

This work is motivated by an interest in understanding the characteristics and advantages of a relatively newly discovered enzyme mechanism involving filament formation. SgrAI is an enzyme responsible for protecting against viral infections in its host bacterium and was one of the first such enzymes shown to utilize such a mechanism. In this work, filament formation by SgrAI is disrupted, and the effects on the speed of the purified enzyme as well as its function in cells are measured. It was found that even small disruptions, which weaken but do not destroy filament formation, eliminate the ability of SgrAI to protect cells from viral infection, its normal biological function. Simulations of enzyme activity were also performed and show how filament formation can greatly speed up an enzyme’s activation compared to that of other known mechanisms, as well as to better localize its action to molecules of interest, such as invading phage DNA.
KEYWORDS: antiphage mechanism, enzyme kinetics, enzyme mechanism, filament forming enzymes, kinetic simulations, phage infection, protein structure-function, restriction endonuclease
ABSTRACT
Here, we investigate an unusual antiviral mechanism developed in the bacterium Streptomyces griseus. SgrAI is a type II restriction endonuclease that forms run-on oligomer filaments when activated and possesses both accelerated DNA cleavage activity and expanded DNA sequence specificity. Mutations disrupting the run-on oligomer filament eliminate the robust antiphage activity of wild-type SgrAI, and the observation that even relatively modest disruptions completely abolish this anti-viral activity shows that the greater speed imparted by the run-on oligomer filament mechanism is critical to its biological function. Simulations of DNA cleavage by SgrAI uncover the origins of the kinetic advantage of this newly described mechanism of enzyme regulation over more conventional mechanisms, as well as the origin of the sequestering effect responsible for the protection of the host genome against damaging DNA cleavage activity of activated SgrAI.
IMPORTANCE This work is motivated by an interest in understanding the characteristics and advantages of a relatively newly discovered enzyme mechanism involving filament formation. SgrAI is an enzyme responsible for protecting against viral infections in its host bacterium and was one of the first such enzymes shown to utilize such a mechanism. In this work, filament formation by SgrAI is disrupted, and the effects on the speed of the purified enzyme as well as its function in cells are measured. It was found that even small disruptions, which weaken but do not destroy filament formation, eliminate the ability of SgrAI to protect cells from viral infection, its normal biological function. Simulations of enzyme activity were also performed and show how filament formation can greatly speed up an enzyme’s activation compared to that of other known mechanisms, as well as to better localize its action to molecules of interest, such as invading phage DNA.
INTRODUCTION
The coevolution of phage and antiphage activities, in what has been called the phage-host arms race, is thought to be among the oldest and largest in scale coevolutionary system on Earth (1, 2, 3). From this system, many useful biomacromolecules have been discovered. For example, the type II restriction endonucleases (REs) have shown great utility in recombinant DNA technology due to their very high sequence specificity and rapid double-stranded DNA cleavage abilities (4). Newer technologies include the CRISPR enzymes, which allow relatively more convenient programming of DNA cleavage specificity (5). Still, the great diversity of REs contrasts with the relatively limited number that have been fully studied, suggesting that much is left to be discovered in this interesting class of enzymes (6). Our studies with the type II RE SgrAI from Streptomyces griseus led us to propose a new mechanism of enzyme regulation involving filament formation (7, 8).
Filament and run-on oligomer formation by noncytoskeletal enzymes is a relatively newly discovered phenomenon, being first described in 2009 to 2010 for such diverse enzymes as Ire1 (the unfolded protein response nuclease-kinase) (9), CTP synthase (10, 11), ACC (acetyl-coenzyme A [CoA] carboxylase) (12), and SgrAI (7). At approximately the same time, large-scale screens for protein localization using fluorescence microscopy showed unexpectedly that many enzymes formed filaments in response to particular metabolic conditions or other stimuli in cells (11, 13–15). The term “run-on oligomer” (ROO) filament is used here to describe an assembly of an enzyme into a filament by the successive addition of enzymes at either end, and which, in principle, could extend indefinitely (8, 16). ROO filament formation by SgrAI was first proposed in 2010 based on behavior in analytical ultracentrifugation and native gels (7) and subsequently using ion-mobility mass spectrometry (17). The enzymatic activity of SgrAI was found to be activated in the ROO and to possess an altered (expanded) DNA sequence specificity (7, 8). The three-dimensional cryoelectron microscopy (cryo-EM) structure of the ROO filament formed by the assembly of SgrAI/DNA complexes shows a left-handed helical arrangement with approximately four DNA-bound dimers of SgrAI per turn (Fig. 1A to D show different views of an individual SgrAI/DNA complex) (8). In the ROO filament helix, both the DNA and SgrAI form stabilizing interactions with neighboring SgrAI/DNA complexes (Fig. 1A, E, and F) (8).
FIG 1.

Run-on oligomer (ROO) filament of SgrAI/DNA complexes and sites of point mutations. (A) The run-on oligomer filament structure with 11 SgrAI/DNA complexes, each colored separately. The red box indicates the area shown in panels E and F. (B) Cartoon and surface rendering of one SgrAI/DNA complex in the same orientation as the salmon-colored complex in panel A. (C) View rotated 180° about a vertical axis relative to that in panel B. (D) View rotated 90° from that in panel C, showing the same orientation as the lightest blue-colored complex in panel A. (E) Zoom-in on the boxed area of panel A, showing the interface between two adjacent SgrAI/DNA complexes within the run-on oligomer filament. Selected residues mutated in this study are indicated. (F) Approximately 90° rotation from panel B, showing the positions of selected residues mutated in this study.
SgrAI is a type II RE cleaving primary sites (CR|CCGGYG, with R indicating A or G, Y indicating C or T, and | indicating cleavage site) and secondary sites (CR|CCGGGG and CR|CCGGY[A or C or T]) in duplex DNA in a magnesium ion-dependent mechanism (18, 19). Its cleavage of secondary sites occurs only under particular conditions, namely, when present on the same DNA molecule as a primary site, or alternatively, when in the presence of high concentrations of both SgrAI and primary site DNA sequences on separate DNA molecules (7, 20–23). The same conditions leading to cleavage of secondary sites by SgrAI also accelerate the cleavage of primary sites by SgrAI over 200-fold (the acceleration of secondary site cleavage is approximately 1,000-fold) (7, 16, 20, 22). Further, under these conditions, SgrAI forms the ROO filament described above, thought to stabilize the activated state of SgrAI (8). The role of this unexpected structure was not known and has been the subject of recent investigations.
Being a relatively newly described enzyme mechanism, several fundamental questions are of interest, including (i) how the ROO filament accelerates the formation of the product of the reaction (i.e., cleaved DNA) without trapping it in the filament, (ii) whether or not the assembly and/or the disassembly of the ROO limits the overall rate of reaction, (iii) what the growth and dissolution mechanism of the ROO filament is (e.g., from the ends only or occurring anywhere in the filament), and (iv) what are the special advantages, if any, of the ROO filament mechanism (over more conventional mechanisms) that evolved due to the particular biological niche of SgrAI. The first three issues were addressed in prior work (24, 25), which showed that ROO filament assembly is rate limiting in in vitro reactions at low concentrations of SgrAI and DNA. It was also found that DNA cleavage is rapid in the ROO filament, faster than dissociation of the ROO filament, making the reaction pathway efficient, since DNA cleavage is much more likely with each addition to the ROO filament prior to its dissociation. However, the relatively rapid dissociation kinetics limits any trapping of cleaved DNA within the filament. As for the growth mechanism, previous work concluded that disassembly of ROO filaments can occur at any junction between adjacent SgrAI/DNA complexes, and assembly must also be possible from two ROO filaments of any size.
In the current work, we address the fourth question of whether there are particular advantages to the ROO filament mechanism, perhaps relating to requirements and challenges of specialized biological niches. First, we show that mutations that disrupt the ROO filament also eliminate the ability of SgrAI to protect a host bacterium from invading phage. We find that even relatively moderate disruptions of ROO filament formation appear to render the protection to nearly that of the parent strain, as if the enzyme were not even present in the cell. This indicates that the speed of SgrAI, in terms of rapid activation and DNA cleavage, is critical to the biological function of SgrAI.
Using kinetic modeling and rate constants derived and determined from prior work (24, 25), we next simulate the in vivo kinetics of SgrAI activity. We also build an alternative, non-ROO model to use side by side in simulations in order to discover any advantages or limitations inherent to the ROO filament mechanism. In order to simulate the reaction in vivo, we estimate concentrations of SgrAI and DNA in the cell and also estimate the local concentration of SgrAI when bound to sites on the same contiguous DNA molecule. Using these concentrations, and the kinetic model and rate constants derived in prior work (24, 25), we discover that while the relatively low association rate constant of SgrAI/DNA complexes into the ROO filament does in fact limit the rate of reaction, it importantly is also the source of the proposed sequestration effect that protects the host genome from the potentially damaging activity of SgrAI. This is because it limits ROO filament formation within the cell to only those SgrAI enzymes bound to the same DNA molecule, meaning in vivo SgrAI would cleave only invading phage DNA and not the host genome. SgrAI, a type II RE, acts as a bacterial defense system to protect its host bacteria from such invading and pathogenic phage. However, the activity of SgrAI must be controlled such that it does not perform damaging DNA cleavages on the host DNA.
