate | 2.70, [0.63, 11.28] | 0.17 | |
| Frequent | 0.87, [0.22, 2.93] | 0.84 | |
| Elation | Sparse | 0, [NA] | 0.99 |
| Moderate | 0.57, [0.13, 1.94] | 0.42 | |
| Frequent | 0.16, [0.03, 0.71] | 0.025 | |
| Hallucinations | Sparse | 0.86, [0.10, 4.98] | 0.88 |
| Moderate | 0.86, [0.21, 2.86] | 0.81 | |
| Frequent | 0.73, [0.27, 1.73] | 0.50 | |
| Irritability | Sparse | 0.74, [0.22, 2.05] | 0.59 |
| Moderate | 1.36, [0.80, 2.26] | 0.24 | |
| Frequent | 0.97, [0.57, 1.59] | 0.92 | |
| Motor | Sparse | 0.91, [0.04, 6.67] | 0.94 |
| Moderate | 0.89, [0.20, 3.06] | 0.86 | |
| Frequent | 1.22, [0.47, 2.86] | 0.66 | |
| Sleep | Sparse | 1.18, [0.48, 2.69] | 0.71 |
| Moderate | 1.05, [0.62, 1.74] | 0.85 | |
| Frequent | 0.94, [0.57, 1.50] | 0.82 | |
| Total NPI | Sparse | 1.03, [0.52, 2.04] | 0.94 |
| Moderate | 0.92, [0.61, 1.39] | 0.70 | |
| Frequent | 0.76, [0.52, 1.09] | 0.14 |
Models adjusted for age, sex, years of education, Braak stage, and CDR-SOB. Reference group: CERAD-NP = None
Agitation, delusions, and depression have one missing case each.
To examine these relationships free of potential age-related effects, we used a conditional logistic regression model in age-matched subgroups of the Braak 0 (controls) and Braak I-II cases corrected for sex, education, CDR-SOB, and CERAD-NP. In agreement with our primary analysis, increased odds were detected for agitation (OR, [95% CI] = 17.6, [1.61, 192.4]; p = 0.019), anxiety (OR, [95% CI] = 2.79, [1.08, 7.14]; p = 0.033), appetite changes (OR, [95% CI] = 2.87, [1.34, 6.17]; p = 0.006), and sleep dysfunction (OR, [95% CI] = 2.95, [1.13, 7.71]; p = 0.028). Although increased odds for depression was not significantly correlated with Braak stages I-II in this secondary analysis, a tendency for increased odds of depression remained (OR, [95% CI] = 3.81 [0.78, 18.5]; p = 0.098). Braak stage I-II did not correlate with higher odds for any of the other seven NPI domains, aligning with the primary analysis.
DISCUSSION
NPS are frequent across all AD stages, adding substantial burden for caregivers and patients [8–21]. By employing a large population-based postmortem sample of cases over 50 years of age, encompassing healthy controls and individuals across the AD spectrum, this study is well-positioned to investigate how NPS correlate with AD pathological hallmarks, particularly in precognitive stages. All cases were free of non-AD pathology, minimizing confounders introduced by co-occurring pathology. We found significant positive associations between NFT burden and several NPS, as early as Braak stages I/II, at which time the NFT are confined to subcortical and allocortical areas and, in most cases, the burden of amyloid-β plaques is very low (Fig. 1) [42, 46, 61]. These results support the hypothesis that NFT-related neurodegeneration constitutes the biological basis of NPS in AD, beginning in pre-cognitive stages.
Fig. 1.
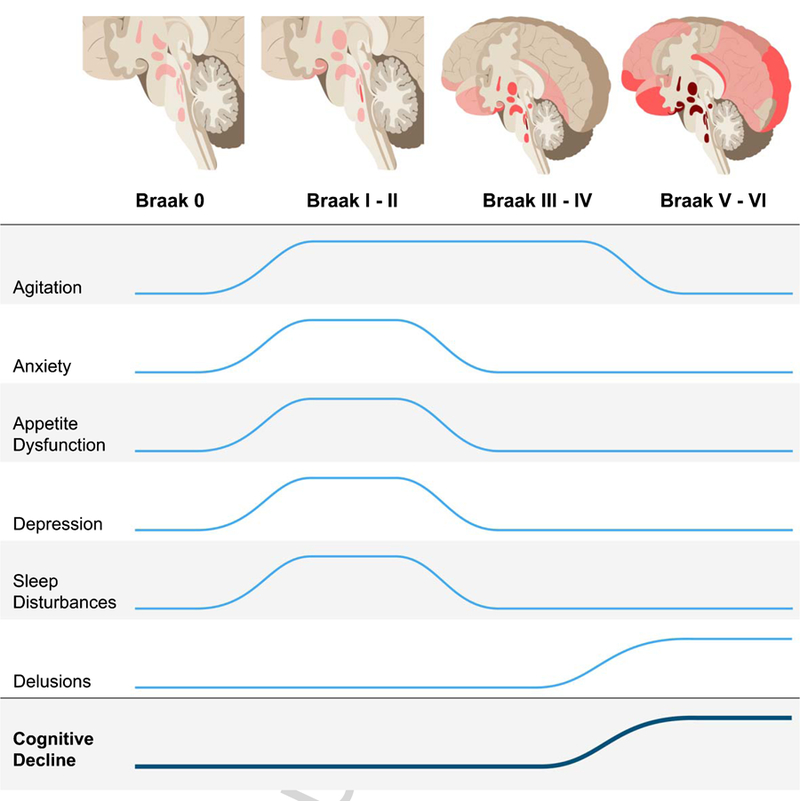
Increased odds for neuropsychiatric symptoms across Alzheimer’s disease neurofibrillary tangle progression. There are significantly higher odds of developing agitation, anxiety, appetite dysfunction, depression, and sleep disturbances during Braak stages I/II, when pathology is restricted to subcortical regions and the transentorhinal cortex, after correcting for age, sex, years of education, Braak stage, and CDR-SOB. After Braak stages I/II, the odds of anxiety, appetite dysfunction, depression, and sleep disturbances are not significant when correcting for CDR-SOB. Agitation remains at significantly increased odds during Braak stages III/IV. Increased odds of delusions are associated with Braak stages V/VI. This illustrates that neuropsychiatric symptoms could possibly be used as a clinical marker in stages preceding the onset cognitive decline. The trajectory for cognitive decline represents median CDR-SOB score (Table 1).
