

Abstract
Mycobacterium tuberculosis (Mtb), the main causative agent of tuberculosis (TB), is naturally resistant to β-lactam antibiotics due to the production of the extended spectrum β-lactamase BlaC. β-Lactam/β-lactamase inhibitor combination therapies can circumvent the BlaC-mediated resistance of Mtb and are promising treatment options against TB. However, still little is known of the exact mechanism of BlaC inhibition by the β-lactamase inhibitors currently approved for clinical use, clavulanic acid, sulbactam, tazobactam, and avibactam. Here, we present the X-ray diffraction crystal structures of the acyl-enzyme adducts of wild-type BlaC with the four inhibitors. The +70 Da adduct derived from clavulanate and the trans-enamine acylation adducts of sulbactam and tazobactam are reported. BlaC in complex with avibactam revealed two inhibitor conformations. Preacylation binding could not be observed because inhibitor binding was not detected in BlaC variants carrying a substitution of the active site serine 70 to either alanine or cysteine, by crystallography, ITC or NMR. These results suggest that the catalytic serine 70 is necessary not only for enzyme acylation but also for increasing BlaC affinity for inhibitors in the preacylation state. The structure of BlaC with the serine to cysteine mutation showed a covalent linkage of the cysteine 70 Sγ atom to the nearby amino group of lysine 73. The differences of adduct conformations between BlaC and other β-lactamases are discussed.
Tuberculosis (TB) is one of the most ancient and deadliest human infectious diseases, and today is still a leading cause of death around the world.5 TB cannot be treated with β-lactam antibiotics because of the intrinsic resistance of the bacterium that causes TB, Mycobacterium tuberculosis (Mtb), caused by the production of the Ambler class A β-lactamase enzyme BlaC. The discovery of the β-lactam analogue clavulanic acid6,7 led to the development of β-lactam/β-lactamase inhibitor combination therapies. β-Lactamase inhibitors bind to β-lactamases and inhibit them, so that the β-lactam antibiotics can exert their bactericidal function. To date, there are four β-lactamase inhibitors approved for clinical use: clavulanic acid, sulbactam, tazobactam, and avibactam (formerly known as NXL104), Figure 1. β-Lactam/β-lactamase combination therapies were shown to be effective in killing Mtb in vivo and in vitro.8−13 BlaC inhibition by clavulanic acid was initially thought to be permanent, but later it was observed that BlaC enzymatic activity recovers very slowly from inhibition, with 50% reached after 14 h.14 Recovery from inhibition by sulbactam and tazobactam was reported to occur after 30 and 45 min, respectively,2 while recovery from avibactam inhibition took 48 h.15 Furthermore, it was shown that the rate of recovery of BlaC activity after clavulanate inhibition is strongly dependent on the ions that are present in the buffer, such as phosphate, acetate, or sulfate, with phosphate and sulfate triggering BlaC recovery, and acetate slowing it down.14 A possible pathway of BlaC inhibition by clavulanate was suggested by Hugonnet and colleagues2 based on mass spectrometry (MS) and crystallographic data (Figure 2). The MS data suggested the formation of four main covalent intermediates corresponding to MS peaks +70, +137, +155, and +199 Da compared to a free enzyme,2,14,16 while BlaC crystal soaking with clavulanic acid and the X-ray crystal structure determination allowed for trapping a covalent adduct corresponding to the +155 Da MS peak (PDB 3CG5, Figure 2d).3 Similar experiments with other Ambler class A β-lactamases also resulted in the visualization of the structures of the +199 inhibitor (PDB 1BLC, Figure 2b), in addition to the +155 Da one.17,18 One novel clavulanate product was modeled in the active site of the BS3 β-lactamase that corresponds to an ethane-imine ester degradation product of clavulanate (PDB 2Y91).4
Figure 1.

Structures of FDA-approved β-lactamase inhibitors.
Figure 2.

(a–h) Proposed mechanism of BlaC inhibition by clavulanate (adapted from refs (1) and (2)). The numbers represent the mass difference (Δ in Da) of the neutral adducts relative to resting state enzyme.
However, despite much research focused on the structural characterization of BlaC19 and its interaction mode with antibiotics20−25 and several inhibitors,2,3,14−16,26,27 the molecular understanding of BlaC inhibition by the main β-lactamase inhibitors is still incomplete. Furthermore, little is known of the mechanism of recognition between BlaC and substrates and inhibitors before the formation of the acyl-enzyme complex. Therefore, we set out to structurally characterize the covalent adducts of BlaC with the β-lactamase inhibitors clavulanic acid, sulbactam, tazobactam, and avibactam by X-ray crystallography and study the preacylation complexes of BlaC with inhibitors by producing two acylation-deficient mutants of BlaC in which the catalytic Ser70 was replaced by either an Ala or a Cys residue. Our results show that BlaC inhibition by β-lactamase inhibitors follows the same reaction mechanism as for classical β-lactamases TEM-1 and SHV-1. Moreover, we show structural evidence for the formation of the inhibitor form of clavulanate corresponding to the +70 Da MS peak and the trans-enamine adducts of sulbactam and tazobactam. Interestingly, the structure of BlaC in complex with avibactam showed two conformations of the covalent adduct, suggesting a stabilization mode of the inhibitor in the active site of BlaC. No interactions could be detected between the inhibitors and the acylation-deficient variants, suggesting the binding is very weak. The structural analysis of the BlaC–inhibitor complexes highlighted several differences with other complexes of β-lactamases and inhibitors that may relate to the higher resistance of BlaC to inhibition and could aid in the design of more potent and specific inhibitors for BlaC.
Materials and Methods
Gene Cloning, Mutagenesis, Protein Production, and Purification
The blaC wild-type gene (Uniprot P9WKD3) from Mycobacterium tuberculosis without the sequence encoding the N-terminal, 42-amino acid signal peptide was cloned in pET28a(+) fused to a C-terminal six histidine tag, as described by Elings et al.14 BlaC S70A and S70C mutants were obtained using the QuikChange method (Agilent), and the presence of the mutations was verified by sequencing. Heterologous expression of blaC gene constructs was done in E. coli BL21 (DE3)pLysS cells as already described.14 Bacteria were disrupted by French-press and the solutions ultracentrifuged at 25000g for 45 min at 4 °C. The supernatant was applied to a 5 mL prepacked HisTrap HP column (GE Healthcare) pre-equilibrated in 25 mM Tris-HCl buffer, pH 8.0, 500 mM NaCl. The column was washed with ten column volumes of 25 mM Tris-HCl buffer, pH 8.0, 500 mM NaCl and 50 mM imidazole. The His-tagged BlaC was eluted with 250 mM imidazole. The eluted protein was loaded on a PD-10 desalting column (GE Healthcare) for fast removal of the imidazole, and further purified by size exclusion chromatography using a Superose 12 10/300 GL column (GE Healthcare) in a final buffer consisting in 25 mM Tris-HCl buffer, pH 8.0, 40 mM NaCl, with or without 1 mM dithiothreitol (DTT). The collected fractions were analyzed by SDS-PAGE, and those containing pure BlaC were pooled, concentrated using a Centriprep centrifugal filter unit (10 kDa cutoff, Millipore), and flash-frozen in liquid nitrogen. Protein concentration was determined by absorbance at 280 nm, using the theoretical extinction coefficient of 29910 M–1 cm–1, calculated using the ProtParam tool on ExPasy (http://web.expasy.org/protparam/).
