

Abstract
Clathrin-mediated endocytosis (CME) is the major endocytic pathway in mammalian cells. It is responsible for the uptake of transmembrane receptors and transporters, for remodeling plasma membrane composition in response to environmental changes, and for regulating cell surface signaling. CME occurs via the assembly and maturation of clathrin-coated pits that concentrate cargo as they invaginate and pinch off to form clathrin-coated vesicles. In addition to the major coat proteins, clathrin triskelia and adaptor protein complexes, CME requires a myriad of endocytic accessory proteins and phosphatidylinositol lipids. CME is regulated at multiple steps—initiation, cargo selection, maturation, and fission—and is monitored by an endocytic checkpoint that induces disassembly of defective pits. Regulation occurs via posttranslational modifications, allosteric conformational changes, and isoform and splice-variant differences among components of the CME machinery, including the GTPase dynamin. This review summarizes recent findings on the regulation of CME and the evolution of this complex process.
Keywords: dynamin, endocytic accessory proteins, signaling, AP2, adaptor protein-2, evolution, endocytic checkpoint
INTRODUCTION
Clathrin-mediated endocytosis (CME) is the major pathway for concentrative uptake of surface receptors (i.e., cargo) and their bound ligands, including nutrient, adhesion, and signaling receptors. It also regulates the surface abundance and hence activity of transmembrane transporters (e.g., for amino acids, glucose, ions, etc.). Thus, CME plays an essential role in controlling cell–cell and cell–substrate interactions, intercellular signaling, and cellular homeostasis. CME is by far the best studied and mechanistically understood endocytic pathway, because of (a) its concentrative properties that make biochemical measurements highly sensitive, (b) the ease of its visualization by electron and fluorescent live-cell microscopy, (c) the multiple interactions between protein partners that can be dissected and manipulated, and (d) its relevance in human health and disease.
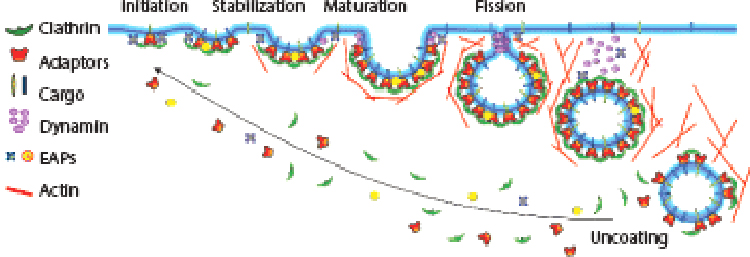
CME occurs through clathrin-coated pits (CCPs) formed by the assembly of the major coat proteins, clathrin triskelia, which comprises three clathrin heavy chains (CHCs), with tightly associated clathrin light chains (CLCs), and the adaptor protein-2 (AP2), a heterotetramer consisting of α, β2, μ2, and σ2 subunits. CME can be dissected into four stages (1–3): (a) initiation, (b) stabilization, (c) maturation, and (d) membrane fission (Figure 1). In subsequent steps, the released clathrin-coated vesicles (CCVs) are rapidly uncoated, undergo multiple homotypic fusion events, and ultimately deliver their cargo to early endosomes. In addition to the major coat proteins, CME requires the activity of numerous endocytic accessory proteins (EAPs) (Table 1) that function as scaffolds, cargo recruiters, membrane curvature generators and sensors, as well as CME regulators. Finally, the large GTPase dynamin, which is recruited at low levels to nascent CCPs and regulates CCP initiation and maturation, assembles into short helical rings around the necks of deeply invaginated CCPs to catalyze membrane fission. Dynamin’s activity as a fission GTPase at late stages of CME has been extensively studied and reviewed elsewhere (4–6). Later in this review, we discuss dynamin’s less appreciated, but equally important, role in regulating early stages of CME.
Figure 1.

Clathrin-mediated endocytosis is regulated by multiple factors at multiple stages. Clathrin-mediated endocytosis is a multistage process involving the initiation and stabilization of nascent CCPs, maturation and curvature generation, and finally dynamin-catalyzed fission. Each step is regulated by multiple inputs. Abbreviations: AAK1, adaptor associated kinase 1; AP2, adaptor protein-2; EAPs, endocytic accessory proteins; PIP, phosphatidylinositol phosphate. Figure modified from Schmid (107).
Table 1.
Key EAPs and their effect on CCPs
 |
Function | Effect on CCP | ||||
|---|---|---|---|---|---|---|
| Major coat proteins | CHC | Coat | KD ↓ initiation | |||
| CLCa | Coat | KD ↓ initiation | ||||
| CLCb | Coat | KD ↓ initiation | ||||
| AP2 | Adaptor | KD ↓ initiation | ||||
| Dyn1 | GTPase | KO no effect; A: ↑ initiation and maturation |
||||
| Dyn2 | GTPase | KD ↓ initiation | ||||
| Easy arriving, pioneer proteins | FCHo1/2 | Scaffold | KD ↓ initiation | |||
| Eps15 | Scaffold | KD ↓ maturation | ||||
| Intersectin | Scaffold | KD ↓ stabilization and maturation | ||||
| NECAP | Adaptor | KD ↓ maturation | ||||
| CALM | Adaptor | KD ↓ maturation | ||||
| Epsin | Adaptor | KD ↓ maturation | ||||
| CCP maturation and fission | Amphiphysin | Curvature | ND | |||
| Endophilin | Curvature | KD ↑ abortive | ||||
| N-WASP | Actin | ND | ||||
| Cortactin | Actin | ND | ||||
| Myosin 1E | Actin | ND | ||||
| Hip1R | Actin | ND | ||||
| SNX9 | Scaffold | KD ↓ maturation | ||||
| Synaptojanin | Lipids | KD ↑ maturation | ||||
| PI3KC2α | Lipids | KD ↓ maturation | ||||
| PIP5K | Lipid | KD ↓ maturation OX ↑ initiation and maturation |
||||
| Uncoating | GAK | Kinase | KD ↑ abortive | |||
| Hsc70 | ATPase | ND | ||||
| OCRL | Lipids | M: ↑ CCVs ↑ U-shaped CCPs |
||||
The effect on CCPs is summarized from several studies described herein. The graphic is adapted from Schmid (107). Abbreviations: A, activation; AP2, adaptor protein-2; CALM, clathrin assembly lymphoid myeloid leukemia; CCP, clathrin-coated pit; CHC, clathrin heavy chain; CLC, clathrin light chain; Dyn, dynamin; EAPs, endocytic accessory proteins; Eps15, EGF-receptor phosphorylation substrate; FCHo1/2, Fer/Cip4 homology domain-only proteins 1/2; GAK, cyclin G associated kinase; Hip1R, Huntingtin interacting protein-1 related; Hsc70, heat shock protein 70 kD; KD, knockdown; KO, knockout; M, mutation; ND, not determined; NECAP, adaptin-ear-binding coat-associated protein; N-WASP, neuralWiskott-Aldrich syndrome protein; OCRL, oculocerebrorenal Lowe syndrome protein; OX, overexpression; PI3KC2α, phosphatidylinositol 3-kinase C2α PIP, phosphatidylinositol phosphate; SNX9, sorting nexin 9.
Because most earlier studies of CME had focused on constitutively internalized nutrient receptors, such as the transferrin receptor (TfnR) and low-density lipoprotein receptor (LDLR), and used bulk and indirect biochemical assays that measure the intracellular accumulation of cargo, CME has until recently been considered a constitutive process. That is, like the circulation of buses according to a set time table irrespective of the number and destinations of passengers, CCVs were thought to form at a fixed rate with receptors and their ligands entering growing CCPs, potentially based on “tickets” that may triage cargo to different pit classes. However, on the basis of the pioneering work of James Keen and colleagues (7), who first imaged CCP dynamics using green fluorescent protein (GFP)-tagged CLCa (GFP-CLCa), and Christien Merrifield, Wolfhard Almers, and colleagues (8), who first applied total internal reflection fluorescence microscopy (TIRFM), researchers are now able to directly monitor CCP initiation, growth, and budding in live cells. Most striking in these early studies were the observed heterogeneity in CCP lifetimes and the finding that a substantial fraction of nascent CCPs failed to mature and aborted, without concomitant cargo uptake (9–11). Together, these and other data, which are the subject of this review, establish that CME is indeed a highly regulated and cargo-driven process. While much has been learned in the past decade, much remains to be discovered regarding the signaling pathways and effectors that regulate CME and their physiological implications.
VISUALIZING AND MEASURING CME
Studies of the regulation of CME have depended on the ability to directly measure the progression from CCP initiation to stabilization, maturation, and finally scission via live-cell microscopy. Biochemical or morphological approaches that measure the intracellular accumulation of receptor-bound ligands cannot discriminate among shifts in the kinetics of one or more of these stages. Moreover, the rate of intracellular accumulation of receptor-bound ligands is not a direct measure of the rate of CCV formation, but instead also depends on (a) the number of receptors on the cell surface, which for many receptors changes depending on their relative rates of uptake and recycling, (b) the effective concentration of the receptor in CCPs, and (c) the rate of recycling of receptor-ligand complexes, which can in some situations be as rapid as the rate of uptake (12).
Among the live-cell imaging approaches for visualizing CME, spinning disk confocal fluorescence microscopy (SDCFM) has been the method of choice for quantifying numbers of molecules recruited to CCPs, as excitation is uniform throughout the illuminated area. However, a major limitation of SDCFM is the highly inefficient use of light inherent to this technology. Consequently, SDCFM requires increased sample illumination for fluorescence excitation, which is accompanied by phototoxicity and a higher rate of photobleaching. Furthermore, the somewhat limited optical sectioning of samples leads to lower signal-to-noise ratios, which renders detection of the early stages of CCP formation less efficient while increasing the detection rate of nonendocytic clathrin-coated structures (CCSs). These drawbacks have been overcome by TIRFM, which itself has some limitations. First, it relies on flat cells that firmly attach to, and spread on, glass: weakly attaching or nonadherent cells are thus less well suited. Nonetheless, TIRFM imaging has been successfully accomplished even in yeast by squeezing cells between the coverslip and glass slide (13, 14). Second, its physiological relevance can be challenged, given that cells are cultured on an artificially stiff surface, which in turn can lead to differences in membrane tension between the ventral and dorsal plasma membrane (PM) (15). However, direct comparisons of the dynamics of diffraction-limited CCPs on the ventral and dorsal surfaces of cells have not shown major differences (9, 16). Moreover, quantitative analysis of molecule numbers is complicated by the fact that the fluorescence excitation intensity decays exponentially as it enters the cell. Hence, signal intensity varies exponentially with the distance of the molecule from the adherent cell surface. Despite these drawbacks, TIRFM has been the imaging modality of choice (10, 17–22). Importantly, the full potential of live-cell imaging relies on the ability to quantitatively analyze CCP dynamics. Early studies manually analyzed CCP growth and lifetimes and hence focused on only a small subset of stereotypically behaving CCPs (8, 23), obscuring their heterogeneous behavior. The development of algorithms (e.g., cmeAnalysis) to comprehensively and accurately detect and track all diffraction-limited CCPs within movies (24, 25) has revealed their heterogeneous dynamic behaviors (26) and enabled accurate determination of the rates of CCP initiation and growth as well as the lifetimes of all CCPs.