An increasing number of studies highlight that early vulnerability of brainstem and hypothalamic nuclei to NFT pathology occurs before involvement of the entorhinal cortex in AD [12, 38–44]. This burden of NFT pathology in subcortical nuclei increases with Braak stage. Taken together with the literature documenting the functional anatomy of these regions, our results showing that AD pathology may manifest first as NPS—agitation, anxiety, appetite dysfunction, depression, and sleep disturbances—are not surprising [12, 14–20, 26, 38–45]. Given the functional correlates of these regions affected by early NFT pathology, we can develop testable hypotheses to guide further research on mechanisms that drive early NPS in AD.
For example, dysregulation of norepinephrine due to early NFT accumulation in the locus coeruleus may contribute to the increased odds for agitation in Braak I–IV [62–64]. Several experimental studies have demonstrated increased agitation in response to dysfunction of norepinephrine-producing neurons, including those of the locus coeruleus [65–67]. In a review of neuroanatomical correlates of NPS in AD, Rosenberg and colleagues highlight studies correlating agitation with volumetric loss in the frontal cortex, anterior and posterior cingulate cortices, insula, amygdala, and hippocampus, rather than subcortical pathology [63]. Other studies detected a correlation between agitation and NFT burden in the orbitofrontal and anterior cingulate cortices in post-mortem samples [48, 52]. While these studies suggest that NFT in these regions contribute to agitation, they were confined to examination of cortical regions in symptomatic individuals. Thus, it is challenging to tease out the contribution of early subcortical and cortical pathology with the patients at more severe disease stages with possible co-occurring pathology.
In fairly large clinicopathological studies, depression has been correlated with an increased burden of cortical plaques and tangles [50, 51]. Similar to agitation, NFT burden in the orbitofrontal and anterior cingulate cortices has also been correlated with depression, but this effect was only seen in later stages [48, 52]. Instead, early NFT-induced dysfunction of neurons in the locus coeruleus and dorsal raphe nucleus is likely driving our findings of increased odds for depression in Braak stages I/II, in line with the monoamine hypothesis of depression [53, 68–70].
Anxiety has been described as an early symptom in AD and a risk factor for dementia [26, 36, 71, 72]. Historically, the norepinephrine-producing locus coeruleus has been viewed as central in modulating networks involved in anxiety [73, 74]. So-called “anxiety cells” have also been identified in CA1 of the hippocampus, an early cortical site to accumulate NFT (Braak stage II) [5, 75]. Thus, both locus coeruleus and hippocampal involvement in Braak stages I/II align with our findings of increased anxiety at these stages [38, 39, 41]. Donovan and colleagues have recently shown that anxiety is an early clinical manifestation of AD [36]. While this investigation was based on neuroimaging measurements of amyloid-β, the findings may be driven by concurrent subcortical NFT pathology, which could not be measured in vivo. Other neuroimaging studies correlate changes in the amygdala, insular cortex, and anterior cingulate cortex (Braak stage III) with several anxiety disorders in symptomatic patients, suggesting that these regions may also contribute to anxiety, in addition to earlier subcortical pathology [76, 77].
The ventromedial and lateral hypothalamus modulate satiety and hunger, respectively, as well as the locus coeruleus [78–83]. These nuclei are known to develop NFT early in AD, which may be driving the higher odds of appetite dysfunction we detect in Braak stages I/II, suggesting that there is a neuropathological basis driving dysfunction related to eating habits and preferences that has been associated with Alzheimer-type dementia [38, 84]. Further work is needed to refine the features of these appetite changes.
Many AD patients experience sleep disturbances even preceding cognitive decline [85–87]. Wakefulness is governed by high firing rates of wake-promoting neurons including neurons of the locus coeruleus, dorsal raphe nucleus, tuberomammillary nucleus, and lateral hypothalamus, all of which are susceptible to NFT in early AD [38, 39, 41, 88–90]. Recent evidence linking poor sleep to exacerbation of cortical pathology suggests existence of a positive-feedback loop leading to an overall increase in pathology [91, 92].