Crystallization and Soaking Conditions
Good quality diffracting crystals of wild-type BlaC were obtained as described by Elings et al.14 BlaC S70A purified in the presence of DTT was concentrated up to 30 mg mL–1 and screened for crystallization in 0.1 M Tris-HCl, pH 7.0–9.0, and 0.1–2 M NH4H2PO4 according to conditions previously used for the crystallization of wild-type BlaC.19 The reservoir solutions (75 μL) were pipetted by a Genesis RS200 robot (Tecan), and the drops (500 nL) were made by an Oryx6 robot (Douglas Instruments). Crystals grew in about one month in several conditions with the best diffracting crystals being in 0.1 M Tris-HCl, pH 7.0, 1.7 M NH4H2PO4. Purified S70A and S70C BlaC solutions without DTT were concentrated to 10 and 15 mg mL–1, respectively. Both proteins were screened for crystallization by sitting-drop vapor-diffusion using the JCSG+ and PACT premier (Molecular Dimensions) screens at 293 K. Initial crystallization plates were prepared using an NT8 robot (Formulatrix) for pipetting the reservoir solution (70 μL) and making the drops (500 nL). BlaC S70A produced first crystal hits in conditions C12 of the JCSG+ screen consisting of 10% w/v PEG1000 and 10% w/v PEG8000. Conditions were further optimized by preparing fresh solutions of PEG1000 and PEG8000 and pipetting 2 μL of crystallization drops with a protein percentage varying from 30 to 70%. Good quality diffracting crystals grew in 20% w/v PEG8000 in drops containing 30% v/v protein and 70% v/v reservoir solution. Crystals of BlaC S70C were obtained by optimization of initial hits in condition JCSG+ C3, which is composed of 0.2 M NH4NO3 and 20% w/v PEG3350. Crystals diffracting to high resolution were obtained in optimized crystallization conditions consisting in 25% w/v PEG3350 using a 1:1 ratio of protein to reservoir solution. The crystals were mounted on cryo-loops and, before flash-cooling in liquid nitrogen, were cryo-protected in a solution consisting of mother liquor and 15% v/v glycerol. Crystals were also soaked in cryo-solutions containing 10 mM of inhibitors clavulanic acid, sulbactam, tazobactam, or avibactam for times varying from a few minutes to 1 h.
X-ray Data Collection and Structure Determination
X-ray data collection was performed at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on beamlines ID29, ID30A-3, and ID30B. The data sets were autoprocessed by the EDNA Autoprocessing package that used XDS28 to integrate the intensities and AIMLESS29 to scale and merge the intensities. The structures were solved by molecular replacement with MOLREP30 from the CCP4 suite31 using the PDB entry 2GDN as a search model. Manual refinement was done using REFMAC32 and Coot.33 For PDB entry 6H28 (BlaC S70A), refinement with anisotropic B-factors was tried but did not improve Rfree and resulted in overfitting, which is attributed to the low completeness at the highest resolution ranges. Data collection and refinement statistics are presented in Table 1.
Table 1. Data Collection and Refinement Statistics (as Reported in the PDB Validation File).
| clavulanate (3 min soak) | clavulanate (10 min soak) | sulbactam | tazobactam | avibactam | S70A DTT | S70A | S70C | |
|---|---|---|---|---|---|---|---|---|
| PDB | 6H2C | 6H2G | 6H2K | 6H2I | 6H2H | 6H2A | 6H28 | 6H27 |
| space group | P1 | P1 | P1 | P1 | P1 | P43212 | P1 | P1211 |
| a (Å) | 39.65 | 39.40 | 39.71 | 39.63 | 39.52 | 108.9 | 39.59 | 39.60 |
| b (Å) | 41.98 | 41.31 | 41.95 | 41.52 | 41.32 | 108.9 | 41.70 | 76.41 |
| c (Å) | 76.92 | 76.22 | 76.93 | 76.42 | 76.36 | 60.5 | 76.76 | 79.32 |
| α (deg) | 78.41 | 75.67 | 105.10 | 103.83 | 104.83 | 90.00 | 101.31 | 90.00 |
| β (deg) | 89.95 | 89.96 | 89.95 | 89.99 | 90.03 | 90.00 | 90.02 | 90.70 |
| γ (deg) | 89.75 | 89.26 | 90.81 | 90.79 | 91.18 | 90.00 | 90.05 | 90.00 |
| observations | 78782 | 14467 | 139386 | 18477 | 202279 | 714649 | 458598 | 389254 |
| unique reflections | 34426 | 9073 | 73926 | 10698 | 51765 | 52631 | 128440 | 58936 |
| completeness (%) | 94.4 | 79.2 | 85.9 | 84.3 | 87.0 | 99.6 | 83.8 | 99.8 |
| Rpim (%), CC(1/2) highest resolution bin | 0.348, 0.709 | 0.122, 0.881 | 0.066, 0.512 | 0.127, 0.949 | 0.652, 0.578 | 0.679, 0.701 | 0.678, 0.452 | 0.538, 0.762 |
| (I/σ(I)) | 4.4 | 5.0 | 8.4 | 9.5 | 5.8 | 1.7 | 7.4 | 9.7 |
| multiplicity | 2.3 | 1.6 | 1.9 | 1.7 | 3.91 | 13.6 | 3.6 | 6.6 |
| resolution range (Å), highest | 41.12–1.93, 1.98–1.93 | 40.03–2.80, 2.87–2.80 | 40.50–1.90, 1.95–1.90 | 40.32–2.72, 2.79–2.72 | 73.82–1.62, 1.66–1.62 | 49.73–2.54, 2.61–2.54 | 40.89–1.19, 1.22–1.19 | 79.31–1.63, 1.67–1.63 |
| R factor (%) | 24.2 | 22.5 | 19.2 | 21.0 | 22.3 | 23.3 | 19.2 | 24.2 |
| Rfree (%) | 28.5 | 29.1 | 23.4 | 26.6 | 25.8 | 25.9 | 22.6 | 28.1 |
| average B, all atoms (Å2) | 14.0 | 28.0 | 12.0 | 25.0 | 15.0 | 64.0 | 14.0 | 17.0 |
| rms Z-Scores | ||||||||
| bond lengths | 0.70 | 0.60 | 0.95 | 0.56 | 0.89 | 0.69 | 0.90 | 0.91 |
| bond angles | 0.89 | 0.80 | 1.02 | 0.77 | 0.99 | 0.91 | 1.06 | 1.06 |
| Number of Atoms | ||||||||
| total | 4210 | 4098 | 4490 | 4084 | 4254 | 6046 | 4322 | 4219 |
| protein (Ch. A, B, C) | 2013, 2013 | 2030, 2013 | 2040, 1993 | 2025, 2005 | 2028, 2016 | 1995, 2014, 1853 | 2063, 2042 | 2033, 2021 |
| ligands | 22 | 10 | 30 | 40 | 51 | |||
| DTT | 32 | |||||||
| glycerol | 6 | 18 | 24 | 6 | ||||
| phosphate | 30 | |||||||
| acetate | 19 | 20 | 20 | 8 | ||||
| Tris | 22 | |||||||
| PEG | 25 | 42 | 21 | 24 | 20 | |||
| water | 112 | 25 | 347 | 6 | 138 | 53 | 187 | 145 |
| Ramachandran Plot (%) | ||||||||
| preferred regions | 98 | 93.8 | 97 | 97 | 98 | 97 | 98 | 98 |
| allowed regions | 2 | 6 | 3 | 3 | 2 | 3 | 2 | 2 |
| outliers | 0 | 0.2 | 0 | 0 | 0 | 0 | 0 | 0 |
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry (ITC) experiments were performed on a MicroCal VP-ITC (Malvern) instrument. Before ITC experiments were performed, purified BlaC S70A was dialyzed overnight at 4 °C in an excess volume of 25 mM Tris-HCl, pH 8.0, 40 mM NaCl. The dialyzed protein was loaded in the sample cell after extensive degassing and determination of protein concentration (32.3 μM) by measuring A280 on a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). Clavulanic acid (Matrix Scientific) was weighed shortly before the experiment and dissolved in the dialysis buffer to a stock concentration of 100 mM. The clavulanate stock solution was diluted in dialysis buffer to experimental concentrations of 1 and 10 mM before degassing and loading in the syringe. The titration protocol consisted of a first 2 μL injection followed by injections of 7 μL with a time spacing of 240 s between injections and a constant stirring at 351 rpm. The temperature was set at 25 °C. Data analysis was done in Origin (OriginLab) using the software provided by Malvern.