Comprehensive analyses of CCP dynamics enabled detection of a subpopulation of short-lived and relatively dim CCPs (9, 10). The fact that their numbers were affected by cargo concentration (9, 10) and by small interfering RNA (siRNA-mediated knockdown of a subset of EAPs) (27) led to the suggestion that these abortive events were reflective of an active fidelity-monitoring process (i.e., an endocytic checkpoint) (10, 27). Using an elegant approach that directly visualizes membrane scission via the sequestration of a chimeric TfnR fused to pH-sensitive superecliptic pHluorin, subsequent studies confirmed that short-lived (<20 s) CCPs fail to undergo scission and to take up cargo (11). However, the existence of abortive CCPs remains controversial, as other studies have failed to detect them (28, 29). Direct comparisons (25) demonstrated that this difference likely reflects the substoichiometric incorporation of fluorescent subunits into clathrin triskelia or AP2 complexes (28, 29), leading to lower sensitivity of detection and inaccuracies in tracking and gap closing (i.e., computational compensation for transient loss of fluctuating weak signals) (25). These hurdles in computer-based image analysis can be overcome by the use of robust fiduciary markers for CCPs (25, 30).
While not discussed herein, also evident in TIRFM movies of CCP dynamics are a subpopulation of larger, more static CCSs, referred to as clathrin plaques (31). Their numbers vary greatly depending on cell type and plating conditions, being especially prevalent in fibroblasts. Clathrin plaques have been implicated in cell adhesion (26, 32–34), as templates for multiple rounds of CCV formation (referred to as nonterminal events) (11, 26), and as signaling platforms (35). They can also serve as platforms for actin-driven endocytosis of large particles (36) or under conditions of high membrane tension (15). This actin-driven membrane invagination is likely most akin to the mode of endocytosis in yeast, in which clathrin coats are restricted to the base of endocytic invaginations and provide a scaffold against which actin assembly can drive invagination (37).
STAGE-SPECIFIC REGULATION OF CME
The ability to measure discrete stages of CCP assembly, maturation, and scission has revealed that each of these stages is subject to regulation, as described in the following sections.
Regulation of Receptor Sorting into CCPs
Transmembrane proteins are recruited into CCPs via short endocytic sorting determinants located within their cytosolic domains (reviewed in 38, 39). Tyrosine-based sequences are the most common and are used by a wide range of transmembrane proteins, including TfnRs, the classical CME cargo that utilize a YXXΦ motif (where Φ is any hydrophobic residue), and members of the LDLR family that bear a [FY]XNPX[YF] motif. These short sorting signals bind directly to the μ2 subunit of AP2 (40) or to other adaptor proteins that encode phosphotryosine binding (PTB) domains (41, 42). Constitutively internalized receptors, such as TfnRs and LDLRs, are clustered in CCPs and are internalized irrespective of ligand occupancy (38, 43, 44).
For many other transmembrane cargo molecules, their interactions with and clustering in CCPs are regulated by ligand binding and/or posttranslational modification. For example, the large family of G protein-coupled receptors (GPCRs) utilizes β-arrestins and/or epsinl as adaptors. Posttranslational modifications of agonist-activated GPCRs (i.e., phosphorylation of serine/threonine residues or ubiquitination, respectively) are recognized by these adaptors to target these receptors to CCPs (45–47). Similarly, despite bearing tyrosine-based sorting motifs equivalent to those found on constitutively internalized cargo, epidermal growth factor receptors (EGFRs) become concentrated into CCPs and are internalized only upon ligand-induced noncovalent dimerization. Formation of dimers leads to receptor autophosphorylation to generate docking sites for multiple SH2 domain–containing downstream effectors. One of these effectors is c-Cbl, a ubiquitin ligase family member that associates with EGFRs, either directly or indirectly via the scaffold protein Grb2 (48, 49), leading to monoubiquitination of the receptor at multiple sites (50), recruitment of the EAP Eps15 (51), and accelerated internalization (52).
Sorting into CCPs can also be negatively regulated. For instance, the protein kinase p56lck interacts with the T cell antigen receptor component CD4 to prevent CD4 loading into CCPs. When T cells are activated by an antigen, CD4 dissociates from p56lck and is recruited to CCPs via a cytoplasmic dileucine motif activated by phosphorylation of a nearby serine residue (53). FcRII-2B, a splice variant of the immunoglobulin Fc receptor, bears an insert in its cytoplasmic domain that links the receptor to the actin cytoskeleton and prevents its recruitment to CCPs (54). Thus, many diverse mechanisms regulate receptor sorting into CCPs.
Irrespective of the mode of cargo recruitment, it has become increasingly clear that cargo–adaptor complexes—either directly or via their multiple interaction partners—can influence multiple early steps of CME, including CCP initiation, CCP maturation, CCP budding, as well as downstream cargo sorting itineraries (9, 10, 55–58).
Regulation of CCP Initiation
The first step of a biochemical pathway is often the focal point for regulation. The same is true for CME. Multiple factors and inputs regulate CCP initiation, many of which impinge on AP2.
Allosteric regulation of AP2 controls CCP initiation, growth, and stabilization.
The heterotetrameric AP2 adaptor complex binds the PM-enriched phosphatidylinositol lipid, PI4,5P2, as well as the endocytic sorting motifs on cargo. AP2, which triggers clathrin assembly and recruits EAPs, is the central node for regulation of CCP initiation (3, 38). AP2 consists of a core comprising the N-terminal domains of the α- and β2-adaptins in complex with the μ2 and σ2 subunits. Long flexible linkers, referred to as hinge regions, connect the C-terminal appendage domains (ADs) of α- and β2-adaptins to the core (59–61). The αAD, and to a lesser extent the β2AD, binds to and recruits numerous EAPs (62–64). The principal binding site for clathrin is encoded within the β2 hinge (65), although a second site exists in the β2AD (66, 67). The μ2 subunit binds tyrosine- and dileucine-based sorting motifs at different sites (61). The core domain has three PI4,5P2 binding sites encoded on the α, β2, and μ2 subunits (60). Quantitative live-cell single-molecule TIRFM imaging has shown that CCP assembly is initiated when two AP2 complexes begin to recruit clathrin triskelia (68) and that siRNA-mediated depletion of AP2 dramatically reduces the number of CCPs at the membrane (69, 70). These findings illustrate the importance of AP2 in CCP initiation and stabilization.
Elegant structural studies of the AP2 core (59, 60) and more recently of the core/β2 hinge region (71) have revealed a series of allosterically regulated AP2 conformational changes that provide the means for complex regulation of CCP initiation, stabilization, and growth (56) (Figure 2a). Cytosolic AP2 exists in a closed conformation, in which the clathrin binding site is buried by interactions between the β2 hinge and the core (60). The PI4,5P2 and cargo binding sites on the μ2 subunit are also buried in this conformation. The interaction of surface-exposed binding sites on both the α- and β2-adaptin with PM-enriched PI4,5P2 is required for CCP nucleation and triggers an allosteric conformational change to an open conformation that exposes the clathrin binding site on the β2 hinge as well as the PI4,5P2 and cargo binding sites of μ2 (56, 60, 71) (Figure 2b). The latter two interactions are required to stabilize nascent CCPs (56). Indeed, overexpression (10) or clustering of TfnR (55) increases the fraction of productive CCPs at the expense of abortive events.
Figure 2.

The central role of AP2 in regulating CCP initiation and stabilization. (a) The allosteric regulation of AP2 conformational changes controls early steps in CME, including: (❶) the recruitment of cytosolic AP2 in the closed conformation to the PM, where (❷) PI4,5P2 binding releases the β2 hinge to nucleate clathrin assembly; (❸) cargo and PI4,5P2 binding to the μ2 subunit, as well as interactions with EAPs, trigger the open conformation to stabilize the growing, nascent CCP and prevent early abortive events; (❹) sustained PI4,5P2 interactions with α, β2, and μ2 binding sites, as well as clathrin-stimulated AAK1 phosphorylation of μ2, supports further cargo recruitment, CCP growth, and (❺) maturation. Boxes correspond to sequential AP2 conformational changes enlarged in panel b. (b) Sequential interactions between PI4,5P2 and α, β2, and μ2 subunits, as well as with pioneer EAPs, trigger and stabilize conformational changes that activate AP2. AAK1, which phosphorylates μ2 to stabilize the open conformation, is activated by assembled clathrin, thus providing a positive feedback loop during CCP maturation. Abbreviations: AAK1, adaptor associated kinase 1; AP2, adaptor protein-2; CALM, clathrin assembly lymphoid myeloid leukemia; CCP, clathrin-coated pit; CME, clathrin-mediated endocytosis; EAPs, endocytic accessory proteins; Eps15, EGF-receptor substrate 15; FCHo, Fer/Cip4 homology domain-only protein; NECAP, adaptin-ear-binding coat-associated protein; PI4,5P2, phosphatidylinositol lipid; PM, plasma membrane.
Endocytic accessory proteins allosterically regulate AP2 and function redundantly in CCP initiation.
Several EAPs (Table 1), including FCHo1/2 (Fer/Cip4 homology domain-only proteins 1/2), Eps15/Eps15R, intersectin, NECAP (adaptin-ear-binding coat-associated protein), and CALM (clathrin assembly lymphoid myeloid leukemia, also known by its gene name, PICALM), are recruited to CCPs at the earliest stages of initiation through multimeric, typically low-affinity interactions between each other and AP2 (11, 72, 73). This subset of EAPs, now collectively referred to as endocytic pioneers (74, 75), initiate and/or stabilize AP2 clusters at the PM and allosterically regulate the AP2 conformational state (Figure 2b) to enhance clathrin recruitment and CCP stabilization and growth (74–76). For example, the curvature-generating, F-BAR (Bin/Amphiphysin/Rvs) domain-containing FCHo1/2 proteins (also known as municins) enhance the efficiency of AP2-mediated CCP initiation by stabilizing its open conformation (74, 76). CALM and its neuron-enriched isoform, AP180 (assembly/adaptor protein 180), function as adaptor proteins for SNAREs (77) that must be incorporated into CCVs to ensure their subsequent targeting and fusion along the endocytic pathway. Depletion of CALM increases the turnover of abortive CCPs and reduces CCP maturation, indicative of an early function during CCP stabilization and maturation (27, 78). The multidomain protein, NECAP, appears to function in coordinating CCP initiation and growth through regulated interactions near the clathrin binding site on the β2 hinge as well as through sequential and synergistic interactions with the αAD, FCHo, and CALM (79). Importantly, these proteins function at substoichiometric levels compared with AP2 (74), presumably by potentiating closed to open conformational changes that are later stabilized by PI4,5P2 and cargo interactions.
