

In vivo–based functional genomic screen identifies DDR2 as an important determinant of efficacy of anti–PD-1 immunotherapy.
Abstract
While a fraction of cancer patients treated with anti–PD-1 show durable therapeutic responses, most remain unresponsive, highlighting the need to better understand and improve these therapies. Using an in vivo screening approach with a customized shRNA pooled library, we identified DDR2 as a leading target for the enhancement of response to anti–PD-1 immunotherapy. Using isogenic in vivo murine models across five different tumor histologies—bladder, breast, colon, sarcoma, and melanoma—we show that DDR2 depletion increases sensitivity to anti–PD-1 treatment compared to monotherapy. Combination treatment of tumor-bearing mice with anti–PD-1 and dasatinib, a tyrosine kinase inhibitor of DDR2, led to tumor load reduction. RNA-seq and CyTOF analysis revealed higher CD8+ T cell populations in tumors with DDR2 depletion and those treated with dasatinib when either was combined with anti–PD-1 treatment. Our work provides strong scientific rationale for targeting DDR2 in combination with PD-1 inhibitors.
INTRODUCTION
Targeting antibodies to programmed cell death protein-1 (PD-1) is an effective treatment across multiple cancer types (1, 2). While a subset of patients receiving these therapies experience favorable responses, many still show disease progression (1–4), highlighting the importance of other mechanisms influencing immune responsiveness in these tumors.
Combining therapies that enhance antitumor immunity has therefore been an area of great interest to the entire cancer community. This is reflected by the number of clinical trials investigating inhibition of PD-1 in combination with a second treatment to enhance response to this relatively new class of anticancer drugs, which has soared from a single new trial in 2009 to over 1100 trials in 2017 (5). While this growth indicates the excitement surrounding immune-focused treatment modalities, it has also markedly outpaced the gathering of preclinical evidence supporting them (4). Furthermore, testing all the possible drug combinations with checkpoint inhibitors may exceed the number of eligible cancer patients who can be enrolled in clinical trials, and this may require lower powered designs (5). Together, these issues highlight the need for an approach to determine rationale therapeutic combinations to apply to patient population.
Here, we describe a critical advance in the rapidly evolving field of cancer immunotherapy using in vivo functional genomics to identify genes whose inhibition potentiates the response to anti–PD-1 immunotherapy. Specifically, we define a novel mechanism whereby targeting the collagen receptor DDR2 (discoidin domain receptor 2) elicits a significantly enhanced response to immune checkpoint blockade with PD-1 inhibitors. We show this combination to be robust across multiple tumor models representing diverse cancer types and present the preclinical data that served as the rational basis for an ongoing clinical trial.
RESULTS
Identification of top candidate genes for enhanced response to anti–PD-1
To assess tumor responses to anti–PD-1 therapy in an immunocompetent animal, we established a syngeneic cell line model of murine bladder cancer derived from chemically induced tumors that are known to have molecular characteristics similar to those found in human bladder cancer (6, 7). Tumors were excised from N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN)–treated C57BL/6 mice and adapted to in vitro cell culture to generate the NA13 cell line (Fig. 1A). Whole-exome sequencing (WES) of NA13 revealed missense mutations in Trp53, Aird1a, and EP300, which are commonly found in BBN-induced mouse bladder tumors and in human bladder cancer datasets (6, 7). The full set of mutations identified through WES is reported in table S1.
Fig. 1. Identification of genes whose depletion enhances anti–PD-1 efficacy.
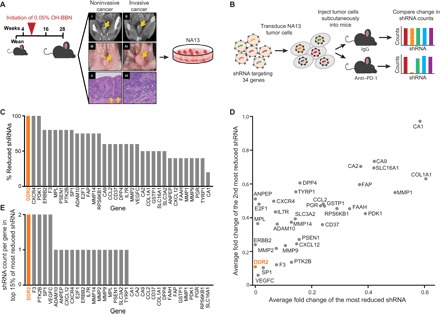
(A) Schematic of how the NA13 cell line was derived. C57BL/6 mice were started on BBN approximately 5 weeks after weaning. Corresponding contrast-enhanced microcomputed tomography scans, necropsy images, and histological preparations collected at 28 weeks. Arrows represent bladder wall in (i) to (iv) and basement membrane in (v). L, lumen of bladder. Images (i), (iii), and (v) are of a non–muscle-invasive bladder tumor, while (ii), (iv), and (vi) are of a muscle-invasive tumor. Tumors were excised and adapted to in vitro cell culture. The NA13 cell line was derived from an invasive tumor and used in our experimental studies. (B) Use of lentiviral pool containing the 34-gene druggable shRNA library to identify genes that, when knocked down, confer enhanced response with anti–PD-1 immunotherapy. (C) Ranking of genes based on the proportion of their reduction in cognate shRNAs relative to the total shRNAs per gene. (D) Normalized fold change of the most reduced shRNA versus the second most reduced shRNA (8). (E) Number of shRNAs targeting each gene that are found in the top 15% of the most reduced shRNAs overall (8).
Using NA13, we performed an in vivo short hairpin RNA (shRNA)–based screen of select druggable targets for which there are U.S. Food and Drug Administration (FDA)–approved drugs and identified genes whose knockdown in tumors was able to potentiate response to anti–PD-1 immunotherapy. First, we injected NA13 cells containing a pooled 34-gene shRNA library (five shRNAs per gene) subcutaneously into syngeneic immunocompetent mice. To identify candidate genes whose depletion led to enhanced cell death mediated by the immune-activating anti–PD-1, we quantified shRNA constructs in tumor samples following therapy using next-generation sequencing and then prioritized genes that were preferentially lost in the anti–PD-1–treated group compared to the immunoglobulin G (IgG)–treated group (Fig. 1B). Following established approaches (8), we determined preferential loss using three different methods: (i) percentage of total shRNAs per gene with reduced counts (Fig. 1C), (ii) ranking genes by their first and second most reduced shRNA based on average fold change (Fig. 1D), and (iii) the number of shRNA constructs per gene identified among the top 15% most depleted shRNAs in the library (Fig. 1E). Assessment using all three methods identified DDR2, a collagen receptor that, when activated, triggers a signaling cascade involving SHP-2, SRC, and MAP (mitogen-activated protein) kinases (9, 10), as the top-ranked gene.