Nuclear Magnetic Resonance (NMR)
The NMR titration of clavulanate into BlaC S70A was performed in two buffering systems. One NMR sample contained 230 μM BlaC S70A in 20 mM Tris-HCl, pH 8.0, 40 mM NaCl, and the other sample contained 140 μM BlaC S70A in 20 mM MES, pH 6.7. NMR spectra were recorded at 25 °C on a Bruker Avance IIIHD NMR spectrometer operating at 20 T (850 MHz), equipped with a TCI cryoprobe. 15N–1H HSQC spectra of BlaC S70A were acquired at each step of the titration with clavulanate. Data were processed using Topspin 3.5 (Bruker Biospin).
Results
Clavulanic Acid
The X-ray crystal structure of BlaC was solved in complex with two covalent clavulanate adducts, obtained by soaking for 3 or 10 min. The structures were refined against diffraction data to a resolution of 1.9 Å for the 3 min soaked crystal, and to 2.8 Å for the one soaked for 10 min. Crystallographic statistics are presented in Table 1. Despite the fact that longer soaking times led to a poorer diffraction and resolution, the overall fold of BlaC was not affected. Both structures show good superposition of the polypeptide chain with the structure of free BlaC (PDB 5OYO), with an average root-mean-square deviation (rmsd) between Cα atoms of 0.30 and 0.29 Å for the 3 min and 10 min soaked structures, respectively. In both structures, positive difference electron density near the catalytic Ser70 clearly indicated the presence of covalent adducts. In the 3 min soaked crystal, a trans-enamine adduct corresponding to the +155 Da MS peak could be modeled in the structure, while in the one soaked for 10 min the covalent inhibitor fragment was a shorter molecule that could be modeled as propionaldehyde ester, corresponding to the MS peak of +70.
Clavulanate trans-Enamine Adduct
A trans-enamine adduct was found in both chains of the asymmetric unit. It has the same chemical structure as the adduct reported in the PDB entry 3CG5.3 The conformation of the trans-enamine inhibitor fragment (Figure 3a) is similar in the two BlaC molecules, with the exception of terminal atoms C7, C8, and O3, which do not interact with any amino acid residues and, thus, show some flexibility (Figure S1a,b). This terminal flexibility of the trans-enamine adducts of clavulanate is also confirmed by the comparison with PDB entry 3CG5 (Figure 3b).
Figure 3.

Structures of the covalent adducts of clavulanate formed by soaking BlaC crystals in a clavulanate solution for 3 and 10 min. (a) Representation of the trans-enamine adduct modeled in chain B of the BlaC structure (light gray ribbon) obtained after 3 min soaking in a clavulanate solution. (b) Superposition of the trans-enamine derivative of clavulanate from chain A (turquoise C) and chain B (gray C) of the BlaC structure presented here with the previously published structure PDB 3CG5 (magenta C).3 (c) In the BlaC structure (pink ribbon) solved after 10 min soaking in a solution containing clavulanate, the electron density suggests the presence of a propionaldehyde ester adduct bound to Ser70, which corresponds to the +70 Da peak identified by MS. (d) Superposition of the propionaldehyde covalent adduct modeled in the structure of BlaC (chain A, pink) presented here and the ethane-imine adduct modeled in PDB 2Y91 (gold).4 In parts a and c, the covalent clavulanate adducts are represented as sticks colored according to atom type (C in green, O in red, and N in blue). Acetate ions and amino acidic residues that form hydrogen bonds (dashed lines) with the adduct or are at a distance < 4 Å are represented as sticks colored according to atom type (C in gray, O in red, and N in blue). Water molecules are indicated with red dots. The 2mFo-DFc electron density map (blue chicken wire with a contour level of 1 σ) is centered on the clavulanate adduct.
Propionaldehyde Ester Adduct of Clavulanate
The structural solution of the BlaC crystal soaked in clavulanate for 10 min showed a different covalent adduct of clavulanate than the trans-enamine intermediate formed in the structure described above. The positive difference electron density clearly suggested the presence of a short covalent inhibitor bound to Ser70 that could be modeled as the +70 Da intermediate proposed by Hugonnet et al.2 The inhibitor is stabilized in the active site by a hydrogen bond between the carboxylic oxygen and the Thr239 backbone nitrogen, and is at distance of <4 Å from essential catalytic residues Ile103 and Asn172 (Figure 3c and Figure S1c,d). Although MS experiments always show the formation of a +70 Da intermediate of BlaC with clavulanate,2,14 the corresponding +70 Da propionaldehyde adduct had never been identified by X-ray crystallography. A +70 Da clavulanate adduct was found in the X-ray crystal structure of the BS3 β-lactamase from Bacillus licheniformis, but the adduct was modeled as an ethane-imine derivative (called CL1 in ref (4)) of clavulanate rather than a propionaldehyde adduct (Figure 3d).4 The authors argued that the C3–N4 bond of the trans-enamine adduct is weakened by the interactions with active site residues of BS3 and breaks with the release of a pentan-3-one-5-ol acid degradation product (CL2). The CL2 molecule is reported to be observed in the structure at a hydrogen bond distance from Ser130 and Thr235, with the carboxylic group in the same position as where the acetate or phosphate ions are in the structure of BlaC (PDB 5OYO and 5NJ2). The weakening and breaking of the trans-enamine adduct would be dependent on the stabilization of the terminal part of the molecule in the acetate/phosphate binding pocket. However, the trans-enamine adduct of clavulanate has never been observed to bind in such a conformation in any of the β-lactamase structures deposited in the PDB so far (PDB codes 1BLC, 2A49, 2H0T, 3CG5). Furthermore, the electron density modeled as CL2 could also be modeled very well as citrate, which was present at a high concentration in the crystallization conditions (Figure S2).4 Thus, the formation of the CL1 and CL2 adducts would still need further validation. So far, the most accredited pathway of β-lactamase inhibition by clavulanate is the one that involves the formation of a propionaldehyde adduct on Ser70. Thus, we believe that the +70 Da adduct is more likely to be propionaldehyde ester.
BlaC in Complex with Sulbactam
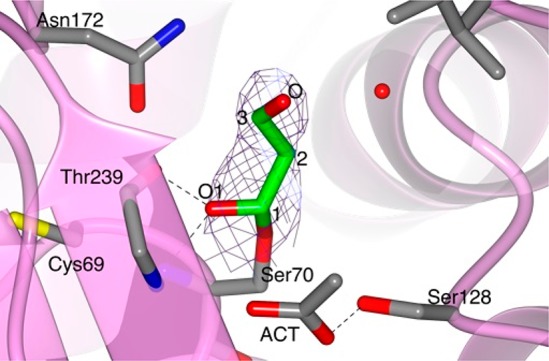
The crystal structure of BlaC in complex with a covalent acyl intermediate derived from sulbactam was obtained by soaking BlaC crystals in a 10 mM sulbactam cryo-solution for 10 min. Soaked crystals diffracted to a final resolution of 1.40 Å, and the structure was refined in space group P1, obtaining a final R-factor of 19.2% and an Rfree of 23.4%. The asymmetric unit contains two protein molecules (chain A and chain B). For both chains, residues 29–293 according to the Ambler numbering convention were modeled, and part of the C-terminal His-tag could be modeled for chain A. The overall fold of BlaC was not affected by sulbactam binding, as shown by an average rmsd of 0.31 Å for Cα atoms of core residues in BlaC in complex with sulbactam and free BlaC (PDB 5OYO). A clear electron density near the catalytic Ser70 signaled the presence of a covalent inhibitor that was modeled as the trans-enamine derivative of sulbactam. Most of the covalent trans-enamine adduct is in the same conformation in both protein chains and is stabilized in the BlaC active site by hydrogen bonds involving the sulfate and carbonyl moieties (Figure 4a,b). The presence of the sulbactam adduct produced the largest effects in the loop region between residues 98–106. Already in the structure of substrate-free BlaC (PDB entry 5OYO)14 some variation was observed for this loop region, which was in a more open conformation in chain B than in chain A. In the BlaC–sulbactam adduct complex, residues 98–106 of chain A are forced into a more open conformation to avoid clashes with the inhibitor. In particular, the side-chain atom CD1 of Ile103 is directed outward relative to the catalytic Ser70, whereas in the free enzyme the side chain of Ile103 is directed toward the active site of the protein (Figure 4c). In chain B of the BlaC–sulbactam structure, the changes involve a larger region, comprising residues 93–103 and 107–116 (Figure 4d).