Dissecting the function of these interactions has been difficult as they are low affinity and, at least in part, redundant. Thus, genetic studies in Caenorhabditis elegans showed that the requirement for FCHo family proteins could be circumvented by mutations in AP2 that destabilize its closed conformation (68, 74, 76). Similarly, overexpression of FCHo overcomes the effects of NECAP depletion (79). Interestingly, a similar situation exists in yeast whereby clathrin- and actin-dependent bulk endocytosis continues in cells even after deletion of all seven early arriving EAPs, including homologs of FCHo, Eps15, and CALM (80); however, selective cargo uptake is impaired. Individually targeting several of these early EAPs to the PM was sufficient to nucleate ectopic endocytic events, indicating considerable flexibility in the mechanisms of CCP initiation (80). Together, these studies suggest that CCP initiation is unlikely to be orchestrated by a stereotypical and linear series of molecular interactions. Nonetheless, this flexibility may provide multiple, potentially cargo-selective and cell-type-specific nodes for regulation of this critical early step.
In many cell types, a residual clathrin/AP2 patch of variable size remains after a CCV pinches off (Figure 3). These are designated nonterminal budding events (11, 26), and the residual CCS can nucleate multiple rounds of CCV formation. Furthermore, CCV formation at so-called hot spots—small membrane areas that are favorable for CCP formation—can be less dependent on bulk levels of AP2 and PI4,5P2 (81), suggesting that residual clathrin patches and hot spots are more potent CCP nucleation sites.
Figure 3.

The endocytic checkpoint hypothesis. A hypothetical endocytic checkpoint monitors the fidelity of CCP maturation. Deficiencies in several endocytic accessory proteins lead to decreased efficiency of CCP maturation and increased rates of turnover of abortive CCPs. Small interfering RNA-mediated knockdown of dynamin-2 decreases the rate of abortive CCP turnover. Based on these data, we speculate that the properties of nascent CCPs, such as coat assembly, curvature generation, and cargo concentration, are monitored by SH3 domain-containing dynamin binding partners that also interact with coat components, cargo, and/or sense curvature and kinetically control dynamin assembly. Together, these interactions function to monitor the fidelity of CCP maturation. Much remains to be done to test this hypothesis and to identify components of the endocytic checkpoint apparatus and the mechanisms to sense and turnover aberrant CCP intermediates. Abbreviations: AD, appendage domain; AP2, adaptor protein-2; CALM, clathrin assembly lymphoid myeloid leukemia; CCP, clathrin-coated pit; CCV, clathrin-coated vesicle; CLCs, clathrin light chains; SH3, Src-homology domain 3; TfnR, transferrin receptor. Figure modified from Schmid (107).
Phosphorylation stabilizes the open conformation of AP2.
In addition to protein and lipid interactions, the open conformation of AP2 can be stabilized by phosphorylation. AAK1 (adaptor-associated kinase 1), a member of the Numb-associated kinase (NAK) family that includes GAK (cyclin G-associated kinase), BIKE/BMP2K (BMP2-inducible kinase), as well as the Ark/Prk kinases in yeast (82), phosphorylates the μ2 subunit and enhances the ability of AP2 to bind membranes (1, 83). AAK1 phosphorylates Thr156 (84), which is located within a flexible linker that is unstructured in the closed conformation but structured in the open conformation. Phosphorylation is thought to stabilize the μ2 subunit in its open conformation, unmasking its PI4,5P2 and cargo binding sites (60). AAK1 also binds to and is activated by assembled clathrin (85), providing the possibility of a positive feedback loop to enhance AP2-driven clathrin assembly (Figure 2b).
Interestingly, siRNA-mediated knockdown of AAK1 has little or no effect on TfnR uptake, perhaps owing to functional redundancy with other NAK family members. However, dependence on AAK1 activity becomes more acute in cells expressing a μ2 mutant defective in PI4,5P2 binding (56). This finding suggests that AAK1 might sense and compensate for factors that perturb AP2 activation and hence CCP maturation.
Regulating CCP Growth and Maturation
Live-cell microscopy in non-neuronal cells has revealed that productive CCPs—those that pinch off from the PM bearing cargo receptors—vary in lifetime from 20 s to >120 s [the rate of CME at the synapse can be orders of magnitude faster (86)]. This range does not appear to be due to differential rates of either clathrin assembly or cargo recruitment, as on average, clathrin (87) and the constitutively internalized TfnR (55) reach maximum levels at CCPs well before scission. Moreover, clustering of TfnR increases the rate and extent of CCP loading but not the rate of CCP maturation (55). Thus, the factors that determine the rate of maturation of individual CCPs remain unknown. Key events occurring during CCP maturation are cargo loading, curvature generation, and phosphatidylinositol phosphate (PIP) conversion (from the PI4,5P2-enriched composition of the PM to the PI3P-enriched composition of early endosomes). As discussed in this section, the mechanisms of curvature generation at CCPs, the role of PIPs, and the role of cargo in regulating CCP maturation remain areas of active research.
Curvature generation during CCP maturation.
Clathrin triskelia are bent, three-legged structures that assemble into polygonal lattices whereby a triskelion sits at each vertex and extends its proximal and distal legs along two adjacent edges of the lattice in three directions. In this way, each edge consists of two proximal legs from adjacent vertices and two distal legs from neighboring vertices (88). Triskelia can flexibly assemble into either hexagons or pentagons. If the polygonal lattice consists only of hexagons, it will be flat. At least 12 pentagons must be incorporated to generate a closed coat. Clathrin and adaptor proteins spontaneously assemble in solution into closed clathrin coats; hence, it has been assumed that clathrin assembly itself can drive curvature formation at CCPs. Indeed, targeting clathrin assembly onto liposomes is sufficient to generate curved coated pits (89, 90). Given the extensive interactions that occur between four triskelia along each edge of the lattice, a predominant model suggests that curvature is introduced by de novo assembly of pentagons at the edges of a growing CCP, rather than by the presumed energetically unfavorable conversion of hexagons to pentagons at some later stage (88). However early quick-freeze deep-etch micrographs of CCPs revealed a diverse collection of curved and flat lattices, including the emergence of curved CCPs from flat lattices and the appearance of heptagons suggested to be intermediates in the conversion of hexagons to pentagons (91). Thus, the precise role of clathrin in curvature generation at CCPs remains unresolved.
Recent studies have shed new light on the mechanism of curvature generation at CCPs. By combining correlative light and quantitative electron tomographic experiments, Kaksonen, Briggs, and colleagues (92) have shown that the clathrin-coated area of pits does not increase with the degree of curvature. Thus, the authors conclude that planar clathrin coats with sufficient area to form a mature CCV are first assembled and subsequently mature into invaginated structures. FRAP (fluorescence recovery after photobleaching) experiments showed rapid rates of clathrin exchange (t1/2 ~2 s) at individual CCPs, even at late stages of maturation as determined by colocalization with dynamin-2. However, the degree of recovery plateaus at ~60%, indicating that only a subset of triskelia, potentially those at the edges, were dynamically exchanging. Importantly, the images used in these analyses were static structures that have been manually, albeit logically, ordered in a temporally related series of intermediates. Moreover, as genome-edited cells that express substoichiometric levels of fluorescently tagged CLCa were used to identify CCPs (92), early intermediates in CCP assembly may not have been detected. Thus, further experiments, ideally using live-cell TIRFM in combination with epifluorescence (epi/TIRFM) to directly and dynamically measure curvature generation at individual CCPs (17, 87), are needed to resolve this issue.
Using a completely different approach, work from our laboratories provides strong evidence that clathrin assembly per se is not sufficient to generate curved CCPs. Through siRNA-mediated knockdown and reconstitution, we replaced the full-length α subunit of AP2 with a C-terminally truncated mutant lacking the AD and hence defective in EAP recruitment. This ΔαAD mutant, which retains the PI4,5P2- and cargo-binding core as well as the clathrin-recruiting β2 subunit, is fully able to rescue CCP assembly as assessed by immunofluorescence; however, upon closer inspection by negative-stain electron microscopy of ripped-off PM fragments, most of the diffraction limited CCPs detected in ΔαAD cells corresponded to flat lattices (25). Live-cell epi/TIRFM revealed that these flat lattices turned over rapidly, within <20 s. Thus, these studies provide strong evidence that EAPs recruited to the αAD are required for efficient generation of curvature at CCPs. While curvature-generating N-BAR (BAR domains encoding an N-terminal amphipathic helix) or ENTH (epsin N-terminal homology) domain-containing EAPs are likely candidates (Table 1), it is also possible that EAPs directly regulate clathrin polymerization into pentagons versus hexagons. Further studies are needed to identify which EAPs are required and their role in curvature generation during CCP maturation.
PIP conversion during CCP maturation.
Organelle identity along the endocytic pathway is, in part, determined by the PIP composition of their limiting membrane: The PM is enriched in PI4,5P2 while early endosomes are enriched in PI3P (93). The interconversion of PIPs is regulated by lipid kinases and phosphatases that add or remove phosphate groups at positions 3, 4, or 5 in the inositol ring. Many components of the CME machinery selectively bind PI4,5P2, which is absolutely required for CCP initiation (94, 95), in part through the recruitment and allosteric activation of AP2, as described above. Paradoxically, however, the PI5-kinases that generate PI4,5P2 are not detected in CCPs (96); rather, PI5-phosphatases including synaptojanin and SHIP2 (inositol-5-phosphatase) are recruited to CCPs (96–98). Assuming they are active, these enzymes would produce PI4P, the preferred substrate for the lipid kinase, PI3KC2α (99), which directly binds to and is stimulated by clathrin (100). PI3KC2α, which is recruited to CCPs and required for their maturation (99), produces PI3,4P2 and may facilitate PIP progression to regulate CCP formation. However, in a more recent study using engineered fusion proteins that encode specific protein and PIP-lipid binding modules capable of sensing changes in PIP composition during CCP maturation by coincidence detection, bursts of PI4P and PI3P production were detected only after scission, when lipid diffusion throughout the PM was curtailed (101).
Initial studies by Haucke and colleagues (99) had also suggested that the PI3,4P2 generated at CCPs was required for recruitment of SNX9 (sorting nexin 9), which in turn recruits dynamin during late stages of CCP maturation. However, a more recent study has shown that SNX9, which encodes a dual lipid-binding PX-BAR domain and a protein-interacting SH3 (Src-homology 3) domain, instead functions to detect the temporal coincidence of PI3P, PI4,5P2, and membrane curvature (102) that occurs only transiently at the necks of deeply invaginated CCPs before fission. Membrane recruitment of SNX9 triggers local actin assembly by recruiting the Arp2/3-activating protein N-WASP (neural Wiskott-Aldrich syndrome protein). To reconcile with previous results (99), Gallop and colleagues (102) showed that the PI4 phosphatase, INNP4, converts PI3,4P2 to PI3P and is itself activated at highly curved membranes and required for actin-dependent CME. Unlike yeast, in which actin assembly is absolutely required for endocytosis, actin is required for CME in mammalian cells only under conditions of high membrane tension or when large cargo is being taken up (15, 36). Thus, one attractive interpretation is that SNX9 might function as a timer activated by a signaling cascade involving the accumulation of PI3P and PI4,5P2 at the curved necks of deeply invaginated but stalled CCPs to recruit actin when other factors are not sufficient to drive vesicle detachment (102). This hypothesis might be more in line with the recent studies by Kirchhausen and colleagues (101) described above. Altogether, the evidence strongly suggests a critical role for PIP conversion in regulating CCP initiation, maturation, fission, and post-fission events.