Comparing simulations of the ROO filament reaction to a non-ROO (binary association) reaction mechanism, we find that the ROO filament mechanism has a significant kinetic advantage over the non-ROO mechanism in the speed of DNA cleavage. Further, the advantage appears to derive from the two distinct ways SgrAI/DNA complexes and ROO filaments can associate (such as at either end of the ROO filament). In addition, investigation of the cleavage of secondary sites by SgrAI, which differ from primary sites by 1 bp, shows that even an accelerated binary reaction (with an increased assembly rate constant) is inferior to the ROO filament mechanism, due to greater host damage resulting from the less effective sequestration found in the non-ROO mechanism.
The phage-host arms race is complex, with diverse mechanisms to evade infection on the part of the host and to evade restriction on part of the phage (3). For example, negative selective pressure should result in the reduction of the number of restriction sites on phage, and genome analyses suggest this is the case (3, 26–29). However, most bacteria contain multiple RM (for restriction-modification) systems, and a positive correlation exists between genome size and the number of such systems (3). The genome size of S. griseus predicts the presence of 4 to 5 RM systems, each possibly containing a unique recognition sequence (3). The evasion of multiple RM systems simultaneously through mutations to eliminate recognition sequences, without affecting genome functions in coding, replication, and expression, would be challenging. The evolutionary pressure exerted on the SgrAI restriction-modification system, which has produced the unusual filament-forming enzyme mechanism, may originate with the relatively large genome of its host, Streptomyces griseus. The larger genome results in many more potential recognition sites which must be methylated by the SgrAI methyltransferase for protection, lest it be cleaved by the SgrAI endonuclease. The surprisingly low DNA cleavage rate of SgrAI (0.1 min−1), as well as the longer and therefore more rarely occurring 8-bp recognition sequence, may both have evolved to reduce this pressure. However, these same properties, the low cleavage rate and rare cleavage site, would also limit the effectiveness of SgrAI against phage infection. The 200- to 1,000-fold activation of DNA cleavage activity, and expansion from 3 DNA recognition sites to 17 upon ROO filament formation, results in many more possible cleavages in the phage genome, which may limit phage infection to a greater extent. For example, phage genomes with more restriction endonuclease cleavage sites are restricted to a greater degree than those with fewer (30–32). The ROO filament stabilizes the activated conformation of SgrAI, yet such stabilization could also in principle be performed by a simple binary mechanism involving the assembly of only two copies of SgrAI. The specific advantage of forming a filament compared to a finite oligomer, such as in a binary system, is indicated by this work to be in both the combined (and competing) properties of speed (faster activation) and sequestration (limiting secondary site cleavage to the invading phage DNA and not the host), resulting from the particular biological niche found in Streptomyces griseus.
RESULTS
DNA cleavage.
Single-turnover DNA cleavage assays were performed to measure the basal rate of DNA cleavage by SgrAI (i.e., that in the absence of activating conditions) as well as under activating conditions (i.e., where ROO filaments normally form, which have accelerated DNA cleavage properties). The basal, unactivated rate of DNA cleavage was measured with an 18-bp DNA containing a single primary recognition site (18-1; see Materials and Methods for the sequence), which does not activate SgrAI but can be cleaved, albeit at a low rate (0.094 ± 0.015 min−1 in the case of wild-type SgrAI). This DNA cleavage rate constant should not be perturbed by the mutations, if the mutations affect only formation of the ROO filament. Table 1 shows this basal DNA cleavage rate constant for the wild type and each mutant of SgrAI. The values range from 0.014 ± 0.003 min−1 (R24E) to 0.14 ± 0.03 min−1 (I59E). Most are within one standard deviation of the wild-type value, and all show measurable activity. Hence, the mutations did not disrupt DNA binding by SgrAI, or the unactivated DNA cleavage activity, as expected.
TABLE 1.
DNA cleavage rate, phage titer, and protein expression level analyses of SgrAI enzymes
| SgrAI enzyme | Phage titera(PFU/μl) | DNA cleavage rate constant(min−1) |
|
|---|---|---|---|
| Unactivatedb | Activatedc | ||
| WT | NPd | 0.094 ± 0.15e | 22 ± 7e |
| T4D | 3.8 × 104 ± 0.5 × 104 | 0.058 ± 0.002 | 0.097 ± 0.009 |
| S6D | 4.3 × 104 ± 1.0 × 104 | 0.03 ± 0.01 | 0.19 ± 0.06 |
| I7E | 4.5 × 104 ± 0.3 × 104 | 0.091 ± 0.008 | 0.47 ± 0.09 |
| R24E | 3.7 × 104 ± 0.6 × 104 | 0.014 ± 0.003 | 0.028 ± 0.004 |
| N25E | 5.4 × 104 ± 0.7 × 104 | 0.05 ± 0.01 | 1.2 ± 0.2 |
| P27W | 6 × 104 ± 1 × 104 | 0.037 ± 0.05f | 0.14 ± 0.01f |
| P27G | 4.6 × 104 ± 0.4 × 104 | 0.06 ± 0.002f | 0.12 ± 0.003f |
| Q34D | 6.4 × 104 ± 1.2 × 104 | 0.04 ± 0.02 | 1.180 ± 0.2 |
| I51E | 4.9 × 104 ± 0.1 × 104 | 0.05 ± 0.01 | 0.26 ± 0.07 |
| S56E | 4.9 × 104 ± 0.3 × 104 | 0.08 ± 0.01g | 0.17 ± 0.03g |
| S56Q | 2.9 × 104 ± 0.3 × 104 | 0.08 ± 0.02g | 5.5 ± 1.8g |
| A57E | 4.6 × 104 ± 1.4 × 104 | 0.098 ± 0.002g | 0.39 ± 0.06g |
| A57Q | NP | 0.09 ± 0.01g | 15 ± 8g |
| I59E | 6.3 × 104 ± 0.2 × 104 | 0.14 ± 0.03 | 0.026 ± 0.006 |
| M62E | 2.6 × 104 ± 0.8 × 104 | 0.021 ± 0.001 | 3.2 ± 0.4 |
| R84E | 3.6 × 104 ± 0.5 × 104 | 0.082 ± 0.005 | 2.6 ± 0.2 |
| R127A | 6 × 104 ± 3 × 104 | 0.04 ± 0.04 | 7.6 ± 0.6 |
| R131A | 5.6 × 104 ± 0.5 × 104 | 0.10 ± 0.01g | 0.28 ± 0.02g |
| R134A | 5.2 × 104 ± 0.8 × 104 | 0.10 ± 0.013g | 0.8 ± 0.2g |
Value for the parent strain [Tuner (DE3) (Novagen, Inc.) with MspI.M] is 5.7 × 104 ± 1.1 × 104 PFU/μl.
Single-turnover DNA cleavage rate constant of 1 nM 32P-labeled 18-1 with 1 μM SgrAI enzyme.
Single-turnover DNA cleavage rate constant of 1 nM 32P-labeled 18-1 with 1 μM SgrAI enzyme and 1 μM PC DNA (activating DNA).
NP, no plaques detected.
From Park et al. (7).
From Park et al. (61).
From Shah et al. (16).
The single-turnover DNA cleavage rate constant under activating conditions was also measured for each SgrAI mutant. These assays include 1 μM PC DNA, which activates SgrAI into forming ROO filaments with accelerated DNA cleavage activity (7, 8, 16). PC DNA is a precleaved 40-bp DNA containing a single primary site sequence (see Materials and Methods for the sequence). Two copies of PC DNA self-anneal to form a 40-bp DNA with nicks (missing the phosphate due the absence of 5′ phosphates on synthetic DNA) at the SgrAI cleavage sites (CR|CCGGYG, where | indicates a cleavage site). This 40-bp DNA binds to SgrAI the same as an uncleaved version would and favors ROO filament formation by forming stabilizing interactions with other SgrAI/DNA complexes (Fig. 1), including those containing the 18-1 DNA. In this way, accelerated cleavage of 18-1 is induced. Mutations which disrupt the ROO filament by removing favorable, or introducing unfavorable, interactions between SgrAI/DNA complexes will result in less activation and therefore a lower DNA cleavage rate under normal activating conditions (i.e., with 1 μM PC DNA). Table 1 shows the results; most mutations severely affect this rate constant (wild type is 22 ± 7 min−1; most mutations reduce this rate constant to ∼1 min−1 or less). Those with intermediate effects on this rate constant include S56Q, M62E, R84E, and R127A (rate constants of 3 to 8 min−1). Only the mutation A57Q gives a wild-type rate constant (within 1 standard deviation of that of wild-type SgrAI, 15 ± 8 min−1), thereby appearing to have minimal effects on disrupting activation (and ROO filament formation) of SgrAI.