Figure 4.

Structure of the covalent trans-enamine adduct formed between sulbactam and BlaC. The modeled adducts in chains A (a) and B (b) of the AU are shown in stick representation colored according to atom type (C in green, O in red, N in blue, and S in yellow). The 2mFo-DFc electron density map (blue chicken wire with a contour level of 1 σ) is centered on the sulbactam adducts. Acetate ions (ACT) and protein residues at a hydrogen bond (dashed lines) distance from the adduct or at a distance < 4 Å are represented as sticks (C in light blue, O in red, and N in blue). Waters are represented as red spheres. BlaC is in ribbon representation (gray). (c and d) Superposition of BlaC in complex with the trans-enamine adduct of sulbactam (chain A, gray ribbon representation) and the structure of free BlaC (PDB 5OYO, lawn green). The presence of sulbactam in the active site of BlaC caused a slight shift of the region between residues 93–116, compared to the structure of free BlaC. (c) In chain A, sulbactam binding forced the side chain of Ile103 into a more open conformation to avoid clashes. (d) In chain B, sulbactam caused a destabilization of the α-helix made by residues 105–113. (e and f) Comparison of BlaC and SHV-1 covalent complexes with sulbactam. (e) Active site of SHV-1 bound to sulbactam (PDB 2A3U). The trans-enamine adduct is in stick representation and is colored according to atom type (C in green, O in red, N in blue, and S in yellow). SHV-1 fold is in ribbon representation (coral), and the residues involved in the stabilization of the inhibitor are represented as sticks (with C colored in gray). Hydrogen bonds are indicated as dashed lines. (f) Superposition of the X-ray crystal structures of BlaC (gray) and SHV-1 (PDB 2A3U, coral) in complex with sulbactam.
The structure of BlaC in complex with the trans-enamine adduct of sulbactam was compared to PDB entries 2A3U and 2H0Y, which show the inhibition of the Ambler class A β-lactamase SHV-1 by the same trans-enamine intermediate of sulbactam as the one modeled in complex with BlaC.18,34 From a chemical point of view, the trans-enamine adducts are identical in both SHV-1 structures and BlaC (Figure 4e), but their conformation in the active site differs, with the sulfate and carboxylic moieties oriented in opposite directions in the two β-lactamases (Figure 4f). In BlaC, the sulfate moiety of the trans-enamine intermediate is stabilized by hydrogen bonds with residues Asn172 and Arg173, whereas in SHV-1 it is the carboxylic moiety that forms hydrogen bonds with the protein, through Asn132 and Asn170. When the sulbactam-bound structures of BlaC and SHV-1 are compared, it is important to bear in mind that both complexes of SHV-1 (PDB 2A3U and 2H0Y) were obtained using SHV-1 mutants with an impaired deacylation process: PDB 2A3U was obtained using a single Glu166Ala SHV-1 mutant and PDB 2H0Y using a double Met69Val/Glu166Ala SHV-1 mutant. Since no experimental data are available of a covalent complex of wild-type SHV-1 with sulbactam, it is not known if the wild-type enzyme can accommodate the same trans-enamine conformation as the one observed in the mutant structures.
BlaC Inhibition by Tazobactam
Similar to the BlaC–sulbactam structure, the BlaC–tazobactam complex was produced by soaking BlaC crystals in a 10 mM tazobactam solution for 10 min. The structure was modeled in space group P1 to a resolution of 2.72 Å for a final R-factor of 21.0% and Rfree of 26.6%. Also in this case, there are two BlaC chains in the asymmetric unit (chains A and B). The X-ray crystal structure of BlaC in complex with tazobactam superposes well to the structure of the free enzyme (PDB 5OYO) with an average rmsd of 0.29 Å for the Cα atoms of corresponding core residues. The covalent adduct was modeled as the trans-enamine derivative of tazobactam (Figure 5a–c). The two modeled inhibitor fragments show good superposition of atoms C5, C6, and C7, but there is a variation in the angle between C3, N4, and C5, which measures 115.8° in chain A and 118.1° in chain B. Furthermore, the triazole ring assumes a different rotation angle in the two chains of the asymmetric unit (Figure 5d). The trans-enamine tazobactam adduct bound to BlaC is chemically identical to the covalent inhibitor fragment formed between tazobactam the Ambler class A β-lactamase SHV-1 (PDB 1VM1),34−37 but the stabilization of the sulfate and triazolyl moieties of the intermediate is different in the two β-lactamases (Figure 5e,f).
Figure 5.

Structure of the covalent BlaC complex with tazobactam. In both chain A (a) and chain B (b) of the AU, the covalent adduct was modeled as a trans-enamine derivative of tazobactam. BlaC is in ribbon representation. The covalent tazobactam adducts are in stick representation colored according to atom type (C in green, O in red, N in blue, and S in yellow). The residues that are at a distance < 4 Å are also represented as sticks with C atoms colored in gray. The 2mFo-DFc electron density map (blue chicken wire with a contour level of 1 σ) is clipped on the covalent adducts and acetate ion (ACT) near Ser128. (c) Closer view of the trans-enamine adduct of chain B with the numbering of the atoms indicated by the labels. (d) Superposition of the trans-enamine adduct modeled in chain A (lilac) and chain B (purple). (e), (f) Structural comparison of the tazobactam trans-enamine adducts bound to SHV-1 (PDB 1VM1, ice blue) and to BlaC (lilac) chain A (e) and chain B (f).
Positive electron density was also found near Ser128 in both chains of the asymmetric unit. Since the BlaC crystals were grown in a buffer containing acetate, and acetate ions were also found at the same site of the protein in the structure of free BlaC (PDB 5OYO), it is reasonable to fit acetate ions also in the structure of BlaC–tazobactam complex (Figure 5a,b). Given the low resolution of the BlaC–tazobactam structure, it is, however, not possible exclude the possibility of a covalent adduct is formed on Ser128.35
BlaC Inhibition by Avibactam
The crystal structure of BlaC in complex with avibactam is presented here at a resolution 1.62 Å. Also the BlaC–avibactam complex was obtained by soaking BlaC crystals in a 10 mM avibactam solution for 10 min. The two BlaC molecules in the asymmetric unit show a good superposition with the structure of free BlaC (PDB 5OYO), with an average rmsd of 0.35 Å for Cα atoms of core residues. In chain A, the electron density map allowed fitting of two conformations of the inhibitor, each with 50% occupancy (Figure 6a). Both conformers in chain A are covalently linked to the catalytic Ser70, but while one has the sulfate moiety interacting with residues Ser128 and Thr239 in the active site, the other one has the sulfate moiety directed outward. In chain B, good fitting of the inhibitor electron density was achieved by modeling a single conformation of the covalent adduct of avibactam with Ser70 with 100% occupancy, in the same orientation as the inward oriented conformer in chain A (Figure 6b). Two loop regions in the protein chains experienced a change in the BlaC–avibactam adduct compared to the free enzyme. In chain A, residues 167–174 were shifted, producing an opening of the active site (Figure 6c). The maximum shift involves residues Asn172 and Arg173, for which the Cα atoms move ∼2 Å compared to the same atoms in the free protein (PDB 5OYO). On the basis of these observations, it is possible to speculate that the entrance of the inhibitor in the active site of BlaC causes a temporary opening of the 167–174 protein region (as in chain A), which subsequently closes back upon complete stabilization of the avibactam covalent adduct (as in chain B). As already observed in other BlaC–inhibitor structures, chain B presents a slight difference in the backbone and side chains coordinates of residues 94–114, in particular of Ile103 (Figure 6d). The X-ray crystal structure of BlaC in a covalent complex with avibactam was first published by Xu et al. (PDB 4DF6).15 In PDB 4DF6, only one conformation of the covalent adduct was found in complex with the enzyme, to which the conformation observed here for chain B is similar, and the backbone of BlaC did not show any big shifts when compared to the structure of the free protein. Recently, Pozzi et al.38 reported a conformation of an avibactam adduct in the lactamase TRU-1 that was similar to that observed for CTX-M-15,39 but different from that in BlaC.15 The conformation in TRU-1 features a short distance between the nitrogen linked to the sulfate and the carbon bonded to Ser70. This proximal conformation was suggested to be primed for release/recyclization, whereas the distal conformation could be a nonactive conformation. One of the conformations observed here in chain A resembles the distal conformation also observed in PDB 4DF6,15 whereas the second one is very different from either the distal or proximal conformation, with sulfur atom swung out of the active site. The distance between the sulfur atoms in the two conformations is 4.6 Å.