Finally, we detected high odds of delusions in subjects at Braak stages V/VI. Delusions manifest in several neurodegenerative conditions including Lewy body disease and frontotemporal dementia, and their anatomical correlates are complex [63, 93–95]. In AD, delusions have been associated with degeneration in the right inferior frontal gyrus, inferior parietal lobe, and claustrum, aligning with our findings of delusions beginning late in Braak stages V/VI [96]. As delusions are infrequent in healthy individuals and remain low until late stages, the odds ratio for delusions at Braak stages V/VI is very high, even after controlling for possible confounders (Table 1). Delusions in AD patients are often considered to be an indication of overlapping Lewy body disease [97]. However, in this series, any cases with α-synuclein positivity were excluded, suggesting that at late stages, delusions may be driven by cortical NFT pathology itself.
Clinical and epidemiological studies suggest a correlation between apathy and Alzheimer-type dementia [8–11, 13, 14, 16, 17, 19–21], but we failed to find such correlations here. Most studies of apathy in AD have been conducted in vivo, without autopsy verification. For this reason, they likely included mixed pathologies, such as Lewy body disease, which can increase the risk of apathy [31, 98, 99]. Inflammatory dysregulation, which is not captured by Braak staging or CERAD-NP, is another possible underpinning of apathy [100–103]. The neuroanatomical basis of apathy in AD has also been suggested to include dysfunction of the anterior cingulate, dorsolateral prefrontal cortex, and left medial frontal cortex [48, 49, 104]. Atrophy in the caudate, temporoparietal junction, temporal gyri, and frontal operculum-anterior insula region, detected by neuroimaging, has been associated with apathy in frontotemporal dementia [105]. Studies of apathy in AD focus on cortical changes and typically do not correct for cognitive status or co-pathologies which may explain the differences with our findings. Further, protein hallmarks such as NFT may only be related to mild apathy symptoms, to which the NPI-informant report may be insensitive, compared to direct clinical assessment.
The negative associations between amyloid-β severity, measured by CERAD-NP, and NPS have been observed previously [48, 49, 51, 106]. There are several possible explanations for this finding. In more advanced neuropathological AD stages, the severe cognitive impairment may overshadow NPS detection by informants. Additionally, in early AD stages, amyloid-β pathology is confined to neocortical regions that are less likely to modulate NPS than the subcortical regions already carrying substantial NFT burden, making possible correlations between amyloid-β and NPS very weak and the results artificial. Evidence that soluble amyloid-β pathology is a better predictor of clinical symptoms than plaques could also explain the negative relationships [107]. It is also possible that our correction of each model for Braak, due to the collinearity between CERAD-NP and Braak stage, could drive the negative relationships we are finding. Nonetheless, these findings add to mounting evidence that NFT, rather than amyloid-β plaque pathology drives the clinical phenotype of AD.
Despite our efforts to minimize weaknesses in the study design, remaining shortcomings should be noted. First, as neuroimaging studies have shown more robust associations between NPS and pathology longitudinally than cross-sectionally, the inherent cross-sectional design of clinicopathological studies prevents us from tracking domain-specific changes and longitudinal relationships among NPS. However, neuropathology remains the gold standard for diagnosing neurodegenerative disease; despite recent advances, methods for staging AD pathological markers in vivo fail to reach the same level of sensitivity and specific pathological prediction. This discrepancy is particularly prominent at early AD stages when tau burden is primarily subcortical. Second, clinicopathological studies such as this one are often criticized for being descriptive or correlational rather than mechanistic. However, clinicopathological studies play an important role in the advancement of medicine by providing the foundation and context for mechanistic studies. Third, despite universal use in clinical and research settings to evaluate NPS, NPI informant reports may not be sensitive to mild symptoms such as dysthymia. Furthermore, NPI scores are informant-dependent and thus, may be imprecise. To minimize potential bias related to this limitation, the BBAS-USP requires informants to have had close weekly contact with the deceased in the six months prior to death. Fourth, some NPS (i.e., hallucinations and elation) were infrequent in our sample, possibly yielding imprecise estimates for the association of these symptoms with neuropathology. Finally, a lack of correlation between amyloid-β plaque scores and NPS may be derived from bias caused by the metric used to assess amyloid-β burden. CERAD-NP measures the highest cortical density of amyloid-β neuritic plaques, which represents a different, albeit related, quality of amyloid-β than the anatomically-defined Braak stage represents for NFT. CERAD-NP scores have a strong ceiling effect and estimate plaque burden in only a small fraction of the brain. Despite this shortcoming, CERAD-NP is sensitive to the earliest brain area to develop neuritic plaques. In our sample, 63% of individuals lack any amyloid neuritic plaques, and the remaining cases represent all CERAD-NP scores. Thus, we would expect to find positive association with amyloid-β burden and NPS in early AD stages if existing. Moreover, extensive literature including clinicopathological and biomarker studies suggests little impact of amyloid-β pathology and several other cognitive metrics [7]. Thal phase is an alternative and perhaps more appropriate metric to use for measuring amyloid-β pathology; however, as this metric was only introduced in 2011, it is unavailable for the majority of our cases. Nevertheless, a need to investigate possible contributions of amyloid-β to NPS further remains.