Residues S56, A57, and R127 are all at a protein-DNA interface occurring between neighboring SgrAI/DNA complexes (Fig. 1). As described previously for S56 and A57 mutations (16), the introduction of a negative charge at the protein interface (i.e., mutations S56E and A57E) creates electrostatic repulsion with the DNA, but mutation to a neutral side chain (i.e., mutations S56Q and A57Q) does not, hence the smaller effects on activated DNA cleavage by those mutations. In the case of R127A, removal of a positive charge at this interface also weakens the ROO filament but perhaps not as effectively as those near R131A and R134A (both with rate constants measured under accelerating conditions of less than 1 min−1) (Table 1). As can be seen in the cryo-EM model of the ROO filament (Fig. 1E), R127 is further from the protein-DNA interface than R131 and R134, providing an explanation for the smaller effect when mutated. M62 and R84 are at a different interface, one formed between the protein chains of two adjacent SgrAI/DNA complexes in the run-on oligomer filament (Fig. 1E and F). R84 and M62 are both more distant from this interface than the other residues mutated in this study. Residues T4, S6, I7, R24, N25, P27, Q34, I51, and I59 are also at the protein-protein interface between adjacent SgrAI enzymes in the ROO filament (Fig. 1E and F), and mutation of these residues results in severe effects on the activation of SgrAI (Table 1).
Phage challenge.
The phage challenge assay uses cells [i.e., Tuner (DE3) Escherichia coli; Novagen, Inc.] expressing wild-type or mutant SgrAI, MspI.M (the MspI methyltransferase with CCGG specificity), and a mutant of the E. coli lambda phage, λJL801, which is incapable of lysogeny (33). The SgrAI protein is expressed from an inducible T7 promoter on a high-copy-number plasmid, while the MspI.M methyltransferase is expressed from its natural promoter, showing nearly consensus −35 and −10 sequences (34, 35) in a plasmid derived from the low-copy-number plasmid pACYC184. The phage challenge assay involves mixing λJL801 phage at different dilutions with cells expressing both MspI.M and wild-type or mutant SgrAI and counting the number of plaques after overnight growth. Each plaque represents a successful infection by λJL801 phage and is quantified as the number of PFU per μl of our stock of purified phage. The parent bacterial strain (having only MspI.M plasmid) gave 5.7 × 104 ± 1.1 × 104 PFU/μl. When cells express wild-type SgrAI, no plaques were found, even using the highest concentration of phage available. In contrast, all mutant SgrAI but one led to the formation of plaques, most near the count found with the parent strain (Table 1). The mutation A57Q, which also had the least effect (if any) on the accelerated (activated) DNA cleavage rate (Table 1), also appeared to completely protect SgrAI from phage infection, showing no plaques even at the highest concentration of phage available (Table 1). Figure 2 plots the plaque titer (PFU/μl) versus the activated DNA cleavage rate constant (min−1) measured for that mutant (or wild-type) SgrAI. As can be seen, the greatest protection (i.e., NP [no plaques observed], at least 104-fold protection relative to the parent strain) is found with the fastest enzymes (wild type and A57Q). Some protection may occur with enzymes showing activation levels at 10% to 35% of that of the wild type (R84E, S56Q, R127A, and M62E), although only slight, if any, protection is found (Fig. 2).
FIG 2.

Plot of phage titer versus activated single-turnover DNA cleavage rate constant. Error bars show ±1 standard deviation (red). Data points for selected mutant or wild-type SgrAI enzyme are labeled (green).
Verification of protein expression.
Cells used in the phage challenge assay were analyzed by Western blotting to confirm expression of SgrAI proteins (Fig. 3). Levels of expression were assessed by determining the relative concentration of protein in each lane and normalizing to that found for wild-type SgrAI (see Materials and Methods). The values were corrected for dilution and the estimate of cells used in each lane (see Table S1 in the supplemental material). Values varied from 29% to 108% (relative to expression of wild-type SgrAI), showing that all mutant SgrAI proteins were expressed. Although the expression level varied, it is uncorrelated with protection against phage measured by the phage challenge assay (Fig. 4).
FIG 3.

Western analysis of wild-type and mutant SgrAI expression levels. The middle gel was spliced to remove irrelevant lanes.
FIG 4.

No correlation is found between plaque titer and protein expression level (levels normalized to that of wild-type SgrAI).
Simulation of in vivo reactions: local concentration and sequestration.
One of the important distinctions between the in vitro and in vivo reactions is the concentration of DNA. In vitro, the concentration of DNA is experimentally controlled, and the average size of the ROO filament and the activation of DNA cleavage by SgrAI are thereby controlled as well. In vivo, DNA concentrations will be limited to one copy of the bacterial genome and one or perhaps more copies of the invading phage DNA. In terms of concentration, this is approximately 3 nM for one copy of DNA per S. griseus cell (Table S2). The estimation of the concentration of SgrAI in the cell is less certain, since the number of copies per cell is unknown, but with 100 copies the concentration would be approximately 300 nM (Table S2). At this concentration, sufficient binding should occur between SgrAI and its recognition site in DNA (equilibrium dissociation constant [KD] of 0.057 ± 0.009 nM) (7); however, at 3 nM SgrAI/DNA complex virtually no ROO assembly (when sites are on separate DNA molecules) is expected, and therefore no activation of SgrAI-mediated DNA cleavage would occur (7). The reason for this, as well as the dependence of the observed DNA cleavage rate constant on the concentration of SgrAI bound to DNA (7), is the very low association rate constant for SgrAI/DNA complexes into the ROO filament (i.e., 1.3 × 105 M−1 s−1) (24, 25), giving a calculated rate of association of only (1.3 × 105 M−1 s−1)(3 × 10−9 M)(3 × 10−9 M) = 1 × 10−12 M/s or 1 × 10−3 nM/s.
However, this low association rate constant, although preventing reactions between SgrAI bound to sites on separate DNA molecules, is sufficiently fast to allow association when on the same DNA molecule, owing to the local concentration effect. When two species, such as SgrAI, are bound to the same molecule, like contiguous DNA, they are constrained in space relative to each other and can act as if their concentrations were much higher. This higher concentration is termed the local concentration and is calculated considering the average distance between the two species, in this case, the two DNA-bound SgrAI enzymes. Table S2 provides the calculations, which include estimation of the average number of SgrAI recognition sites within a typical phage genome, as well as their distance apart in base pairs. A radius of gyration was used to estimate the average distance in angstroms between them and then used as the diameter of a sphere for a volume to be calculated (36). Given the number of DNA-bound SgrAI (two in this example) and the volume of the sphere, a concentration can be calculated (80 nM). This means that although their actual concentration is 3 nM each (each bound to one molecule of DNA in the cell, i.e., one phage genome), they occupy the same space two separate complexes would occupy if at 80 nM. Note that this concentration would be even greater for secondary-site sequences, which occur much more frequently (with 14 different sequences) and hence are closer together. At 80 nM, much faster association is expected, (1.3 × 105 M−1s−1)(80 × 10−9 M)(80 × 10−9 M) = 8 × 10−10 M/s, or 0.8 nM/s, 800 times faster than the association of complexes on separate DNA molecules (i.e., phage and host genomes). Both the relatively low association rate constant (1.3 × 105 M−1 s−1) and the requirement for an association step between SgrAI/DNA complexes before DNA cleavage are responsible for the control of SgrAI activation by the local concentration effect, which results in sequestration. Sequestration of activated SgrAI on the DNA that contains the primary sites is important, since although primary sites on the host DNA are protected from SgrAI by methylation, in principle secondary sites could be cleaved by activated SgrAI, causing damage to the host genome. Hence, this elegant mechanism may have evolved to ensure sequestration of activated SgrAI (which cleaves secondary as well as primary recognition sites) on only the invading phage DNA (see below).
Simulation of in vivo reactions of SgrA with phage DNA.
Because cleavage of sites on the same DNA molecule is different in some respects from the experimental system used in our prior kinetic investigations (25), a new kinetic model was created to simulate the cleavage of SgrAI recognition sites on phage (and/or host) DNA (Table S3). This model differs in using one type of DNA site rather than two, which can both be cleaved and also result in activation of other SgrAI/DNA complexes when bound by SgrAI. Previous kinetic models (24, 25) use two types of DNA, the reporter DNA (i.e., 18-1), which can be cleaved, and an activator DNA (i.e., PC DNA), as also used in the single-turnover DNA cleavage reactions described above. In the in vivo case, and considering only primary sites, only one type of site will be found, and that site can both be cleaved by SgrAI as well as activate it by stimulating ROO filament formation. Hence, the model for activity of SgrAI on phage DNA contains only one type of DNA recognition site, which is capable of activating SgrAI (by inducing filamentation) and can do so whether or not the bound DNA is cleaved (16).