Figure 6.

BlaC in complex with avibactam. The avibactam carbamyl adducts in chains A (a, red) and B (b, teal) of the AU of BlaC crystals are shown. The structure of BlaC is in ribbon representation, and the residues involved in binding and stabilization of the covalent adduct are represented as sticks (C in gray, O in red, N in blue, and S in yellow). The covalent adduct of avibactam is also represented as sticks with C colored in green. (a) Active site of chain A. Avibactam could be modeled in two conformations. (b) In chain B, the avibactam-derived covalent inhibitor was found only in one conformation, in which the sulfate moiety is hydrogen bonded to Ser128, Thr237, and Thr239. In both images, the 2mFo-DFc electron density map (blue chicken wire with a contour level of 1 σ) is centered on the avibactam adducts. (c and d) Comparison of the structures of BlaC in complex with avibactam and of free BlaC (PDB 5OYO). Both structures are in ribbon representation. (c) Close view of the shifted loop of chain A (residues 167–174) in the structure of BlaC in complex with avibactam (lawn green) superposed to free BlaC (PDB 5OYO, coral). The side chains of residues 167–174 are shown as sticks colored according to atom type (O in red, N in blue, and C in lawn green and coral for BlaC–avibactam and free BlaC, respectively). (d) Detail of the structural superposition of chain B of BlaC–avibactam (green) with chain B of free BlaC (red).
BlaC S70A
A first batch of BlaC S70A was purified in a buffer containing DTT, and the protein was screened for crystallization. Crystals grew in approximately one month in 0.1 M Tris-HCl, pH 7.6, 1.7 M ammonium phosphate. The structural solution was found by molecular replacement using wild-type BlaC (PDB 2GDN) as a search model. The X-ray crystal structure of BlaC S70A was refined against diffraction data to a final resolution of 2.54 Å. Three protein chains could be modeled in the asymmetric unit of the crystal, but the extra electron density was present for possibly a fourth chain of a contaminant or a degradation product of BlaC S70A itself. However, it was not possible to model this electron density and also submitting the data to contaminants search tools like ContaMiner40 did not help to model the extra density present in the crystal. Nevertheless, the electron density was good for chains A and B, and most of chain C, so that the structure was refined including these three chains in the model. Because of the unmodelled density, final R-factors remained relatively high, but still in acceptable limits for the resolution of the data: the R-factor was 23.3% and Rfree 25.9%. The overall fold of BlaC was not affected by the mutation of Ser70 to Ala, and the structure of the mutant can be superposed to that of wild-type BlaC with a rmsd for Cα atoms of 0.61 Å (Figure S3a). Extra electron density was present in the active site of the enzyme in all three chains of the crystal. DTT, glycerol, and Tris molecules were modeled in each density, and the ligand showing the best fit was refined in the final model. In chain A, the best fit was obtained by modeling a molecule of Tris and a phosphate ion, each with a partial occupancy of 50% (Figure S3b). In chains B and C, DTT showed the best fit (Figure S3c,d). Although DTT tends to rapidly oxidize in solution and form a circular product, reduced DTT was found in the active site of BlaC S70A. Presence of reduced DTT in protein crystals has been reported, and more than 200 structures containing DTT have been deposited in the PDB (http://www4.rcsb.org/ligand/DTT).41−44
A second crystal form of BlaC S70A was obtained after purifying the protein in the absence of DTT. Crystals grew in PEG3350 and diffracted to a resolution of 1.19 Å. The overall X-ray crystal structure of BlaC S70A superposes well with the structure of wild-type BlaC (PDB 5OYO), as shown by an average rmsd of 0.35 Å between the Cα atoms of the two structures (Figure S4a). The active site residues also superpose well between the mutant and wild-type structure, with the exception of the catalytic Ser70 that was an Ala in the mutant (Figure S4b). Again, a clear electron density was found in the active site of the protein, which, in this case, could be modeled as polyethylene glycol (PEG, Figure S4c,d). To look at the Michaelis complexes between BlaC and β-lactamase inhibitors, both forms of BlaC S70A crystals were soaked in cryo-protecting solutions containing either clavulanic acid, sulbactam, tazobactam, or avibactam. However, none of the inhibitors could be modeled in the electron density. Sulbactam was peculiar in that it led to rapid dissolution of the crystals of BlaC S70A grown in the absence of DTT, and no extra density could be found in the active site of the enzyme even after a few seconds of soaking.
Since no complex could be determined between BlaC S70A and clavulanate by crystal soaking, clavulanate binding to BlaC S70A was tested in solution by isothermal titration calorimetry (ITC). ITC experiments were done by titrating 10 mM clavulanate into 32 μM BlaC S70A. ITC traces did not reveal any binding events (Figure S5a). However, since ITC requires binding constants in the nM to low μM range in order to detect binding, a technique more sensitive to low affinities was also used to reveal clavulanate binding to BlaC S70A. Clavulanate was titrated into 15N–1H labeled BlaC S70A, and HSQC-NMR spectra were recorded at each titration step. HSCQ-NMR data analysis only revealed very small chemical shift perturbations (CSPs) of a few residues at high inhibitor concentrations, and a KD could not be calculated (Figure S5b).
BlaC S70C
Good quality crystals of BlaC S70C were obtained that diffracted to 1.6 Å resolution. Two identical protein molecules were found in the asymmetric unit that differed by an rmsd of 0.39 Å. The overall structure of the S70C mutant is not affected by the mutation, and it superposes well to that of wild-type BlaC (PDB 5OYO) with an average rmsd of 0.31 Å (Figure S4e,f). The structure of BlaC S70C shows one peculiarity in the active site. Cys70 is oriented toward the nearby residue Lys73, yielding a distance between the Sγ of Cys70 and Nζ of Lys73 of 1.45 and 1.44 Å in chains A and B, respectively. One continuous electron density connects the Cys and Lys side chains, indicating the presence of a covalent bond (Figure 7a). Despite being quite rare, a chemical bond between Cys and Lys side chains has been reported before in the literature, and it is known as a sulfenamide bond.45−47 Mostly, circular sulfenamide bonds were observed between an oxidized Cys and an amide of the protein backbone, as seen in the protein tyrosine phosphatases (PTPs)47,48 and in the Bacillus subtilis transcription factor OhrR.49 In two other cases reported in the literature, a sulfenamide bond was observed between a Cys and the side-chain amine of Lys.45,46 In particular, Rodkey and co-workers solved the X-ray crystal structure of the S70C mutant of the β-lactamases SHV-1 in its substrate-free state and in Michaelis complex with sulbactam.46 Interestingly, substrate-free SHV-1 S70C (PDB 4FD8) did not show the formation of the sulfenamide bond, but it was present in the sulbactam preacylation structure (PDB 4FH2). On the basis of the structures of several substrate-free and sulbactam-complexed structures, the authors suggested that the Cys70–Lys73 sulfenamide bond is not formed in solution because both residues are important for substrate acylation. However, our structure of substrate-free BlaC S70C clearly shows a covalent bond between Cys70 and Lys73 side chains. Moreover, both crystallographic monomers of substrate-free BlaC S70C show extra electron density in the active site that could be modeled as polyethylene glycol (PEG, Figure 7a). The position of PEG in both BlaC S70C chains corresponds to the binding site of sulbactam as shown in the inhibitor preacylation complex with SHV-1 S70C (PDB 4FH2) (Figure 7b). Unfortunately, we were unable to establish whether the bond is formed upon protein production or during crystal growth/X-ray irradiation. The mass difference before and after bond formation is only 2 Da, which is close to the error margin of intact protein MS. The covalently modified peptides could not be detected after trypsin digestion and peptide MS analysis of fresh BlaC S70C.
