

ABSTRACT
The interaction between tumor cells and macrophages is crucial in promoting tumor invasion and metastasis. In this study, we examined a novel mechanism of intercellular communication, namely membranous actin-based tunneling nanotubes (TNTs), that occurs between macrophages and tumor cells in the promotion of macrophage-dependent tumor cell invasion. The presence of heterotypic TNTs between macrophages and tumor cells induced invasive tumor cell morphology, which was dependent on EGF–EGFR signaling. Furthermore, reduction of a protein involved in TNT formation, M-Sec (TNFAIP2), in macrophages inhibited tumor cell elongation, blocked the ability of tumor cells to invade in 3D and reduced macrophage-dependent long-distance tumor cell streaming in vitro. Using an in vivo zebrafish model that recreates macrophage-mediated tumor cell invasion, we observed TNT-mediated macrophage-dependent tumor cell invasion, distant metastatic foci and areas of metastatic spread. Overall, our studies support a role for TNTs as a novel means of interaction between tumor cells and macrophages that leads to tumor progression and metastasis.
KEY WORDS: Tunneling nanotubes, Cell communication, Macrophages, Tumor cell streaming, Invasion
Summary: A novel mechanism of intercellular communication through tunneling nanotubes (TNTs) on macrophage-mediated tumor cell invasion reveals TNTs as a potential therapeutic target.
INTRODUCTION
The process of metastasis of breast adenocarcinoma starts with the escape of epithelial cells from the primary tumor by breaking through the basement membrane, invading into the underlying stromal tissue and migrating towards the blood and lymphatic vessels. Tumor cells then intravasate into vessels where they circulate and eventually extravasate into secondary organs (Chaffer and Weinberg, 2011; Valastyan and Weinberg, 2011; Weigelt et al., 2005). Throughout the metastatic cascade, tumor cells interact with other cell types within the tumor microenvironment, including macrophages (Hanahan and Coussens, 2012). This interaction promotes tumor cell acquisition of unique characteristics that allow them to spread into distant sites. Studies in breast cancer mouse models have shown that macrophages are essential partners for tumor cell invasion, intravasation into the blood vessels and extravasation into secondary sites (Denning et al., 2007; Roussos et al., 2011; Sidani et al., 2006). In addition, tumor cells migrate alongside macrophages directionally along extracellular fibers towards blood vessels in a process referred to as multicellular streaming, which is observed in vivo (Harney et al., 2015; Patsialou et al., 2013; Roussos et al., 2011) and can be mimicked in vitro (Leung et al., 2017; Sharma et al., 2012). Eventually, both cell types reach the blood vessel, where macrophages aid in the process of tumor cell intravasation into the blood circulation at intravasation doorways called tumor microenvironments of metastasis (TMEMs) (Harney et al., 2015; Pignatelli et al., 2014). Therefore, it is highly important to comprehensively characterize various mechanisms of tumor cell–macrophage interactions.
Previous studies done in our lab and others have described the importance of the interaction between macrophages and breast cancer cells within the tumor microenvironment (Ishihara et al., 2013; Park et al., 2014). Macrophages interact with tumor cells through the production of epidermal growth factor (EGF), which binds to the epidermal growth factor receptor (EGFR) on tumor cells. Tumor cells in turn secrete colony stimulating factor 1 (CSF-1), which attracts macrophages through colony stimulating factor 1 receptor (CSF-1R) (Goswami et al., 2005; Wyckoff et al., 2004). In fact, functional blocking of macrophages in vivo significantly reduces tumor cell migration and invasion (Patsialou et al., 2013). Recent studies have indicated that direct contact between macrophages and tumor cells can induce tumor cell invadopodia formation important for tumor cell intravasation (Pignatelli et al., 2016, 2014; Roh-Johnson et al., 2014). While the knowledge of cellular communication via secreted soluble factors, exosomes and microvesicles has cast light on distant tumor cell and tumor–stromal interactions (Hoshino et al., 2015; El Andaloussi et al., 2013), direct contact with non-malignant macrophages within the complex and dense heterogeneous tumor matrix is still greatly underappreciated.
Recently a novel mechanism of intercellular communication through long membranous tunneling nanotubes (TNTs) has been identified in many cell types (Abounit and Zurzolo, 2012; Rustom et al., 2004; Watkins and Salter, 2005) including macrophages and various cancer cells (Hanna et al., 2017; Hase et al., 2009; Onfelt et al., 2006; Osswald et al., 2015; Watkins and Salter, 2005). TNTs are thin (70–800 nm wide) membranous structures connecting cells, which can be several cell diameters in length. This allows connected cells to act in a synchronized manner over long distances, with some interactions on the scale of hundreds of microns away (Osswald et al., 2015; Watkins and Salter, 2005). In contrast to soluble factors that diffuse and decrease over distance, TNTs propagate signals through a network of cells that remain strong and robust despite the distance traveled (Chauveau et al., 2010; Wang et al., 2012). We and others have recently reviewed the importance of TNTs in immune cell function and coordination during immune responses (Ariazi et al., 2017; Baker, 2017; McCoy-Simandle et al., 2016). M-Sec, also known as TNFAIP2 (tumor necrosis factor α-induced protein), has been identified as a potential marker for TNTs. M-Sec interacts with the small GTPase RALA and serves as a key factor for TNT formation and function, particularly in macrophages (Hanna et al., 2017; Hase et al., 2009; Ohno et al., 2010). However, it is important to note that signaling mechanisms for TNT formation may vary depending on the cell type or model used. For instance, actin regulators CDC42 and RAC1 are important for macrophage TNT biogenesis (Hanna et al., 2017); TNTs between neuronal cells are negatively regulated by CDC42 through IRSp53 (also known as BAIAP2) and VASP (Delage et al., 2016). Therefore, additional studies are needed to identify specific TNT regulatory factors depending on the cell type being studied.
TNTs have been suggested to play an important role in tumor microenvironments. In human malignant pleural mesothelioma for instance, TNTs can provide a conduit for intercellular transfer of cellular contents (Lou et al., 2012). In addition, transfer of microRNAs (miRNAs) has been shown to occur between cancer cells and stromal cells in vitro in both osteosarcoma and ovarian cancer (Thayanithy et al., 2014a). Moreover, TNTs connecting astrocytoma tumor cells observed in vivo have been shown to form a multicellular network that correlates with prognostic features of malignant brain tumors (Osswald et al., 2015). In addition, TNTs can mediate the transport of material between malignant and stromal cells, with potential effects on gene expression and contributing to cancer progression (Lou et al., 2017a, 2018). Taken together, these studies establish the importance of TNTs in the tumor microenvironment. Given the ability of macrophages to form TNTs and the importance of macrophages in the tumor microenvironment, we predicted that an interaction between tumor cells and macrophages via TNTs may be critical for cellular networking and coordination within the tumor microenvironment that leads to tumor progression and metastasis. However, despite the recent findings on the importance of TNTs in intercellular signaling, little is known about the mechanism of heterotypic TNT formation and function, particularly between macrophages and tumor cells.
In this study, we show that the formation of heterotypic TNTs between macrophages and breast tumor cells results in stimulation of an invasive tumor cell phenotype with enhanced directional tumor cell streaming alongside macrophages towards the endothelium and increased tumor cell invasion in vitro as well as in vivo using a zebrafish model.
RESULTS
Formation of heterotypic TNTs between tumor cells and macrophages
TNTs have been previously reported to mediate specific direct cell–cell contact between macrophages (Hanna et al., 2017; Hase et al., 2009) as well as between tumor cells (Ady et al., 2014; Lou et al., 2012, 2017b; Osswald et al., 2015). Given the important role of macrophages in breast cancer progression and the existence of TNTs in both contexts, we investigated whether heterotypic TNTs form between macrophages and tumor cells. To address this question, we used RAW/LR5 macrophages, a subline of RAW 264.7 cells in which we have recently characterized the formation and function of TNTs (Hanna et al., 2017), together with rat mammary adenocarcinoma cell line MTLn3 and human breast adenocarcinoma cell line MDA-MB-231. We utilized super-resolution 3-dimensional illumination microscopy (3D-SIM) in order to visualize the thin TNT structures. In each condition, one cell type was labeled to distinguish heterotypic from homotypic cell pairs connected by TNTs. Heterotypic TNTs were identified as long thin structures containing actin that connected a macrophage and a tumor cell. Importantly, these heterotypic TNT connections were not attached to the substrate and were only visible in upper planes, as shown in Fig. 1. Consistent with these actin-containing structures being membranous nanotubes, they were surrounded by plasma membrane as indicated by labeling with fluorescent wheat germ agglutinin (WGA) in MTLn3 cells (Fig. 1A). Podosomes, which are actin-rich structures in macrophages (Mφ), and actin-rich stress fibers in the tumor cells (TC), are only visible at the bottom planes (see Fig. 1A; Movies 1, 2). Heterotypic TNTs (indicated by yellow arrows in Fig. 1) are only visible in the upper planes above the surface, unlike other actin-rich structures, which are usually found in the bottom portions of cells. In addition, homotypic TNTs (indicated by white arrows in Movies 2 and 4) are often observed between two macrophages or two tumor cells (see Movie 2). Similar TNT structures were also detected between macrophages and MDA-MB-231 tumor cells (Fig. 1C; Movies 3,4). Using WGA as a membrane label, we then determined the average diameter using at least four points along the length of individual heterotypic TNTs. Quantitation of 3D-SIM images demonstrates that heterotypic TNTs, whether between macrophages and MTLn3 or MDA-MB-231 tumor cells, have a similar average diameter of ∼500 nm, albeit with a broad range in individual diameters (Fig. 1B,D). Therefore, the criteria of long thin actin-filled structures above the substrate and connecting two cells were used to identify TNTs throughout the current study.
Fig. 1.