A role for cargo, in particular signaling receptors, in regulating CCP maturation.
An elegant series of studies on GPCR endocytosis has provided strong evidence that endocytic cargo, in particular signaling receptors, can control the rate of maturation of the CCPs in which they reside. GPCRs are by far the largest class of signaling receptors. The binding of small-molecule agonists triggers GPCR clustering and conformational changes that activate intracellular trimeric G proteins. Signaling is terminated by phosphorylation of GPCRs and the recruitment of β-arrestins, which serve as adaptors that target GPCRs to CCPs (103). The first evidence that ligand-activated GPCRs cluster in biochemically and functionally distinct CCPs was provided by von Zastrow and colleagues (104). Subsequent studies showed that as yet uncharacterized interactions between PDZ-domain ligands encoded at the C terminus of a subset of GPCRs (e.g., the β1 and β2 adrenergic receptors) and the actin cytoskeleton were both necessary and sufficient to delay CCP maturation and the recruitment of dynamin (57). An even more complex role for ubiquitination in the cargo-selective control of CCP maturation was revealed in studies of the μ-opioid neuropeptide receptor, MOR (46). MOR is constitutively ubiquitinated in resting cells. Upon ligand activation, MOR is further ubiquitinated and recruited to CCPs that mature more slowly than the bulk population. Mutating all cytoplasmic lysine residues in MOR to prevent ubiquitination further delays CCP maturation, whereas restoring lysine residues (Lys94/96) and ubiquitination in the first cytoplasmic loop of the seven transmembrane receptor triggers more rapid CCP maturation. The authors suggest that ubiquitination at Lys94/96 releases a brake on CCP maturation and provide evidence that this more rapid CCP maturation requires the ubiquitin-dependent recruitment of epsin1. Given the diversity of GPCRs, it is likely that other mechanisms of GPCR-regulated CCP maturation remain to be discovered.
A second example of signaling receptors selectively regulating their own CME and hence signaling was recently provided in studies of TRAIL (tumor necrosis factor α-related apoptosis-inducing ligand)-induced uptake of death receptors (DRs) (105). While uptake of TRAIL–DR requires both clathrin and AP2, surprisingly, CME of TRAIL–DR did not require the ubiquitously expressed isoform of dynamin, Dyn2, but instead required Dyn1, previously considered a neuron-specific isoform. In contrast, and as expected, CME of TfnR in the same cells required Dyn2, but not Dyn1. As described in more detail below, at the synapse, Dyn1 is negatively regulated by phosphorylation and rapidly dephosphorylated by the Ca2+-dependent phosphatase calcineurin to trigger rapid synaptic vesicle endocytosis and recycling (106). Similarly, Dyn1 activation downstream of DRs was found to be calcineurin dependent (105). DRs activated endoplasmic reticulum (ER)–localized ryanodine receptors, thereby releasing Ca2+ to activate calcineurin and subsequently Dyn1. The Dyn1-dependent uptake of TRAIL-DRs suppressed their apoptotic signaling.
Recent studies have shown that Dyn1, although highly expressed in neurons, is in fact broadly expressed at low levels (107) but inactive due to phosphorylation by the constitutively active kinase, GSK3β (glycogen synthase kinase 3β). GSK3β activity is itself regulated by inhibitory phosphorylation downstream of numerous kinases (108), including Akt (also known as protein kinase B). Strikingly, acute inhibition of GSK3β results in enhanced rates of TfnR uptake due to increased rates of CCP initiation and more rapid CCP maturation (109). These effects of GSK3β inhibition on CME and CCP dynamics are dependent on Dyn1 but not on Dyn2. Interestingly, the rate of TfnR uptake in Akt-driven non-small cell lung cancer cells is sensitive to Akt inhibition and to siRNA-mediated depletion of Dyn1. The effects of Akt inhibition on CME are dependent on Dyn1 expression in these cells (109). Thus, while still unexplored, other signaling receptors could trigger signaling cascades that inhibit GSK3β or activate calcineurin to activate Dyn1 and regulate CME in either a cargo-selective or a global manner. Indeed, systematic analyses have revealed a plethora of kinases and other signaling pathways that impinge on CME (110, 111), highlighting the importance of regulating this key endocytic pathway.
AN ENDOCYTIC CHECKPOINT
As described in the preceding section, each stage of CME is subject to regulation by cargo, phosphatidylinositol lipids, kinases, and interactions among accessory proteins. However, CME does not always proceed in a stereotypical manner, and live-cell imaging has shown that as many as 50% of nascent CCPs fail to mature into cargo-loaded CCVs and instead are aborted (9–11). Statistical modeling of CCP lifetimes revealed three kinetically distinct subpopulations, which were designated early abortive, late abortive, and productive, with time constants of ~5 s, ~15 s, and ~80 s, respectively (10). Quantification of events during the first 5 s of CCP assembly by single-molecule TIRFM revealed the recruitment of no more than 4–6 AP2 complexes and correspondingly <6 triskelia (68); thus, the early abortive events likely reflect failed attempts at CCP nucleation. Indeed, more recent analyses, based on the intensity of GFP-CLCa measured by TIRFM (25), have distinguished these dim and transient clathrin structures from bona fide CCPs. Continued growth of these early nucleating events in CCP assembly requires a sufficiently high concentration of AP2 complexes (29), the ability of both α and β subunits to bind PI4,5P2 (56), FCHo (68), and potentially other endocytic pioneers (75) (Figure 2).
A second subpopulation of abortive pits (20–30%) are able to recruit clathrin and cargo but nonetheless fail to complete maturation to form productive CCVs (9–11, 55, 56). The fraction of late abortive pits is reduced, with a corresponding increase in productive CCPs, when cargo (i.e., TfnR) concentration is increased by either overexpression (10) or clustering (55). Several manipulations have also been shown to increase the fraction of late abortive pits, including siRNA-mediated knockdown of CALM and epsin (27); defects in PI4,5P2 binding of either the α, β2, or μ2 subunits of AP2 (56); and clustering of PDZ ligand-bearing GPCRs that delay CCP maturation (57).
A striking example of defects in CCP maturation leading to an increase in the fraction of abortive pits comes from our previously described analysis of cells expressing the truncated ΔαAD AP2 mutant, defective in EAP recruitment. Although TfnR uptake and CCP assembly are unperturbed in these ΔαAD cells (25, 70), electron microscopic (EM) analysis revealed that the vast majority of diffraction-limited CCPs were small (~100 nm), flat lattices. Importantly, live-cell, near-coincident epi/TIRFM revealed that most of these flat structures were turned over in ≤20 s (25). Given the numerous AP2 and clathrin interactions involved in assembly of these otherwise full-sized CCPs, we suggested that active, energy-dependent mechanisms would be needed to sense and disassemble these aberrant structures. These and the studies described above have led us to propose the endocytic checkpoint hypothesis (Figure 3), which suggests that fidelity-monitoring mechanisms exist to sense and actively turn over aberrant CCPs.
Further analysis of endocytosis in the ΔαAD cells provided strong evidence that the endocytic checkpoint indeed monitors fidelity. As mentioned above, while aberrant flat CCPs are detected and turned over by the endocytic checkpoint machinery, a subpopulation of CCPs bypasses this checkpoint, gains curvature, and pinches off to restore near-normal rates of TfnR uptake. This finding raised two questions: What are the functional consequences of bypassing the endocytic checkpoint? And what might be the compensatory mechanism that restores CME in these cells? To address the first question, analysis of subsequent trafficking along the early endocytic pathway in ΔαAD cells revealed numerous defects. These included (a) delayed homotypic fusion of nascent endocytic vesicles; (b) accumulation of peripheral endosomes bearing the early endosome associated scaffold proteins APPL1 and EEA1; (c) reduced endosome acidification, suggesting a delay in endosome maturation; and (d) more rapid recycling of TfnR from early endosomes to the PM (109). While the molecular mechanisms responsible for these defects remain to be determined, they presumably reflect a deficit in fidelity monitoring, resulting in an altered molecular composition of mature CCVs. As to the latter question, surprisingly, these cells were found to activate Dyn1, resulting in increased rates of CCP initiation and more rapid and dysregulated maturation of productive CCPs (109). Dyn1 activation was dependent on Akt and on APPL1, the scaffolds of which activated Akt on early endosomal membranes (112). Together, these Dyn1-dependent effects, perhaps in combination with other as yet unidentified compensatory mechanisms, are sufficient to restore TfnR uptake despite the profound defect in CCP maturation in ΔαAD cells.
In addition to demonstrating the high degree of plasticity in the mechanisms governing CME, the data discussed above provide strong evidence for the existence of an endocytic checkpoint, but many questions remain. Most pressing are the following: What are the defects in CCP maturation sensed by the checkpoint apparatus? What are the components of the checkpoint machinery that sense these defects? How is this information relayed to yet other, unidentified components of the machinery that trigger disassembly of aberrant or stalled CCPs? As described in the following section, dynamin, in an isoform-specific manner, may be an integral component of the endocytic checkpoint.
DYNAMIN: A KEY REGULATOR OF CME
The small Arf and Sar-family GTPases are essential for the assembly and budding of coat protein complexes I or II (COPI, COPII)-coated vesicles from the ER and Golgi complex, as well as for AP1-CCVs from the trans-Golgi network (113). Arf and Sar are also thought to regulate cargo loading of COPI- and COPII-coated vesicles, respectively (113). In contrast, endocytic CCV formation does not appear to require an Arf/Sar-family GTPase. Instead, the large GTPase dynamin, best studied for its role in catalyzing membrane fission at late stages of CME, also plays a key role in regulating multiple early stages of CCP maturation (25, 109). Several recent reviews have discussed dynamin’s well-studied role in membrane fission (4–6), which involves its higher order assembly into rings and spirals around the necks of deeply invaginated CCPs. Here, we focus on its less appreciated role, likely performed in its unassembled tetrameric state, as a key regulator of CME.