For comparison to a non-ROO filament mechanism, equations for what we refer to as a Binary mechanism are included to allow side-by-side comparisons between the two mechanisms. The same rate constants (derived from prior work [25]) were used for analogous steps in both kinetic mechanisms (Table S4). Finally, to simulate the in vivo case, the DNA concentrations used were 80 nM to mimic the local concentration of sites when present on the same contiguous DNA and 3 nM when simulating reactions occurring on separate DNA molecules. Due to software limitations, the ROO filament model was limited to a size of 3 SgrAI/DNA complexes; however, this is not unrealistic given the predicted number of primary sites on the typical phage DNA (Table S2). Dissociation of cleaved DNA from SgrAI is considered reversible in this simulation and is discussed further below.
Figure 5A shows the results when the ROO filament mechanism (red and blue) is compared to the closed-ended non-ROO Binary mechanism (green and pink). At 80 nM SgrAI/DNA (E/DNA for the Binary reaction, E being the hypothetical enzyme in the Binary mechanism), both ROO filament and Binary mechanisms show robust DNA cleavage. However, the ROO filament mechanism has a very clear advantage (approximately 2-fold) in the rate of cleaving DNA (Fig. 5A, compare the red dotted lines to the green dotted lines of the Binary model). Some cleaved DNA is released (∼25%, solid red and green lines, from the ROO filament and Binary models, respectively) (Fig. 5A). Some of the cleaved DNA is still held in ROO filaments or Binary assemblies (Fig. 5A, blue and pink lines, respectively). The remainder of the cleaved DNA is bound to SgrAI but is not in an assembly.
FIG 5.

Model of phage DNA cleavage and comparison of ROO and binary mechanisms. Cleaved DNA bound to SgrAI or the binary enzyme is shown in dotted red (ROO mechanism) or green (binary mechanism). Cleaved DNA released from SgrAI or the binary enzyme is shown in solid lines (red, ROO mechanism; green, Binary). Concentrations of ROO filaments (with cleaved and uncleaved DNA) are blue, and Binary complexes are purple. See Tables S3 and S4 for model details and rate constants. (A) Simulation of DNA sites in phage DNA. Starting concentration was 80 nM DNA, the estimated local concentration of SgrAI bound to two primary sites present on the same DNA molecule. (B) As for panel A, but the Binary non-ROO mechanism is now set to allow for two ways to form assemblies. (C) As for panel A, but with rebinding of cleaved DNA set to a lower off-rate constant to mimic the lower concentrations of DNA free in the cell (see the text for details). (D) As for panel A, but with 3 nM DNA to mimic reactions between separate molecules in the cell (i.e., host genome and phage DNA).
To investigate the origin of the advantage of the ROO filament mechanism, the Binary mechanism was altered to have one feature unique to the ROO filament mechanism, namely, the two ways each SgrAI/DNA complex (or E/DNA in the case of the Binary reaction) can come together (Fig. 6, compare the red complexes of the ROO mechanism to the green ones of the Binary mechanism). Most (but not all) of the advantage of the ROO model is lost (Fig. 5B). This implies that the run-on nature of the ROO filament (where SgrAI/DNA complexes may bind to either end of a filament or even a single SgrAI/DNA complex) is the origin of the kinetic advantage over the Binary mechanism.
FIG 6.
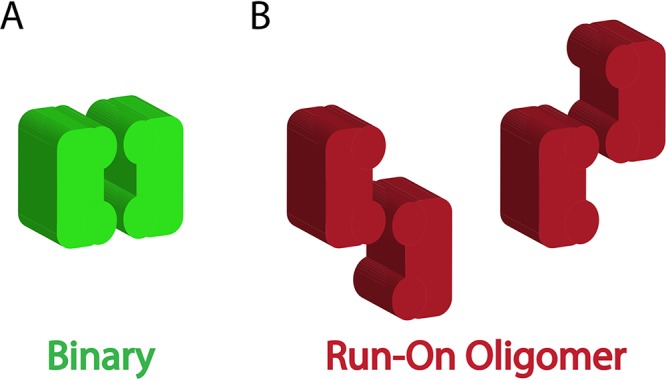
Possible association patterns in a simple Binary association and the run-on oligomer filament mechanism. (A) Only one type of association is found with the closed-ended Binary model. (B) Two ways are found for enzyme complexes to associate in the ROO filament mechanism.
The simulations of Fig. 5A and B allowed for rebinding of the cleaved DNA; however, the simulation did not allow for the inclusion of the change in relative concentrations (local to actual). The initial concentration was set to 80 nM to simulate the local concentration; however, upon cleavage and dissociation, local concentration effects no longer apply. The actual concentration of DNA in the cell was estimated at 3 nM (for 1 copy per cell, as described above). The KD for SgrAI binding to PC DNA (7) in the presence of 10 mM Mg2+ has been measured to be 14 nM. To simulate the change in concentration from 80 nM to 3 nM actual concentration, the rebinding rate constant was made 10-fold lower. Figure 5C shows the result. The total amount of cleaved sites with time is not affected by this change (Fig. 5C, dotted lines); however, more of the cleaved DNA is shown free of SgrAI, as expected. This leads to lower final ROO filament and Binary complex concentrations (Fig. 5C, blue and purple lines). This simulation actually overestimates the amount of ROO and Binary assemblies, since those would also change from local to actual concentrations upon separation of the DNA sites to which they bind, although this change in concentration was not included in the simulation. Further, cleaved DNA is expected to dissociate into the two cleaved products (an estimated KD of the self-association of 375 nM [24]), limiting reassociation to SgrAI.
To simulate DNA cleavage on separate DNA molecules (i.e., SgrAI bound to a site on a phage DNA and a site on the bacterial host genome), the simulation shown in Fig. 5D was performed. In this case, the initial DNA concentration was set to 3 nM. Only a small amount of DNA cleavage is seen in 200 s, the time it takes to cleave nearly 90% of the sites shown in Fig. 5A, indicating that minimal cleavage of sites on the host are due to SgrAI activated by binding to sites on the phage DNA. Further, primary sites on the host DNA would be methylated and thereby protected and unable to be cleaved via this pathway. Therefore, the sequestration effect is likely more important for protecting from cleavage of secondary sites on the host, since those would require ROO filament formation with SgrAI bound to unmethylated primary sites, only available on the invading DNA (see below for further discussion).
Finally, Fig. 5D also shows that the sequestration effect is not unique to the ROO filament mechanism, as the non-ROO Binary mechanism (Fig. 5D, green) also shows very low DNA cleavage at the low concentrations of DNA in the cell but rapid DNA cleavage at the estimated local concentration when sites are present on the same contiguous DNA molecule (Fig. 5A, green).
Simulations with secondary-site DNA sequences.
To investigate cleavage of secondary sites, a modified kinetic model was created (Table S5). Secondary sites differ from primary ones in one base pair, at either the 7th or 8th position of the recognition sequence (primary sites include CRCCGGYG, where R is A or G and Y is C or T, and secondary sites include CRCCGGGG and CRCCGGYX, where X is A, C, or T and underlining indicates secondary site sequence substitutions). Cleavage of secondary sites by SgrAI is nearly undetectable unless assembly into an ROO filament with SgrAI (bound to a primary site) occurs (7, 16). In that case, the observed rate constant for cleavage of secondary-site DNA is generally ∼2-fold lower than that of the primary site and is dependent on the concentration of SgrAI bound to primary site DNA (hence, the cleavage rate is also limited by ROO filament assembly). To model the cleavage of secondary sites by SgrAI, two changes were made to the kinetic model. First, assembly of SgrAI/secondary-site DNA complexes into the ROO filament (or Binary complex) was set to 2-fold lower than that of SgrAI/primary site complexes. Second, an additional equation is included to allow for the two types of Binary complexes: that with two SgrAI/primary site DNA complexes and that with one primary site- and one secondary site-bound SgrAI (the ROO mechanism already allows for both types of associations). Tables S6 and S7 give the rate constants and concentrations of species used in the simulations. In addition, using the same logic as that shown in Table S2, the local concentration of secondary sites relative to primary sites was calculated to be 1 μM (for the nearest pairs of sites).