3D-SIM imaging of heterotypic TNTs between tumor cells and macrophages. Tunneling nanotubes (TNTs) are long thin structures above the surface connecting two cells and contain actin. (A) 3D-SIM image of TNTs connecting a RAW/LR5 macrophage (Mφ) with rat mammary adenocarcinoma cell line, MTLn3 (tumor cell, TC), fixed and stained for WGA (green) to visualize the membrane and F-actin (magenta). Upper plane shows heterotypic TNT (yellow arrows) connecting the macrophage with a tumor cell. Orthogonal view shows the xz planes of the separate channels showing the TNT between the two cell types. Scale bar: 4 μm. (B) Mean±s.e.m. distribution of TNT diameters from SIM images between RAW/LR5 macrophage and MTLn3 tumor cells. n=15 TNTs. (C) 3D-SIM image of a co-culture between RAW/LR5 macrophages and human breast adenocarcinoma cells, MDA-MB-231, fixed and stained for WGA (green) and F-actin (magenta). Heterotypic TNTs connecting the two cell types (yellow arrows) are visible in the upper planes. Orthogonal views show the xz planes of the separate channels. Scale bar: 3 μm. (D) Mean±s.e.m. distribution of TNT diameters from SIM images between RAW/LR5 macrophage and MDA-MB-231 tumor cells. n=25 TNTs.
Live-cell imaging of heterotypic TNT formation between tumor cells and macrophages
TNTs are known to be very fragile and sensitive to fixation and subsequent processing for imaging (Watkins and Salter, 2005). Therefore, in order to minimize breakage and reduce fixation artifacts, we opted to use a live-cell imaging approach to further characterize these heterotypic structures. We generated cell lines stably expressing fluorescently-tagged plasma membrane markers with GFP in RAW/LR5 macrophages or mCherry–CAAX in MTLn3 and MDA-MB-231 tumor cells. To confirm that the ability to form heterotypic TNTs was not cell-line specific, we co-cultured murine primary bone marrow-derived macrophages (BMMs) overnight with mCherry–CAAX MTLn3 tumor cells and examined whether heterotypic TNTs were formed between the two cell types (Fig. 2A). TNT formation was quantified after an overnight co-culture and the percentage of TNT connections specifically between a macrophage and tumor cell was determined (Fig. 2B, gray bar) and compared to a culture of tumor cells alone (Fig. 2B, white bar). While it was apparent that tumor cells produced a basal level of homotypic TNTs (∼10%), there were twice as many TNT connections present between macrophages and tumor cells. One of the unique functions of TNTs is that they can mediate the transfer of multiple cargos including cytoplasmic components, signaling molecules, vesicles and organelles (Islam et al., 2012; Plotnikov et al., 2010; Thayanithy et al., 2014b). Therefore, in order to verify that these heterotypic structures were functional TNTs, we labeled RAW/LR5 macrophages with DiI (red) prior to co-culture with MTLn3 tumor cells labeled with GFP–CAAX and the transfer of labeled membranes and/or vesicles was determined using microscopy and flow cytometry. DiI-labeled material was observed to be transferred to GFP–CAAX MTLn3 tumor cells connected to macrophages through TNTs (Fig. S1) while no material was detected in tumor cells not connected to macrophages (Fig. S1, lower panels). This indicated that TNTs with macrophages mediated the transfer of DiI-labeled material to tumor cells. Using flow cytometry assays, the transfer of DiI-labeled material from macrophages into GFP–CAAX-expressing tumor cells was determined as the number of double positive cells. To confirm whether this transfer was dependent on the ability of macrophages to form TNTs, we utilized a macrophage cell line with reduced ability in TNT production generated previously by decreasing endogenous levels of M-Sec (Hanna et al., 2017). Following transduction with shM-Sec, these RAW/LR5 macrophages have stably reduced levels of M-Sec protein (Fig. 2C, inset), which is one of the known markers that localizes to TNTs in macrophages and is important for their formation and function (Hanna et al., 2017; Hase et al., 2009). Indeed, using shM-Sec RAW/LR5s, the transfer of DiI-labeled material was significantly reduced (Fig. 2C), indicating that these heterotypic TNTs were functional in wild-type RAW/LR5 macrophages.
Fig. 2.

Heterotypic TNTs between tumor cells and macrophages are functional and can be quantified using live-cell imaging. (A) Co-culture between primary bone marrow-derived macrophages (BMMs) and MTLn3 mCherry–CAAX tumor cells, showing the formation of TNTs between the different cell type (heterotypic). Right panel shows a zoomed area where a TNT is connecting both cell types, arrows indicate that TNT is composed of tumor cell (red arrowhead) and BMM (white arrowhead) membrane. Scale bars: 10 μm (main images), 5 μm (inset). (B) Mean±s.e.m. number of TNTs after overnight co-culture represented as the percentage of TNTs formed between BMMs and MTLn3 tumor cells (gray bar) for a population of n>64 cells for each experiment and compared to TNTs formed between tumor cells alone (clear bar). (C) RAW/LR5 macrophages are labeled with DiI prior to co-culture with GFP–CAAX MTLn3 tumor cells. Using flow cytometry, we quantified the population of DiI-positive GFP-expressing MTLn3 cells, indicating the transfer of DiI-labeled material in the case of co-culture with control or shM-Sec (TNT deficient) macrophages. Data are the mean±s.e.m. of 3 independent experiments. Inset shows western blot analysis of M-Sec levels in shControl and shM-Sec RAW/LR5 s (full blots can be seen in Fig. S2). (D) Mean±s.e.m. percentage of TNTs between mCherry–CAAX-labeled tumor cells and GFP–CAAX-labeled control and shM-Sec or shWASP RAW/LR5 macrophages. (E,G) Live-cell images of TNTs between MTLn3 tumor cells (E) or MDA-MB-231 tumor cells (G) stably expressing mCherrry–CAAX, and macrophages expressing GFP–CAAX plasmid to label the cell membrane. Left panels: A TNT is formed between a tumor cell (red) and a RAW/LR5 macrophage (green). Panels (i) show a zoomed area of the TNT formed by wrapping of membrane from both cell types. Panels (ii) and (iii) show individual channels. Right panels: A TNT is formed solely from a macrophage. Panels (i) shows a zoomed area of the TNT formed of macrophage membrane. Panels (ii) and (iii) show individual channels. Scale bars: 10 μm. (F) Mean±s.e.m. percentage of TNTs formed between mCherry–CAAX-labeled MDA-MB-231 tumor cells and GFP–CAAX-labeled control and shM-Sec RAW/LR5 macrophages. Data in all graphs are represented as histograms representing the overall mean of at least three independent experiments. Individual values of the number of TNT connections for each independent experiment in each case are indicated as dots. *P<0.05; **P<0.02; ***P<0.003; ns, not significant by Student's t-test.
Using live-cell imaging, we quantified the percentage of heterotypic TNTs formed between the two cell types. Indeed, a significant percentage of heterotypic TNTs formed in co-cultures between GFP–CAAX RAW/LR5 macrophages and mCherry–CAAX MTLn3 tumor cells (Fig. 2D). The percentage of heterotypic TNTs was significantly reduced in co-cultures of MTLn3 tumor cells and two TNT-defective macrophage cell lines transduced with shM-Sec or shWASP, compared to co-cultures of MTLn3 tumor cells with control macrophages (Fig. 2D, gray bar versus white bars). This result indicated that heterotypic TNT formation was dependent on the ability of macrophages to generate TNTs. However, like our other identified regulators of macrophage TNTs, WASP is highly important for other actin-based macrophage functions including phagocytosis, podosome formation and chemotaxis (Rougerie et al., 2013). Also, we have shown that WASP is important in multiple ways in macrophage-mediated tumor cell invasion (Ishihara et al., 2013). In order not to complicate the interpretation of the role of these heterotypic TNTs in the macrophage–tumor cell interaction, we opted to not use WASP for additional experiments but employed the shM-Sec cell line as a specific disruptor of macrophage TNTs since the shM-Sec cell line does not exhibit defects in phagocytosis, chemotaxis or podosome-mediated degradation (Hanna et al., 2017). Examining these heterotypic TNTs more closely, we observed that they were either composed of membrane from both cell types (Fig. 2E, left panels i–iii) or solely contained macrophage membrane (Fig. 2E, right panels i–iii). Similarly, we quantified heterotypic TNTs between GFP–CAAX RAW/LR5 macrophages and mCherry–CAAX MDA-MB-231 tumor cells as shown in Fig. 2F, and a significant decrease in heterotypic TNTs was observed in the presence of shM-Sec macrophages compared to control macrophages (Fig. 2F, gray bar versus white bars). These TNTs are also observed to be composed of membrane from both cell types (Fig. 2G, left panels i–iii) or initiated solely from the macrophage towards the tumor cell (Fig. 2G, right panels i–iii).
Given the presence of membrane from both cells in the heterotypic TNTs, time-lapse imaging was performed to determine the contribution of each cell type in TNT formation. After overnight culturing, heterotypic TNTs were present and were often maintained over 30 min during imaging (Movie 5). In addition, it was observed that macrophages initiated the formation of TNT-like protrusions towards the tumor cells (Fig. 3A; Movie 6). These protrusions are referred to as ‘TNT-like’ because although they meet the criteria of TNTs by being membranous and suspended above the surface, it is unknown whether these protrusions can mediate the transfer of material, particularly within the imaging time-frame. In many cases, macrophages generated protrusions that stimulated TNT-like protrusions from the tumor cells and these protrusions appear to intertwine and/or interact at the tip of the TNT-like protrusion extending from both cell types (Fig. 3B; Movie 7). Consistent with both cell types contributing to heterotypic TNT formation, dual-label imaging of these heterotypic TNTs indicated that membranes from both cell types can be present in these structures using either MTLn3 cells or human MD-MBA-231 cells (Fig. 2E,G). Overall, results demonstrate that heterotypic TNTs between macrophages and tumor cells are initiated by the macrophage but often involve membrane protrusion from both cell types.
Fig. 3.