Figure 7.

Structure of the active site of BlaC S70C. (a) The crystal structure of BlaC S70C showing the presence of continuous 2mFo-DFc electron density connecting the side chains of Cys70 and Lys73. The proximity of the S and N atoms suggests the presence of a covalent sulfenamide bond. Positive difference mFo-DFc electron density was found in the active site of both protein chains in the crystal, which was modeled as polyethylene glycol (PEG). (b) Superposition of the X-ray crystal structures of BlaC S70C (pale crimson) and SHV-1 S70C mutant in complex with sulbactam (PDB 4FH2, teal). Sulbactam lies in the same position as the PEG molecule modeled in the active site of BlaC S70C.
Discussion
In the present work, two clavulanate acylation products were identified by soaking BlaC crystals in clavulanate for 3 and 10 min. The first identified inhibitor fragment was a classical trans-enamine derivative of clavulanate, like the one modeled in PDB 3CG5.3 A longer soaking time allowed for trapping of a new BlaC–clavulanate complex that had not been observed by X-ray crystallography yet, but had been identified on the basis on MS data (+70 Da peak). The modeled inhibitor fragment is a 3-oxopropanoic acid with an ester bond with the catalytic Ser70. Our crystallographic data thus showed that BlaC is only transiently inhibited by clavulanic acid, and the +155 Da trans-enamine adduct can be hydrolyzed into a +70 Da propionaldehyde inhibitor. The +70 Da inhibitor can be further hydrolyzed releasing an active enzyme, as suggested by kinetics and MS data.14,15 The inhibition of β-lactamases by mechanism-based inhibitors can also result in the formation of a nonhydrolyzable adduct on Ser128 (Ser130 in the classical Ambler numbering) that permanently inhibits the enzyme.2,35,50 A positive electron density near Ser128 was present in all BlaC structures presented in this work and could be modeled as an acetate ion, as previously shown for free BlaC (PDB 5OYO).14 The orientation of the acetate is very conserved in all BlaC structures presented here, but it showed some displacement in the structure of BlaC bound to the +70 Da propionaldehyde inhibitor. In the latter structure, it is also possible to model a covalent adduct bound to Ser128 without affecting the R-factor and the Rfree of the model. However, the low resolution of the data set did not allow to confidently model a covalent inhibitor bound to Ser128, but it cannot be excluded that it was present in some BlaC molecules of the crystal. Similar observations were done for the structure of BlaC in covalent complex with tazobactam. The importance of Ser130 (Ser128 in BlaC) as an additional nucleophile for the covalent binding of inhibitors together with Ser70 also emerged from X-ray crystallographic studies of two clinically derived mutants of TEM, TEM-32 (PDB 1LI0), and TEM-34 (PDB 1LI9).51 TEM-32 and TEM-34 carry point mutations Met69Ile and Met69Val, respectively, and are both more resistant to β-lactam antibiotics and β-lactamase inhibitors. Wang and colleagues showed that the mutation of Met69 causes small variations in the Ser130 side chain that could be responsible for the higher resistance of these clinical TEM species by affecting the irreversible inhibition of the enzymes.51 The mutation of Met69 was also studied in the Ambler class A β-lactamase SHV-1 using a deacylation impaired Glu166Ala mutant of SHV-1.18,35,36 The structures of wild-type SHV-1, Glu166Ala SHV-1, and Met69Val/Glu166Ala SHV-1 in complex with tazobactam (PDB 1VM1, 1RCJ, and 2H10, respectively) showed that the trans-enamine intermediate of tazobactam assumes a different conformation in the wild-type enzyme compared to the two mutants (Figure 8). In both SHV-1 mutants, the carboxylic moieties of the tazobactam as well as sulbactam trans-enamine intermediates discussed above are in a comparable orientation, while in wild-type SHV-1 the orientation of the carboxylic moiety of the tazobactam trans-enamine intermediate is in a position more similar to the carboxylic moiety of the sulbactam trans-enamine intermediate found in BlaC. Thus, it is reasonable to think that the Glu166Ala mutation is already sufficient to cause the different orientation of the trans-enamine adducts, and it is not known what would be the effect of the single Met69Val mutation. The Glu166Ala mutation causes a reduction of the negative charge in the active site of SHV-1, which could allow the carboxylic moieties of the acyl intermediates to face the active site of the protein, like in PDB 2A3U and 2H0Y for sulbactam, and PDB 1RCJ and 2H10 for tazobactam. Such a conformation of the trans-enamine adducts would be destabilized by the more negative charge in the wild-type active site.
Figure 8.

Comparison of the X-ray crystal structures of BlaC and SHV-1 in complex with the trans-enamine adducts of sulbactam and tazobactam. (a) Superposition of wild-type SHV-1 in complex with tazobactam (PDB 1VM1, gold) with Glu166Ala SHV-1 bound to tazobactam (PDB 1RCJ, ice blue). (b) Superposition of BlaC in complex with sulbactam (chain A, gray) with wild-type SHV-1 bound to tazobactam (PDB 1VM1, light blue).
In recent years, the emergence of inhibitor-resistant β-lactamases has triggered the development of a second generation of inhibitors, including avibactam, with a non-β-lactam chemistry so as to avoid hydrolysis of the inhibitor.1 Here, the structure of BlaC in complex with avibactam was presented. One important feature of avibactam compared to β-lactam-based inhibitors is the presence of the sulfate moiety, which is stabilized in the active site by hydrogen bonds with Ser128, Thr237, and Thr239. Interestingly, this same site of the protein was found to bind phosphate both by crystallography (PDB 5NJ2) and by solution NMR studies.14 As suggested by Xu et al.,15 the higher stabilization of the avibactam carbamyl adduct might be at the basis of the more stable inhibition of BlaC by avibactam compared to clavulanate, sulbactam, or tazobactam. However, as shown by Xu and colleagues,15 avibactam has an extremely low affinity for BlaC, which might preclude its use in the treatment of TB in combination with β-lactam antibiotics. A low affinity for antibiotics and inhibitors had already been reported in previous studies.2,19 Thus, the major challenge for the future is the design of new inhibitors that combine the high stability of the avibactam covalent adduct with an enhanced affinity for BlaC. To this end, the study of the preacylation complexes is essential for understanding the factors driving binding of BlaC to inhibitors and substrates. In our study, two mutants of the β-lactamase BlaC from Mtb were produced, in which the active site Ser70 was replaced by an Ala or a Cys residue, obtaining BlaC S70A and S70C, respectively. Both mutants were crystallized in their resting state, and the X-ray crystal structures were solved. However, soaking of two different crystal forms of BlaC S70A and of BlaC S70C in solutions containing 10 mM of either clavulanic acid, sulbactam, tazobactam, or avibactam did not result in the formation of a preacylation complex with the enzymes. Interestingly, both BlaC S70A and BlaC S70C crystals contained solvent molecules in the active site, which could be modeled as either reduced DTT or PEG molecules. ITC and NMR titrations showed that the affinity of clavulanic acid for BlaC S70A is very low (high mM range), suggesting that the hydroxyl group of Ser70 could fulfill a dual role of catalysis and substrate binding. The role of Ser residues in driving β-lactamase-substrate recognition was also highlighted by Helfand and colleagues,52 who showed that the substitution of Ser130 with a Gly in the SHV-1 β-lactamase resulted in a weaker affinity (higher Km) for antibiotic substrates.