Simulations were carried out at 1 μM DNA concentration (Fig. 7A and B, solid lines) to simulate cleavage of secondary sites in the phage DNA and 3 nM (Fig. 7A and B, dotted lines) to model cleavage of secondary sites on the host genome. In these simulations, it is assumed that the only source of primary sites (which induce the formation of assemblies, i.e., ROO filaments or Binary complexes) are from the phage DNA. Orange lines represent total DNA cleavage with the ROO filament mechanism, and light blue represents that for the Binary. Figure 7A shows the results when the two mechanisms have the same rate constants. Cleavage at 1 μM DNA (representing phage DNA) is faster (2-fold) in the ROO filament mechanism (Fig. 7A, orange solid line), and very little cleavage occurs with 3 nM DNA (representing host DNA) in both mechanisms (Fig. 7A, dotted lines), demonstrating the sequestration of activated DNA cleavage away from the host DNA. Although very little, Fig. 7A shows that some cleavage of the host DNA (orange and blue dotted lines) is predicted. Upon cleavage of primary sites in phage, the phage DNA should dissociate from SgrAI and separate into fragments and undergo degradation (37), with little reassociation with SgrAI and into ROO filaments (Fig. 5C, blue line), ending the threat to host DNA. At 100 s, the time it takes for most primary sites in phage to be cleaved (Fig. 5A, red dotted line), ∼4% of host secondary sites are predicted to be cleaved as well (Fig. 7A, orange dotted line). There are ∼2,500 predicted secondary sites in S. griseus, and 4% is ∼100 secondary sites. Studies have shown that such autoimmunity (i.e., cleavage of host DNA by REs) does occur in RM systems, particularly in the case of more efficient endonucleases, and that double-stranded breaks in the host genome are repaired via the SOS/RecA repair pathway (38). This autoimmunity is tolerated under rapid growth conditions and high nutrient availability, although it is less tolerated when resources are limited (38–40).
FIG 7.

Total cleaved secondary site DNA from the ROO filament and Binary mechanisms with the same and different assembly rate constants. (A) Solid lines simulate cleavage on phage DNA, and dotted lines simulate cleavage of sites on host DNA due to activation by phage DNA. Light blue, total cleavage sites from Binary mechanism; orange, total cleaved sites from ROO filament mechanism. Equations, rate constants, and concentrations are given in Tables S5 to S7. (B) As for panel A, but with rate constants of the Binary mechanism increased 4.6-fold to give the same rate of cleavage on the phage DNA as the ROO filament mechanism. More cleavage of host DNA occurs with the Binary model (compare light blue dotted line to orange dotted line).
Figure 7B shows the results when the assembly rate constant of the Binary mechanism is increased until cleavage at 1 μM DNA (solid lines) matches that of the ROO filament mechanism. Now the Binary mechanism shows much greater cleavage at 3 nM (Fig. 7B, light blue dotted line) than the ROO filament mechanism (Fig. 7B, orange dotted line). Hence, these simulations show that the ROO filament mechanism is also superior to the Binary mechanism in sequestering activated DNA cleavage away from the host DNA. Although DNA cleavage by the ROO filament mechanism is twice as fast as that by the Binary mechanism when utilizing all the same rate constants (Fig. 7A and Table S6), a 4.6-fold increase in the rate constant for assembly of enzyme-DNA complexes into binary complexes is necessary to achieve the same rate of secondary site cleavage by the two mechanisms (Fig. 7B, rate constants k3 and k4, and Table S6). This is likely due to the competition for association that occurs between the two types of enzyme complexes (bound to primary or secondary sites) that necessarily occurs in the Binary mechanism and the fact that enzymes bound to primary sites preferentially self-assemble rather than associating with enzymes bound to secondary sites (Fig. 8, left). Competition is not necessary in the ROO filament mechanism, since any complex can assemble at either end of the ROO filament (Fig. 8, right).
FIG 8.
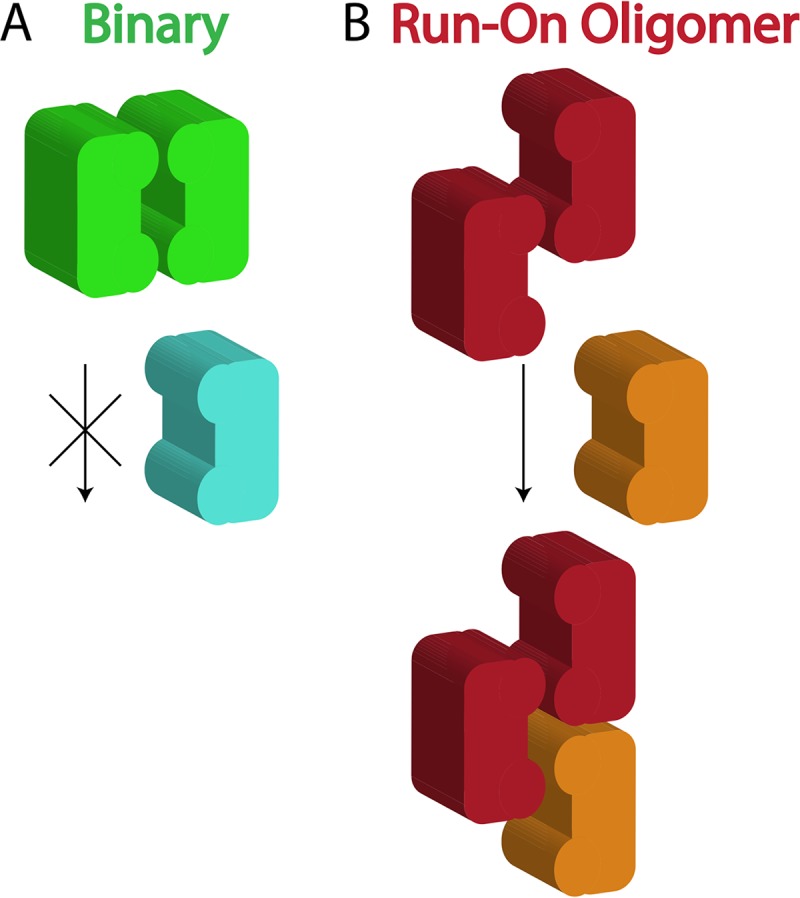
Competition occurs in the Binary mechanism for assemblies but not in ROO filament mechanism. SgrAI bound to primary site (green and red), binding to a second copy of the same or to SgrAI bound to a secondary site (light blue or orange).
DISCUSSION
Compared to other bacterial immune proteins, SgrAI is unusual in several respects (its low unactivated DNA cleavage rate and allosteric activation via filament formation with sequence specificity expansion), although it exhibits similar DNA cleavage rates when activated. For example, the unactivated SgrAI DNA cleavage rate is very low (0.1 min−1) but similar to those of other type II REs under activating conditions (22 min−1, compared to 20 min−1 and 36 min−1 for the type II REs EcoRI and EcoRV, respectively [7, 41, 42]). Note that the single-turnover DNA cleavage rate constant measures all steps from DNA binding to DNA cleavage; however, global kinetic data fitting indicates that the chemical cleavage step (once within the filament) is faster in SgrAI, estimated at 48 min−1 (25). Cas9, a bacterial immune defense enzyme from the Streptococcus pyogenes CRISPR system and of interest in genome engineering applications (43, 44), has been found to have single-turnover DNA cleavage rate constants of 60 min−1 and 30 min−1 for cleavage by the two endonuclease domains (HNH and RuvC, respectively) (45). Cas9, however, remains tightly bound to the cleaved product DNA, while SgrAI (and other type II REs) rapidly dissociate the cleaved DNA product (>24 min−1) following dissociation from the ROO filament (1.8 min−1), freeing it to perform multiple rounds of enzymatic turnover (25, 45, 46).
Here, we include both experiments and simulations to show that the ROO filament mechanism of SgrAI possesses unique characteristics that appear to have evolved to perform the requirements of its biological niche. First, based on the cryo-EM structural model of the ROO filament formed by the assembly of SgrAI/DNA complexes (8), point mutations were designed with the intention of disrupting interfaces and thereby destabilizing this assembly. As a result, the DNA cleavage properties of SgrAI, which are stimulated 200- to 1,000-fold (7, 16) in the ROO filament, are predicted to be hampered by these mutations, since they weaken filament formation. In fact, most mutations diminished the activated DNA cleavage rate (Table 1), as predicted. Further, they did not affect the basal, unactivated DNA cleavage rate, showing that the mutations do not disrupt DNA binding or normal, unactivated DNA cleavage by SgrAI when not in the ROO filament. Hence, these mutations must disrupt ROO filament formation, since activation seen under conditions where wild-type SgrAI forms the ROO filament are not found.