This study also offers several considerable strengths. We studied a unique population-based clinicopathological series free of biases typically found in convenience samples that are usually enriched for later AD stages and oldest-old individuals. 28% of our large series of individuals over 50 years of age lacked cortical AD changes and another 44% had AD-tau changes limited to the limbic areas and low amyloid-β burden, which is unprecedented. Moreover, due to the comprehensive postmortem examination, we were able to exclude all cases with non-AD pathology to isolate the independent effects of AD-tau and AD-amyloid-β deposition, a feat not achievable by other available methods. Additionally, in the population sampled, the rates of antipsychotic and antidepressant use are low, which could otherwise mask NPS [108].
In this study, we identify increased risk for five NPS associated with NFT pathology, four of which were associated with early stages (Fig. 1). Our results strongly support the hypothesis that NPS are part of the AD clinical spectrum and are a manifestation of NFT-related neurodegeneration. Further research is needed to clarify the relationships between subcortical pathology and early NPS, as well as determine the prevalence of specific NPS subtypes. By understanding the pathophysiology driving early symptoms, better strategies can be developed to diagnose, target, and effectively treat AD before significant cognitive decline in advanced stages. In similar fashion, neuropathological evidence that Parkinson’s disease develops in non-dopaminergic nuclei, preceding substantia nigra involvement, recharacterized Parkinson’s disease as a multi-systemic disease with important non-motor components that precede motor symptoms. Such a change in paradigm opened room for improved management of non-motor symptoms and encouraged basic and clinical research to properly treat these symptoms. The positive outcomes from this revised view of Parkinson’s disease illustrate the importance of understanding the relationships between NPS and AD pathology and will hopefully encourage the field to follow suit.
ACKNOWLEDGMENTS
This study was supported financially by NIH Grant #R01AG040311 and the John Douglas French Alzheimer’s Foundation. Additional financial support was provided by FMUSP LIM-22, FAPESP, CAPES, and Hospital Israelita Albert Einstein, São Paulo. Dr. Grinberg mentoring effort is supported by NIH Grant #K24AG053435. Dr. Resende is an Atlantic Fellow for Equity in Brain Health at the Global Brain Health Institute and thanks the program for their support on this study. Dr. Gatchel has received funding from the BrightFocus Foundation and the Alzheimer’s Association and thanks them for their support.
We thank Shayna Herns (University of California, San Diego School of Medicine) for helpful editorial feedback, the families of the brain donors, the physicians, and staff of the São Paulo Autopsy service for their unconditional support, and the Brazilian Aging Brain Study Group for their assistance with data collection.
Footnotes
Authors’ disclosures available online (www.j-alz.com/manuscript-disclosures/18-0688r1).
REFERENCES
- [1].Morris JC (2005) Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord 19, 163–165. [DOI] [PubMed] [Google Scholar]
- [2].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on Aging, Alzheimer’s Association (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41, 17–24. [DOI] [PubMed] [Google Scholar]
- [4].Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. [DOI] [PubMed] [Google Scholar]
- [5].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [6].Scholl M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ (2016) PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Suemoto CK, Ferretti-Rebustini RE, Rodriguez RD, Leite RE, Soterio L, Brucki SM, Spera RR, Cippiciani TM, Farfel JM, Chiavegatto Filho A, Naslavsky MS, Zatz M, Pasqualucci CA, Jacob-Filho W, Nitrini R, Grinberg LT (2017) Neuropathological diagnoses and clinical correlates in older adults in Brazil: A cross-sectional study. PLoS Med 14, e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van der Linde RM, Dening T, Stephan BC, Prina AM, Evans E, Brayne C (2016) Longitudinal course of behavioural and psychological symptoms of dementia: Systematic review. Br J Psychiatry 209, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aalten P, Verhey FR, Boziki M, Bullock R, Byrne EJ, Camus V, Caputo M, Collins D, De Deyn PP, Elina K, Frisoni G, Girtler N, Holmes C, Hurt C, Marriott A, Mecocci P, Nobili F, Ousset PJ, Reynish E, Salmon E, Tsolaki M, Vellas B, Robert PH (2007) Neuropsychiatric syndromes in dementia. Results from the European Alzheimer Disease Consortium: Part I. Dement Geriatr Cogn Disord 24, 457–463. [DOI] [PubMed] [Google Scholar]
- [10].Assal F, Cummings JL (2002) Neuropsychiatric symptoms in the dementias. Curr Opin Neurol 15, 445–450. [DOI] [PubMed] [Google Scholar]