Knockdown of DDR2 sensitizes tumor cells to anti–PD-1 therapy
To validate the screening results, we knocked down DDR2 in the NA13 cell line and tracked tumor growth in the presence and absence of anti–PD-1 treatment. Stable shRNA-mediated knockdown of DDR2 was confirmed by quantitative polymerase chain reaction (qPCR; fig. S1A) and immunoblot (Fig. 2A). The combination of DDR2 depletion and treatment with anti–PD-1 was highly effective in controlling NA13 tumors (Fig. 2B). Because DDR2 alterations (overexpression, amplification, and mutations) are known to drive more aggressive phenotypes in several types of cancer (11–19), we tested the generalizability of our NA13 results using breast, melanoma, and colon cancer cell lines. Knockdown of DDR2 in B16F10, a melanoma cell line, was confirmed (Fig. 2C and fig. S1B). Anti–PD-1 treatment of mice was effective at reducing pulmonary metastases only in shDDR2 B16F10 cells (Fig. 2, D to F). A marked reduction or elimination of visible tumors on the lung surface was observed (Fig. 2, D and E). Upon histologic evaluation, both shControl and shDDR2 tumors treated with IgG presented with high tumor infiltration, areas of focal inflammation and necrosis, and loss of bronchial epithelium (fig. S2). No visual differences were observed with respect to tumor size, suggesting that the tumor numbers are what account for the observed increased tumor burden in the lungs. In contrast, the mice bearing shDDR2 tumors treated with anti–PD-1 exhibited minimal tumor burden (fig. S2). Single-gene validation with shDDR2 in the mammary tumor cell line E0771 showed a similar effect, where knockdown of DDR2 (Fig. 2G and fig. S1C) rendered the tumors more sensitive to anti–PD-1 treatment (Fig. 2, H and I).
Fig. 2. In vivo evaluation of the therapeutic efficacy of targeting DDR2 combined with anti–PD-1 immunotherapy.
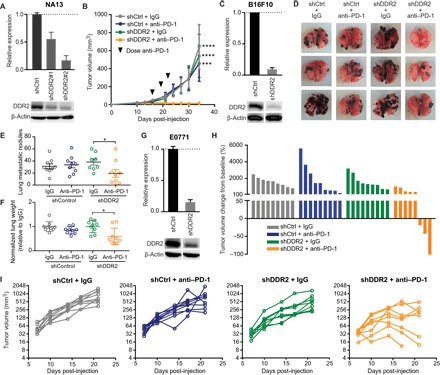
(A) Immunoblot of NA13 cells transduced with two different DDR2 shRNAs, with graph showing densitometric analysis of DDR2 protein levels. (B) Subcutaneous tumor growth in syngeneic mice receiving NA13 shDDR2 #1 cells stably expressing shControl (shCtrl) or shDDR2 (n = 4 to 5 mice per group). Mean ± SEM. ***P < 0.001, ****P < 0.0001. (C) Immunoblot of B16F10 cells with shControl or shDDR2 construct. (D) Representative images of murine pulmonary lung metastases at 22 days following intravenous (tail vein) inoculation of B16F10 cells. (E) Quantification of the number of metastatic B16F10 lung nodules (n = 9 mice per group). Mean ± SEM. *P < 0.05. (F) Lung weight of mice bearing B16F10 lung metastases (n = 9 mice per group). Mean ± SEM. *P < 0.05. (G) Immunoblot of E0771 cells with shControl or shDDR2 construct. (H) Waterfall plot showing change in E0771 mammary fat pad tumor volume compared to baseline before treatment. (I) E0771 mammary tumor volume as a function of time for each mouse. n = 8 to 9 mice per group.
RNA from mice bearing shControl and shDDR2 NA13 tumors treated with anti–PD-1 were analyzed using RNA sequencing (RNA-seq) and then gene set enrichment analysis (GSEA) to discern gene and pathway differences (20, 21). This analysis revealed a strong immune response in the shDDR2 tumors treated with anti–PD-1. Up-regulation of immune-related pathways was specific to the shDDR2 tumors treated with anti–PD-1 compared to IgG treatment; we did not find this strong immune response in shControl tumors treated with anti–PD-1 (table S2). We also observed a strong T cell signature with multiple T cell receptor signaling pathways significantly enriched in the former group (Fig. 3A). Cytometry by time-of-flight (CyTOF) analysis of these tumors revealed increased CD8+ T cell infiltration into shDDR2 tumors treated with anti–PD-1 (Fig. 3B), which was not seen in the spleen (fig. S3). No changes were observed in PD-1 expression levels on the tumor-infiltrating T cells across treatment groups (fig. S4). These observations through both analysis at the cellular and gene expression levels suggest a strong T cell presence following treatment of shDDR2- or dasatinib-treated tumors with anti–PD-1.
Fig. 3. Transcriptomic and CyTOF analysis of tumors.
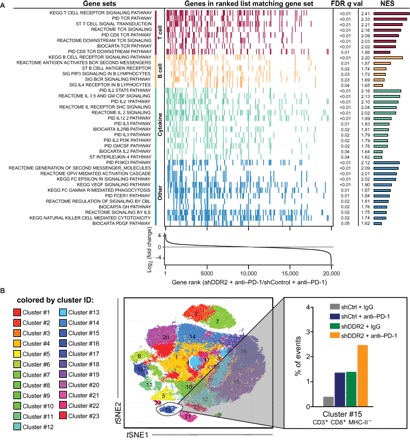
(A) RNA-seq analysis was performed comparing shControl and shDDR2 NA13 tumors grown in syngeneic mice treated with anti–PD-1. Summarized GSEA results for significantly [false discovery rate (FDR) < 0.01] up-regulated, immune-related gene sets from the canonical pathways v6.1 gene set collection (21). Gene sets are grouped and colored according to primary function. The colored tick marks represent a gene for a given gene set. Genes are ranked according to differential expression of the RNA-seq data. FDR-corrected q values and normalized enrichment scores (NES) are reported for gene set. (B) PhenoGraph-defined cellular distribution and clustering, as defined by tSNE1 (t-distributed stochastic neighbor embedding 1) and tSNE2, colored by cluster ID for all treatment conditions. The frequency of cluster #15 (defined as CD3+ CD8+ MHCII−) for each experimental condition is shown. Data show all normalized viable single cells, subjected to the PhenoGraph algorithm.
Dasatinib and anti–PD-1 combination therapy enhances tumor control
DDR2 is a target of multiple FDA-approved kinase inhibitors (22). Dasatinib, the most potent of these inhibitors, has been studied in multiple clinical trials, including lung cancer patients with activating DDR2 mutations (16). To further evaluate the efficacy of targeting DDR2, we tested the combination of dasatinib and anti–PD-1. Whereas therapeutic blockade of PD-1 or DDR2 alone had little or no effect on NA13 tumors, treatment with the combination of dasatinib and anti–PD-1 showed a significant reduction in tumor burden (Fig. 4A). Similar results were seen with MC38 colon cancer model (Fig. 4, B and C) and 1956 sarcoma model (Fig. 4D).
Fig. 4. Therapeutic efficacy of combined pharmacologic inhibition of DDR2 and anti–PD-1 immunotherapy.