Dynamin Regulates Early, Rate-Limiting Stages of CME
Dynamin is a large, multidomain GTPase (Figure 4) comprising an N-terminal G domain; an internal, membrane-binding pleckstrin homology domain (PHD); a C-terminal proline/arginine-rich domain (PRD) that binds SH3 domain-containing partners; and a stalk region formed by folding two α-helical domains, the middle domain and the GTPase effector domain (GED), that flank the PHD (6). Dynamin’s role in endocytosis was first revealed when it was found to be the mammalian homolog of Drosophila shibire (114, 115). Temperature-sensitive shibire mutations result in paralysis owing to a block in endocytosis at the synapse and defects in synaptic vesicle recycling (116, 117). Early EM studies of neuromuscular junctions of shibirets flies revealed the accumulation of Ω-shaped endocytic profiles, many of which exhibited electron-dense collars at their necks, suggesting a role for shibire/dynamin in membrane fission (116). While Drosophila species and most metazoans encode only a single dynamin isoform, vertebrate genomes encode three isoforms, which are ~70% identical but differentially expressed. Dyn1 and Dyn3 are enriched in neurons, while Dyn2 is ubiquitously expressed. Overexpression of dominant-negative mutants of Dyn1 defective in GTP binding and hydrolysis blocks CME at intermediate stages, before the formation of deeply invaginated CCPs (118, 119). Consistent with these findings, the earliest cryo-immuno EM studies of endogenous dynamin localization in non-neuronal cells showed that it could be detected throughout the clathrin lattice of all CCPs at all stages of invagination (120, 121) and is thus positioned to play an early role in CME. Notably, a more recent study using Dyn2-specific antibodies localized Dyn2 to the periphery of nascent CCPs (122). This difference could be attributed to the antibodies used or, potentially more interesting, it may reflect differential distribution, and hence functions, of Dyn1 versus Dyn2 during early stages of CCP maturation.
Figure 4.

Regulation of dynamin by posttranslational modifications, intramolecular interactions, and splice variants. The GTPase dynamin consists of five functional domains and exists as a tetramer in solution (PDB ID 53AF; 152). Dynamin is regulated by: (a) phosphorylation of the PRD, which modulates SH3 domain binding; (b) SH3 domain-containing partners that bind the PRD and can either stimulate or inhibit dynamin assembly and/or GTPase activity; (c) intramolecular interactions between the PHD and stalk that prevent higher order assembly; and (d) splice variants within the PRD that alter interactions with kinases or phosphatases or splice variants within the middle domain that affect allosteric conformational changes and modulate the effects of SH3 domain interactions on dynamin assembly. Dynamin is a key regulator of CME; therefore, regulation of dynamin can impact multiple stages of CME. Abbreviations: CME, clathrin-mediated endocytosis; GED, GTPase effector domain; PDB, Protein Data Bank; PHD, pleckstrin homology domain; PRD, proline/arginine-rich domain; SH3, Src-homology 3 domain.
The first evidence for a regulatory role for dynamin in CME derived from the surprising finding that CME was accelerated in cells overexpressing Dyn1 mutants that are defective in selfassembly, and therefore in assembly stimulated GTPase activity, but otherwise are fully active in the unassembled state (123, 124). This observation, recently confirmed by others (125), provided strong evidence for a role for unassembled dynamin in regulating early, rate-limiting steps in CME. Subsequent studies of CCP dynamics measured using TIRFM showed that siRNA-mediated knockdown of Dyn2 slowed both the rate of maturation of CCPs and the rate of turnover of abortive CCPs (10), providing further evidence that dynamin regulates early stages of CME.
Even the earliest stage in CME, namely CCP initiation, can be regulated by dynamin. As described above, activation of Dyn1 by either acute inhibition of GSK3β or as a compensatory mechanism for defects in CCP maturation (109) results in increases in the rates of both CCP initiation and maturation. The mechanisms by which dynamin controls these early stages of CME remain a mystery, although the colocalization of Dyn2 together with the endocytic pioneers, Eps15 and FCHo2, at the periphery of nascent CCPs is noteworthy (122).
Isoform-Specific Roles for Dynamin in Regulating CME
Despite the high degree of structural and functional similarity between Dyn1 and Dyn2, the two isoforms have distinct biochemical properties. Whereas Dyn1 is able on its own to release vesicles from planar artificial lipid templates, Dyn2 cannot (126). Instead, Dyn2 works synergistically with other curvature-generating, SH3 domain-containing proteins to catalyze fission (127). Both isoforms are able to catalyze fission of preformed membrane tubules (126). Thus, in simple terms Dyn1 is a potent curvature generator, while Dyn2 is a curvature sensor. These biochemical differences can be directly linked to differences in the PHDs (126). Indeed, a single amino acid change within a hydrophobic, membrane-inserting variable loop in the PHD (Tyr600 in Dyn1, Leu600 in Dyn2) is sufficient to account for their differential curvature-generating properties (126). In vivo analyses of Dyn1/Dyn2 chimeras expressed in Dyn1/Dyn2 knockout fibroblasts also revealed differences in their recruitment to CCPs. Dyn2 is more efficiently recruited to CCPs than Dyn1, and this difference is encoded in their PRDs (126).
The differential biochemical properties of Dyn1 and Dyn2 may account for their tissue-specific functions. Thus, Dyn1, which can be rapidly activated by Ca2+/calcineurin-dependent dephosphorylation (106) and is a more powerful curvature generator (126), is ideally suited for its role in rapid synaptic vesicle recycling during neurotransmission, which under some conditions occurs even in the absence of clathrin (86). In non-neuronal cells, Dyn2 is targeted to nascent CCPs and requires other curvature-generating EAPs, including amphiphysin, endophilin, and/or SNX9, to trigger its assembly around the highly curved necks of fully matured CCPs to catalyze their release. In addition to membrane curvature, Dyn2 can be regulated by intramolecular interactions between the PHD and the stalk (128) (Figure 4). Thus, we speculate that unassembled, CCP-associated Dyn2 could function as a kinetic timer receiving input from binding partners that sense coat assembly, cargo, and/or membrane curvature and that Dyn2 assembles only after CCP maturation is complete to catalyze membrane fission. This model is, in part, based on a well-described, GTPase-dependent fidelity monitoring system that functions in cotranslational translocation across the ER. In this case, cross talk among cargo (i.e., ribosome-nascent chain complexes), two GTPases [i.e., the signal recognition particle (SRP) and its receptor], and the membrane translocon controls the kinetics of assembly of the two GTPases and their rates of GTP hydrolysis, providing a proofreading mechanism that discriminates between authentic and nonauthentic cargos and ensures the fidelity and progression of the multistep cotranslocation process (129).
Much work is needed to test this hypothesis for dynamin function. However, it is interesting to note that disease-causing mutants of Dyn2 map to regions that regulate its self-assembly (128, 130). Moreover, the binding of different SH3 domains to dynamin’s PRD can either enhance or inhibit its assembly and GTPase activities (127, 131).
Regulation of Dynamin
Consistent with the central role for dynamin in regulating CME, dynamin itself is subject to regulation at multiple levels. At the synapse, Dyn1 regulation is well understood (132). In the resting state, Dyn1 is constitutively inactivated by GSK3β-mediated phosphorylation at S774, which depends on upstream phosphorylation at S778; both of these sites are located within the PRD. These two serines are not conserved in Dyn2. Dyn1 is rapidly dephosphorylated when an action potential triggers Ca2+ influx and activation of the Ca2+-dependent phosphatase calcineurin. As described above, GSK3β and calcineurin also regulate Dyn1 activity in non-neuronal cells. Whether Dyn2 function is regulated by posttranslational modifications has not been investigated.
Phosphorylation of the PRD regulates interaction of Dyn1 with its many SH3 domain–containing binding partners (133) (Figure 4). In turn, interactions of SH3 domain–containing proteins can have multiple and complex regulatory effects on both Dyn1 and Dyn2, including altering GTPase activity (127, 131), stabilizing the oligomeric conformation (131), and targeting to CCPs (134). Interestingly, a comprehensive study of the effects of 13 SH3 domains on Dyn1 revealed that despite equivalent binding levels, the SH3 domain interactions conferred differential effects on Dyn1 assembly and GTPase activity (131). Moreover, effects on GTPase activity did not always correlate with increases in self-assembly and vice versa. Thus, the many SH3 domain–containing dynamin binding partners, which also variably encode domains that bind to other coat constituents, cargo, actin regulators, and/or sense curvature, are positioned to act as sensors of CCP maturation feeding into dynamin, thereby regulating its activity (Figure 3).
Interestingly, Dyn1 (but not Dyn2) has multiple splice variants within the PRD that encode inserts or alternate C termini (135) (Figure 4). This provides ample opportunity for splice variant–specific regulation of Dyn1. For example, the variant Dyn1xa includes a canonical Dyrk1A phosphorylation site, Ser857, which is shown to modulate amphiphysin I and Grb2 interactions (136). Additionally, calcineurin binds preferentially to the sequence PRITISDP, which is limited to the variant Dyn1xb (137, 138) and facilitates dephosphorylation for activity-dependent bulk endocytosis at the synapse. The differential PRD splicing also affects the phosphorylation state and subcellular localization of Dyn1 (139). Neither the expression pattern of these Dyn1 splice variants nor the effects of phosphorylation on Dyn1 function in either neuronal or non-neuronal cells has been fully explored.
Both Dyn1 and Dyn2 have conserved splice variants in their middle domains (135), encompassing the α2 helix (Figure 4) shown to link nucleotide binding to global conformational changes in the dynamin tetramer (128). Interestingly, the GTPase and assembly activities of these different splice variants are differentially affected by SH3 domain binding (131), suggesting that this middle domain region functions in the allosteric regulation of dynamin GTPase and assembly activities through PRD interactions.
Altogether, these studies suggest complex patterns of posttranslational and allosteric regulation of Dyn1 and Dyn2 that likely impinge on their role in regulating CME.
Dynamin-1 as a Nexus Between Signaling and Endocytosis
Although it is well-known that CME can modulate the signaling properties of cell surface receptors (e.g., receptor tyrosine kinases and GPCRs) (140, 141), recent studies have shown that signaling, in turn, can modulate CME. This cross talk may be specifically adapted in cancer cells to enhance tumor progression and metastasis (107). As a key regulator of CME, Dyn1 appears to function as a nexus for cross talk between signaling and endocytosis, and interestingly, Dyn1 is upregulated in many cancer cell lines (105). As described above, a signaling cascade downstream of activated TRAIL–DR leads to activation of Dyn1, Dyn1-dependent CME of the TRAIL-DR complexes, suppression of their apoptotic signaling, and enhanced resistance to TRAIL in a number of cancer cell lines (105). Dyn1 is also activated downstream of the oncogenic kinase Akt, which phosphorylates and inactivates GSK3β (109). Alterations in CME in cancer cells—for example, by upregulation of CLCb (the normally neuronally enriched isoform of clathrin light chain) (142)—can activate Dyn1 through an Akt-dependent mechanism. Interestingly, as in the ΔαAD mutant cells (109), APPL1-positive endosomes accumulate in the cell periphery in CLCb overexpressing cells, in a Dyn1-dependent manner (142). APPL1 functions as a scaffold to activate Akt; hence, a feedback loop, mediated by Dyn1, is established among signaling receptors, CME, and APPL1 endosomes. The potential role of Dyn1 in regulating CME and signaling in non-neuronal cells in human health and disease, especially in cancer cell progression, remains to be explored.