Live-cell imaging of heterotypic TNT formation between tumor cells and macrophages. (A) Time-lapse imaging of GFP–CAAX RAW/LR5 macrophages in co-culture with mCherry–CAAX MTLn3 tumor cells showing TNT-like protrusion initiated from macrophage towards a tumor cell (white arrows, lower panels). Upper and middle panels show individual channels. See Movies 5 and 6. (B) Time-lapse imaging of GFP–CAAX RAW/LR5 macrophages in co-culture with mCherry–CAAX MTLn3 tumor cells showing TNT-like protrusions extending from both cell types where they appear to intertwine and/or interact at the tip of the TNT-like protrusion (white arrows, lower panels). See Movie 7. Duration of original sequence at least 30 min. Magnification 60×, 2×2 binning. Frame interval: 10 s. Scale bars: 2 μm.
Tumor cells respond to macrophage interaction by acquiring a more invasive phenotype in an EGF–EGFR dependent manner
Next, we explored tumor cell response to TNT interaction with macrophages. Previous studies by our group and others have demonstrated that macrophages induce tumor cells to acquire a more invasive phenotype as measured by tumor cell elongation (Goswami et al., 2005; Ishihara et al., 2013). Consistent with previously published data, our results showed a 60% increase in tumor cell elongation in the presence of control macrophages (Fig. 4A, upper panels; Fig. 4B, black bars). In order to eliminate the ability of macrophages to secrete factors, particularly EGF, we used a broad-spectrum matrix metalloproteinase inhibitor (GM6001) that we have previously shown to block the shedding of EGF from the surface of macrophages (Ishihara et al., 2013). This inhibited the ability of tumor cells to elongate in response to macrophages when not in contact with macrophages (Fig. 4B, gray bars; compare MTLn3 alone to MTLn3+control macrophages with no TNT contact). However, tumor cells directly connected to macrophages via TNTs were able to bypass the requirement for secreted EGF and still elongated to a similar extent to the control conditions without GM6001 (Fig. 4B, gray bars). In addition, the presence of TNT-deficient shM-Sec macrophages failed to induce tumor cell elongation (Fig. 4B). Control experiments were performed that verified the specificity of M-Sec in TNT formation and that it did not affect other normal macrophage functions including podosome formation and phagocytosis (data not shown). In addition, using Boyden transwell assays, we tested the chemotaxis of shM-Sec macrophages and their ability to migrate towards CSF-1, a factor secreted by tumor cells. Our results showed a more than 13-fold increase compared to control with no CSF-1 in the lower chamber (data not shown, data from n=2 experiments). Importantly, since EGF mediates the activation of tumor cells, we verified that shM-Sec macrophages have normal EGF secretion. In fact, similar to previous studies (Ishihara et al., 2013), conditioned medium taken from shM-Sec macrophages significantly stimulated a 2-fold increase in tumor cell area in an upshift experiment, which was not statistically different from the increase induced by conditioned medium taken from control macrophages [2-fold increase ±0.07 (s.e.m.), P=0.279 compared to control; data not shown]. Hence, these data confirm that the role of M-Sec is specific to TNT formation in macrophages. To more specifically inhibit metalloprotease-dependent EGF shedding in macrophages, we used a RAW/LR5 macrophage cell line transduced with shMT1-MMP to reduce expression levels of MT1-MMP, a master regulator metalloprotease, in co-culture with tumor cells (Fig. 4C). The presence of shMT1-MMP macrophages failed to induce tumor cell elongation except when directly in contact with a tumor cell through TNTs (Fig. 4D). These results suggest that macrophage interaction with tumor cells through TNTs could bypass the requirement for macrophage factors secreted into the conditioned media.
Fig. 4.

Tumor cells respond to macrophage interaction by acquiring a more invasive phenotype in an EGF–EGFR dependent manner. (A) MTLn3 tumor cells cultured alone or in the presence of control RAW/LR5 macrophages (Mφs). To eliminate the role of secreted factors and focus on the direct TNT contact, cells were treated with either DMSO or the global MMP protease inhibitor GM6001. Cells were fixed and stained for F-actin (green) and DAPI (blue). Arrow indicates a TNT between the two cell types. (B) Mean±s.e.m. tumor cell elongation measured as the length-to-width ratio in the co-culture with control or shM-Sec (TNT deficient) RAW/LR5 macrophages. In the presence of GM6001, the elongation index was measured for tumor cells with no contact with macrophages or with TNT contact with macrophages. n=3 independent experiments. *P≤0.01, **P≤0.001, ***P≤0.0001 by Student's t-test. (C) mCherry–CAAX MTLn3 tumor cells cultured alone or in the presence of control or shMT1-MMP RAW/LR5 macrophages (green). Yellow arrows in insets indicate TNTs between tumor cells and macrophages. (D) Mean±s.e.m. tumor cell elongation index in co-culture with control or shMT1-MMP RAW/LR5 macrophages, in which case, elongation index was measured for tumor cells with no contact with macrophages or with TNT contact with macrophages. Inset shows western blot analysis (full blots can be seen in Fig. S2) of MT1-MMP levels in shControl and shMT1-MMP RAW/LR5 cells. n=3 independent experiments. Statistical significance is compared to control MTLn3 alone or indicated above the bracket comparing two adjacent bars. *P≤0.01, **P≤0.001 by Student's t-test. Scale bars: 10 μm.
Next, we investigated whether tumor cell response to TNT interaction with macrophages is mediated through localized EGF–EGFR signaling. Therefore, we tested the effect of blocking EGFR signaling on macrophage-induced tumor cell elongation. Our results showed that when EGFR signaling is inhibited using IRESSA, even tumor cells connected to a macrophage through TNTs could not respond and elongate (Fig. 5A,B). Additionally, when the fraction of elongated cells were determined for each condition, it was apparent that the fraction of elongated cells not connected to a macrophage through TNTs was also returned to basal levels by the inhibition of soluble EGF with GM6001, whereas cells with TNT contact remain elongated. Consistent with the role for EGF in tumor cell elongation, the fraction of elongated cells was not significantly different from the basal levels regardless of the presence of a macrophage TNT (Fig. 5C). This was not due to a defect in macrophage tumor cell TNT formation since the number of heterotypic TNTs formed in the presence of either GM6001 or IRESSA was not reduced (P values of 0.87 and 0.47, respectively. n=4). In order to further confirm the dependence of tumor cell elongation on EGF–EGFR in response to TNT interaction with macrophages, we utilized a non-metastatic rat tumor cell line (MTC) that is closely related to the MTLn3 cell line employed. These cells express significantly lower levels of EGFR on their cell surface and this has been correlated with their reduced invasion and metastatic capacity (Bailly et al., 1998; Segall et al., 1996) (Fig. 5D, lane 1 versus 3). Co-cultures of RAW/LR5 macrophages with MTCs did not show a significant increase in tumor cell elongation even when they are directly connected to a macrophage through TNTs (Fig. 5E). Interestingly, when EGFR expression was rescued in these cells by expression of human EGFR (Bailly et al., 1998; Segall et al., 1996) (Fig. 5D), the co-culture showed a significant increase in tumor cell elongation when in contact with macrophages through TNTs (Fig. 5F). This was accompanied by an increase in heterotypic TNTs (24%±0.9%, mean±s.e.m.) between the tumor cells and macrophages. This further suggests that the mechanism of TNT-mediated interaction between tumor cells and macrophages occurs through EGF–EGFR signaling.
Fig. 5.

EGFR signaling is important for tumor cell response to TNT interaction with macrophages. (A) Co-culture between MTLn3 tumor cells and RAW/LR5 macrophages (Mφs) in the presence of DMSO control or the EGFR inhibitor IRESSA, in order to inhibit tumor cell EGFR-dependent response. Yellow arrows indicate heterotypic TNTs between a tumor cell and a macrophage. Scale bars: 10 μm (main images), 5 μm (inset). (B) Mean±s.e.m. tumor cell elongation index in co-culture with control RAW/LR5 macrophages, in the presence of DMSO control or the EGFR inhibitor IRESSA, in which case, elongation index was measured for tumor cells with no contact with macrophages or with TNT contact with macrophages. n=3 independent experiments. Statistical significance is compared to control MTLn3 alone or indicated above the brackets comparing adjacent bars with. **P≤0.01 by Student's t-test. (C) Mean±s.e.m. percentage of elongated cells, one s.e.m. above the mean of MTLn3s alone. n=3 independent experiments. Statistical significance is compared to control MTLn3 alone. (D) Representative western blot analysis of the relative expression levels of EGFR in MTLn3, MTC and EGFR-expressing MTC (MTC-EGFR) tumor cells using anti-rat/mouse antibody (top) or anti-human antibody (bottom). β-actin was used as a loading control. Full blots can be seen in Fig. S2. (E,F) Mean±s.e.m. tumor cell elongation index of non-invasive MTC (E) or MTC-EGFR (F) tumor cells in co-culture with RAW/LR5 macrophages. Elongation index was measured for tumor cells with no contact with macrophages or with TNT contact with macrophages. n=3 independent experiments. **P≤0.01; ns, non-significant by Student's t-test compared to tumor cell alone control.
Collectively, these data suggest that tumor cells directly connected to macrophages via heterotypic TNTs acquire an elongated phenotype in the absence of soluble signals. In addition, this TNT interaction can promote macrophage-dependent tumor cell functions through localized EGF–EGFR signaling.
TNT interaction with macrophages promotes tumor cell invasion
Following on from our demonstration above that the heterotypic TNT interaction with macrophages promoted a more elongated phenotype in tumor cells, we hypothesized that this interaction may be important for tumor cell invasion. Therefore, we performed 3D invasion assays as done previously (Goswami et al., 2005), using control or TNT-defective (shM-Sec) macrophages to evaluate the effect of TNT formation on tumor cell invasion (Fig. 6A). Results showed that while control macrophages enhanced the ability of tumor cells to invade, the presence of TNT-defective macrophages failed to induce tumor cell invasion into the collagen layer (Fig. 6B, black bars). Interestingly, tumor cell invasion was not affected in the presence of GM6001 inhibitor (Fig. 6B, gray bars), which eliminated the release of EGF, suggesting that soluble EGF was not required for macrophage-induced tumor cell invasion. Similarly, co-culture of control macrophages induced invasion of MDA-MB-231 cells, which was significantly reduced in the presence of TNT-defective (shM-Sec) macrophages (Fig. 6C). These data suggest that TNTs are important for tumor cell invasion in vitro even in the presence of macrophage-secreted EGF.