- [11].Lanctôt KL, Amatniek J, Ancoli-Israel S, Arnold SE, Ballard C, Cohen-Mansfield J, Ismail Z, Lyketsos C, Miller DS, Musiek E, Osorio RS, Rosenberg PB, Satlin A, Steffens D, Tariot P, Bain LJ, Carrillo MC, Hendrix JA, Jurgens H, Boot B (2017) Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimers Dement (N Y) 3, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, Shah RC, Schneider JA, Arnold SE, Bennett DA (2013) Brainstem aminergic nuclei and late-life depressive symptoms. JAMA Psychiatry 70, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, Cedarbaum J, Brashear R, Miller DS (2011) Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement 7, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Edwards ER, Spira AP, Barnes DE, Yaffe K (2009) Neuropsychiatric symptoms in mild cognitive impairment: Differences by subtype and progression to dementia. Int J Geriatr Psychiatry 24, 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D, Duara R, Kukull WA, Chui H, Edeki T, Griffith PA, Friedland RP, Bachman D, Farrer L (2003) Depression as a risk factor for Alzheimer disease: The MIRAGE study. Arch Neurol 60, 753–759. [DOI] [PubMed] [Google Scholar]
- [16].Rajan KB, Wilson RS, Weuve J, Barnes LL, Evans DA (2015) Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 85, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH (2011) Toward defining the pre-clinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ganguli M, Du Y, Dodge HH, Ratcliff GG, Chang CC (2006) Depressive symptoms and cognitive decline in late study. Arch Gen Psychiatry 63, 153–160. [DOI] [PubMed] [Google Scholar]
- [19].Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA (2006) Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology 27, 143–153. [DOI] [PubMed] [Google Scholar]
- [20].Onyike CU, Sheppard JM, Tschanz JT, Norton MC, Green RC, Steinberg M,Welsh-Bohmer KA, Breitner JC, Lyketsos CG (2007) Epidemiology of apathy in older adults: The Cache County Study. Am J Geriatr Psychiatry 15, 365–375. [DOI] [PubMed] [Google Scholar]
- [21].Savva GM, Zaccai J, Matthews FE, Davidson JE, McKeith I, Brayne C, Medical Research Council Cognitive F, Ageing S (2009) Prevalence, correlates and course of behavioural and psychological symptoms of dementia in the population. Br J Psychiatry 194, 212–219. [DOI] [PubMed] [Google Scholar]
- [22].Li XL, Hu N, Tan MS, Yu JT, Tan L (2014) Behavioral and psychological symptoms in Alzheimer’s disease. Biomed Res Int 2014, 927804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kessing LV, Andersen PK (2004) Does the risk of developing dementia increase with the number of episodes in patients with depressive disorder and in patients with bipolar disorder? J Neurol Neurosurg Psychiatry 75, 1662–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D (2006) Depression and risk for Alzheimer disease: Systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry 63, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gualtieri CT, Johnson LG (2008) Age-related cognitive decline in patients with mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 32, 962–967. [DOI] [PubMed] [Google Scholar]
- [26].Acosta I, Borges G, Aguirre-Hernandez R, Sosa AL, Prince M, 10/66 Dementia Research Group (2017) Neuropsychiatric symptoms as risk factors of dementia in a Mexican population: A 10/66 Dementia Research Group study. Alzheimers Dement 14, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yaffe K (2018) Modifiable risk factors and prevention of dementia: What is the latest evidence? JAMA Intern Med 178, 281–282. [DOI] [PubMed] [Google Scholar]
- [28].Bensamoun D, Guignard R, Furst AJ, Derreumaux A, Manera V, Darcourt J, Benoit M, Robert PH, David R (2016) Associations between neuropsychiatric symptoms and cerebral amyloid deposition in cognitively impaired elderly people. J Alzheimers Dis 49, 387–398. [DOI] [PubMed] [Google Scholar]
- [29].Boublay N, Schott AM, Krolak-Salmon P (2016) Neuroimaging correlates of neuropsychiatric symptoms in Alzheimer’s disease: A review of 20 years of research. Eur J Neurol 23, 1500–1509. [DOI] [PubMed] [Google Scholar]
- [30].Chung S, Weber F, Zhong P, Tan CL, Nguyen TN, Beier KT, Hormann N, Chang WC, Zhang Z, Do JP, Yao S, Krashes MJ, Tasic B, Cetin A, Zeng H, Knight ZA, Luo L, Dan Y (2017) Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 545, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Anor CJ, O’Connor S, Saund A, Tang-Wai DF, Keren R, Tartaglia MC (2017) Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia. Neurodegener Dis 17, 127–134. [DOI] [PubMed] [Google Scholar]