The findings described in this section point to dynamin as a key regulator of CME and suggest profound isoform-specific differences in the effects of dynamin in regulating CME. Further studies are needed to elucidate the isoform-specific mechanisms by which dynamins regulate CCP initiation and maturation.
EVOLUTION OF THE REGULATION OF CME
Nature’s first step toward evolution of the endomembrane system most likely began with endocytosis. According to the protocoatamer model, primordial endocytic compartments predate the nucleus (143). Consequently, the endocytic machinery is evolutionarily ancient and highly conserved (144, 145). For instance, clathrin appeared very early in the evolution of eukaryotic life and shares a high degree of sequence homology across taxa, including >50% identity between yeast and humans (146). Despite the early, evolutionary appearance of clathrin-dependent endocytosis, a recent proteomic and phylogenetic analysis of a subset of common CME proteins has revealed that many were gradually incorporated during the evolution of single and multicellular eukaryotes (147). In addition, an expansion of paralogs occurred in vertebrates, providing additional opportunities for evolving complexity. Importantly, this expansion of the endocytic machinery suggests an evolving specialization of CME with regard to several aspects of its regulation, including cargo sorting, the role of AP2 complexes in CCP initiation, and the requirement for dynamin. The specialization of CME coincided with the evolution of intracellular communication in multicellular eukaryotes and the emergence and diversification of PM receptors (148).
Most notable among the evolutionary additions to the CME machinery is the late appearance of true dynamins, which encode both a PHD and PRD. Dynamins first appeared in holozoans (i.e., multicellular organisms and their closest single-cell relatives; e.g., choanoflagellates) (147, 149) and are absent in fungi and ciliates. This observation further supports an early regulatory role for dynamin, rather than an evolutionarily obligate role in membrane fission. The appearance of dynamin SH3 domain–containing partners, SNX9 and amphiphysin, overlaps with the emergence of dynamins, highlighting their evolved role in recruiting dynamin to the PM through SH3–PRD interaction (147). True dynamins undoubtedly evolved from the more ancient dynamin-related proteins (DRPs), which lack a PHD and PRD and are involved in mitochondrial inheritance. While DRPs have been recruited to function in endocytosis in some ciliates and amoebae, extensive phylogenetic analysis suggests that these represent sporadic innovations (150). We speculate that addition of the PHD to ancient DRPs conferred their ability to efficiently catalyze fission, while the PRD conferred regulatory potential.
As described above, the allosteric regulation of AP2 conformational changes by interactions with PI4,5P2, tyrosine-based cargo sorting motifs, and pioneer EAPs plays a central role in regulating CCP initiation. Thus, it is interesting that although conserved across evolution, AP2 complexes are not required for endocytosis in yeast (151). Moreover, residues in α and μ2 subunits of metazoan AP2 that stabilize the closed conformation, as well as a critical residue in μ2 required for tyrosine-based cargo binding, are divergent in fungi, suggesting evolution of the allosteric regulation of AP2 in metazoans. Consistent with this, the otherwise conserved FCHo proteins, which also regulate CCP initiation in fungi, lack the AP2 activating region that facilitates the closed-open conformational switch (76, 147).
Finally, an expansion of tissue-specific or cargo-specialized adaptor molecules, such as AP180; stonin; the LDLR adaptor protein, ARH; and Numb, as well as the incorporation of cargo-binding domains (e.g., for ubiquitin) into EAPs, such as Eps15, occurred throughout the evolution of CME (147). Thus, more complex, potentially cargo-selective regulation of CME emerged concurrently with multicellularity, the need for intercellular communication, and divergence of cell surface receptors, and this complexity continued to expand in vertebrate lineages.
CONCLUSIONS
CME controls not only nutrient uptake but also how cells communicate with each other and with their environment; hence, CME should be a highly regulated process. Perhaps because research on CME has focused on the uptake of abundant and constitutively internalized nutrient receptors, studies on the regulation of CME have lagged behind our understanding of the fundamental mechanisms driving CCV formation. However, attention is shifting toward identifying regulatory mechanisms with the more recent appreciation of the roles of specific cargo receptors, signaling pathways, and the GTPase dynamin in regulating CME. Future studies are needed to identify which signaling pathways impinge on CME and how—and by what mechanisms—cargo, dynamin, and other factors regulate CME. Results from these studies will likely shed new light on the importance of regulating CME in health and the consequences of dysregulated CME in disease.
FUTURE ISSUES.
Although TIRFM has proven invaluable in measuring CCP dynamics, distinguishing abortive events from rapidly maturing productive CCPs remains a challenge. Identifying unambiguous molecular markers of abortive versus productive CCPs is an urgent need.
While we understand the biochemical properties of EAPs, the functional and temporal hierarchy of their activities in CCV formation remain poorly understood and in many cases controversial. Further studies on EAP function in CCV formation in vivo and in vitro are needed.
Whether abortive CCPs reflect an active process or instead stochastic disassembly of unstable CCP intermediates remains to be established. Distinguishing these possibilities requires identification of the endocytic checkpoint machinery that senses, signals, and actively turns over aberrant CCP intermediates.
Still only a handful of cargo molecules and signaling receptors have been studied with regard to their effects on CCP dynamics. Increasing this number will certainly reveal a diversity of mechanisms regulating CME.
Systematic screens have shown that numerous signaling proteins impinge on CME. Defining their targets, the stage(s) at which they regulate CME, and their mechanisms of action will be an active area of research.
The downstream effectors and molecular mechanisms that account for the unexpected isoform-specific functions of Dyn1 and Dyn2 in regulating CME in non-neuronal cells need to be identified and elucidated.
ACKNOWLEDGMENTS
We thank current and past members of the Schmid laboratory for their scientific contributions and current laboratory members for careful reading of the manuscript and helpful discussions. We are grateful for almost 30 years of combined support from the National Institute of General Medical Sciences and the National Institute of Mental Health for the basic research conducted in the Schmid laboratory. Our work on CME and dynamin has been funded by National Institutes of Health grants GM42455 and MH61345 to S.L.S. and GM73165, a collaborative dual–primary investigator grant, to S.L.S. and G.D. P.H.C. was partly supported by Taiwan National Science Council grant 103–2917-I-564–029, and S.S. was supported by Welch Foundation grant I-1823 to S.L.S.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Conner SD, Schmid SL. 2002. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol 156:921–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson MS. 2015. Forty years of clathrin-coated vesicles. Traffic 16:1210–38 [DOI] [PubMed] [Google Scholar]
- 3.Schmid EM, McMahon HT. 2007. Integrating molecular and network biology to decode endocytosis. Nature 448:883–88 [DOI] [PubMed] [Google Scholar]
- 4.Antonny B, Burd C, De Camilli P, Chen E, Daumke O, et al. 2016. Membrane fission by dynamin: what we know and what we need to know. EMBO J. 35:2270–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morlot S, Roux A. 2013. Mechanics of dynamin-mediated membrane fission. Annu. Rev. Biophys 42:629–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmid SL, Frolov VA. 2011. Dynamin: functional design of a membrane fission catalyst. Annu. Rev. Cell Dev. Biol 27:79–105 [DOI] [PubMed] [Google Scholar]
- 7.Gaidarov I, Santini F, Warren RA, Keen JH. 1999. Spatial control of coated pit dynamics in living cells. Nat. Cell Biol 1:1–7 [DOI] [PubMed] [Google Scholar]
- 8.Merrifield CJ, Feldman ME, Wan L, Almers W. 2002. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol 4:691–98 [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, et al. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591–605 [DOI] [PubMed] [Google Scholar]
- 10.Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, et al. 2009. Cargo and dynamin regulate clathrin-coated pit maturation. PLOS Biol. 7:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor MJ, Perrais D, Merrifield CJ. 2011. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLOS Biol. 9:e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol 5:121–32 [DOI] [PubMed] [Google Scholar]
- 13.Burston HE, Maldonado-Baez L, Davey M, Montpetit B, Schluter C, et al. 2009. Regulators of yeast endocytosis identified by systematic quantitative analysis. J. Cell Biol 185:1097–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaksonen M, Toret CP, Drubin DG. 2005. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell 123:305–20 [DOI] [PubMed] [Google Scholar]
- 15.Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. 2011. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol 13:1124–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kural C, Akatay AA, Gaudin R, Chen BC, Legant WR, et al. 2015. Asymmetric formation of coated pits on dorsal and ventral surfaces at the leading edges of motile cells and on protrusions of immobile cells. Mol. Biol. Cell 26:2044–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saffarian S, Kirchhausen T. 2008. Differential evanescence nanometry: live-cell fluorescence measurements with 10-nm axial resolution on the plasma membrane. Biophys. J 94:2333–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keyel PA, Watkins SC, Traub LM. 2004. Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J. Biol. Chem 279:13190–204 [DOI] [PubMed] [Google Scholar]
- 19.Mattheyses AL, Atkinson CE, Simon SM. 2011. Imaging single endocytic events reveals diversity in clathrin, dynamin and vesicle dynamics. Traffic 12:1394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merrifield CJ, Perrais D, Zenisek D. 2005. Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell 121:593–606 [DOI] [PubMed] [Google Scholar]
- 21.Rappoport JZ, Simon SM. 2003. Real-time analysis of clathrin-mediated endocytosis during cell migration. J. Cell Sci. 116:847–55 [DOI] [PubMed] [Google Scholar]
- 22.Grassart A, Cheng AT, Hong SH, Zhang F, Zenzer N, et al. 2014. Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. J. Cell Biol 205:721–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rappoport JZ, Taha BW, Lemeer S, Benmerah A, Simon SM. 2003. The AP-2 complex is excluded from the dynamic population of plasma membrane-associated clathrin. J. Biol. Chem 278:47357–60 [DOI] [PubMed] [Google Scholar]
- 24.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, et al. 2008. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5:695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguet F, Antonescu CN, Mettlen M, Schmid SL, Danuser G. 2013. Advances in analysis of low signal-to-noise images link dynamin and AP2 to the functions of an endocytic checkpoint. Dev. Cell 26:279–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lampe M, Vassilopoulos S, Merrifield C. 2016. Clathrin coated pits, plaques and adhesion. J. Struct. Biol. 196:48–56 [DOI] [PubMed] [Google Scholar]