Fig. 6.
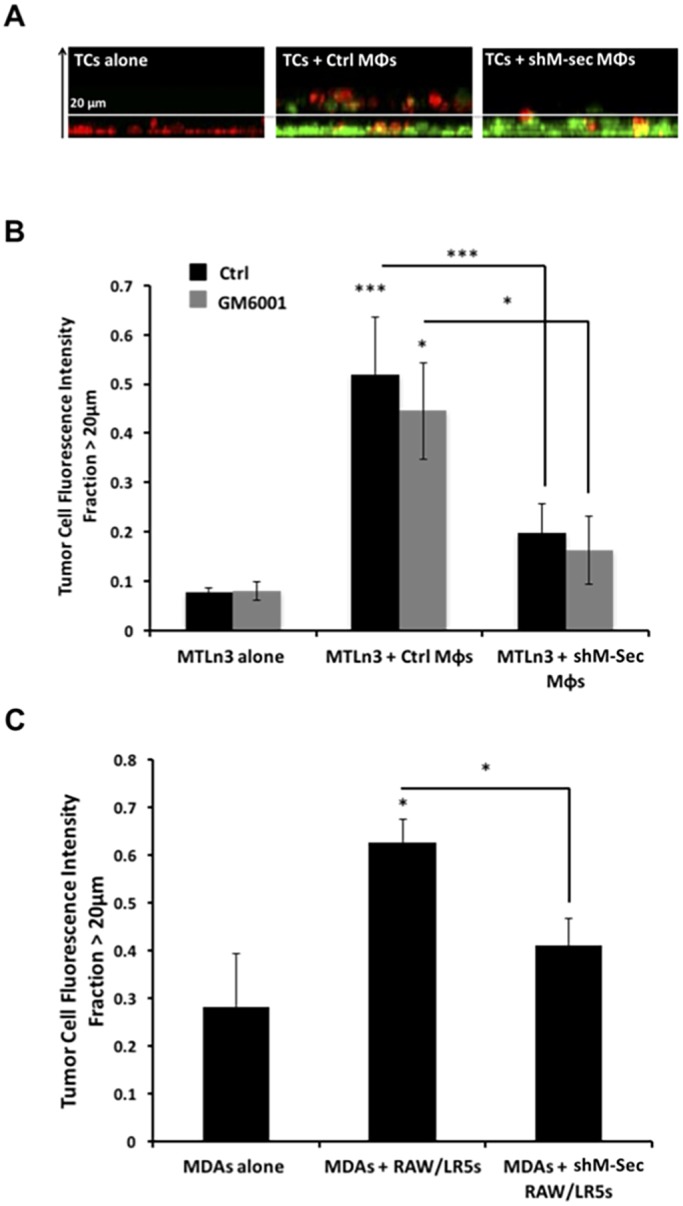
TNTs are important in promoting tumor cell invasion in vitro. (A) Tumor cells (TCs, red) were either plated alone or in co-culture with control or shM-Sec (TNT deficient) RAW/LR5 macrophages (Mφs, green) overnight then overlaid with a layer of collagen (5.8 mg/ml) and incubated for 24 h. (B) Mean±s.e.m. fraction of untreated (black bars) MTLn3 tumor cells that crossed above 20 μm was measured as the fluorescent intensity of tumor cells (red) that have crossed above the 20 μm optical slice relative to the total tumor cell fluorescence intensity. The same experiment was repeated in the presence of GM6001 inhibitor, which blocks macrophage EGF secretion (gray bars). n=3 independent experiments. Statistical significance is compared to control MTLn3 alone or indicated above the brackets comparing adjacent bars. *P<0.05 by Student's t-test. (C) Same experiment as in B, performed using human MDA-MB-231 tumor cells and the mean±s.e.m. fraction of tumor cell fluorescent intensity >20 μm is measured. n=3 independent experiments. Statistical significance is compared to control alone or indicated above the brackets comparing adjacent bars. *P<0.05, ***P<0.003 by Student's t-test.
TNTs are important for directional tumor cell streaming towards the endothelium
Other studies have shown that following the initial step of invasion from the primary tumor, cells then directionally migrate along with macrophages towards the endothelium in a process known as streaming in vivo (Patsialou et al., 2013). This sustained directional migration towards the endothelium can be tested in vitro using a modified one-dimensional (1D) assay (Leung et al., 2017; Sharma et al., 2012). This assay uses patterned lines coated with fibronectin, with human umbilical vein endothelial cells (HUVECs) coated onto Sephadex beads placed on the one end. This mimics cell streaming in vivo from the primary tumor towards the blood vessel. Previous studies have shown that while tumor cells close to the endothelium can migrate directionally, cells that are >500 μm away require the presence of macrophages to sustain their directional migration (Leung et al., 2017). Therefore, we investigated whether the heterotypic TNT interaction is important for sustained tumor cell migration towards blood vessels using the in vitro 1D assay as we have done previously. We observed that TNTs formed between MTLn3 tumor cells and primary mouse BMMs using scanning electron microscopy (SEM) (Fig. 7A). Moreover, using membrane-labeled mCherry–CAAX MTLn3 tumor cells (red) and GFP–CAAX RAW/LR5 macrophages (green) we were able to monitor tumor cell migration along fibronectin-coated strips towards the endothelial-coated bead under live-cell imaging (Fig. 7B; Movies 8–10). We also observed heterotypic TNTs between the two cell types (Fig. 7B). Further analysis of tumor cell directionality and persistence showed a significant increase in directional migration of tumor cells in the presence of control macrophages, which is significantly reduced in the presence of TNT-defective (shM-Sec) macrophages to similar levels to tumor cells alone (Fig. 7C). These results suggest that heterotypic TNTs between macrophages and tumor cells are required for tumor cell streaming beyond a distance of 500 μm to directionally migrate towards the endothelium.
Fig. 7.

TNTs are important for directional tumor cell streaming towards the endothelium. (A) Fixed images using scanning electron microscopy (SEM) of the 1D assay where MTLn3 tumor cells were cultured alone on fibronectin coated strips (left panel) or in co-culture with murine primary bone marrow-derived macrophages (BMMs) (middle panel). Right panel: zoomed image of the boxed area showing a TNT between the two cell types. (B) Live-cell imaging of co-culture between mCherry–CAAX MTLn3 tumor cells (red) and GFP–CAAX RAW/LR5 macrophages (green) plated on fibronectin (FN)-coated strips (magenta). Arrow indicates a TNT connecting the two cell types. Inset shows the corresponding phase image. (C) Analysis of the persistent directional migration of tumor cells >500 μm away from the endothelium composed of HUVEC-coated beads (black bar). Tumor cell persistent directional migration is also calculated in co-culture with control or shM-Sec RAW/LR5 macrophages (gray bars). Sustained directionality is calculated as the net path length over the total path length during the course of >8 h movie. Data represent mean±s.e.m. of three independent experiments. *P<0.05; **P<0.02; ns, non-significant by Student's t-test.
Effect of TNTs on tumor cell invasion in vivo
Our results so far have shown that TNT interactions between tumor cells and macrophages play a role in enhancing the invasive phenotype of tumor cells as well as promoting cellular invasion and sustained directional migration towards the endothelium in vitro. We next wanted to determine whether this TNT interaction was also important in vivo. Given that there are no optimal mouse models available to study TNTs and considering the difficulty in imaging TNTs in mice, we opted to use zebrafish to study their role in promoting tumor invasion and metastasis in vivo. We employed a zebrafish model that has been previously used by others to verify the requirement for tumor-associated macrophages in breast cancer cell metastasis (Wang et al., 2015). Tumor cells alone or with macrophages were injected into the pervitelline space of day 2 embryos, and macrophage-dependent tumor cell spread was measured four days following injection (Fig. 8A). The tumor area formed after 4 days was significantly larger in the presence of control macrophages compared to the level induced by TNT-defective macrophages, which was similar to that of tumor cells injected alone (Fig. 8B). We also analyzed the number of tumor cell foci disseminated from the primary site of injection, as well as the maximal distance traveled by these foci. Results showed that tumor cell invasion was significantly enhanced in the presence of control macrophages, while significantly reduced in the presence of TNT-defective macrophages (Fig. 8C,D). Hence, these results show that in addition to promoting tumor cell invasion in vitro, the interaction with macrophages via TNTs plays an important role in tumor cell invasion in vivo in zebrafish.
Fig. 8.

Effect of TNTs on tumor cell invasion in vivo. (A) MTLn3 tumor cells labeled with CMPTX CellTracker Red either injected alone or co-injected with RAW/LR5 macrophages labeled with CMFDA CellTracker Green. 4 days post-injection, zebrafish embryos were anesthetized and imaged by fluorescent microscopy. Representative images are shown of the following conditions: MTLn3s alone, MTLn3s with control RAW/LR5 macrophages (Mφs) or MTLn3s with shM-Sec (TNT deficient) RAW/LR5 macrophages. (B–D) Images were analyzed for the mean±s.e.m. area of tumor formed (B), mean±s.e.m. number of disseminated metastatic foci (C), and the mean±s.e.m. maximal distance of metastatic foci from the primary tumor (D). n>40 embryos/group from three different experiments. **P<0.02; ***P<0.003; ns, non-significant by Student's t-test.