The mutant SgrAI enzymes were then tested for their ability to protect host cells from invading phage DNA (i.e., the phage challenge assay). Western blot analyses were performed to verify expression of mutant proteins (Fig. 3). Expression of wild-type and mutant SgrAI was induced overnight, and then cells were mixed with a modified form of lambda phage incapable of forming lysogens (λJL801) (33) so that all productive infections can be counted by counting plaques formed from cell lysis. The parent host strain, without SgrAI, showed levels of phage infection of 5.7 × 104 ± 1.1 × 104 PFU/μl with our stock of purified λJL801; however, the expression of wild-type SgrAI in these cells resulted in complete protection from infection as far as we could measure, as no plaques were found, even with the highest concentrations of phage tested (this represents a >10,000-fold protection relative to that of the parent strain, within ranges seen with other RM systems [47]). In the case of the mutant SgrAI enzymes, all but one allowed phage infection to proceed, indicating dramatically diminished protection. Many SgrAI mutants showed plaque numbers within error for the unprotected parent strain (Table 1), and these mutant versions of SgrAI also showed the lowest levels of activated DNA cleavage (Fig. 2). Some mutants with intermediate levels of activation (i.e., activated DNA cleavage rate constants 3- to 8-fold lower than that of the wild type but still accelerated relative to the basal DNA cleavage rate), such as R84E, M62E, R127A, and S56Q, may have shown some protection from phage infection, although the protection is very weak. Only A57Q, which showed activated DNA cleavage within error for that of the wild type, gave complete protection from phage infection (Table 1 and Fig. 2). Hence, mutations that disrupt the ROO filament of SgrAI disrupt accelerated DNA cleavage, and importantly, these mutations also diminish the enzyme’s ability to protect its host cell from phage infection. We find that rather than a linear relationship between the activated DNA cleavage rate of SgrAI and protective ability, a step function better describes the plot in Fig. 2, with the most active of the affected mutations (R127A, at 35% of wild-type activity) still completely ineffective at protecting its host cell against phage infection. This suggests that wild-type SgrAI is faster than the R127A mutant in order to be effective against phage infection.
Biological relevance of the phage challenge assay.
The phage challenge assay was used to measure the ability of SgrAI (wild-type and mutant) enzymes to protect host cells from phage infection. Because phage of the natural host of SgrAI, Streptomyces griseus, have not been well characterized, this assay was instead performed in E. coli. Further, the cognate SgrAI methyltransferase has not been identified; therefore, the methyltransferase MspI.M was used instead to create a functional RM system in E. coli. MspI.M methylates the C-5 position of the first C in CCGG sequences; hence, it protects both primary and secondary site sequences within the E. coli genome. It is expressed from a plasmid derived from the low-copy-number plasmid pACYC184 and from its natural promoter, which shows near consensus with −35 and −10 sequences (34, 35). In contrast, SgrAI proteins are expressed from a high-copy-number vector with an inducible T7 promoter giving high levels of expression (48). Relative expression levels of R (endonuclease) and M (methyltransferase) enzymes are important, as too much R leads to autoimmunity (cleavage of host DNA) (38). Too little risks poor antiphage activity due to slower cleavage (from low levels of R) and higher possibilities of escape via methylation of the invading phage DNA by the methyltransferase. Interestingly, studies show that some autoimmunity occurs in natural systems with some REs and is repaired efficiently via the SOS/RecA pathway under nutrient-rich conditions but not nutrient-limited conditions (38–40). The RM system used here is more likely to be tilted toward the R enzyme due to the overexpression of SgrAI proteins compared to the constitutive expression of MspI.M. Even so, most mutant SgrAI enzymes were unable to show any protection against phage infection.
The second key element of the assay is the phage, and lambda phage λJL801 was chosen due to its well-studied biology and the inability of this mutant to form lysogens. Lambda phage contains 6 primary sites and 32 secondary sites in its 50-kb genome, similar in size to two known phage of Streptomyces coelicolor, R4 and PhiC31 (50 and 40 kb, respectively) (49, 50). Hence, the phage challenge assay developed here recapitulates the necessary elements to test the ability of SgrAI to protect its host organism from phage infection.
Simulation of in vivo activity of SgrAI.
Previous work used kinetic modeling to investigate the ROO filament mechanism of SgrAI and, when applied to in vitro data, allowed for the extraction of microscopic rate constants for each step of the reaction mechanism (24, 25). Using those mechanistic models and experimentally determined rate constants, simulations were performed here to model reactions as they may occur in vivo. To better mimic the in vivo reaction, only a single type of recognition site is used in this simulation rather than the two types of DNA used in in vitro reactions (i.e., the reporter and the activator DNA). Also, to better mimic the in vivo reaction, DNA concentrations used were those estimated for the concentration of a single copy of DNA in the cell (3 nM) and that estimated for the local concentration of recognition sites with respect to each other on the same molecule of DNA (80 nM for primary to primary sites, 1 μM for primary to secondary sites). In addition, to compare the ROO filament mechanism to non-ROO mechanisms, we have also constructed a simple Binary model. In the Binary model, activation occurs when two hypothetical enzymes (E), each bound to a DNA site, associate into a dimer. All rate constants are otherwise identical in the simulations with both mechanisms. Finally, simulations to predict cleavage rates of secondary site sequences were also performed.
Advantages of the ROO mechanism.
Finally, the question of advantage of the ROO filament mechanism is addressed by comparing DNA cleavage kinetics via both the ROO filament and non-ROO (Binary) mechanisms. Clearly, the ROO filament mechanism has the advantage (Fig. 5A, red versus green solid and dashed lines) in the rate of cleavage of DNA, and cleavage and release of DNA, under these reaction conditions. Such an advantage may be necessary where speed is required, such as in the race against viral replication (and methylation by the SgrAI methyltransferase) of invading DNA. The rate-limiting assembly of SgrAI/DNA complexes into the ROO filament, which is required to provide sequestration of activated SgrAI, may be a limiting parameter for speed but is compensated for by this kinetic advantage of the ROO mechanism, explaining why a simple Binary mechanism is inferior (although it also possesses the sequestration effect). The origin of the kinetic advantage is found to be due to the two ways SgrAI enzymes may come together (Fig. 6). Hence, the formation of a large ROO filament is not necessary for this kinetic advantage. Indeed, allowing the Binary (non-ROO) mechanism two ways for enzyme association to occur reduced most of the advantage of the ROO filament mechanism (Fig. 5B). A small additional advantage derives from the third or more enzyme additions to the ROO filament. Our results with mutants that disrupt the ROO filament of SgrAI show that the very fast DNA cleavage performed by activated SgrAI is critical for protection against phage infection, since even the fastest affected mutant, R127A, with 35% of WT activity, is completely ineffective (Fig. 2).
Why not merely evolve a faster binary reaction?
Secondary sites differ from primary sites in one base pair, occurring at either the 7th or 8th position of the 8-bp recognition sequence. These sites are not appreciably cleaved by SgrAI without assembly into ROO filaments composed of SgrAI bound to primary site DNA (7, 16). Primary sites on the host DNA are methylated and thereby are protected from cleavage by SgrAI, but secondary sites likely are not; therefore, they may be susceptible to cleavage by activated SgrAI. Hence, the need for sequestration is most relevant to the prevention of cleavage of secondary site sequences on the host DNA. We hypothesized that the ROO filament mechanism has evolved to increase its ability to sequester the DNA cleavage activity of SgrAI to the same copy of DNA containing the activating primary sites, which in vivo would be the invading phage DNA.
This sequestration would serve to protect the host DNA from damaging cleavage at secondary site sequences. The simulations show that the ROO filament mechanism is 2-fold faster than the Binary one due to the two ways assembly may occur (Fig. 6) each time an SgrAI/DNA complex adds to the ROO filament. In fact, the simulations show that when the Binary reaction is allowed a higher (4.6-fold) assembly rate constant such that its accelerated DNA cleavage rate matches that of the ROO filament mechanism (Fig. 7B, solid lines), greater cleavage of secondary sites on the host DNA compared to that of the ROO filament mechanism is predicted (Fig. 7B, light blue dotted line). Thus, these simulations predict that the ROO filament mechanism is superior in both rapid DNA cleavage and in sequestration. The origin of both advantages derives from the multiple ways SgrAI/DNA complexes can assemble in the ROO filament (Fig. 6 and 8). Secondary site-bound SgrAI (Fig. 8, orange) enzymes need not compete with primary site-bound SgrAI (Fig. 8, red) for association into ROO filaments, as assembly interfaces are always available. In contrast, the Binary complex associates more strongly when both SgrAI/DNA complexes contain primary site sequences (Fig. 8, green), and SgrAI bound to secondary site DNA (Fig. 8, light blue) must compete with this complex for association with SgrAI/primary site DNA complexes.
Conclusions.
The ROO filament mechanism of SgrAI appears to have evolved out of phage-host competition, or arms race, one of the oldest evolution-coevolution systems in evolutionary history (1), to accommodate specific challenges. These include reduced activity on the host genome through both a lower DNA cleavage rate and a longer (hence, rarer) recognition site, as well as the ability to become rapidly activated on DNA containing multiple unmethylated primary sites, such as invading DNA, through enzyme assembly into ROO filaments. SgrAI must sequester its DNA cleavage activity on the invading DNA, and it does so by having a low association rate constant that limits assembly into the ROO filament to only those SgrAI enzymes bound to sites on the same molecule of DNA. However, speed is critical to performing SgrAI’s biological role (as evidenced by the mutant study described here), hence ROO filament formation allows for multiple ways for the enzymes to assemble, thereby increasing the rate of this step significantly. The ability to form longer ROO filaments provides for additional speed, and relatively fast dissociation prevents trapping of what may be limiting amounts of SgrAI in the cell (25). Merely increasing the assembly rate constant in a Binary mechanism does not match the advantage of the ROO filament mechanism, and loss of sequestration results. Hence, the ROO filament mechanism is superior to the Binary (nonfilament) mechanism in both speed and sequestration, both of which are important to the biological function of SgrAI.