- 27.Mettlen M, Stoeber M, Loerke D, Antonescu CN, Danuser G, Schmid SL. 2009. Endocytic accessory proteins are functionally distinguished by their differential effects on the maturation of clathrin-coated pits. Mol. Biol. Cell 20:3251–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, et al. 2011. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat. Cell Biol 13:331–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong SH, Cortesio CL, Drubin DG. 2015. Machine-learning-based analysis in genome-edited cells reveals the efficiency of clathrin-mediated endocytosis. Cell Rep. 12:2121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mettlen M, Danuser G. 2014. Imaging and modeling the dynamics of clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol 6:a017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saffarian S, Cocucci E, Kirchhausen T. 2009. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLOS Biol. 7:e1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batchelder EM, Yarar D. 2010. Differential requirements for clathrin-dependent endocytosis at sites of cell-substrate adhesion. Mol. Biol. Cell 21:3070–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maupin P, Pollard TD. 1983. Improved preservation and staining of HeLa cell actin filaments, clathrin-coated membranes, and other cytoplasmic structures by tannic acid-glutaraldehyde-saponin fixation. J. Cell Biol 96:51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elkhatib N, Bresteau E, Baschieri F, Rioja AL, van Niel G, et al. 2017. Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science 356:eaal4713. [DOI] [PubMed] [Google Scholar]
- 35.Grove J, Metcalf DJ, Knight AE, Wavre-Shapton ST, Sun T, et al. 2014. Flat clathrin lattices: stable features of the plasma membrane. Mol. Biol. Cell 25:3581–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cureton DK, Massol RH, Whelan SP, Kirchhausen T. 2010. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLOS Pathog. 6:e1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skruzny M, Brach T, Ciuffa R, Rybina S, Wachsmuth M, Kaksonen M. 2012. Molecular basis for coupling the plasma membrane to the actin cytoskeleton during clathrin-mediated endocytosis. PNAS 109:E2533–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traub LM. 2009. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol 10:583–96 [DOI] [PubMed] [Google Scholar]
- 39.Bonifacino JS, Traub LM. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem 72:395–447 [DOI] [PubMed] [Google Scholar]
- 40.Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, et al. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269:1872–75 [DOI] [PubMed] [Google Scholar]
- 41.Maurer ME, Cooper JA. 2006. The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH. J. Cell Sci 119:4235–46 [DOI] [PubMed] [Google Scholar]
- 42.Garuti R, Jones C, Li WP, Michaely P, Herz J, et al. 2005. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J. Biol. Chem 280:40996–1004 [DOI] [PubMed] [Google Scholar]
- 43.Hopkins CR, Miller K, Beardmore JM. 1985. Receptor-mediated endocytosis of transferrin and epidermal growth factor receptors: a comparison of constitutive and ligand-induced uptake. J. Cell Sci. Suppl 3:173–86 [DOI] [PubMed] [Google Scholar]
- 44.Trowbridge IS, Collawn JF, Hopkins CR. 1993. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell Biol 9:129–61 [DOI] [PubMed] [Google Scholar]
- 45.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. 2002. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 41:3321–28 [DOI] [PubMed] [Google Scholar]
- 46.Henry AG, Hislop JN, Grove J, Thorn K, Marsh M, von Zastrow M. 2012. Regulation of endocytic clathrin dynamics by cargo ubiquitination. Dev. Cell 23:519–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller WE, Lefkowitz RJ. 2001. Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr. Opin. Cell Biol 13:139–45 [DOI] [PubMed] [Google Scholar]
- 48.Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, et al. 2002. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 21:303–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertelsen V, Sak MM, Breen K, Rodland MS, Johannessen LE, et al. 2011. A chimeric pre-ubiquitinated EGF receptor is constitutively endocytosed in a clathrin-dependent, but kinase-independent manner. Traffic 12:507–20 [DOI] [PubMed] [Google Scholar]
- 50.de Melker AA, van der Horst G, Borst J. 2004. c-Cbl directs EGF receptors into an endocytic pathway that involves the ubiquitin-interacting motif of Eps15. J. Cell Sci 117:5001–12 [DOI] [PubMed] [Google Scholar]
- 51.Regan-Klapisz E, Sorokina I, Voortman J, de Keizer P, Roovers RC, et al. 2005. Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM-UBL interaction. J. Cell Sci 118:4437–50 [DOI] [PubMed] [Google Scholar]
- 52.Confalonieri S, Salcini AE, Puri C, Tacchetti C, Di Fiore PP. 2000. Tyrosine phosphorylation of Eps15 is required for ligand-regulated, but not constitutive, endocytosis. J. Cell Biol 150:905–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pitcher C, Honing S, Fingerhut A, Bowers K, Marsh M. 1999. Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the CD4 endocytosis signal by serine phosphorylation. Mol. Biol. Cell 10:677–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miettinen HM, Matter K, Hunziker W, Rose JK, Mellman I. 1992. Fc receptor endocytosis is controlled by a cytoplasmic domain determinant that actively prevents coated pit localization. J. Cell Biol 116:875–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu AP, Aguet F, Danuser G, Schmid SL. 2010. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol 191:1381–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kadlecova Z, Spielman SJ, Loerke D, Mohanakrishnan A, Reed DK, Schmid SL. 2017. Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J. Cell Biol 216:167–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puthenveedu MA, von Zastrow M. 2006. Cargo regulates clathrin-coated pit dynamics. Cell 127:113–24 [DOI] [PubMed] [Google Scholar]
- 58.Mettlen M, Loerke D, Yarar D, Danuser G, Schmid SL. 2010. Cargo- and adaptor-specific mechanisms regulate clathrin-mediated endocytosis. J. Cell Biol 188:919–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell 109:523–35 [DOI] [PubMed] [Google Scholar]
- 60.Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, et al. 2010Alarge-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141:1220–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirchhausen T, Owen D, Harrison SC. 2014. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol 6:a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mishra SK, Hawryluk MJ, Brett TJ, Keyel PA, Dupin AL, et al. 2004. Dual engagement regulation of protein interactions with the AP-2 adaptor α appendage. J. Biol. Chem 279:46191–203 [DOI] [PubMed] [Google Scholar]
- 63.Schmid EM, Ford MG, Burtey A, Praefcke GJ, Peak-Chew SY, et al. 2006. Role of the AP2 β-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLOS. Biol 4:e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Praefcke GJ, Ford MG, Schmid EM, Olesen LE, Gallop JL, et al. 2004. Evolving nature of the AP2 α-appendage hub during clathrin-coated vesicle endocytosis. EMBO J. 23:4371–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shih W, Gallusser A, Kirchhausen T. 1995. A clathrin-binding site in the hinge of the β2 chain of mammalian AP-2 complexes. J. Biol. Chem 270:31083–90 [DOI] [PubMed] [Google Scholar]
- 66.Owen DJ, Vallis Y, Pearse BMF, McMahon HT, Evans PR. 2000. The structure and function of the ß2-adaptin appendage domain. EMBO J. 19:4216–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edeling MA, Mishra SK, Keyel PA, Steinhauser AL, Collins BM, et al. 2006. Molecular switches involving the AP-2 β2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev. Cell 10:329–42 [DOI] [PubMed] [Google Scholar]
- 68.Cocucci E, Aguet F, Boulant S, Kirchhausen T. 2012The first five seconds in the life of a clathrin-coated pit. Cell 150:495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hinrichsen L, Harborth J, Andrees L, Weber K, Ungewickell EJ. 2003. Effect of clathrin heavy chain- and α-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J. Biol. Chem 278:45160–70 [DOI] [PubMed] [Google Scholar]
- 70.Motley AM, Berg N, Taylor MJ, Sahlender DA, Hirst J, et al. 2006. Functional analysis of AP-2 α and μ2 subunits. Mol. Biol. Cell 17:5298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelly BT, Graham SC, Liska N, Dannhauser PN, Honing S, et al. 2014. Clathrin adaptors. AP2 controls clathrin polymerization with a membrane-activated switch. Science 345:459–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, et al. 2010. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science 328:1281–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritter B, Philie J, Girard M, Tung EC, Blondeau F, McPherson PS. 2003. Identification of a family of endocytic proteins that define a new α-adaptin ear-binding motif. EMBO Rep. 4:1089–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Umasankar PK, Ma L, Thieman JR, Jha A, Doray B, et al. 2014. A clathrin coat assembly role for the muniscin protein central linker revealed by TALEN-mediated gene editing. eLife 3:e04137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma L, Umasankar PK, Wrobel AG, Lymar A, McCoy AJ, et al. 2016. Transient Fcho1/2Eps15/RAP-2 nanoclusters prime the AP-2 clathrin adaptor for cargo binding. Dev. Cell 37:428–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hollopeter G, Lange JJ, Zhang Y, Vu TN, Gu M, et al. 2014. The membrane-associated proteins FCHo and SGIP are allosteric activators of the AP2 clathrin adaptor complex. eLife 3:e03648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller SE, Sahlender DA, Graham SC, Honing S, Robinson MS, et al. 2011. The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 147:1118–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller SE, Mathiasen S, Bright NA, Pierre F, Kelly BT, et al. 2015. CALM regulates clathrin-coated vesicle size and maturation by directly sensing and driving membrane curvature. Dev. Cell 33:163–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ritter B, Murphy S, Dokainish H, Girard M, Gudheti MV, et al. 2013. NECAP 1 regulates AP-2 interactions to control vesicle size, number, and cargo during clathrin-mediated endocytosis. PLOS Biol. 11:e1001670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brach T, Godlee C, Moeller-Hansen I, Boeke D, Kaksonen M. 2014. The initiation of clathrin-mediated endocytosis is mechanistically highly flexible. Curr. Biol 24:548–54 [DOI] [PubMed] [Google Scholar]
- 81.Nunez D, Antonescu C, Mettlen M, Liu A, Schmid SL, et al. 2011. Hotspots organize clathrin-mediated endocytosis by efficient recruitment and retention of nucleating resources. Traffic 12:1868–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smythe E, Ayscough KR. 2003. The Ark1/Prk1 family of protein kinases. Regulators of endocytosis and the actin skeleton. EMBO Rep. 4:246–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ricotta D, Conner SD, Schmid SL, von Figura K, Honing S. 2002. Phosphorylation of the AP2 μ subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J. Cell Biol 156:791–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olusanya O, Andrews PD, Swedlow JR, Smythe E. 2001. Phosphorylation of threonine 156 of the μ2 subunit of the AP2 complex is essential for endocytosis in vitro and in vivo. Curr. Biol 11:896–900 [DOI] [PubMed] [Google Scholar]
- 85.Conner SD, Schroter T, Schmid SL. 2003. AAK1-mediated μ2 phosphorylation is stimulated by assembled clathrin. Traffic 4:885–90 [DOI] [PubMed] [Google Scholar]
- 86.Watanabe S, Rost BR, Camacho-Perez M, Davis MW, Sohl-Kielczynski B, et al. 2013. Ultrafast endocytosis at mouse hippocampal synapses. Nature 504:242–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loerke D, Mettlen M, Schmid SL, Danuser G. 2011. Measuring the hierarchy of molecular events during clathrin-mediated endocytosis. Traffic 12:815–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kirchhausen T 1993. Coated pits and coated vesicles—sorting it all out. Curr. Opin. Struct. Biol 3:182–88 [Google Scholar]
- 89.Dannhauser PN, Ungewickell EJ. 2012. Reconstitution of clathrin-coated bud and vesicle formation with minimal components. Nat. Cell Biol 14:634–39 [DOI] [PubMed] [Google Scholar]