DISCUSSION
Intercellular communication between tumor and stromal cells within the tumor microenvironment is crucial to the progression of invasive cancers (Liotta and Kohn, 2001). However, the mechanism by which signal transduction occurs between such cell types within the dense heterogeneous tumor tissue remains poorly understood. Macrophages are the main host cells present within the breast tumor matrix and interact with tumor cells starting at the early stages during cancer development, promoting tumor cell growth and invasion (Condeelis and Pollard, 2006; Noy and Pollard, 2014; Pollard, 2008).
In addition to soluble secreted factors, exosomes and microvesicles have been extensively studied as purveyors of long-distance communication between cells. However, relying on diffusion within the dynamic 3D tumor matrix found in a physiological context may prevent signals from reaching potential target cells with high specificity and certainty (Francis and Palsson, 1997; Pap et al., 2011; Simons and Raposo, 2009). Therefore, the primary aim of this study was to investigate the novel role of TNTs as a means of extending the range of direct cell–cell communication between macrophages and tumor cells in tumor cell invasion. Previous studies have shown that direct contact with macrophages induces the formation of actin-rich tumor cell invadopodium structures. Unlike TNTs, invadopodia are present at the ventral surface of tumor cells and are required for tumor cell invasion and transendothelial migration (Roh-Johnson et al., 2014; Yamaguchi et al., 2006). In addition, previous findings have shown that direct contact between a macrophage and a tumor cell promotes MenaINV expression, causing MenaINV-dependent sensitization of tumor cells to growth factor signals (Eddy et al., 2017; Weidmann et al., 2016) and TMEM-dependent tumor cell intravasation across an endothelial barrier (Pignatelli et al., 2016). Here, we investigate yet another means of macrophage–tumor cell interaction occurring through TNTs. Our results indicate that TNTs form between tumor cells and macrophages, which leads to tumor cell activation as evidenced by tumor cells developing a more migratory phenotype. Our previous studies have demonstrated that EGF is the factor required for macrophage promotion of tumor cell elongation (Goswami et al., 2005) and blocking the shedding of EGF from the cell surface blocks tumor cell elongation (Ishihara et al., 2013). The current study demonstrates that TNT contact could bypass the requirement for soluble EGF, yet occurs in an EGFR-dependent manner. This suggests that direct contact through TNTs could mediate EGF–EGFR signaling, similar to the previously observed induction of tumor cell protrusions in response to EGF that has been immobilized on a bead (Kempiak and Segall, 2004). This is consistent with previous studies showing that TNTs can mediate receptor–ligand interactions, particularly between immune cells (Chauveau et al., 2010). In addition, the TNT interaction between cells was also required for macrophage-mediated tumor cell invasion even when soluble EGF was present. This suggests that membrane-bound EGF may be mobilized along the TNT membrane where it interacts with EGFR on the tumor cell surface. In fact, Chang et al. (2017) have demonstrated the movement of EGFR molecules on actin-based intercellular connections between HeLa cells using an optical trap combined with single-molecule imaging. This EGF–EGFR surface interaction may lead to a prolonged activation of EGF since EGFR cannot be downregulated through internalization.
Alternatively, there have been reports on crosstalk between EGFR and Notch signaling in breast cancer as a compensatory mechanism used by cancer cells to propagate survival, proliferation, metastasis and resistance to EGFR-targeted inhibition (Baker et al., 2014; Dai et al., 2009). Previous findings showed that the direct physical contact between a macrophage and tumor cell induces Notch signaling, resulting in the induction of MenaINV expression that enhances EGF sensitivity, intravasation and dissemination (Eddy et al., 2017; Pignatelli et al., 2016). Overall, these studies suggest that direct cell–cell contact through TNTs can activate EGF–EGFR signaling, leading to tumor cell invasion independently of soluble EGF.
Following invasion, macrophage–tumor cell pairing is required for efficient tumor cell migration and directional streaming towards the circulatory system. In fact, blocking macrophage function in vivo using clodronate significantly reduces this organized and directional migration pattern of tumor cells while increasing more random migration (Patsialou et al., 2009). However, cell streaming is only partially blocked by inhibiting the secreted CSF-1 or EGF factors (Patsialou et al., 2009; Roussos et al., 2011). This indicates that other means of communication can contribute to cell streaming including other secreted factors as well as direct cell–cell contact between the two cell types. Reconstituting this process in vitro using a 1D assay has shown that tumor cells and macrophages can spontaneously form pairs on micropatterened surfaces (Sharma et al., 2012) and migrate directionally to HGF secreted by endothelial cells (Leung et al., 2017). While tumor cells alone migrate up to a distance of 500 μm from the endothelial-coated bead, the presence of macrophages extends tumor cell response beyond the 500 μm range (Leung et al., 2017). This last study further extends on the other cited studies demonstrating that the macrophage–tumor cell interaction through TNTs is important in sustaining tumor cell ability to directionally migrate towards the endothelial bead, particularly beyond the 500 μm range (Fig. 7). This suggests that TNTs could provide a direct signal that promotes long-distance tumor cell streaming towards blood vessels as observed in vivo.
Intravital imaging has been shown to be a powerful method to directly visualize these multicellular dynamics within the tumor microenvironment in live mice at a single-cell resolution (Entenberg et al., 2015; Harney et al., 2015; Timpson et al., 2011) However, to date, there are no optimal mouse models available to study TNTs and their role in promoting tumor invasion and metastasis in vivo. In this current study, we used a zebrafish system which allowed us to test our hypothesis that TNTs are important for tumor invasion and progression. In fact, a study by Teng et al. (2013) showed that the zebrafish model can faithfully recapitulate the metastatic potential observed in vitro using cells from prostate, colon, pancreas and breast, including human MDA-MB-231 cells. Cells were subsequently seen in the vasculature throughout the fish body. Also, their results recapitulated the invasion and metastatic behavior of MCF10A seen in vitro as well as in vivo (Teng et al., 2013). However, this study was done in the absence of added macrophages. In this current study, our results showed that the co-injection of tumor cells with control macrophages promotes tumor cell spreading and metastasis away from the site of injection (Fig. 8). This is in accordance with previous studies using the same model to study the role of tumor-associated macrophage (TAM)-mediated tumor cell dissemination and metastasis (Wang et al., 2015). Interestingly, the authors found a positive correlation between metastasis in human patients and TAM-mediated tumor cell dissemination and metastasis using this zebrafish model, although the mechanism remains unidentified. Consistent with our in vitro studies, our in vivo findings demonstrate that TNT-defective macrophages with reduced physical interaction with tumor cells significantly reduced tumor cell dissemination and spreading (Fig. 8). Collectively, our findings provide evidence that TNT-mediated intercellular communication is important for tumor cell invasion, not only in vitro, but also in a zebrafish model in vivo. Additional work will be required to determine the role of TNTs in invasion, intravasation and metastasis in breast cancer in mice where the tumor microenvironment is known to determine the malignant phenotype of the primary tumor (Leung et al., 2017). Therefore, targeting TNT-mediated communication with macrophages could potentially not only inhibit tumor cell activation but also prevent tumor cells from efficiently migrating towards the blood vessels. In particular, it will be important to determine the importance of TNTs in the function of TMEM, the intravasation doorway in breast tumors that are predictive of distant metastatic relapse in breast cancer patients (Karagiannis et al., 2017; Pignatelli et al., 2014; Rohan et al., 2014; Sparano et al., 2017).
A number of other studies have visualized TNTs in tumors from human patient explants with various examples of different cancer types including mesothelioma (Lou et al., 2012), ovarian cancer, osteosarcoma (Thayanithy et al., 2014a) and laryngeal carcinomas (Antanavičiu¯te˙ et al., 2014; Xu et al., 2009). However, whether the observed TNTs originate from the tumor cells or are connections between tumor and stromal cells could not be determined in these studies. In addition, transfer of microRNAs (miRNAs) has been shown in osteosarcoma and ovarian cancer to stromal cells in vitro (Thayanithy et al., 2014a). More interestingly, a study by Osswald et al. (2015) has revealed the presence of TNTs in malignant brain tumors in a live animal model. These TNTs can mediate intercellular transfer of chemoresistance and invasion factors. Our study suggests an additional role for TNTs in cancer by mediating the macrophage–tumor cell interaction in breast cancer. Consequently, TNTs represent an attractive target for combination therapy, where disrupting their formation and maintenance via pharmacological inhibitors or other methods could represent a promising approach for cancer treatment. Therefore, we believe that targeting these cellular structures using specific inhibitors, while still underdeveloped, represents an important strategic approach for selective and more effective therapy.
MATERIALS AND METHODS
Cell lines, transfections and plasmids
All cells were cultured and maintained in a 5% CO2 and 37°C incubator. Murine RAW/LR5 monocyte/macrophages were cultured in RPMI 1640 medium (Mediatech) supplemented with 10% heat-inactivated newborn calf serum (Sigma-Adrich) and antibiotics (100 units/ml penicillin, 100 µg/ml streptomycin, Sigma-Adrich). Membrane-labeled RAW/LR5s were generated through stable transfection using a DNA construct containing a GFP–CAAX plasmid cloned into a pcDNA3.1 vector as described in Abou-Kheir et al. (2008). Following transfection using Fugene HD (Promega), cells were selected in the presence of G418 (Invitrogen). The shM-Sec and shWASP RAW/LR5 cell lines were generated previously using stable transduction with short hairpin RNA (shRNA) constructs directed against either WASP and M-Sec (Dovas et al., 2009; Hanna et al., 2017). Rat mammary adenocarcinoma MTLN3 cells were maintained at 37°C in a 5% CO2 incubator and were cultured in αMEM (GIBCO) supplemented with 5% heat-inactivated fetal bovine serum (Sigma-Adrich) and antibiotics (100 units/ml penicillin, 100 µg/ml streptomycin, Sigma-Adrich). Membrane-labeled MTLn3s were generated by stable transfection using a DNA construct containing a mCherry–CAAX plasmid cloned into a pcDNA3.1 vector. Following transfection using Lipofectamine 2000, cells were selected in the presence of G418. Human adenocarcinoma MDA-MB-231s cells were maintained at 37°C in a 5% CO2 incubator and were cultured in DMEM (Corning) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Adrich) and antibiotics (100 units/ml penicillin, 100 µg/ml streptomycin, Sigma-Adrich). Membrane-labeled MDA-MB-231s were generated by stable transfection using a DNA construct containing a mCherry–CAAX plasmid cloned into a pcDNA3.1 vector. Following transfection using Lipofectamine 2000, cells were selected in the presence of G418. HUVEC endothelial cells were grown in EGM-2 SingleQuot Kit media (Lonza) and used at passage 4–6. Murine bone marrow-derived macrophages (BMMs) were isolated from wild-type mice, prepared as previously described (Stanley, 1997), and grown in 15% FBS/αMEM media supplemented with 360 ng/ml recombinant human CSF-1 (generously provided by E. Richard Stanley, Albert Einstein College of Medicine).