- [32].Gatchel JR, Donovan NJ, Locascio JJ, Becker JA, Rentz DM, Sperling RA, Johnson KA, Marshall GA, Alzheimer’s Disease Neuroimaging Inititative (2017) Regional 18F-fluorodeoxyglucose hypometabolism is associated with higher apathy scores over time in early Alzheimer disease. Am J Geriatr Psychiatry 25, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gatchel JR, Donovan NJ, Locascio JJ, Schultz AP, Becker JA, Chhatwal J, Papp KV, Amariglio RE, Rentz DM, Blacker D, Sperling RA, Johnson KA, Marshall GA (2017) Depressive symptoms and tau accumulation in the inferior temporal lobe and entorhinal cortex in cognitively normal older adults: A pilot study. J Alzheimers Dis 59, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Osorio RS, Gumb T, Pomara N (2014) Soluble amyloid-beta levels and late-life depression. Curr Pharm Des 20, 2547–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pomara N, Bruno D, Sarreal AS, Hernando RT, Nierenberg J, Petkova E, Sidtis JJ, Wisniewski TM, Mehta PD, Pratico D, Zetterberg H, Blennow K (2012) Lower CSF amyloid beta peptides and higher F2-isoprostanes in cognitively intact elderly individuals with major depressive disorder. Am J Psychiatry 169, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Donovan NJ, Locascio JJ, Marshall GA, Gatchel J, Hanseeuw BJ, Rentz DM, Johnson KA, Sperling RA, Harvard Aging Brain Study (2018) Longitudinal association of amyloid beta and anxious-depressive symptoms in cognitively normal older adults. Am J Psychiatry 175, 530–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rüb U, Stratmann K, Heinsen H, Turco DD, Seidel K, Dunnen W, Korf HW (2016) The brainstem tau cytoskeletal pathology of alzheimer’s disease: A brief historical overview and description of its anatomical distribution pattern, evolutional features, pathogenetic and clinical relevance. Curr Alzheimer Res 13, 1178–1197. [DOI] [PubMed] [Google Scholar]
- [38].Stratmann K, Heinsen H, Korf HW, Del Turco D, Ghebremedhin E, Seidel K, Bouzrou M, Grinberg LT, Bohl J, Wharton SB, den Dunnen W, Rub U (2016) Precortical phase of Alzheimer’s disease (AD)-related tau cytoskeletal pathology. Brain Pathol 26, 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, Leite RP, Diehl Rodriguez R, Mejia MB, Rub U, Farfel JM, de Lucena Ferretti-Rebustini RE, Nascimento CF, Nitrini R, Pasquallucci CA, Jacob-Filho W, Miller B, Seeley WW, Heinsen H, Grinberg LT (2017) Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 43, 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Andres-Benito P, Fernandez-Duenas V, Carmona M, Escobar LA, Torrejon-Escribano B, Aso E, Ciruela F, Ferrer I (2017) Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol Appl Neurobiol 43, 373–392. [DOI] [PubMed] [Google Scholar]
- [41].Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite RE, Rodriguez RD, Mejia MB, Suemoto CK, Ferretti-Rebustini RE, Polichiso L, Nascimento CF, Seeley WW, Nitrini R, Pasqualucci CA, Jacob Filho W, Rueb U, Neuhaus J, Heinsen H, Grinberg LT (2017) Locus coeruleus volume and cell population changes during Alzheimer’s disease progression:A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 13, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121, 171–181. [DOI] [PubMed] [Google Scholar]
- [43].Zweig RM, Ross CA, Hedreen JC, Steele C, Cardillo JE, Whitehouse PJ, Folstein MF, Price DL (1988) The neuropathology of aminergic nuclei in Alzheimer’s disease. Ann Neurol 24, 233–242. [DOI] [PubMed] [Google Scholar]
- [44].German DC, White CL, Sparkman DR (1987) Alzheimer’s disease: Neurofibrillary tangles in nuclei that project to the cerebral cortex. Neuroscience 21, 305–312. [DOI] [PubMed] [Google Scholar]
- [45].Frosch MP (2017) Tau aggregates: Where, when, why and what consequences? Neuropathol Appl Neurobiol 43, 371–372. [DOI] [PubMed] [Google Scholar]
- [46].Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969. [DOI] [PubMed] [Google Scholar]
- [47].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Contributors (2018) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tekin S, Mega MS, Masterman DM, Chow T, Garakian J, Vinters HV, Cummings JL (2001) Orbitofrontal and anterior cingulate cortex neurofibrillary tangle burden is associated with agitation in Alzheimer disease. Ann Neurol 49, 355–361. [PubMed] [Google Scholar]
- [49].Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL (2006) Neuropathologic correlates of apathy in Alzheimer’s disease. Dement Geriatr Cogn Disord 21, 144–147. [DOI] [PubMed] [Google Scholar]
- [50].Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, Gorman JM, Haroutunian V (2006) Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry 63, 161–167. [DOI] [PubMed] [Google Scholar]
- [51].Rapp MA, Schnaider-Beeri M, Purohit DP, Perl DP, Haroutunian V, Sano M (2008) Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry 16, 168–174. [DOI] [PubMed] [Google Scholar]
- [52].Guadagna S, Esiri MM, Williams RJ, Francis PT (2012) Tau phosphorylation in human brain: Relationship to behavioral disturbance in dementia. Neurobiol Aging 33, 2798–2806. [DOI] [PubMed] [Google Scholar]
- [53].Förstl H, Burns A, Luthert P, Cairns N, Lantos P, Levy R (1992) Clinical and neuropathological correlates of depression in Alzheimer’s disease. Psychol Med 22, 877–884. [DOI] [PubMed] [Google Scholar]
- [54].Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J (1994) The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 44, 2308–2314. [DOI] [PubMed] [Google Scholar]
- [55].Grinberg LT, Ferretti RE, Farfel JM, Leite R, Pasqualucci CA, Rosemberg S, Nitrini R, Saldiva PH, Filho WJ, Brazilian Aging Brain Study Group (2007) Brain bank of the Brazilian aging brain study group - a milestone reached and more than 1,600 collected brains. Cell Tissue Bank 8, 151–162. [DOI] [PubMed] [Google Scholar]