- 90.Takei K, Haucke V, Slepnev V, Farsad K, Salazar M, et al. 1998. Generation of coated intermediates of clathrin-mediated endocytosis on protein-free liposomes. Cell 94:131–41 [DOI] [PubMed] [Google Scholar]
- 91.Heuser J 1980. Three-dimensional visualization of coated vesicle formation in fibroblasts. J. Cell Biol 84:560–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Avinoam O, Schorb M, Beese CJ, Briggs JA, Kaksonen M. 2015. Endocytic sites mature by continuous bending and remodeling of the clathrin coat. Science 348:1369–72 [DOI] [PubMed] [Google Scholar]
- 93.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–57 [DOI] [PubMed] [Google Scholar]
- 94.Jost M, Simpson F, Kavran JM, Lemmon MA, Schmid SL. 1998. Phosphatidylinositol-4,5-bisphosphate is required for endocytic coated vesicle formation. Curr. Biol 8:1399–402 [DOI] [PubMed] [Google Scholar]
- 95.Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, et al. 2007. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. PNAS 104:3793–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Antonescu CN, Aguet F, Danuser G, Schmid SL. 2011. Phosphatidylinositol-(4,5)-bisphosphate regulates clathrin-coated pit initiation, stabilization, and size. Mol. Biol. Cell 22:2588–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perera RM, Zoncu R, Lucast L, De Camilli P, Toomre D. 2006. Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. PNAS 103:19332–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakatsu F, Perera RM, Lucast L, Zoncu R, Domin J, et al. 2010. The inositol 5-phosphatase SHIP2 regulates endocytic clathrin-coated pit dynamics. J. Cell Biol 190:307–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, et al. 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499:233–37 [DOI] [PubMed] [Google Scholar]
- 100.Gaidarov I, Smith ME, Domin J, Keen JH. 2001. The class IIphosphoinositide3-kinase C2αis activated by clathrin and regulates clathrin-mediated membrane trafficking. Mol. Cell 7:443–49 [DOI] [PubMed] [Google Scholar]
- 101.He K, Marsland R, Upadhyayula S, Song E, Dang S, et al. 2017. Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 552:410–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Daste F, Walrant A, Holst MR, Gadsby JR, Mason J, et al. 2017. Control of actin polymerization via the coincidence of phosphoinositides and high membrane curvature. J. Cell Biol 216:3745–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moore CAC, Milano SK, Benovic JL. 2007. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol 69:451–82 [DOI] [PubMed] [Google Scholar]
- 104.Cao TT, Mays RW, von Zastrow M. 1998. Regulated endocytosis of G-protein-coupled receptors by a biochemically and functionally distinct subpopulation of clathrin-coated pits. J. Biol. Chem 273:24592–602 [DOI] [PubMed] [Google Scholar]
- 105.Reis CR, Chen PH, Bendris N, Schmid SL. 2017. TRAIL-death receptor endocytosis and apoptosis are selectively regulated by dynamin-1 activation. PNAS 114:504–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Smillie KJ, Cousin MA. 2005. Dynamin I phosphorylation and the control of synaptic vesicle endocytosis. Biochem. Soc. Symp 72:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmid SL. 2017. Reciprocal regulation of signaling and endocytosis: implications for the evolving cancer cell. J. Cell Biol 216:2623–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Medina M, Wandosell F. 2011. Deconstructing GSK-3: the fine regulation of its activity. Int. J. Alzheimer’s Dis 2011:479249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reis CR, Chen PH, Srinivasan S, Aguet F, Mettlen M, Schmid SL. 2015. Crosstalk between Akt/GSK3β signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J. 34:2132–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liberali P, Snijder B, Pelkmans L. 2014. A hierarchical map of regulatory genetic interactions in membrane trafficking. Cell 157:1473–87 [DOI] [PubMed] [Google Scholar]
- 111.Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, et al. 2005. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436:78–86 [DOI] [PubMed] [Google Scholar]
- 112.Schenck A, Goto-Silva L, Collinet C, Rhinn A, Giner A, et al. 2008. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133:486–97 [DOI] [PubMed] [Google Scholar]
- 113.Pucadyil TJ, Schmid SL. 2009. Conserved functions of membrane active GTPases in coated vesicle formation. Science 325:1217–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen MS, Obar RA, Schroeder CC, Austin TW, Poodry CA, et al. 1991. Multiple forms of dynamin are encoded by shibire, a Drosophila gene involved in endocytosis. Nature 351:583–86 [DOI] [PubMed] [Google Scholar]
- 115.van der Bliek AM, Meyerowitz EM. 1991. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 351:411–14 [DOI] [PubMed] [Google Scholar]
- 116.Koenig JH, Ikeda K. 1989. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J. Neurosci 9:3844–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Poodiy CA, Hall L, Suzuki DT. 1973. Developmental properties of shibirets1 a pleiotropic mutation affecting larval and adult locomotion and development. Dev. Biol 32:373–86 [DOI] [PubMed] [Google Scholar]
- 118.Damke H, Binns DD, Ueda H, Schmid SL, Baba T. 2001. Dynamin GTPase domain mutants block endocytic vesicle formation at morphologically distinct stages. Mol. Biol. Cell 12:2578–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. 1993Mutations in human dynamin block an intermediate stage in coated vesicle formation. J. Cell Biol 122:553–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Damke H, Baba T, Warnock DE, Schmid SL. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol 127:915–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Warnock DE, Baba T, Schmid SL. 1997. Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol. Biol. Cell 8:2553–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sochacki KA, Dickey AM, Strub MP, Taraska JW. 2017. Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat. Cell Biol 19:352–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sever S, Damke H, Schmid SL. 2000. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol 150:1137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sever S, Muhlberg AB, Schmid SL. 1999. Impairment of dynamin’s GAP domain stimulates receptor-mediated endocytosis. Nature 398:481–86 [DOI] [PubMed] [Google Scholar]
- 125.Faelber K, Posor Y, Gao S, Held M, Roske Y,et al. 2011. Crystal structure of nucleotide-free dynamin. Nature 477:556–60 [DOI] [PubMed] [Google Scholar]
- 126.Liu YW, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, Schmid SL. 2011. Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. PNAS 108:E234–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Neumann S, Schmid SL. 2013. Dual role of BAR domain-containing proteins in regulating vesicle release catalyzed by the GTPase, dynamin-2. J. Biol. Chem 288:25119–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Srinivasan S, Dharmarajan V, Reed DK, Griffin PR, Schmid SL. 2016. Identification and function of conformational dynamics in the multidomain GTPase dynamin. EMBO J. 35:443–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang X, Shan S-O. 2014. Fidelity of cotranslational protein targeting by the signal recognition particle. Annu. Rev. Biophys 43:381–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kenniston JA, Lemmon MA. 2010. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 29:3054–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Krishnan S, Collett M, Robinson PJ. 2015. SH3 domains differentially stimulate distinct dynamin I assembly modes and G domain activity. PLOS ONE 10:e0144609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clayton EL, Sue N, Smillie KJ, O’Leary T, Bache N, et al. 2010. Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat. Neurosci 13:845–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Solomaha E, Palfrey HC. 2005. Conformational changes in dynamin on GTP binding and oligomerization reported by intrinsic and extrinsic fluorescence. Biochem. J 391:601–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shpetner HS, Herskovits JS, Vallee RB. 1996. A binding site for SH3 domains targets dynamin to coated pits. J. Biol. Chem 271:13–16 [DOI] [PubMed] [Google Scholar]
- 135.Cao H, Garcia F, McNiven MA. 1998. Differential distribution of dynamin isoforms in mammalian cells. Mol. Biol. Cell 9:2595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang Y, Chen-Hwang MC, Dolios G, Murakami N, Padovan JC, et al. 2004. Mnb/Dyrk1A phosphorylation regulates the interaction of dynamin 1 with SH3 domain-containing proteins. Biochemistry 43:10173–85 [DOI] [PubMed] [Google Scholar]
- 137.Xue J, Graham ME, Novelle AE, Sue N, Gray N, et al. 2011. Calcineurin selectively docks with the dynamin Ixb splice variant to regulate activity-dependent bulk endocytosis. J. Biol. Chem 286:30295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bodmer D, Ascano M, Kuruvilla R. 2011. Isoform-specific dephosphorylation of dynamin1 by calcineurin couples neurotrophin receptor endocytosis to axonal growth. Neuron 70:1085–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chan LS, Hansra G, Robinson PJ, Graham ME. 2010. Differential phosphorylation of dynamin I isoforms in subcellular compartments demonstrates the hidden complexity of phosphoproteomes. J. Proteom. Res 9:4028–37 [DOI] [PubMed] [Google Scholar]
- 140.Di Fiore PP, von Zastrow M. 2014. Endocytosis, signaling, and beyond. Cold Spring Harb. Perspect. Biol 6:a016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sorkin A, von Zastrow M. 2009. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol 10:609–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen PH, Bendris N, Hsiao YJ, Reis CR, Mettlen M, et al. 2017. Crosstalk between CLCb/Dyn1-mediated adaptive clathrin-mediated endocytosis and epidermal growth factor receptor signaling increases metastasis. Dev. Cell 40:278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jekely G 2007. Origin of eukaryotic endomembranes: a critical evaluation of different model scenarios. Adv. Exp. Med. Biol 607:38–51 [DOI] [PubMed] [Google Scholar]
- 144.Wideman JG, Leung KF, Field MC, Dacks JB. 2014. The cell biology of the endocytic system from an evolutionary perspective. Cold Spring Harb. Perspect. Biol 6:a016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rout MP, Field MC. 2017. The evolution of organellar coat complexes and organization of the eukaryotic cell. Annu. Rev. Biochem 86:637–57 [DOI] [PubMed] [Google Scholar]
- 146.Royle SJ. 2006. The cellular functions of clathrin. Cell. Mol. Life Sci 63:1823–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dergai M, Iershov A, Novokhatska O, Pankivskyi S, Rynditch A. 2016. Evolutionary changes on the way to clathrin-mediated endocytosis in animals. Genome Biol. Evol 8:588–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ben-Shlomo I, Yu Hsu S, Rauch R, Kowalski HW, Hsueh AJW. 2003. Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci. Signal Transduct. Knowl. Environ 187:RE9. [DOI] [PubMed] [Google Scholar]
- 149.Liu Y-W, Su AI, Schmid SL. 2012The evolution of dynamin to regulate clathrin-mediated endocytosis: speculations on the evolutionarily late appearance of dynamin relative to clathrin-mediated endocytosis. Bioessays 34:643–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elde NC, Morgan G, Winey M, Sperling L, Turkewitz AP. 2005. Elucidation of clathrin-mediated endocytosis in tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLOS Genet. 1:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang KM, D’Hondt K, Riezman H, Lemmon SK. 1999. Clathrin functions in the absence of heterotetrameric adaptors and AP180-related proteins in yeast. EMBO J. 18:3897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Reubold TF, Faelber K, Plattner N, Posor Y, Ketel K, et al. 2015. Crystal structure of the dynamin tetramer. Nature 525:404–8 [DOI] [PubMed] [Google Scholar]