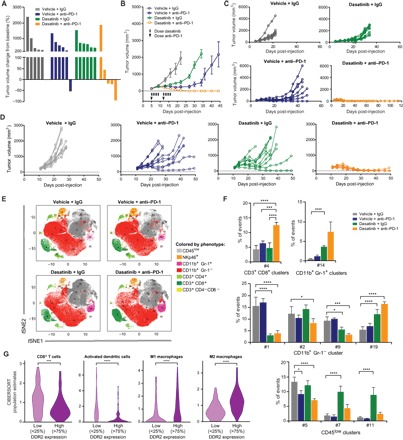
(A) Waterfall plot of NA13 tumor volume in response to dasatinib and anti–PD-1 treatment relative to pretreatment baseline (before anti–PD-1 treatment). n = 5 to 6 mice per group. (B) Average MC38 tumor volume in response to dasatinib and anti–PD-1. (C) Individual tumor volumes of mice in (B). n = 8 mice per group. (D) Individual tumor volumes of mice injected with the 1956 sarcoma cell line in response to dasatinib and anti–PD-1 treatment. Each line represents a single mouse. n = 10 mice per group. (E) PhenoGraph-defined cellular distribution and clustering, as defined by tSNE1 and tSNE2, colored by cellular phenotype for all treatment conditions of MC38 tumors. Data show all normalized viable single cells, subjected to the PhenoGraph algorithm. (F) Frequency of all statistically significant PhenoGraph-identified clusters compared to vehicle + IgG–treated organized according to cluster phenotypic designation. Mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001. (G) Relative abundance of tumor-infiltrating immune cell populations determined by the CIBERSORT methodology (24) in bladder cancer patients from RNA-seq data in TCGA (n = 433) as a function of DDR2 expression. ***P < 0.001, ****P < 0.0001.
Immune profiling of tumors shows enhanced presence of CD8+ T cells
CyTOF analysis of MC38 tumors in mice receiving dasatinib and anti–PD-1 showed a significant increase in both splenic and tumor-infiltrating CD8+ T cells (Fig. 4, E and F, and fig. S5). Because these findings with dasatinib and anti–PD-1 combination reflect a similar pattern as seen in the shDDR2 and anti–PD-1 combination, this suggests a direct role of tumor DDR2 expression in mediating this immune response (Fig. 3B). While dasatinib and anti–PD-1 treatment increased CD8+ T cells in both the tumor and spleen, treatment of shDDR2 tumors with anti–PD-1 led to increased CD8+ T cells only in the tumor. In both cases, the observed increase in CD8+ T cells is unique to the combination therapy and is suggestive of a specific immune response to tumor antigens because of both treatments. The presence of CD8+ T cells in the tumor microenvironment and the expansion of preexisting, tumor antigen–specific T cell clones are also critical and predictive of a favorable response with anti–PD-1 therapy (23).
To identify associations between DDR2 expression and inferred immune infiltration in human tumors, we estimated the relative abundance of immune populations using the CIBERSORT (24) method and bulk tumor RNA-seq data from The Cancer Genome Atlas (TCGA; fig. S6). Low DDR2 expression is associated with increased CD8+ T cells and activated dendritic cell infiltration (Fig. 4G), which has been shown to be predictive of better patient outcome (25–31). These findings support our own experimental findings with shDDR2- and dasatinib-treated tumors, with PD-1 blockade exhibiting an increased presence of CD8+ T cells (Fig. 4F). Human tumors with low DDR2 expression also exhibit reduced levels of macrophages (Fig. 4G), whose presence has been shown to negatively affect the efficacy of anti–PD-1(32). These findings suggest that patients with tumors low in DDR2 expression have a tumor microenvironment more likely to favorably respond to anti–PD-1 treatment.
DISCUSSION
While immune checkpoint–based therapies have been shown to be successful across multiple tumor types, many patients still progress on these therapies. Combining approved checkpoint inhibitor antibodies with each other and with other approved cancer therapeutics is a natural attempt to improve efficacy of the former but, as discussed above, has major practical and scientific limitations. Our study focused on identifying drug target genes, using a functional genomics–based approach that could be targeted in conjunction with anti–PD-1 to increase immune response and tumor clearance. DDR2, our top candidate gene, was validated by both shRNA-mediated knockdown and pharmacological inhibition with dasatinib. In both cases, targeting DDR2 alongside anti–PD-1 therapy proved to be very efficacious across multiple different cancer types in preclinical in vivo models.
DDR2 is a receptor tyrosine kinase that is activated by fibrillar collagen, thus playing a key role in the regulation of collagen-cell interactions (33, 34). Alterations to DDR2, including overexpression, amplification, and mutations, have been reported across broad cancer types and, in many cases, are known to drive a more aggressive phenotype (11–19). While DDR2 is inhibited by several FDA-approved receptor tyrosine kinase inhibitors, dasatinib was chosen for our study due to its status as the most potent inhibitor of DDR2 both in vitro and in vivo and the prevalence of its use in clinical trials for tumors with DDR2-specific activating mutations (19, 22). We use dasatinib as an example of an immediately applicable, widely available drug, which also happens to target our gene of interest, DDR2. We recognize that a possible disadvantage with dasatinib could be the multitargeted nature of this drug (e.g., SRC and ABL) (35, 36), and so, the effects seen with dasatinib cannot be parsed out specifically to account for the effects due to DDR2 and that of the other targets. While the inhibition of these additional kinases is known to affect critical immune cell subsets (e.g., BTK and FMS) (37, 38), inhibition of DDR2 likely plays a prominent role in the efficacy of dasatinib due to the observations from our DDR2 genetic knockdown experiments and the work of other groups (15, 16, 19). DDR2 is involved in the remodeling of the extracellular matrix during morphogenesis and tissue repair, as well as differentiation and proliferation (39, 40). DDR2 is a critical regulator of the mesenchymal stem cell phenotype, type I collagen–related functions, and migration (12). DDR2 expression in both tumor and stromal cells has been shown to play a critical role in cancer progression and metastasis formation. DDR2 in the basal epithelium of tumor cells controls the formation of invasion of tumor organoids, while in the stroma (cancer-associated fibroblasts), it is critical for extracellular matrix production and the organization of collagen fibers through cancer-associated fibroblasts (41). Future work will look at parsing dasatinib’s combinatorial effect with checkpoint inhibitors on DDR2 versus that of it’s other known targets especially since dasatinib has been shown to reduce T regulatory cell numbers (42).
Our screen approach builds a more immediately translational framework compared to genome-wide screens that have aimed to better understand the mechanisms of resistance to immunotherapy. Manguso et al. (43) completed a pooled in vivo genetic screen that identified known and previously unknown immune evasion molecules. Their loss-of-function screen identified PTPN2 as a potential gene that could be targeted to enhance immunotherapy (43). Patel et al. (8) identified genes, with a focus on APLNR, which are a prerequisite for an effective response to immunotherapy using a genomic screen with a T cell–focused approach. Loss of APLNR reduced the efficacy of T cell–based immunotherapies (8). Our work further expands upon the use of functional genomics–based screens to find gene targets that can improve current immunotherapies. While these other screens used CRISPR-Cas9 technology for complete knockout of a gene, we chose an shRNA-based approach, which, in most cases, has incomplete knockdown of a gene and mimics drug-mediated inhibition more faithfully.