As already observed in the S70C mutant of SHV-1,46 a sulfenamide bond was clearly present between the side chains of Cys70 and Lys73 in the structure of BlaC S70C. Rodkey et al. showed that the sulfenamide bond was not present in the substrate-free structure of SHV-1 S70C and would only form in the preacylation complex.46 However, here, we showed that apo BlaC S70C already contains the sulfenamide bond and it is not clear when and how this bond is formed. By comparing SHV-1 S70C crystal structures in the resting state, where no sulfenamide bond was formed, and in the preacylation complex with sulbactam, where the sulfenamide bond was fully formed, the authors also brought evidence in support of the hypothesis that Lys73 deprotonates upon formation of the preacylation complex.46 However, our structure of substrate-free BlaC S70C shows the presence of the sulfenamide bond between Cys70 and Lys73. The protonation state of Lys73 in the resting state is thus still unclear, but recent ultrahigh resolution crystallography data indicate that the lysine is capable of accepting a hydrogen from Ser130, suggesting that it can act as a base.53
Acknowledgments
We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b01085.
Structures of the covalent adducts formed, analysis of the PDB 2Y91 structure, structure of BlaC S70A crystals prepared in the presence of DTT, structures of BlaC S70A and S70C prepared in the absence of DTT, and BlaC–clavulanate interaction studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Drawz S. M.; Bonomo R. A. (2010) Three decades of b-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201. 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J. E.; Blanchard J. S. (2007) Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. 10.1021/bi701506h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L. W.; Hugonnet J. E.; Blanchard J. S. (2008) Structure of the covalent adduct formed between Mycobacterium tuberculosis beta-lactamase and clavulanate. Biochemistry 47, 5312–5316. 10.1021/bi8001055. [DOI] [PubMed] [Google Scholar]
- Power P.; Mercuri P.; Herman R.; Kerff F.; Gutkind G.; Dive G.; Galleni M.; Charlier P.; Sauvage E. (2012) Novel fragments of clavulanate observed in the structure of the class A beta-lactamase from Bacillus licheniformis BS3. J. Antimicrob. Chemother. 67, 2379–2387. 10.1093/jac/dks231. [DOI] [PubMed] [Google Scholar]
- WHO . (2018) Global Tuberculosis Report 2018, ISBN 9789241565646.
- Brown A. G.; Butterworth D.; Cole M.; Hanscomb G.; Hood J. D.; Reading C.; Rolinson G. N. (1976) Naturally-occurring beta-lactamase inhibitors with antibacterial activity. J. Antibiot. 29, 668–669. 10.7164/antibiotics.29.668. [DOI] [PubMed] [Google Scholar]
- Reading C.; Cole M. (1977) Clavulanic acid: a beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 11, 852–857. 10.1128/AAC.11.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H. F.; Moreau D.; Yajko D.; Miick C.; Wagner C.; Hackbarth C.; Kocagoz S.; Rosenberg E.; Hadley W. K.; Nikaido H. (1995) Can penicillins and other beta-lactam antibiotics be used to treat tuberculosis. Antimicrob. Agents Chemother. 39, 2620–2624. 10.1128/AAC.39.12.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England K.; Boshoff H. I. M.; Arora K.; Weiner D.; Dayao E.; Schimel D.; Via L. E.; Barry C. E. (2012) Meropenem-clavulanic acid shows activity against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 56, 3384–3387. 10.1128/AAC.05690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Wang Y. F.; Lu J.; Pang Y. (2016) In vitro activity of beta-lactams in combination with beta-lactamase inhibitors against multidrug-resistant Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 60, 393–399. 10.1128/AAC.01035-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacon A. H.; van der Merwe L.; Barnard M.; von Groote-Bidlingmaier F.; Lange C.; García-Basteiro A. L.; Sevene E.; Ballell L.; Barros-Aguirre D. (2016) β-Lactams against tuberculosis — New trick for an old dog?. N. Engl. J. Med. 375, 393–394. 10.1056/NEJMc1513236. [DOI] [PubMed] [Google Scholar]
- Hugonnet J.-E.; Tremblay L. W.; Boshoff H. I.; Barry C. E.; Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar N.; Dubée V.; Ballell L.; Cuinet G.; Hugonnet J.-E.; Signorino-Gelo F.; Barros D.; Arthur M.; McKinney J. D. (2015) Rapid cytolysis of Mycobacterium tuberculosis by faropenem, an orally bioavailable β-lactam antibiotic. Antimicrob. Agents Chemother. 59, 1308–1319. 10.1128/AAC.03461-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elings W.; Tassoni R.; van der Schoot S. A.; Luu W.; Kynast J. P.; Dai L.; Blok A. J.; Timmer M.; Florea B. I.; Pannu N. S.; Ubbink M. (2017) Phosphate promotes the recovery of Mycobacterium tuberculosis beta-lactamase from clavulanic acid inhibition. Biochemistry 56, 6257–6267. 10.1021/acs.biochem.7b00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Hazra S.; Blanchard J. S. (2012) NXL104 irreversibly inhibits the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 51, 4551–4557. 10.1021/bi300508r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroka D.; Li de la Sierra-Gallay I.; Dubee V.; Triboulet S.; van Tilbeurgh H.; Compain F.; Ballell L.; Barros D.; Mainardi J. L.; Hugonnet J. E.; Arthur M. (2015) Hydrolysis of clavulanate by Mycobacterium tuberculosis beta-lactamase BlaC harboring a canonical SDN motif. Antimicrob. Agents Chemother. 59, 5714–5720. 10.1128/AAC.00598-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. C. H.; Herzberg O. (1992) Inhibition of beta-lactamase by clavulanate - trapped intermediates in cryocrystallographic studies. J. Mol. Biol. 224, 1103–1113. 10.1016/0022-2836(92)90472-V. [DOI] [PubMed] [Google Scholar]
- Padayatti P. S.; Helfand M. S.; Totir M. A.; Carey M. P.; Carey P. R.; Bonomo R. A.; van den Akker F. (2005) High resolution crystal structures of the trans-enamine intermediates formed by sulbactam and clavulanic acid and E166A SHV-1 beta-lactamase. J. Biol. Chem. 280, 34900–34907. 10.1074/jbc.M505333200. [DOI] [PubMed] [Google Scholar]
- Wang F.; Cassidy C.; Sacchettini J. C. (2006) Crystal structure and activity studies of the Mycobacterium tuberculosis beta-lactamase reveal its critical role in resistance to beta-lactam antibiotics. Antimicrob. Agents Chemother. 50, 2762–2771. 10.1128/AAC.00320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiler C.; Fisher A. C.; Boock J. T.; Marrichi M. J.; Wright L.; Schmidpeter P. A. M.; Blankenfeldt W.; Pavelka M.; Delisa M. P. (2013) Directed evolution of Mycobacterium tuberculosis beta-lactamase reveals gatekeeper residue that regulates antibiotic resistance and catalytic efficiency. PLoS One 8, e73123 10.1371/journal.pone.0073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores A. R.; Parsons L. M.; Pavelka M. S. (2005) Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology (London, U. K.) 151, 521–532. 10.1099/mic.0.27629-0. [DOI] [PubMed] [Google Scholar]