As discussed in prior work (24, 25), recognition of the filament-forming enzyme mechanisms has until recently been largely limited to the cytoskeletal ATPases and GTPases such as actin and tubulin. However, large-scale screening using newer imaging technologies has allowed for the observation of filament formation by metabolic and other enzymes previously unknown to form such structures (11, 13, 14). The details of the roles of the filaments in enzyme activity have been investigated in only a few such enzymes (9, 12, 51–54), and here we provide the most detailed kinetic and mechanistic investigation of an enzyme filament mechanism to date. The ROO filament of SgrAI no doubt has unique features compared to the other systems, for example, SgrAI is unlikely to form the large-scale filaments seen in many of the fluorescence microscopy studies, which persist over minutes to hours, yet common features likely also result. The detailed structures of run-on oligomers and filaments of most filament-forming enzymes are also not known; exceptions include SgrAI, CTP synthase, acetyl-CoA carboxylase (55), and Ire1, the unfolded response nuclease-kinase (8, 9, 51, 52). Our kinetic investigations complement our structural work (8) in elucidating the features of the SgrAI ROO filament that make it unique, optimized for its biological niche, and advantageous over other mechanisms.
MATERIALS AND METHODS
Protein purification.
Wild-type and mutant SgrAI proteins were expressed with a C-terminal His tag in Tuner (DE3) E. coli, which also contained the pLysS plasmid (Novagen, Inc.) and the MspI.M expression plasmid (pBAK.MspI) (34, 56). MspI.M methylates at the C-5 position of the first cytosine of CCGG sequences and is expressed from its natural promoter in plasmid pBAK.MspI, a derivative of pACYC184 with the natural coding sequence of MspI.M (34, 35). Mutagenesis was performed as described previously (16), and all expression vectors were sequenced fully in the SgrAI gene to verify the point mutation. The proteins were purified using Talon metal affinity resin (Clontech, Inc.) followed by ion-exchange fast-performance liquid chromatography (FPLC) using heparin resin (GE Healthcare Life Sciences). First, the cell lysate was incubated with Talon resin in lysis buffer (50 mM sodium phosphate buffer, pH 8.0 at room temperature [RT], 800 mM NaCl, 10 mM imidazole, and 1 mM β-mercaptoethanol [BME]) for 30 min to overnight. The unbound cell lysate was washed away using wash buffer (50 mM sodium phosphate buffer [pH 8.0 at RT], 300 mM NaCl, 20 mM imidazole, and 1 mM BME), followed by high-salt wash buffer (50 mM sodium phosphate buffer [pH 8.0 at RT], 2 M NaCl, 20 mM imidazole, and 1 mM BME). Finally, the protein was eluted using elution buffer (50 mM sodium phosphate buffer [pH 8.0 at RT], 300 mM NaCl, 250 mM imidazole, and 1 mM BME). For ion exchange FPLC purification, the protein was excessively dialyzed into heparin A buffer (50 mM Tris-HCl [pH 8.0 at RT], 50 mM NaCl, 0.1 mM EDTA, 10 mM BME) and then purified using heparin FF chromatography (GE Healthcare Biosciences) and a gradient of heparin B buffer (50 mM Tris-HCl [pH 8.0 at RT], 1 M NaCl, 0.1 mM EDTA, 10 mM BME). Purity of the protein was confirmed using SDS-PAGE. The purified protein was then aliquoted into single-use aliquots, flash frozen in liquid nitrogen, and stored at −80°C.
DNA preparation.
The oligonucleotides were made synthetically and purified using C18 reverse-phase high-performance liquid chromatography or denaturing PAGE (57). The concentration was measured spectrophotometrically, with an extinction coefficient calculated from standard values for the nucleotides (58). The self-complementary DNA strands, or equimolar quantities of complementary DNA, were annealed by heating to 90°C for 10 min at a concentration of 0.1 to 1 mM, followed by slow cooling to 4°C for 4 to 5 h in a thermocycler or heat block. Sequences of the DNA used are given in Table 2 (recognition sequences are shown in boldface; a vertical line [|] marks the SgrAI-specific cleavage site).
TABLE 2.
DNA sequences used in the single-turnover DNA cleavage assays
| Name | Sequencea |
|---|---|
| PC-top | 5′-GATGCGTGGGTCTTCACA-3′ |
| PC-bot | 3′-CTACGCACCCAGAAGTGTGGCC-5′ |
| 18-1-top | 5′-AAGTCCA|CCGGTGGACTT-3′ |
| 18-1-bot | 3′-TTCAGGTGGCC|ACCTGAA-5′ |
Recognition sequences are shown in boldface; a vertical line (|) marks the SgrAI-specific cleavage site.
The 5′ 32P end labeling of DNA was performed with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Perkin-Elmer, Inc.), followed by removal of excess ATP using P-30 spin columns (Bio-Rad Laboratories, Inc.).
Western analysis to measure protein expression levels.
Western blot analyses were performed with lysates from cells used in the phage challenge assays, with the optical density (OD) of the cells noted before pelleting 1 ml of overnight growth, reconstituting in protein loading buffer (25 mM Tris-HCl, pH 6.8, 2% SDS, 6% glycerol, 0.1 M dithiothreitol [DTT], 0.004% bromophenol blue), heating to 90°C for 5 min, and centrifuging at 10,000 rpm for 10 min. SDS-PAGE was performed on the samples followed by transfer to polyvinylidene difluoride (PVDF) membrane (ThermoFisher Scientific) after soaking in Towbin buffer (25 mM Tris, 192 mM glycine, 20% methanol, 0.1% SDS). Blots were blocked with 3% bovine serum albumin (BSA) in PBST (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl, 0.2% Tween), followed by probing with horseradish peroxidase-labeled primary antibody (mouse anti-His monoclonal antibody; MA1-135; ThermoFisher, Inc.) at a 1:1,000 dilution and then washed 3 times in PBST. For visualization of bands, the blot was soaked in chemiluminescence solution (made from mixing equal parts of 0.78 mg/ml luminol, 0.95 mg/ml p-iodophenol, 0.1 M Tris-HCl, pH 9.35, with 0.03% H2O2 in 0.1 M Tris-HCl, pH 9.35) for 1 min, followed by imaging using a Chemidoc scanner (Bio-Rad Laboratories, Inc.). Bands corresponding to SgrAI proteins were integrated using Image Lab (Bio-Rad Laboratories, Inc.), corrected for dilution and OD, and normalized to levels quantitated for wild-type SgrAI.
DNA cleavage assays.
Single-turnover kinetic measurements of DNA cleavage were performed using 32P-labeled oligonucleotides substrates (1 nM) under conditions of excess enzyme (1 μM SgrAI dimer), with and without the addition of unlabeled PC DNA. All DNA cleavage reactions were performed at 37°C in 20 mM Tris-acetic acid (HOAc) (pH 8.0 at RT), 50 mM KOAc, 10 mM Mg(OAc)2, and 1 mM DTT. Five-μl aliquots were withdrawn at specific time intervals after mixing the enzyme and labeled DNA (100 μl total reaction volume), quenched by addition to 5 μl of quench solution (80% formamide, 50 mM EDTA, 1 mg/ml xylene cyanol dye, and 1 mg/ml bromophenol blue dye), and electrophoresed on denaturing polyacrylamide gels (20% acrylamide-bisacrylamide [19:1 ratio], 4 M urea, 89 mM Tris base, 89 mM boric acid, 2 mM EDTA). Autoradiography of gels was performed without drying using a phosphor image plate exposed at 4°C for 12 to 17 h. Densitometry of phosphor image plates was performed with a phosphorimager (GE Healthcare Life Sciences, Pittsburgh, PA, USA, or Bio-Rad, Inc., Hercules, CA, USA) and integration using Image Lab software (Bio-Rad, Inc., Hercules, CA, USA). The percentage of product formed as a function of time was determined by integrating the density of both cleaved and uncleaved DNA bands and normalizing to the total amount cleaved. The percentage of cleaved DNA was then fit to a single exponential function to determine the single-turnover rate constant of DNA cleavage using Kaleidagraph (Synergy Software, Reading, PA, USA), namely, percent cleaved DNA = C1 + C2 × (1 – e−kt), where C1 is a constant fitting the baseline, C2 is the total percentage of DNA predicted to be cleaved by SgrAI, k is the cleavage rate constant, and t is the length of incubation in minutes.
Phage challenge assay.