Our results reveal important insights into the effects of targeting DDR2, a collagen receptor, in combination with PD-1 inhibition. The preclinical efficacy of the DDR2 and PD-1–targeted combination provided compelling evidence for the initiation of, and support for, the FRACTION-Lung trial (NCT02750514), which evaluates nivolumab in combination with dasatinib. Last, our results provide a strong rationale for further investigation into the molecular mechanisms by which DDR2 and increased collagen signaling from the tumor microenvironment support immune evasion. It will be of interest to determine whether targeting collagen signaling, which is a common feature of many tumor types, represents a viable strategy to enhance the activity of immune checkpoint inhibitors.
MATERIALS AND METHODS
Mouse cancer cell lines
NA13 cell line was isolated and cultured from BBN carcinogen–induced bladder tumor of C57BL/6 female mice. C57BL/6 mice (7 to 8 weeks old) were treated with 0.05% BBN in water for 12 weeks. After 12 weeks of BBN treatment, mice were kept for an additional 12 weeks on the regular water. Mice were then sacrificed, and a part of the bladder tumor was washed in phosphate-buffered saline (PBS) and minced into small pieces with RPMI 1640 medium supplemented with 20% fetal bovine serum (FBS), 1 mM sodium pyruvate, and 1% antibiotic/antimycotic solution. The culture dish was then kept in a CO2 incubator with 5% CO2 and 95% humidity to allow the tumor cells to attach and grow. After 2 days in culture, the excess floating tumor tissue was removed, and cells were further maintained in culture for 6 to 8 weeks before freezing in liquid N2. B16F10 was obtained from the American Type Culture Collection through the University of Colorado Tissue Culture Core. B16F10 was a gift from T. Lyons (University of Colorado). MC38 and 1956 were provided by Bristol-Myers Squibb. All cell lines were authenticated and tested to be mycoplasma-free. E0771 was maintained in RPMI 1640 medium with 5% FBS. B16F10 was maintained in Dulbecco’s modified Eagle’s medium with 10% FBS. MC38 was maintained in RPMI 1640 medium supplemented with 25 mM Hepes and 10% fetal calf serum. All cells were grown at 37°C in a humidified atmosphere (5% CO2).
WES of NA13
Genomic DNA (gDNA) was extracted from the NA13 cell line using the Puregene Cell and Tissue Kit (Qiagen). Exome capture was performed using the SureSelect XT Mouse All Exon Kit (Agilent Technologies). Novogene (https://en.novogene.com/) performed WES using the Illumina NovaSeq 6000 platform. Paired-end 150–base pair (bp) reads were sequenced at a depth of 100×. Reads from the raw FASTQ file were aligned to the mouse reference genome (MM9) using BWA (v0.7.8-R455) (44). The BAM file generated from BWA was sorted using SAMtools (v1.0) (45), and Picard (v1.111) was used to mark duplicates and recalibrate base quality scores. Single-nucleotide polymorphisms (SNPs) and small indels were called according to the GATK command line tools (v3.8.0) (46), including HaplotypeCaller, SelectVariants, and VariantFiltration programs using default parameters. We lifted over the mouse MM9 coordinates to genome version MM10 using the University of California, Santa Cruz Genome Browser LiftOver Tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). To characterize the alterations on gene location and impact on the associated protein, we annotated the NA13 SNPs and small indels using SnpEff (47).
Library design and analysis
A target gene list began with a listing of all cancer clinical trials available in clinicaltrials.gov. Using this portal, the word cancer was entered in the “Condition/Disease” field. We then parsed this file to count most common drugs and biologicals, selected those with readily available inhibitors, and then determined which of these are relatively specific for their targets and have mouse homologs. The custom pooled mouse shRNA library was prepared by the University of Colorado Functional Genomics Shared Resource using MISSION shRNA lentiviral transduction particles (Sigma-Aldrich). Each gene was targeted by an average of five distinct shRNA The RNA Consortium (TRC) constructs for a total of 169 shRNAs. NA13 cells were transfected with lentivirus and underwent puromycin selection (1.5 μg/ml) 48 hours later. Cells underwent selection for 4 days before expansion in RPMI 1640 medium supplemented with 10% FBS and 1 mM sodium pyruvate. Cells were expanded for at least 2 days before subcutaneous hind flank injection into 8-week-old C57BL/6 female mice. Once tumors reached 50 to 100 mm3, mice were injected with 50 μg of mouse anti–PD-1 antibody (IgG1-D265A) or isotype control (IgG1; clone 4F7, Bristol-Myers Squibb) every 3 days for a total of three doses. Mice were euthanized once tumors reached 500 mm3. Tumor was harvested, snap-frozen in liquid N, and stored at −80°C until DNA isolation. gDNA was isolated from each tumor using the Gentra Puregene Kit (Valencia, CA, USA). Custom dual-indexed primers (48, 49) were used to amplify the shRNA constructs. One microgram of total gDNA was used in each normal goat serum library PCR with Herculase II Fusion Enzyme (Agilent Technologies, Cedar Creek, TX, USA) for 26 cycles. The 290-bp amplicon of interest from each reaction was purified using SPRIselect beads (Beckman Coulter, Brea, CA, USA). Each sample was quantified using the Qubit dsDNA HS Assay Kit (Invitrogen, Eugene, OR, USA) and KAPA Library Quant Kit for Illumina-based sequencers (Cape Town, South Africa) before equimolar pooling for sequencing. Sequencing was performed on an Illumina HiSeq4000 (San Diego, CA, USA) at 1 × 151 bp through the University of Colorado Microarray and Sequencing Core. Deconvolution of samples was dependent on the unique dual indices based on Illumina P5 and P7 sequences.
shRNA counts were normalized using the following equation
where CN is the normalized count, AT is the averaged total count across samples, and ST is the total shRNA count of the normalized sample. Treatment and control samples were normalized separately using separate (treatment- and control-specific) total count averages. Outliers were identified using Grubbs test. The shRNAs were counted by comparing the shRNA library against the obtained sequences and searching for exact sequence match. This method assumed that sequencing errors are equally distributed among nucleotides and shRNAs. The percentage of reduced shRNAs was calculated by comparing the mean read count of the checkpoint blockade–treated sample to the IgG control–treated sample for the same shRNA. A reduction was considered to be true if the read counts were 80% or less of that of the IgG control group. The percentage of reduced shRNAs was then determined on the basis of the number of shRNAs that were reduced compared to the total shRNAs per a given gene.
qPCR and immunoblotting (Western blot analysis)
NA13, B16F10, and E0771 cell lines were homogenized using QIAshredder (Qiagen), followed by RNA extraction using the RNeasy Plus Mini Kit with gDNA Eliminator (Qiagen). Complementary DNA (cDNA) was synthesized using iScript Reverse Transcription Supermix (Bio-Rad). qPCR for DDR2 was performed using 5′-TGGCATGAGCAGAAACCTGT-3′ (forward) and 5′-ACTTGCCGTGGTGAATTTGC-3′ (reverse) primers, with iQ SYBR Green Supermix (Bio-Rad) run on a QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems). To determine the changes in mRNA expression as measured by quantitative reverse transcription PCR, the ΔΔCt method was used. Expression was normalized to internal control β-actin, and the level of gene knockdown was determined by comparing with control cells.