- [56].O’Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, Lupo PJ, Reisch JS, Doody R, Texas Alzheimer’s Research Consortium (2008) Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: A Texas Alzheimer’s research consortium study. Arch Neurol 65, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Morris JC (1993) The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- [58].Ferretti REL, Damim AE, Brucki SMD, Morillo LS, Perroco T, Campora F, Moreira EG, Balbino ES, Lima MCA, Battela C, Ruiz L, Grinberg LT, Farfel JM, Leite REP, Suemoto CZ, Pasqualucci CA, Jacob-Filho W, Nitrini R (2010) Post-moretem diagnosis of dementia by informant interview. Dement Neuropsychol 4, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- [61].Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18, 351–357. [DOI] [PubMed] [Google Scholar]
- [62].Geerts H, Roberts P, Spiros A, Carr R (2013)A strategy for developing new treatment paradigms for neuropsychiatric and neurocognitive symptoms in Alzheimer’s disease. Front Pharmacol 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rosenberg PB, Nowrangi MA, Lyketsos CG (2015) Neuropsychiatric symptoms in Alzheimer’s disease: What might be associated brain circuits? Mol Aspects Med 43–44, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Weinshenker D (2018) Long road to ruin: Noradrenergic dysfunction in neurodegenerative disease. Trends Neurosci 41, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Beleslin DB, Samardzic R, Krstic SK, Micic D, Terzic B (1981) Comparison of behavioral changes in cats treated with intracerebroventricular 6-hydroxydopamine and reserpine. Brain Res Bull 6, 285–287. [DOI] [PubMed] [Google Scholar]
- [66].Nakamura K, Thoenen H (1972) Increased irritability: A permanent behavior change induced in the rat by intraventricular administration of 6-hydroxydopamine. Psychopharmacologia 24, 359–372. [DOI] [PubMed] [Google Scholar]
- [67].Sorenson CA, Gordon M (1975) Effects of 6-hydroxydopamine on shock-elicited aggression, emotionality and maternal behavior in female rats. Pharmacol Biochem Behav 3, 331–335. [DOI] [PubMed] [Google Scholar]
- [68].Moret C, Briley M (2011) The importance of nore-pinephrine in depression. Neuropsychiatr Dis Treat 7, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455, 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Belmaker RH, Agam G (2008) Major depressive disorder. N Engl J Med 358, 55–68. [DOI] [PubMed] [Google Scholar]
- [71].Gallagher D, Coen R, Kilroy D, Belinski K, Bruce I, Coakley D, Walsh B, Cunningham C, Lawlor BA (2011) Anxiety and behavioural disturbance as markers of prodromal Alzheimer’s disease in patients with mild cognitive impairment. Int J Geriatr Psychiatry 26, 166–172. [DOI] [PubMed] [Google Scholar]
- [72].Seignourel PJ, Kunik ME, Snow L, Wilson N, Stanley M (2008) Anxiety in dementia: A critical review. Clin Psychol Rev 28, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Charney DS, Redmond DE Jr. (1983) Neurobiological mechanisms in human anxiety. Evidence supporting central noradrenergic hyperactivity. Neuropharmacology 22, 1531–1536. [DOI] [PubMed] [Google Scholar]
- [74].Charney DS, Grillon C, Bremner JD (1998) The neurobiological basis of anxiety and fear: Circuits, mechanisms, and neurochemical interactions (Part I). Neuroscientist 4, 35–44. [Google Scholar]
- [75].Jimenez JC, Su K, Goldberg AR, Luna VM, Biane JS, Ordek G, Zhou P, Ong SK, Wright MA, Zweifel L, Paninski L, Hen R, Kheirbek MA (2018) Anxiety cells in a hippocampal-hypothalamic circuit. Neuron 97, 670–683.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Holzschneider K, Mulert C (2011) Neuroimaging in anxiety disorders. Dialogues Clin Neurosci 13, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sehlmeyer C, Schoning S, Zwitserlood P, Pfleiderer B, Kircher T, Arolt V, Konrad C (2009) Human fear conditioning and extinction in neuroimaging: A systematic review. PLoS One 4, e5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Talakoub O, Paiva RR, Milosevic M, Hoexter MQ, Franco R, Alho E, Navarro J, Pereira JF, Popovic MR, Savage C, Lopes AC, Alvarenga P, Damiani D, Teixeira MJ, Miguel EC, Fonoff ET, Batistuzzo MC, Hamani C (2017) Lateral hypothalamic activity indicates hunger and satiety states in humans. Ann Clin Transl Neurol 4, 897–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Neary NM, Goldstone AP, Bloom SR (2004) Appetite regulation: From the gut to the hypothalamus. Clin Endocrinol (Oxf) 60, 153–160. [DOI] [PubMed] [Google Scholar]
- [80].Vettor R, Fabris R, Pagano C, Federspil G (2002) Neuroendocrine regulation of eating behavior. J Endocrinol Invest 25, 836–854. [DOI] [PubMed] [Google Scholar]
- [81].Vilberg TR, Beatty WW (1975) Behavioral changes following VMH lesions in rats with controlled insulin levels. Pharmacol Biochem Behav 3, 377–384. [DOI] [PubMed] [Google Scholar]
- [82].Ahima RS, Antwi DA (2008) Brain regulation of appetite and satiety. Endocrinol Metab Clin North Am 37, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bresch A, Rullmann M, Luthardt J, Becker GA, Patt M, Ding YS, Hilbert A, Sabri O, Hesse S (2017) Hunger and disinhibition but not cognitive restraint are associated with central norepinephrine transporter availability. Appetite 117, 270–274. [DOI] [PubMed] [Google Scholar]