- Hazra S.; Kurz S. G.; Wolff K.; Nguyen L.; Bonomo R. A.; Blanchard J. S. (2015) Kinetic and structural characterization of the interaction of 6-methylidene penem 2 with the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 54, 5657–5664. 10.1021/acs.biochem.5b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J. E.; Tremblay L. W.; Boshoff H. I.; Barry C. E.; Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L. W.; Fan F.; Blanchard J. S. (2010) Biochemical and structural characterization of Mycobacterium tuberculosis beta-Lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49, 3766–3773. 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L. V.; Xu H.; Blanchard J. S. (2010) Structures of the Michaelis complex (1.2 Å) and the covalent acyl intermediate (2.0 Å) of cefamandole bound in the active sites of the Mycobacterium tuberculosis beta-lactamase K73A and E166A mutants. Biochemistry 49, 9685–9687. 10.1021/bi1015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz S. G.; Hazra S.; Bethel C. R.; Romagnoli C.; Caselli E.; Prati F.; Blanchard J. S.; Bonomo R. A. (2015) Inhibiting the beta-lactamase of Mycobacterium tuberculosis (Mtb) with novel boronic acid transition-state inhibitors (BATSIs). ACS Infect. Dis. 1, 234–242. 10.1021/acsinfecdis.5b00003. [DOI] [PubMed] [Google Scholar]
- Soroka D.; Ourghanlian C.; Compain F.; Fichini M.; Dubee V.; Mainardi J.-L.; Hugonnet J.-E.; Arthur M. (2016) Inhibition of beta-lactamases of mycobacteria by avibactam and clavulanate. J. Antimicrob. Chemother. 72, 1081–1088. 10.1093/jac/dkw546. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. (2013) How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. 10.1107/S0021889897006766. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Totir M. A.; Padayatti P. S.; Helfand M. S.; Carey M. P.; Bonomo R. A.; Carey P. R.; van den Akker F. (2006) Effect of the inhibitor-resistant M69V substitution on the structures and populations of trans-enamine beta-lactamase intermediates. Biochemistry 45, 11895–11904. 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzin A. P.; Nukaga M.; Nukaga Y.; Hujer A.; Bonomo R. A.; Knox J. R. (2001) Inhibition of the SHV-1 beta-lactamase by sulfones: Crystallographic observation of two reaction intermediates with tazobactam. Biochemistry 40, 1861–1866. 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]
- Padayatti P. S.; Helfand M. S.; Totir M. A.; Carey M. P.; Hujer A. M.; Carey P. R.; Bonomo R. A.; van den Akker F. (2004) Tazobactam forms a stoichiometric trans-enamine intermediate in the E166A variant of SHV-1 beta-lactamase: 1.63 angstrom crystal structure. Biochemistry 43, 843–848. 10.1021/bi035985m. [DOI] [PubMed] [Google Scholar]
- Sun T.; Bethel C. R.; Bonomo R. A.; Knox J. R. (2004) Inhibitor-resistant class A beta-lactamases: Consequences of the Ser130-to-Gly mutation seen in apo and tazobactam structures of the SHV-1 variant. Biochemistry 43, 14111–14117. 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- Pozzi C.; Di Pisa F.; De Luca F.; Benvenuti M.; Docquier J. D.; Mangani S. (2018) Atomic-resolution structure of a Class C -lactamase and its complex with avibactam. ChemMedChem 13, 1437–1446. 10.1002/cmdc.201800213. [DOI] [PubMed] [Google Scholar]
- Lahiri S. D.; Mangani S.; Durand-Reville T.; Benvenuti M.; De Luca F.; Sanyal G.; Docquier J.-D. (2013) Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: Avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC beta-lactamases. Antimicrob. Agents Chemother. 57, 2496–2505. 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungler A.; Momin A.; Diederichs K.; Arold S. T. (2016) ContaMiner and ContaBase: a webserver and database for early identification of unwantedly crystallized protein contaminants. J. Appl. Crystallogr. 49, 2252–2258. 10.1107/S1600576716014965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel G.; Buhrman G.; Mattos C. (2012) Shift in the equilibrium between on and off states of the allosteric switch in Ras-GppNHp affected by small molecules and bulk solvent composition. Biochemistry 51, 6114–6126. 10.1021/bi300509j. [DOI] [PubMed] [Google Scholar]
- Kiema T. R.; Harijan R. K.; Strozyk M.; Fukao T.; Alexson S. E. H.; Wierenga R. K. (2014) The crystal structure of human mitochondrial 3-ketoacyl-CoA thiolase (T1): insight into the reaction mechanism of its thiolase and thioesterase activities. Acta Crystallogr., Sect. D: Biol. Crystallogr. 70, 3212–3225. 10.1107/S1399004714023827. [DOI] [PubMed] [Google Scholar]
- Lin J. S.; Pozharski E.; Wilson M. A. (2017) Short carboxylic acid-carboxylate hydrogen bonds can have fully localized protons. Biochemistry 56, 391–402. 10.1021/acs.biochem.6b00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch L. B.; Martini V. P.; Iulek J.; Huggan J. K.; Lee J. H.; Gibson C. L.; Smith T. K.; Suckling C. J.; Hunter W. N. (2010) Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the Leishmaniases. J. Med. Chem. 53, 221–229. 10.1021/jm901059x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M. J.; Li Y.; Leichert L. I. O.; Xu Z. H.; Jakob U. (2015) Does the transcription factor NemR use a regulatory sulfenamide bond to sense bleach?. Antioxid. Redox Signaling 23, 747–754. 10.1089/ars.2015.6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodkey E. A.; Drawz S. M.; Sampson J. M.; Bethel C. R.; Bonomo R. A.; van den Akker F. (2012) Crystal structure of a preacylation complex of the beta-lactamase inhibitor sulbactam bound to a sulfenamide bond-containing thiol-beta-lactamase. J. Am. Chem. Soc. 134, 16798–16804. 10.1021/ja3073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Groen A.; Lemeer S.; Jans A.; Slijper M.; Roe S. M.; den Hertog J.; Barford D. (2007) Reversible oxidation of the membrane distal domain of receptor PTP alpha is mediated by a cyclic sulfenamide. Biochemistry 46, 709–719. 10.1021/bi061546m. [DOI] [PubMed] [Google Scholar]
- Lee C.; Shin J.; Park C. (2013) Novel regulatory system nemRA-gloA for electrophile reduction in Escherichia coli K-12. Mol. Microbiol. 88, 395–412. 10.1111/mmi.12192. [DOI] [PubMed] [Google Scholar]
- Gray M. J., Wholey W. Y., and Jakob U. (2013) Bacterial responses to reactive chlorine species. In Annu. Rev. Microbiol. (Gottesman S., Ed.), Vol. 67, pp 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. P. A.; Aplin R. T.; Schofield C. J. (1996) Inhibition of TEM-2 beta-lactamase from Escherichia coli by clavulanic acid: Observation of intermediates by electrospray ionization mass spectrometry. Biochemistry 35, 12421–12432. 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- Wang X. J.; Minasov G.; Shoichet B. K. (2002) The structural bases of antibiotic resistance in the clinically derived mutant beta-lactamases TEM-30, TEM-32, and TEM-34. J. Biol. Chem. 277, 32149–32156. 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- Helfand M. S.; Bethel C. R.; Hujer A. M.; Hujer K. M.; Anderson V. E.; Bonomo R. A. (2003) Understanding resistance to beta-lactams and beta-lactamase inhibitors in the SHV beta-lactamase - Lessons from the mutagenesis of Ser-130. J. Biol. Chem. 278, 52724–52729. 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- Lewandowski E. M.; Lethbridge K. G.; Sanishvili R.; Skiba J.; Kowalski K.; Chen Y. (2018) Mechanisms of proton relay and product release by Class A beta-lactamase at ultrahigh resolution. FEBS J. 285, 87–100. 10.1111/febs.14315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.