Whole-cell extracts were prepared from NA13, B16F10, and E0771 cell lines using radioimmunoprecipitation assay buffer [50 mM tris (pH 8.0), 150 mM NaCl, 0.1% Triton X-100, 0.1% SDS, and protease inhibitors (Roche)]. Following total protein quantification, lysates were separated by SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (Bio-Rad) membrane using a transfer apparatus according to the manufacturer’s instruction (Bio-Rad). After incubation with 5% nonfat milk in TBST [25 mM tris, 150 mM NaCl. 0.1% Tween 20 (pH 7.5)] for 1 hour, the membrane was probed with antibodies against DDR2 (AF2538; 1:1000; R&D Systems) or anti–β-actin AC-74 (1:20,000; Sigma Chemical Co.) monoclonal antibodies overnight. Membranes were washed three times for 10 min with TBST and incubated with a 1:5000 dilution of horseradish peroxidase–conjugated anti-goat (SC-2020, Santa Cruz Biotechnology) or anti-mouse (Bio-Rad) antibodies for 2 hours. Blots were washed three times with TBST and incubated in SuperSignal West Pico PLUS Chemiluminescent Substrate for 5 min at room temperature (Thermo Fisher Scientific). The blots were imaged using ChemiDoc imaging system according to the manufacturer’s instruction (Bio-Rad).
In vivo studies
All experiments were approved by the University of Colorado Denver Animal Care and Use Committee and carried out according to approved protocols. Female C57BL/6 mice (Charles River) were received at 6 weeks old and allowed to acclimate for at least 1 week in sterile microisolator cages with constant temperature and humidity. Mice had free access to food and water.
For NA13, mice were injected with 1 × 106 cells in 100 μl of sterile PBS subcutaneously in the right hind flank. For the E0771 mammary tumor model, mice were injected with 5 × 104 cells in the third left thoracic mammary fat pad. For the 1956 sarcoma and MC38 colon carcinoma model, 6- to 8-week-old mice were injected subcutaneously on the hind flank with 1 × 106 cells. All tumor cells were harvested in log-phase growth on the day of injection.
Mice were monitored twice weekly for tumor development. Measurements commenced from when the tumor was first palpable. Tumor size was determined using an electronic caliper to measure the length and width and calculated by (L × W2)/2, where L is the largest diameter measurement of the tumor and W is the shorter perpendicular tumor measurement. Animals were randomized into treatment groups, ensuring similar average tumor volumes among the groups, weighed, and identified via ear punch. For treatment randomization, MC38 tumors were allowed to grow to 75 to 200 mm3 (tumors outside the range were excluded), and animals were evenly distributed to various treatment and control groups.
Dasatinib was synthesized by Bristol-Myers Squibb Laboratories (Princeton, NJ), as previously described (50). Dasatinib was formulated in 80 mM citrate buffer and administered orally to mice by body weight at a dose of 30 mg/kg (1956, initiated on day 11 after tumor injection; MC38, initiated on day 5 after tumor injection for 10 doses) or 60 mg/kg (NA13, initiated on day 16 after tumor injection for three doses). Mouse anti–PD-1 antibody (IgG1-D265A) and isotype control (IgG1; clone 4F7) were produced by Bristol-Myers Squibb Laboratories (Redwood City, CA) and were formulated in PBS and administered intraperitoneally at a dose of 50 μg per mouse (NA13, initiated on day 16 for three doses; E0771, initiated on day 7 for three doses), 100 μg per mouse (B16F10, initiated on day 8 for three doses), or 200 μg per mouse (1956, initiated on day 11; MC38, initiated on day 5 for two doses). The health of the mice was closely monitored, and mice were immediately euthanized if any signs of distress were observed or tumor volume exceeded 15 mm in diameter.
For the B16F10-induced pulmonary metastases model, mice were intravenously challenged with 2 × 105 B16F10 cells in 100 μl of sterile PBS. Treatment with anti–PD-1 commenced on day 8, and mice were euthanized on day 22. Lungs were harvested, formalin-fixed, paraffin-embedded with 5-μm sections, cut, and hematoxylin and eosin–stained by the University of Colorado Histology Core. Lungs were analyzed by an American College of Veterinary Pathologists board-certified veterinary pathologist.
Single-cell mass cytometry by time of flight
NA13 tumors were collected after final measurement day. Tumors were weighed, mechanically dissociated, and enzymatically digested using Liberase DL (Roche) in Hanks’ balanced salt solution for 30 min at 37°C. The solution was passed through a 70-μm cell strainer (Fisherbrand) to obtain a single-cell suspension. Cells were counted using a TC-20 counter (Bio-Rad) with trypan blue exclusion.
Metal-conjugated antibodies were purchased from Fluidigm and used at manufacturer-recommended concentrations (table S4). Live tumor and spleen cells were washed with Maxpar PBS (Fluidigm) and counted on the Vi-CELL Cell Viability Analyzer (Beckman Coulter), normalizing to a final count of 2 × 106 to 3 × 106 cells per sample in RPMI 1640 medium (Corning Cellgro). Live versus dead staining was achieved by resuspending cells in a solution of 25 mM cisplatin (Fluidigm) in RPMI 1640 medium and incubating for 1 min at room temperature. Samples were quenched with Maxpar Cell Staining Buffer (Fluidigm), spun down at 500g for 5 min, and then fixed by resuspending 1.6% paraformaldehyde (PFA) in PBS for 10 min at room temperature. The cells were washed once with Maxpar Cell Staining Buffer to remove fixative from the solution and once with Maxpar Barcode Perm Buffer to begin cell permeabilization for barcoding, centrifuging at 800g at 22°C per spin. Barcoding was performed as per the manufacturer’s recommendations with palladium metal barcoding reagents (Fluidigm), where samples were eventually pooled together, with spleen samples pooled separately from tumor samples. Treatment with anti-mouse FcγII/FCγIII-blocking antibody 2.4G2 (prepared in-house) for 10 min was followed by surface staining at the manufacturer’s recommended antibody staining concentrations (Fluidigm). The cells were stained for 30 min at room temperature, then washed with Maxpar Cell Staining Buffer, and permeabilized in 4× FoxP3 Fix/Perm Buffer overnight at 4°C. The cells were washed once with 1× FoxP3 Fix/Perm Buffer, and intracellular staining was performed for 2 hours at 4°C following the manufacturer’s protocol (Fluidigm) and then washed twice with 1× FoxP3 Perm Buffer. The cells were incubated in 1.6% PFA in PBS with 100 nM iridium nucleic acid intercalator (Fluidigm) overnight and washed twice in double-distilled H2O before analysis on a Helios mass cytometer (CyTOF, Fluidigm) at an event rate of 400 to 500 events per second.