- [84].Kai K, Hashimoto M, Amano K, Tanaka H, Fukuhara R, Ikeda M (2015) Relationship between eating disturbance and dementia severity in patients with Alzheimer’s disease. PLoS One 10, e0133666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Moran M, Lynch CA, Walsh C, Coen R, Coakley D, Lawlor BA (2005) Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med 6, 347–352. [DOI] [PubMed] [Google Scholar]
- [86].Rothman SM, Mattson MP (2012) Sleep disturbances in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med 14, 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mander BA, Winer JR, Walker MP (2017) Sleep and human aging. Neuron 94, 19–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Gotter AL, Forman MS, Harrell CM, Stevens J, Svetnik V, Yee KL, Li X, Roecker AJ, Fox SV, Tannenbaum PL, Garson SL, Lepeleire ID, Calder N, Rosen L, Struyk A, Coleman PJ, Herring WJ, Renger JJ, Winrow CJ (2016) Orexin 2 receptor antagonism is sufficient to promote NREM and REM sleep from mouse to man. Sci Rep 6, 27147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Takahashi K, Lin JS, Sakai K (2006) Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci 26, 10292–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhao LZ, Zhang GL, Gao J, Zhang JX, Zhong MK, Zhang J (2003) Role of serotonergic neurons in dorsal raphe nuclei in regulation of sleep. Zhongguo Ying Yong Sheng Li Xue Za Zhi 19, 175–178. [PubMed] [Google Scholar]
- [91].Lucey BP, Holtzman DM (2015) How amyloid, sleep and memory connect. Nat Neurosci 18, 933–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (2009) Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Shinagawa S, Naasan G, Karydas AM, Coppola G, Pribadi M, Seeley WW, Trojanowski JQ, Miller BL, Grinberg LT (2015) Clinicopathological study of patients with C9ORF72-associated frontotemporal dementia presenting with delusions. J Geriatr Psychiatry Neurol 28, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].McKeith I, Cummings J (2005) Behavioural changes and psychological symptoms in dementia disorders. Lancet Neurol 4, 735–742. [DOI] [PubMed] [Google Scholar]
- [95].Reeves SJ, Gould RL, Powell JF, Howard RJ (2012) Origins of delusions in Alzheimer’s disease. Neurosci Biobehav Rev 36, 2274–2287. [DOI] [PubMed] [Google Scholar]
- [96].Bruen PD, McGeown WJ, Shanks MF, Venneri A (2008) Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer’s disease. Brain 131, 2455–2463. [DOI] [PubMed] [Google Scholar]
- [97].Klatka LA, Louis ED, Schiffer RB (1996) Psychiatric features in diffuse Lewy body disease: A clinicopathologic study using Alzheimer’s disease and Parkinson’s disease comparison groups. Neurology 47, 1148–1152. [DOI] [PubMed] [Google Scholar]
- [98].Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- [99].Rahimi J, Kovacs GG (2014) Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 6, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].D’Anna L, Abu-Rumeileh S, Fabris M, Pistis C, Baldi A, Sanvilli N, Curcio F, Gigli GL, D’Anna S, Valente M (2017) Serum interleukin-10 levels correlate with cerebrospinal fluid amyloid beta deposition in Alzheimer disease patients. Neurodegener Dis 17, 227–234. [DOI] [PubMed] [Google Scholar]
- [101].Guimaraes HC, Levy R, Teixeira AL, Beato RG, Caramelli P (2008) Neurobiology of apathy in Alzheimer’s disease. Arq Neuropsiquiatr 66, 436–443. [DOI] [PubMed] [Google Scholar]
- [102].Ruhe HG, Mason NS, Schene AH (2007) Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: A meta-analysis of monoamine depletion studies. Mol Psychiatry 12, 331–359. [DOI] [PubMed] [Google Scholar]
- [103].Eurelings LS, Richard E, Eikelenboom P, van Gool WA, Moll van Charante EP (2015) Low-grade inflammation differentiates between symptoms of apathy and depression in community-dwelling older individuals. Int Psychogeriatr 27, 639–647. [DOI] [PubMed] [Google Scholar]
- [104].Apostolova LG, Akopyan GG, Partiali N, Steiner CA, Dutton RA, Hayashi KM, Dinov ID, Toga AW, Cummings JL, Thompson PM (2007) Structural correlates of apathy in Alzheimer’s disease. Dement Geriatr Cogn Disord 24, 91–97. [DOI] [PubMed] [Google Scholar]
- [105].Eslinger PJ, Moore P, Antani S, Anderson C, Grossman M (2012) Apathy in frontotemporal dementia: Behavioral and neuroimaging correlates. Behav Neurol 25, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Karran E, De Strooper B (2016) The amyloid cascade hypothesis: Are we poised for success or failure? J Neurochem 139(Suppl 2), 237–252. [DOI] [PubMed] [Google Scholar]
- [107].DaRocha-Souto B, Scotton TC, Coma M, Serrano-Pozo A, Hashimoto T, Sereno L, Rodriguez M, Sanchez B, Hyman BT, Gomez-Isla T (2011) Brain oligomeric beta-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol 70, 360–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Blay SL, Fillenbaum GG, Pitta JC, Peluso ET (2014) Factors associated with antidepressant, anxiolytic, and other psychotropic medication use to treat psychiatric symptoms in the city of Sao Paulo, Brazil. Int Clin Psychopharmacol 29, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]