TNT quantitation
TNT formation was monitored following the co-culture of GFP–CAAX-labeled RAW/LR5 with mCherry–CAAX-labeled MTLn3 cells in a 1:1 ratio overnight in MatTek dishes (MatTek Corporation) in αMEM growth medium. Cells were briefly washed with PBS buffer and then imaged live. For TNT quantitation, in order to be counted as a TNT connection, at least one thin membranous structure was required to be present connecting two cells and a portion of the structure must not be adherent to the substratum, and have a minimum length of 8 µm. Cells with no TNTs were required to be within one cell body length of another cell without touching any other cell to be counted as negative. A total of 128 cells for every experiment were quantified and the numbers of TNTs formed were presented as dot plots with individual values of the number of TNT connections for each independent experiment, as well as the overall mean of all experiments±s.e.m. Experiments were repeated at least three times.
Transfer of labeled material
To test functionality of TNTs, RAW/LR5 cells were stained with DiI (Invitrogen) according to the manufacturer's instructions. These labeled cells were incubated with MTLn3 cells expressing GFP–CAAX at a 1:1 ratio overnight. Cells were washed three times with PBS, detached from the plate, and analyzed for transfer of DiI material into GFP-positive cells using a FACSCalibur flow cytometer (BD Biosciences). Control experiments where DiI-labeled cells were fixed before co-culture showed no transfer of labeled material, suggesting the transfer of material is not due to phagocytic activity of recipient cells.
Immunofluorescence, super-resolution microscopy (3D-SIM) and scanning electron microscopy (SEM)
Cells plated in MatTek dishes were fixed in 3.7% formaldehyde in PBS buffer for 10 min at 37°C. To visualize TNTs, cells were then stained with wheat germ agglutinin with Alexa Fluor 488 conjugate (Molecular Probes, Invitrogen) at a concentration of 1 μg/ml in HBSS buffer for 10 min at room temperature. Cells were then permeabilized in 0.2% Triton X-100 (in buffer with divalent; BWD) for 5 min. F-actin was visualized by staining with Alexa Fluor 647-labelled phalloidin (Molecular Probes, Invitrogen). Coverslips were mounted in SlowFade Diamond Antifade mountant (Molecular Probes, Invitrogen). Multichannel structured illumination microscopy (SIM) images were acquired using a Nikon Structured Illumination N-SIM system on an inverted Nikon ECLIPSE Ti-E equipped with a 100×1.49 NA objective. Multicolor fluorescence was generated using diode lasers (488, 561 and 647 nm). Acquisition was performed with electron-multiplying CCD cameras (Andor iXon3 DU897) 512×512 pixel frame size. Z-stack images were imaged with a step size of 0.12 µm. Three reconstruction parameters (Illumination Modulation Contrast, High Resolution Noise Suppression and Out of Focus Blur Suppression) were extensively tested to generate consistent images across experiments without abnormal features or artifacts and producing the best Fourier transforms. The images were processed using Nikon Elements software. 3D reconstruction was performed with Imaris software (Bitplane). TNT width quantitation was performed using the WGA channel where diameters were measured over at least four different points within individual heterotypic TNTs and the mean width was determined. Analysis and quantitation of TNT widths were performed with ImageJ (National Institutes of Health, http://imagej.nih.gov/ij/).
For SEM imaging, cells grown on patterned substrate were fixed with 2.5% buffered glutaraldehyde, dehydrated through a graded series of ethanol, critical point-dried using liquid carbon dioxide in a Tousimis 790 Critical Point Dryer (Rockville), sputter-coated with chromium in an Electron Microscopy Sciences EMS150T-ES coating unit, imaged in a Zeiss Supra 40 field emission scanning electron microscope using an accelerating voltage of 2 kV.
Live-cell imaging of heterotypic TNTs
TNT formation was monitored by plating GFP–CAAX labeled RAW/LR5 macrophages and mCherry–CAAX tumor cells to MatTek dishes in 1:1 density ratio (1×105 total cells per 35 mm dish). Cells were cultured in tumor cell growth media overnight. To image TNTs, imaging was performed in BWD supplemented with 5% FBS. The MatTek dish was then mounted on a heated stage maintained at 37°C for time-lapse imaging. Following temperature stabilization, images were acquired at 10 s intervals for 30 min at 60× magnification. Analysis of captured images was performed using Metamorph (Molecular Devices) and ImageJ software.
2D elongation assay
A co-culture of control or shM-Sec RAW/LR5 cells and MTLn3 cells at 1:1 ratio was grown in MatTek dishes in αMEM with 5% FBS and 100 U/ml penicillin and 100 µg/ml streptomycin and cultured overnight. In the case of inhibitor treatment, GM6001 (Enzo Life Science) was added to the co-culture at a concentration of 25 μM. IRESSA was added to the co-culture at a concentration of 1 μM. The elongation index was determined as described previously (Ishihara et al., 2013). Briefly, cell perimeters were traced using ImageJ and the ratio of the major axis over the minor axis of the fit ellipse was measured. At least 30 cells per experiment were analyzed for each case in three independent experiments. Images were taken using 60× objective on an Olympus IX71 microscope coupled to a Sensicam cooled CCD camera.
3D in vitro invasion
3D in vitro invasion assays were performed and quantified as described (Goswami et al., 2005). Briefly, 8×104 Cell Tracker Red-labeled MTLn3 or MDA-MB-231 cells were plated on MatTek dishes in the presence or absence of 4×105 of the indicated RAW/LR5 cells and grown in αMEM containing 15% FBS, 0.02 mg/ml asparagine, antibiotics and 10,000 units/ml CSF-1 for 16 h. Cells were overlaid with 5.8 mg/ml type I collagen, incubated for 24 h and fixed. Z-stack projections were generated from images acquired at 1 μm steps on a Zeiss 5 Live DuoScan confocal microscope (20×0.8NA air objective). To quantify the MTLn3 cell invasion, fluorescence in z-sections from 20 µm into the collagen and above was added up and divided by the sum of fluorescence in all the z-sections. Data are reported as the fraction of tumor cell fluorescence from an average of five independent fields per experiment.
In vitro 1D assay
In vitro 1D assay was performed as described previously (Leung et al., 2017). Briefly, Custom 170 μm (#1.5) thick, 20×20 mm2 coverslips micro-patterned by micro-photolithography with parallel linear 2.5 μm stripes of fibronectin labeled with 650 nm dye (CYTOO, Grenoble, France) were fitted into a 4-well CYTOO chamber. For the HUVEC bead preparation, 1×106 HUVEC cells were mixed with 500 Sephadex beads in 1.5 ml of EGM-2 media in a FACs tube. The cell–bead mixture was incubated for 4 h at 37°C, shaking the tube every 20 min. After 4 h, the beads were transferred into a 5 mm dish with EGM-2 media and left overnight. The next morning, the beads were labeled with Celltracker dye. The beads were flushed off the surface of the dish and transferred into a 15 ml conical tube. When the beads settled, the media was replaced and the cells were allowed to equilibrate for 30 min before use. 5×103 mCherry-labeled MTLn3 tumor cells were plated per well in the CYTOO Chamber. Cells were allowed to attach overnight at 37°C in the 5% CO2 incubator. The day of the experiment, 1×103 GFP–CAAX control or shM-Sec RAW/LR5 cells and 10–15 HUVEC beads were plated per well. Time-lapse images were obtained on the wide-field DeltaVision microscope equipped with a Photometrics CoolSnap HQ2 CCD camera and NanoMotion III stage. Imaging was performed at 37°C with a 20× air objective, NA=0.4. Images were acquired every 10 min for up to 10 h. Image analysis was performed as described previously (Leung et al., 2017). Briefly, images were assembled using the LOCI plug-in and the ‘Merge Channels’ command in ImageJ. A montage of adjacent fields totaling approximately 1000 μm of continuous stripe length was analyzed. The montage was rotated so that the HUVEC endothelial beads were always on the left. Tumor cell centroids were tracked for a minimum of 30 frames. Directionality was calculated as net path length divided by the total path length.
Zebrafish tumor model
Zebrafish were obtained from the establish colony maintained by the Developmental and Molecular Biology Zebrafish Core at Albert Einstein College of Medicine. Breeding and husbandry practices were performed under standard operating procedures described in the zebrafish SOP approved by the IACUC. Twenty-four hours after fertilization (hpf) zebrafish embryos were incubated in aquarium water containing 0.2 mmol/l 1-phenyl-2-thio-urea (PTU) to prevent pigmentation. At 48 hpf, the zebrafish embryos were dechorionated with a pair of sharp-tip forceps and anesthetized with 0.04 mg/ml of tricaine to immobilize them. Anesthetized embryos were moved onto a modified agarose gel for microinjection. Before injection, tumor cells or macrophages were labeled using CellTracker Red (CMPTX) or CellTracker Green (CMFDA) according to manufacturer's instructions (Life Technologies). Approximately 300–500 fluorescently labeled tumor cells or a mixture of equal numbers of 150–250 tumor cells and 150–250 fluorescently labeled macrophages were resuspended in serum-free DMEM (Hyclone) and 5 nl of tumor cell solution were injected into the perivitelline space (PVS) of each embryo using non-filamentous borosilicate glass capillaries needles. After injection, the zebrafish embryos were immediately transferred into PTU aquarium water and kept at 28°C for 4 days after injection. Embryos were then checked for tumor invasion and metastasis using an Olympus IX71 microscope coupled to a Sensicam cooled CCD camera.