MC38 tumors were collected in RPMI 1640 medium and then enzymatically dissociated using Mouse Tumor Dissociation Kit and gentleMACS Dissociator (Miltenyi Biotec) according to the manufacturer’s protocol. The cells were stained with a panel of metal isotope–conjugated anti-mouse antibodies (DVS Sciences; table S5).
Data were analyzed with PhenoGraph. The 1.0.153 version of R studio was downloaded from the official R website (www.r-project.org/). Release 3.6 of the cytofkit package was downloaded from Bioconductor (https://bioconductor.org/packages/release/bioc/manuals/cytofkit/man/cytofkit.pdf) and opened in R. Manually gated singlet (19Ir + 193Ir +), viable (195Pt +) events were imported into cytofkit, subjected to PhenoGraph analysis, and clustered on the basis of 30 markers (insert antibody table), with the following settings: merge method: “min” (100,380 events total, 25,095 events from each file), transformation: cytofAsinh, cluster method: Rphenograph, visualization method: tSNE (t-distributed stochastic neighbor embedding), and cellular progression: NULL. PhenoGraph identified 23 unique clusters. These results were visualized via the R package “Shiny,” where labels, dot size, and cluster color were customized. Clusters were colored according to phenotype based on the median expression of various markers. The frequency of each cluster was determined via csv files generated by the algorithm. All graphs were made using GraphPad Prism 7. P values were calculated using the two-way analysis of variance (ANOVA) with multiple comparisons, with statistical analyses subjected to multiple testing correction.
Tumor expression analysis (RNA-seq)
Tumors were mechanically disassociated with a homogenizer (Omni International) and then passed through a QIAshredder (Qiagen), gDNA eliminator columns (Qiagen), and RNeasy Plus Mini Kit, as per the manufacturer’s instructions, for RNA extraction. Library preparation and sequencing were performed by Novogene. Quality and quantity of RNA were analyzed using NanoDrop, agarose gel electrophoresis, and Agilent 2100. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. mRNA was first fragmented randomly by fragmentation buffer, followed by NEB library preparation (New England Biolabs). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H−), with second-strand cDNA synthesized using DNA polymerase I and RNase H. Double-stranded cDNA was purified using AMPure XP beads (Beckman Coulter). Exonuclease was used to eliminate overhangs on the double-stranded cDNA. 3′ ends were adenylated, and NEBNext adaptors were ligated in preparation for hybridization. Library fragments were purified with AMPure XP system (Beckman Coulter). Sequencing was performed by Novogene on an Illumina platform paired-end 150 bp with 20 million reads per sample.
Transcript quantification was performed using RSEM (v1.2.31) (51) with default parameters and Bowtie 2 (v2.1.0) as the read aligner (52). Reads were mapped directly to mouse transcripts and summarized at the gene level using annotations from Ensembl r91, genome build GRCm38.p5. Quantification of genes as expected counts was compiled. Differential expression was performed using voom function in the limma R package (53). Genes with an average expected count less than five were removed, normalization factors were calculated, and comparisons between groups were made using the voom function using default parameters.
The curated gene set canonical pathways (C2.CP) was downloaded from the MSigDB collection of genes sets (v6.1) (21). Human genes were mapped and then replaced with mouse genes using the vertebrate homology mapping from the Mouse Genome Informatics resources (www.informatics.jax.org/downloads/reports/HOM_AllOrganism.rpt). The updated C2.CP gene set with mouse genes and the log2-transformed fold change ranked list of genes from the RNA-seq experiment comparing control and shDDR2 NA13 tumors grown in syngeneic mice treated with anti–PD-1 were used as input to GSEA (20, 21). GSEA was run in preranked mode with default parameters.
To identify immune-related gene sets that were significantly up-regulated, we further filtered the full set of GSEA results, based on the Jaccard distance (number of genes in the intersection divided by the number of genes in the union of any two gene sets). This quantifies the overlap in genes between two gene sets. We selected gene sets that shared ≥0.25, were significantly up-regulated (false discovery rate < 0.01), and were immune related. From the comparison of control and shDDR2 NA13 tumors grown in syngeneic mice treated with anti–PD-1, we found 39 gene sets that met these criteria. These are the gene sets shown in Fig. 3. Results from CIBERSORT (24) using TCGA bladder cancer gene expression (54) were downloaded from The Cancer Immune Atlas (TCIA) and processed according to TCIA protocols (55).
Supplementary Material
Acknowledgments
Funding: This work was supported by the Canadian Institutes of Health Research Fellowship (M.M.T.), the Boettcher Foundation (J.C.Co.), and NIH CA075115 (D.T.). We thank the University of Colorado Cancer Center Genomics Shared Resource, Functional Genomics Shared Resource, Biostatistics and Bioinformatics Shared Resource, Protein Production/Monoclonal Antibody/Tissue Culture Shared Resource, and Flow Cytometry Shared Resource, each supported in part by the Cancer Center Support Grant (P30CA046934). Author contributions: M.M.T., F.Y.F.L., E.S., R.F.G., J.E.D., and D.T. designed experiments. M.M.T., F.Y.F.L., R.T.J., E.S., R.F.G., B.C., K.M., J.Y., E.M., H.A.A.-H., N.A., and A.C.-D. prepared samples and acquired data. M.M.T., F.Y.F.L., R.T.J., E.S., R.F.G., H.C., H.A.A.-H., A.I.R., L.K.J., T.L.N., E.T.C., and J.C.Co. analyzed and interpreted data. M.M.T. wrote the manuscript in consultation with F.Y.F.L., R.T.J., A.K.K., E.S., R.F.G., B.C., H.A.A.-H., A.R., N.A., T.L.N., J.C.Co., A.J.K., and D.T. J.C.Ca., E.T.C., J.C.Co., A.J.K., and D.T. supervised the study. Competing interests: D.T., M.M.T., and J.E.D. are inventors on a provisional patent application related to this work filed by the University of Colorado (no. PCT/US2018/043268, filed on 23 July 2018). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav2437/DC1
Fig. S1. shRNA-mediated knockdown of DDR2 expression.
Fig. S2. Histological comparison of lungs from B16F10 tumor–bearing mice.
Fig. S3. Spleen from NA13 tumor–bearing mice analyzed using CyTOF with PhenoGraph algorithm.
Fig. S4. Analysis of PD-1 expression on tumor-infiltrating T cells.
Fig. S5. Spleen from MC38 tumor–bearing mice analyzed using CyTOF with PhenoGraph algorithm.
Fig. S6. Quantification of immune cell populations in bladder cancer patients with varying DDR2 expression levels.
Table S1. NA13 mutation analysis.
Table S2. Gene set enrichment analysis.
Table S3. Thirty-four–gene shRNA library.
Table S4. CyTOF antibody panel used for the analysis of spleen and tumors from mice implanted with the NA13 cell line.
Table S5. CyTOF antibody panel used for the analysis of tumors from mice implanted with the MC38 cell line.