Data analysis
For every experiment, n-value was greater or equal to three independent experiments (n≥3). Results were considered statistically different when two-tailed analysis performed using a Student’s t-test resulted in differences between two means with a P-value of less than or equal to 0.05 (P≤0.05). Error bars signify standard error of the mean (±s.e.m.).
Supplementary Material
Acknowledgements
We would like to thank Peng Guo for his assistance and help with the 3D-SIM imaging and analysis. We would like to thank Louis Hodgson for the use of his microscope to perform time-lapse live-imaging. We would like to thank Michael Cammer for his assistance with image analysis and processing. We would like to thank Mr Clinton DePaolo, Operations Director of the Zebrafish Core Facility at Albert Einstein College of Medicine, for his assistance with the zebrafish experiments. This work is in partial fulfillment of the PhD requirements for S.J.H.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.J.H., K.M.-S., D.C.; Methodology: S.J.H., K.M.-S., D.C.; Validation: S.J.H., K.M.-S., D.C.; Formal analysis: S.J.H., K.M.-S., E.L., D.C.; Investigation: S.J.H., K.M.-S., E.L., D.C.; Resources: E.L., D.C.; Data curation: S.J.H., K.M.-S., E.L., A.G., D.C.; Writing - original draft: S.J.H., K.M.-S., E.L., J.C., D.C.; Writing - review & editing: S.J.H., K.M.-S., E.L., A.G., J.C., D.C.; Visualization: S.J.H., K.M.-S., E.L., D.C.; Supervision: D.C.; Project administration: S.J.H., J.C., D.C.; Funding acquisition: S.J.H., K.M.-S., J.C., D.C.
Funding
This work was supported by National Institutes of Health grants R01 GM071828 to D.C., P01 CA100324 to S.J.H., J.C. and D.C., P01 CA150344 to J.C. and F99 CA212451 (National Cancer Institute) to S.J.H. K.M.-S. was funded under a National Institutes of Health IRACDA fellowship where the content is solely the responsibility of the authors and does not necessarily represent the official views supporting K12GM102779. The 3D-SIM Nikon super resolution microscope used in this study is part of the Analytical Imaging Facility at Albert Einstein College of Medicine supported by National Cancer Institute cancer center grant P30CA013330 and shared instrumentation grant (SIG) 1S10OD18218-1. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.223321.supplemental
References
- Abou-Kheir W., Isaac B., Yamaguchi H. and Cox D. (2008). Membrane targeting of WAVE2 is not sufficient for WAVE2-dependent actin polymerization: a role for IRSp53 in mediating the interaction between Rac and WAVE2. J. Cell Sci. 121, 379-390. 10.1242/jcs.010272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abounit S. and Zurzolo C. (2012). Wiring through tunneling nanotubes–from electrical signals to organelle transfer. J. Cell Sci. 125, 1089-1098. 10.1242/jcs.083279 [DOI] [PubMed] [Google Scholar]
- Ady J. W., Desir S., Thayanithy V., Vogel R. I., Moreira A. L., Downey R. J., Fong Y., Manova-Todorova K., Moore M. A. and Lou E. (2014). Intercellular communication in malignant pleural mesothelioma: properties of tunneling nanotubes. Front. Physiol. 5, 400 10.3389/fphys.2014.00400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antanavičiūtė I., Rysevaitė K., Liutkevičius V., Marandykina A., Rimkutė L., Sveikatienė R., Uloza V. and Skeberdis V. A. (2014). Long-distance communication between laryngeal carcinoma cells. PLoS ONE 9, e99196 10.1371/journal.pone.0099196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi J., Benowitz A., De Biasi V., Den Boer M. L., Cherqui S., Cui H., Douillet N., Eugenin E. A., Favre D., Goodman S. et al. (2017). Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front. Mol. Neurosci. 10, 333 10.3389/fnmol.2017.00333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly M., Yan L., Whitesides G. M., Condeelis J. S. and Segall J. E. (1998). Regulation of protrusion shape and adhesion to the substratum during chemotactic responses of mammalian carcinoma cells. Exp. Cell Res. 241, 285-299. 10.1006/excr.1998.4031 [DOI] [PubMed] [Google Scholar]
- Baker M. (2017). How the internet of cells has biologists buzzing. Nature 549, 322-324. 10.1038/549322a [DOI] [PubMed] [Google Scholar]
- Baker A. T., Zlobin A. and Osipo C. (2014). Notch-EGFR/HER2 bidirectional crosstalk in breast cancer. Front. Oncol. 4, 360 10.3389/fonc.2014.00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer C. L. and Weinberg R. A. (2011). A perspective on cancer cell metastasis. Science 331, 1559-1564. 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- Chang M., Oh J., Kim Y., Hohng S. and Lee J.-B. (2017). Extended depth of field for single biomolecule optical imaging-force spectroscopy. Opt. Express 25, 32189-32197. 10.1364/OE.25.032189 [DOI] [PubMed] [Google Scholar]
- Chauveau A., Aucher A., Eissmann P., Vivier E. and Davis D. M. (2010). Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells. Proc. Natl. Acad. Sci. USA 107, 5545-5550. 10.1073/pnas.0910074107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. and Pollard J. W. (2006). Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124, 263-266. 10.1016/j.cell.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Dai J., Ma D., Zang S., Guo D., Qu X., Ye J. and Ji C. (2009). Cross-talk between Notch and EGFR signaling in human breast cancer cells. Cancer Invest. 27, 533-540. 10.1080/07357900802563036 [DOI] [PubMed] [Google Scholar]
- Delage E., Cervantes D. C., Pénard E., Schmitt C., Syan S., Disanza A., Scita G. and Zurzolo C. (2016). Differential identity of Filopodia and Tunneling Nanotubes revealed by the opposite functions of actin regulatory complexes. Sci. Rep. 6, 39632 10.1038/srep39632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning T. L., Wang Y.-C., Patel S. R., Williams I. R. and Pulendran B. (2007). Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086-1094. 10.1038/ni1511 [DOI] [PubMed] [Google Scholar]
- Dovas A., Gevrey J.-C., Grossi A., Park H., Abou-Kheir W. and Cox D. (2009). Regulation of podosome dynamics by WASp phosphorylation: implication in matrix degradation and chemotaxis in macrophages. J. Cell Sci. 122, 3873-3882. 10.1242/jcs.051755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy R. J., Weidmann M. D., Sharma V. P. and Condeelis J. S. (2017). Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27, 595-607. 10.1016/j.tcb.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Andaloussi S., Mäger I., Breakefield X. O. and Wood M. J. A. (2013). Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347-357. 10.1038/nrd3978 [DOI] [PubMed] [Google Scholar]
- Entenberg D., Rodriguez-Tirado C., Kato Y., Kitamura T., Pollard J. W. and Condeelis J. (2015). In vivo subcellular resolution optical imaging in the lung reveals early metastatic proliferation and motility. Intravital 4, 1-11. 10.1080/21659087.2015.1086613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis K. and Palsson B. O. (1997). Effective intercellular communication distances are determined by the relative time constants for cyto/chemokine secretion and diffusion. Proc. Natl. Acad. Sci. USA 94, 12258-12262. 10.1073/pnas.94.23.12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S., Sahai E., Wyckoff J. B., Cammer M., Cox D., Pixley F. J., Stanley E. R., Segall J. E. and Condeelis J. S. (2005). Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 65, 5278-5283. 10.1158/0008-5472.CAN-04-1853 [DOI] [PubMed] [Google Scholar]
- Hanahan D. and Coussens L. M. (2012). Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309-322. 10.1016/j.ccr.2012.02.022 [DOI] [PubMed] [Google Scholar]
- Hanna S. J., McCoy-Simandle K., Miskolci V., Guo P., Cammer M., Hodgson L. and Cox D. (2017). The role of Rho-GTPases and actin polymerization during macrophage tunneling nanotube biogenesis. Sci. Rep. 7, 8547 10.1038/s41598-017-08950-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney A. S., Arwert E. N., Entenberg D., Wang Y., Guo P., Qian B.-Z., Oktay M. H., Pollard J. W., Jones J. G. and Condeelis J. S. (2015). Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 5, 932-943. 10.1158/2159-8290.CD-15-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase K., Kimura S., Takatsu H., Ohmae M., Kawano S., Kitamura H., Ito M., Watarai H., Hazelett C. C., Yeaman C. et al. (2009). M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 11, 1427-1432. 10.1038/ncb1990 [DOI] [PubMed] [Google Scholar]
- Hoshino A., Costa-Silva B., Shen T.-L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder S. et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329-335. 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara D., Dovas A., Hernandez L., Pozzuto M., Wyckoff J., Segall J. E., Condeelis J. S., Bresnick A. R. and Cox D. (2013). Wiskott-Aldrich syndrome protein regulates leukocyte-dependent breast cancer metastasis. Cell Rep. 4, 429-436. 10.1016/j.celrep.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. N., Das S. R., Emin M. T., Wei M., Sun L., Westphalen K., Rowlands D. J., Quadri S. K., Bhattacharya S. and Bhattacharya J. (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759-765. 10.1038/nm.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis G. S., Pastoriza J. M., Wang Y., Harney A. S., Entenberg D., Pignatelli J., Sharma V. P., Xue E. A., Cheng E., D'Alfonso T. M. et al. (2017). Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 9, eaan0026 10.1126/scitranslmed.aan0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempiak S. J. and Segall J. E. (2004). Stimulation of cells using EGF-coated magnetic beads. Sci. STKE 2004, pl1 10.1126/stke.2182004pl1 [DOI] [PubMed] [Google Scholar]
- Leung E., Xue A., Wang Y., Rougerie P., Sharma V. P., Eddy R., Cox D. and Condeelis J. (2017). Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 36, 2680-2692. 10.1038/onc.2016.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta L. A. and Kohn E. C. (2001). The microenvironment of the tumour-host interface. Nature 411, 375-379. 10.1038/35077241 [DOI] [PubMed] [Google Scholar]
- Lou E., Fujisawa S., Morozov A., Barlas A., Romin Y., Dogan Y., Gholami S., Moreira A. L., Manova-Todorova K. and Moore M. A. S. (2012). Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 7, e33093 10.1371/journal.pone.0033093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou E., Gholami S., Romin Y., Thayanithy V., Fujisawa S., Desir S., Steer C. J., Subramanian S., Fong Y., Manova-Todorova K. et al. (2017a). Imaging tunneling membrane tubes elucidates cell communication in tumors. Trends Cancer 3, 678-685. 10.1016/j.trecan.2017.08.001 [DOI] [PubMed] [Google Scholar]
- Lou E., O'Hare P., Subramanian S. and Steer C. J. (2017b). Lost in translation: applying 2D intercellular communication via tunneling nanotubes in cell culture to physiologically relevant 3D microenvironments. FEBS J. 284, 699-707. 10.1111/febs.13946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou E., Zhai E., Sarkari A., Desir S., Wong P., Iizuka Y., Yang J., Subramanian S., McCarthy J., Bazzaro M. et al. (2018). Cellular and molecular networking within the ecosystem of cancer cell communication via tunneling nanotubes. Front. Cell Dev. Biol. 6, 95 10.3389/fcell.2018.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy-Simandle K., Hanna S. J. and Cox D. (2016). Exosomes and nanotubes: control of immune cell communication. Int. J. Biochem. Cell Biol. 71, 44-54. 10.1016/j.biocel.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R. and Pollard J. W. (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49-61. 10.1016/j.immuni.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H., Hase K. and Kimura S. (2010). M-Sec: emerging secrets of tunneling nanotube formation. Commun. Integr. Biol. 3, 231-233. 10.4161/cib.3.3.11242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onfelt B., Nedvetzki S., Benninger R. K. P., Purbhoo M. A., Sowinski S., Hume A. N., Seabra M. C., Neil M. A. A., French P. M. W. and Davis D. M. (2006). Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 177, 8476-8483. 10.4049/jimmunol.177.12.8476 [DOI] [PubMed] [Google Scholar]
- Osswald M., Jung E., Sahm F., Solecki G., Venkataramani V., Blaes J., Weil S., Horstmann H., Wiestler B., Syed M. et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93-98. 10.1038/nature16071 [DOI] [PubMed] [Google Scholar]
- Pap E., Pállinger E. and Falus A. (2011). The role of membrane vesicles in tumorigenesis. Crit. Rev. Oncol. Hematol. 79, 213-223. 10.1016/j.critrevonc.2010.07.015 [DOI] [PubMed] [Google Scholar]
- Park H., Dovas A., Hanna S., Lastrucci C., Cougoule C., Guiet R., Maridonneau-Parini I. and Cox D. (2014). Tyrosine phosphorylation of Wiskott-Aldrich syndrome protein (WASP) by Hck regulates macrophage function. J. Biol. Chem. 289, 7897-7906. 10.1074/jbc.M113.509497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A., Wyckoff J., Wang Y., Goswami S., Stanley E. R. and Condeelis J. S. (2009). Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 69, 9498-9506. 10.1158/0008-5472.CAN-09-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A., Bravo-Cordero J. J., Wang Y., Entenberg D., Liu H., Clarke M. and Condeelis J. S. (2013). Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2, e25294 10.4161/intv.25294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J., Goswami S., Jones J. G., Rohan T. E., Pieri E., Chen X., Adler E., Cox D., Maleki S., Bresnick A. et al. (2014). Invasive breast carcinoma cells from patients exhibit MenaINV- and macrophage-dependent transendothelial migration. Sci. Signal. 7, ra112 10.1126/scisignal.2005329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J., Bravo-Cordero J. J., Roh-Johnson M., Gandhi S. J., Wang Y., Chen X., Eddy R. J., Xue A., Singer R. H., Hodgson L. et al. (2016). Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/MenaINV-initiated invadopodium formation. Sci. Rep. 6, 37874 10.1038/srep37874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov E. Y., Khryapenkova T. G., Galkina S. I., Sukhikh G. T. and Zorov D. B. (2010). Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp. Cell Res. 316, 2447-2455. 10.1016/j.yexcr.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Pollard J. W. (2008). Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 84, 623-630. 10.1189/jlb.1107762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh-Johnson M., Bravo-Cordero J. J., Patsialou A., Sharma V. P., Guo P., Liu H., Hodgson L. and Condeelis J. (2014). Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 33, 4203-4212. 10.1038/onc.2013.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohan T. E., Xue X., Lin H.-M., D'Alfonso T. M., Ginter P. S., Oktay M. H., Robinson B. D., Ginsberg M., Gertler F. B., Glass A. G. et al. (2014). Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J. Natl. Cancer Inst. 106, dju136 10.1093/jnci/dju136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougerie P., Miskolci V. and Cox D. (2013). Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol. Rev. 256, 222-239. 10.1111/imr.12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos E. T., Goswami S., Balsamo M., Wang Y., Stobezki R., Adler E., Robinson B. D., Jones J. G., Gertler F. B., Condeelis J. S. et al. (2011). Mena invasive (Mena(INV)) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin. Exp. Metastasis 28, 515-527. 10.1007/s10585-011-9388-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A., Saffrich R., Markovic I., Walther P. and Gerdes H. H. (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007-1010. 10.1126/science.1093133 [DOI] [PubMed] [Google Scholar]
- Segall J. E., Tyerech S., Boselli L., Masseling S., Helft J., Chan A., Jones J. and Condeelis J. (1996). EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin. Exp. Metastasis 14, 61-72. 10.1007/BF00157687 [DOI] [PubMed] [Google Scholar]
- Sharma V. P., Beaty B. T., Patsialou A., Liu H., Clarke M., Cox D., Condeelis J. S. and Eddy R. J. (2012). Reconstitution of in vivo macrophage-tumor cell pairing and streaming motility on one-dimensional micro-patterned substrates. Intravital 1, 77-85. 10.4161/intv.22054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidani M., Wyckoff J., Xue C., Segall J. E. and Condeelis J. (2006). Probing the microenvironment of mammary tumors using multiphoton microscopy. J. Mammary Gland Biol. Neoplasia 11, 151-163. 10.1007/s10911-006-9021-5 [DOI] [PubMed] [Google Scholar]
- Simons M. and Raposo G. (2009). Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 21, 575-581. 10.1016/j.ceb.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Sparano J. A., Gray R., Oktay M. H., Entenberg D., Rohan T., Xue X., Donovan M., Peterson M., Shuber A., Hamilton D. A. et al. (2017). A metastasis biomarker (MetaSite Breast™ Score) is associated with distant recurrence in hormone receptor-positive, HER2-negative early-stage breast cancer. NPJ Breast Cancer 3, 42 10.1038/s41523-017-0043-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley E. R. (1997). Murine bone marrow-derived macrophages. Methods Mol. Biol. 75, 301-304. 10.1385/0-89603-441-0:301 [DOI] [PubMed] [Google Scholar]
- Teng Y., Xie X., Walker S., White D. T., Mumm J. S. and Cowell J. K. (2013). Evaluating human cancer cell metastasis in zebrafish. BMC Cancer 13, 453 10.1186/1471-2407-13-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayanithy V., Babatunde V., Dickson E. L., Wong P., Oh S., Ke X., Barlas A., Fujisawa S., Romin Y., Moreira A. L. et al. (2014a). Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 323, 178-188. 10.1016/j.yexcr.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayanithy V., Dickson E. L., Steer C., Subramanian S. and Lou E. (2014b). Tumor-stromal cross talk: direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 164, 359-365. 10.1016/j.trsl.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpson P., McGhee E. J. and Anderson K. I. (2011). Imaging molecular dynamics in vivo–from cell biology to animal models. J. Cell Sci. 124, 2877-2890. 10.1242/jcs.085191 [DOI] [PubMed] [Google Scholar]
- Valastyan S. and Weinberg R. A. (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275-292. 10.1016/j.cell.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Bukoreshtliev N. V. and Gerdes H.-H. (2012). Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 7, e47429 10.1371/journal.pone.0047429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Cao Z., Zhang X.-M., Nakamura M., Sun M., Hartman J., Harris R. A., Sun Y. and Cao Y. (2015). Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 75, 306-315. 10.1158/0008-5472.CAN-14-2819 [DOI] [PubMed] [Google Scholar]
- Watkins S. C. and Salter R. D. (2005). Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 23, 309-318. 10.1016/j.immuni.2005.08.009 [DOI] [PubMed] [Google Scholar]
- Weidmann M. D., Surve C. R., Eddy R. J., Chen X., Gertler F. B., Sharma V. P. and Condeelis J. S. (2016). Mena(INV) dysregulates cortactin phosphorylation to promote invadopodium maturation. Sci. Rep. 6, 36142 10.1038/srep36142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B., Peterse J. L. and van't Veer L. J. (2005). Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591-602. 10.1038/nrc1670 [DOI] [PubMed] [Google Scholar]
- Wyckoff J., Wang W., Lin E. Y., Wang Y., Pixley F., Stanley E. R., Graf T., Pollard J. W., Segall J. and Condeelis J. (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022-7029. 10.1158/0008-5472.CAN-04-1449 [DOI] [PubMed] [Google Scholar]
- Xu W., Santini P. A., Sullivan J. S., He B., Shan M., Ball S. C., Dyer W. B., Ketas T. J., Chadburn A., Cohen-Gould L. et al. (2009). HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 10, 1008-1017. 10.1038/ni.1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Pixley F. and Condeelis J. (2006). Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 85, 213-218. 10.1016/j.ejcb.2005.10.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.