REFERENCES AND NOTES
- 1.Chen L., Han X., Anti–PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Invest. 125, 3384–3391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchbinder E. I., Desai A., CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 39, 98–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rotte A., Jin J. Y., Lemaire V., Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 29, 71–83 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Chen D. S., Mellman I., Elements of cancer immunity and the cancer–immune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Schmidt C., The benefits of immunotherapy combinations. Nature 552, S67–S69 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Williams P. D., Lee J. K., Theodorescu D., Molecular credentialing of rodent bladder carcinogenesis models. Neoplasia 10, 838–846 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fantini D., Glaser A. P., Rimar K. J., Wang Y., Schipma M., Varghese N., Rademaker A., Behdad A., Yellapa A., Yu Y., Sze C. C.-L., Wang L., Zhao Z., Crawford S. E., Hu D., Licht J. D., Collings C. K., Bartom E., Theodorescu D., Shilatifard A., Meeks J. J., A carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 37, 1911–1925 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel S. J., Sanjana N. E., Kishton R. J., Eidizadeh A., Vodnala S. K., Cam M., Gartner J. J., Jia L., Steinberg S. M., Yamamoto T. N., Merchant A. S., Mehta G. U., Chichura A., Shalem O., Tran E., Eil R., Sukumar M., Guijarro E. P., Day C.-P., Robbins P., Feldman S., Merlino G., Zhang F., Restifo N. P., Identification of essential genes for cancer immunotherapy. Nature 548, 537–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leitinger B., Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. J. Biol. Chem. 278, 16761–16769 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Leitinger B., Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 310, 39–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki S., Ueda M., Iguchi T., Kaneko M., Nakayama H., Watanabe T., Sakamoto A., Mimori K., DDR2 expression is associated with a high frequency of peritoneal dissemination and poor prognosis in colorectal cancer. Anticancer Res. 37, 2587–2591 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez M. E., Martin E. E., Anwar T., Arellano-Garcia C., Medhora N., Lama A., Chen Y.-C., Tanager K. S., Yoon E., Kidwell K. M., Ge C., Franceschi R. T., Kleer C. G., Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Rep. 18, 1215–1228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai M.-C., Li W.-M., Huang C.-N., Ke H.-L., Li C.-C., Yeh H.-C., Chan T.-C., Liang P.-I., Yeh B.-W., Wu W.-J., Lim S.-W., Li C.-F., DDR2 overexpression in urothelial carcinoma indicates an unfavorable prognosis: A large cohort study. Oncotarget 7, 78918–78931 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurashige J., Hasegawa T., Niida A., Sugimachi K., Deng N., Mima K., Uchi R., Sawada G., Takahashi Y., Eguchi H., Inomata M., Kitano S., Fukagawa T., Sasako M., Sasaki H., Sasaki S., Mori M., Yanagihara K., Baba H., Miyano S., Tan P., Mimori K., Integrated molecular profiling of human gastric cancer identifies DDR2 as a potential regulator of peritoneal dissemination. Sci. Rep. 6, 22371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terai H., Tan L., Beauchamp E. M., Hatcher J. M., Liu Q., Meyerson M., Gray N. S., Hammerman P. S., Characterization of DDR2 inhibitors for the treatment of DDR2 mutated nonsmall cell lung cancer. ACS Chem. Biol. 10, 2687–2696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammerman P. S., Sos M. L., Ramos A. H., Xu C., Dutt A., Zhou W., Brace L. E., Woods B. A., Lin W., Zhang J., Deng X., Lim S. M., Heynck S., Peifer M., Simard J. R., Lawrence M. S., Onofrio R. C., Salvesen H. B., Seidel D., Zander T., Heuckmann J. M., Soltermann A., Moch H., Koker M., Leenders F., Gabler F., Querings S., Ansén S., Brambilla E., Brambilla C., Lorimier P., Brustugun O. T., Helland Å., Petersen I., Clement J. H., Groen H., Timens W., Sietsma H., Stoelben E., Wolf J., Beer D. G., Tsao M. S., Hanna M., Hatton C., Eck M. J., Janne P. A., Johnson B. E., Winckler W., Greulich H., Bass A. J., Cho J., Rauh D., Gray N. S., Wong K.-K., Haura E. B., Thomas R. K., Meyerson M., Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 1, 78–89 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barker K. T., Martindale J. E., Mitchell P. J., Kamalati T., Page M. J., Phippard D. J., Dale T. C., Gusterson B. A., Crompton M. R., Expression patterns of the novel receptor-like tyrosine kinase, DDR, in human breast tumours. Oncogene 10, 569–575 (1995). [PubMed] [Google Scholar]
- 18.Nemoto T., Ohashi K., Akashi T., Johnson J. D., Hirokawa K., Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology 65, 195–203 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Beauchamp E. M., Woods B. A., Dulak A. M., Tan L., Xu C., Gray N. S., Bass A. J., Wong K.-k., Meyerson M., Hammerman P. S., Acquired resistance to dasatinib in lung cancer cell lines conferred by DDR2 gatekeeper mutation and NF1 loss. Mol. Cancer Ther. 13, 475–482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mootha V. K., Lindgren C. M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., Groop L. C., PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day E., Waters B., Spiegel K., Alnadaf T., Manley P. W., Buchdunger E., Walker C., Jarai G., Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 599, 44–53 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Tumeh P. C., Harview C. L., Yearley J. H., Shintaku I. P., Taylor E. J. M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., West A. N., Carmona M., Kivork C., Seja E., Cherry G., Gutierrez A. J., Grogan T. R., Mateus C., Tomasic G., Glaspy J. A., Emerson R. O., Robins H., Pierce R. H., Elashoff D. A., Robert C., Ribas A., PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu-Trantien C., Loi S., Garaud S., Equeter C., Libin M., de Wind A., Ravoet M., Buanec H. L., Sibille C., Manfouo-Foutsop G., Veys I., Haibe-Kains B., Singhal S. K., Michiels S., Rothé F., Salgado R., Duvillier H., Ignatiadis M., Desmedt C., Bron D., Larsimont D., Piccart M., Sotiriou C., Willard-Gallo K., CD4+ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Invest. 123, 2873–2892 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L., Conejo-Garcia J. R., Katsaros D., Gimotty P. A., Massobrio M., Regnani G., Makrigiannakis A., Gray H., Schlienger K., Liebman M. N., Rubin S. C., Coukos G., Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348, 203–213 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Pagès F., Berger A., Camus M., Sanchez-Cabo F., Costes A., Molidor R., Mlecnik B., Kirilovsky A., Nilsson M., Damotte D., Meatchi T., Bruneval P., Cugnenc P.-H., Trajanoski Z., Fridman W.-H., Galon J., Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 353, 2654–2666 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Sato E., Olson S. H., Ahn J., Bundy B., Nishikawa H., Qian F., Jungbluth A. A., Frosina D., Gnjatic S., Ambrosone C., Kepner J., Odunsi T., Ritter G., Lele S., Chen Y.-T., Ohtani H., Old L. J., Odunsi K., Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 18538–18543 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naito Y., Saito K., Shiiba K., Ohuchi A., Saigenji K., Nagura H., Ohtani H., CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 58, 3491–3494 (1998). [PubMed] [Google Scholar]
- 30.Nakano O., Sato M., Naito Y., Suzuki K., Orikasa S., Aizawa M., Suzuki Y., Shintaku I., Nagura H., Ohtani H., Proliferative activity of intratumoral CD8+ T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 61, 5132–5136 (2001). [PubMed] [Google Scholar]
- 31.Sharma P., Shen Y., Wen S., Yamada S., Jungbluth A. A., Gnjatic S., Bajorin D. F., Reuter V. E., Herr H., Old L. J., Sato E., CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma. Proc. Natl. Acad. Sci. U.S.A. 104, 3967–3972 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peranzoni E., Lemoine J., Vimeux L., Feuillet V., Barrin S., Kantari-Mimoun C., Bercovici N., Guérin M., Biton J., Ouakrim H., Régnier F., Lupo A., Alifano M., Damotte D., Donnadieu E., Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. U.S.A. 115, E4041–E4050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson J. D., Edman J. C., Rutter W. J., A receptor tyrosine kinase found in breast carcinoma cells has an extracellular discoidin I-like domain. Proc. Natl. Acad. Sci. U.S.A. 90, 5677–5681 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shrivastava A., Radziejewski C., Campbell E., Kovac L., McGlynn M., Ryan T. E., Davis S., Goldfarb M. P., Glass D. J., Lemke G., Yancopoulos G. D., An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1, 25–34 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Shah N. P., Tran C., Lee F. Y., Chen P., Norris D., Sawyers C. L., Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Weisberg E., Manley P. W., Cowan-Jacob S. W., Hochhaus A., Griffin J. D., Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer 7, 345–356 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Hantschel O., Rix U., Schmidt U., Bürckstümmer T., Kneidinger M., Schütze G., Colinge J., Bennett K. L., Ellmeier W., Valent P., Superti-Furga G., The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. U.S.A. 104, 13283–13288 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brownlow N., Mol C., Hayford C., Ghaem-Maghami S., Dibb N. J., Dasatinib is a potent inhibitor of tumour-associated macrophages, osteoclasts and the FMS receptor. Leukemia 23, 590–594 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Márquez J., Olaso E., Role of discoidin domain receptor 2 in wound healing. Histol. Histopathol. 29, 1355–1364 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Olaso E., Labrador J.-P., Wang L., Ikeda K., Eng F. J., Klein R., Lovett D. H., Lin H. C., Friedman S. L., Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J. Biol. Chem. 277, 3606–3613 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Corsa C. A. S., Brenot A., Grither W. R., Van Hove S., Loza A. J., Zhang K., Ponik S. M., Liu Y., DeNardo D. G., Eliceiri K. W., Keely P. J., Longmore G. D., The action of discoidin domain receptor 2 in basal tumor cells and stromal cancer associated fibroblasts is critical for breast cancer metastasis. Cell Rep. 15, 2510–2523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hekim C., Ilander M., Yan J., Michaud E., Smykla R., Vähä-Koskela M., Savola P., Tähtinen S., Saikko L., Hemminki A., Kovanen P. E., Porkka K., Lee F. Y. F., Mustjoki S., Dasatinib changes immune cell profiles concomitant with reduced tumor growth in several murine solid tumor models. Cancer Immunol. Res. 5, 157–169 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Manguso R. T., Pope H. W., Zimmer M. D., Brown F. D., Yates K. B., Miller B. C., Collins N. B., Bi K., LaFleur M. W., Juneja V. R., Weiss S. A., Lo J., Fisher D. E., Miao D., Van Allen E., Root D. E., Sharpe A. H., Doench J. G., Haining W. N., In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Durbin R., Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Philippakis A. A., del Angel G., Rivas M. A., Hanna M., McKenna A., Fennell T. J., Kernytsky A. M., Sivachenko A. Y., Cibulskis K., Gabriel S. B., Altshuler D., Daly M. J., A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cingolani P., Platts A., Wang L. L., Coon M., Nguyen T., Wang L., Land S. J., Lu X., Ruden D. M., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregory M. A., D’Alessandro A., Alvarez-Calderon F., Kim J., Nemkov T., Adane B., Rozhok A. I., Kumar A., Kumar V., Pollyea D. A., Wempe M. F., Jordan C. T., Serkova N. J., Tan A. C., Hansen K. C., DeGregori J., ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. U.S.A. 113, E6669–E6678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan K. D., Padilla-Just N., Henry R. E., Porter C. C., Kim J., Tentler J. J., Eckhardt S. G., Tan A. C., DeGregori J., Espinosa J. M., ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat. Chem. Biol. 8, 646–654 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lombardo L. J., Lee F. Y., Chen P., Norris D., Barrish J. C., Behnia K., Castaneda S., Cornelius L. A. M., Das J., Doweyko A. M., Fairchild C., Hunt J. T., Inigo I., Johnston K., Kamath A., Kan D., Klei H., Marathe P., Pang S., Peterson R., Pitt S., Schieven G. L., Schmidt R. J., Tokarski J., Wen M.-L., Wityak J., Borzilleri R. M., Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 47, 6658–6661 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Law C. W., Chen Y., Shi W., Smyth G. K., voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robertson A. G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A. D., Hinoue T., Laird P. W., Hoadley K. A., Akbani R., Castro M. A. A., Gibb E. A., Kanchi R. S., Gordenin D. A., Shukla S. A., Sanchez-Vega F., Hansel D. E., Czerniak B. A., Reuter V. E., Su X., Carvalho B. S., Chagas V. S., Mungall K. L., Sadeghi S., Pedamallu C. S., Lu Y., Klimczak L. J., Zhang J., Choo C., Ojesina A. I., Bullman S., Leraas K. M., Lichtenberg T. M., Wu C. J., Schultz N., Getz G., Meyerson M., Mills G. B., McConkey D. J., Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540–556.e25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z., Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav2437/DC1
Fig. S1. shRNA-mediated knockdown of DDR2 expression.
Fig. S2. Histological comparison of lungs from B16F10 tumor–bearing mice.
Fig. S3. Spleen from NA13 tumor–bearing mice analyzed using CyTOF with PhenoGraph algorithm.
Fig. S4. Analysis of PD-1 expression on tumor-infiltrating T cells.
Fig. S5. Spleen from MC38 tumor–bearing mice analyzed using CyTOF with PhenoGraph algorithm.
Fig. S6. Quantification of immune cell populations in bladder cancer patients with varying DDR2 expression levels.
Table S1. NA13 mutation analysis.
Table S2. Gene set enrichment analysis.
Table S3. Thirty-four–gene shRNA library.
Table S4. CyTOF antibody panel used for the analysis of spleen and tumors from mice implanted with the NA13 cell line.
Table S5. CyTOF antibody panel used for the analysis of tumors from mice implanted with the MC38 cell line.