Tuner (DE3) E. coli (Novagen) cells were transformed with (pBAK.MspI) (coding for MspI.M methyltransferase expression; New England Biolabs, Inc.) and pET.21a_SgrAIR (coding for His-tagged SgrAI expression, wild type or mutants), grown overnight in 6 ml LB culture with 50 μg/ml ampicillin, 30 μg/ml kanamycin, 0.2% maltose, and 1 mM MgSO4, and then grown overnight with induction in LB (with 0.4 mM isopropyl-β-d-thiogalactopyranoside). Cells were chilled to 4°C and centrifuged at low speed (4,000 rpm for 10 min), supernatant was removed, and the cells were resuspended in 1/10 volume of TMG (10 mM Tris-HCl, pH 8, 10 mM MgSO4, 0.01% gelatin, sterile). Top agar (10 g/liter agar, 10 g/liter NaCl, 10 g/liter tryptone) was prepared and aliquoted (3 ml) into screw-cap culture tubes, autoclaved, and kept at 50°C prior to use. Phage (λJL801, with the first 4 codons of the cI protein-coding region deleted to result in a purely lytic form of lambda phage without lysogeny [33]) was prepared from large-scale infections of Tuner (DE3) (without MspI.M or SgrAI) and kept in TMG at 4°C. Tenfold dilutions of the phage solution were prepared in TMG. For plating cells with each dilution of phage, 0.1 ml of the cells and 1 μl of the phage solution were added to the side of a tube containing the top agar, vortexed gently to mix, and quickly poured onto the top of prewarmed agar plates (20 g/liter agar, 10 g/liter NaCl, 10 g/liter tryptone in 100-mm by 15-mm dishes). Plates were incubated overnight at 37°C and plaques counted to give PFU per μl of phage stock. Triplicate measurements were done for each SgrAI protein (wild type or mutant) at the best dilution of phage (giving plaques between 25 and 250 per plate).
The phage stock was prepared by infecting the parent strain [Tuner (DE3)] with λJL801 (provided by J. Little [33]) and plating in top agar as described above. Plates were incubated for 2 h with 3 ml of TMG at 37°C and gentle shaking to elute the phage particles. One ml of eluate was then transferred to a 1.5-ml tube and 50 μl of chloroform added, followed by brief vortexing. The samples were then centrifuged for 10 min at 10,000 rpm, and the aqueous fraction was transferred to a new 1.5-ml tube with 30 μl chloroform added. Concentration (in terms of PFU per μl) was measured as described above for the phage challenge assay, and the same stock was used in all tests.
In vivo DNA cleavage reaction simulations.
Kintek Global Kinetic Explorer (version 6.2.170301) (Kintek Global Kinetic Explorer Corp.) (59, 60) was used for the simulations. Equations for modeling are given in the supplemental material. Rate constants are also given in the supplemental material and are those derived from prior work (25). Modeling of cleavage of primary sites in phage and in host DNA (as a result of activation via primary sites on phage DNA) used the equilibria found in Table S3 in the supplemental material. Two mechanisms are present in this case, that for the ROO filament mechanism and that for the Binary mechanism. The Binary mechanism is simpler and involves DNA (site) binding by the hypothetical enzyme E to create the enzyme/DNA complex R. Two Rs may associate to give the Binary complex RR. DNA cleavage only occurs in this Binary complex and is symbolized by the conversion of R to X. This occurs independently for the two R complexes in the Binary complex RR. The Binary complexes with cleaved DNA may also dissociate before or after cleavage occurs, but dissociation of cleaved DNA from X only occurs when X is isolated from the Binary complex. The forward and reverse rate constants for each equilibrium are numbered and the values given in Table S4 (and are those derived from fitting experimental data of SgrAI reactions from prior work [25]). The ROO filament mechanism is more complicated, although software limitations prevented the modeling of ROO filaments longer than 3 SgrAI/DNA complexes long, similar to the predicted number of primary sites in the typical Streptomyces phage (Table S2). Identical nomenclature is used in Table S3 for this mechanism; however, complexes of R and X include those of size 3.
The kinetic model used for simulating the cleavage of secondary sites is given in Table S5, and corresponding rate constants and starting concentrations are given in Tables S6 and S7. Slightly different nomenclature is used for species to denote the difference between SgrAI/DNA (or E/DNA) complexes: P for that bound to primary site DNA and S for that bound to secondary site DNA. In this model, complexes with primary site DNA (i.e., P) may self-associate and may also associate with complexes containing secondary site DNA (i.e., S). Complexes with secondary sites do not self-associate, consistent with experimental observations of SgrAI activity (7, 16). To reduce the number of equilibria in the modeling, only cleavage of secondary sites is considered. This is realistic, since even cleaved primary sites will bind SgrAI and induce ROO filament formation (8). This allows modeling up to an ROO size of 4 (in the ROO filament mechanism). Again, cleavage of secondary site DNA in complexes is symbolized by the conversion of S to X (Table S5). To estimate rate constants for complexes with secondary site DNA, a preliminary fitting of single-turnover DNA cleavage data was performed (16). As with primary site DNA, the apparent DNA cleavage rate constants of secondary site cleavage by SgrAI are dependent on the concentration of SgrAI bound to primary site DNA; hence, they are also rate limited by the association step (k4 in Table S5) in ROO filament formation, but this limitation is approximately 2-fold lower. Hence, the rate constant k4 was set to 2-fold lower than that for complexes with only primary site DNA (i.e., k3). All other rate constants were held the same for complexes with secondary site DNA as those with primary site DNA. This assumes that the only effect of secondary site DNA on SgrAI is on the rate of assembly of ROO and not on its dissociation or DNA cleavage.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge John Little, University of Arizona, for his kind gift of λJL801.
Research reported in this publication was supported by the National Science Foundation under grant no. MCB-1410355 (to N.C.H.), the Office of the Director, National Institutes of Health, under award number S10OD013237 (to C.K.P.), and the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM008659 (to J.L.S.). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NIH, or NSF.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01647-18.
REFERENCES
- 1.Stern A, Sorek R. 2011. The phage-host arms race: shaping the evolution of microbes. Bioessays 33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nechaev S, Severinov K. 2008. The elusive object of desire–interactions of bacteriophages and their hosts. Curr Opin Microbiol 11:186–193. doi: 10.1016/j.mib.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasu K, Nagaraja V. 2013. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol Mol Biol Rev 77:53–72. doi: 10.1128/MMBR.00044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pingoud A, Wilson GG, Wende W. 2014. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res 42:7489–7527. doi: 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terns MP. 2018. CRISPR-based technologies: impact of RNA-targeting systems. Mol Cell 72:404–412. doi: 10.1016/j.molcel.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park CK, Stiteler AP, Shah S, Ghare MI, Bitinaite J, Horton NC. 2010. Activation of DNA cleavage by oligomerization of DNA-bound SgrAI. Biochemistry 49:8818–8830. doi: 10.1021/bi100557v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyumkis D, Talley H, Stewart A, Shah S, Park CK, Tama F, Potter CS, Carragher B, Horton NC. 2013. Allosteric regulation of DNA cleavage and sequence-specificity through run-on oligomerization. Structure 21:1848–1858. doi: 10.1016/j.str.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. 2009. The unfolded protein response signals through high-order assembly of Ire1. Nature 457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. 2010. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat Cell Biol 12:739–746. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noree C, Sato BK, Broyer RM, Wilhelm JE. 2010. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J Cell Biol 190:541–551. doi: 10.1083/jcb.201003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim CW, Moon YA, Park SW, Cheng D, Kwon HJ, Horton JD. 2010. Induced polymerization of mammalian acetyl-CoA carboxylase by MIG12 provides a tertiary level of regulation of fatty acid synthesis. Proc Natl Acad Sci U S A 107:9626–9631. doi: 10.1073/pnas.1001292107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O'Connell JD, Mirrielees J, Ellington AD, Marcotte EM. 2009. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci U S A 106:10147–10152. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. 2009. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc Natl Acad Sci U S A 106:7858–7863. doi: 10.1073/pnas.0901781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JL. 2010. Intracellular compartmentation of CTP synthase in Drosophila. J Genet Genomics 37:281–296. doi: 10.1016/S1673-8527(09)60046-1. [DOI] [PubMed] [Google Scholar]
- 16.Shah S, Sanchez J, Stewart A, Piperakis MM, Cosstick R, Nichols C, Park CK, Ma X, Wysocki V, Bitinaite J, Horton NC. 2015. Probing the run-on oligomer of activated SgrAI bound to DNA. PLoS One 10:e0124783. doi: 10.1371/journal.pone.0124783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, Shah S, Zhou M, Park CK, Wysocki VH, Horton NC. 2013. Structural analysis of activated SgrAI-DNA oligomers using ion mobility mass spectrometry. Biochemistry 52:4373–4381. doi: 10.1021/bi3013214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tautz N, Kaluza K, Frey B, Jarsch M, Schmitz GG, Kessler C. 1990. SgrAI, a novel class-II restriction endonuclease from Streptomyces griseus recognizing the octanucleotide sequence 5ʹ-CR/CCGGYG-3ʹ. Nucleic Acids Res 18:3087. doi: 10.1093/nar/18.10.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bilcock DT, Daniels LE, Bath AJ, Halford SE. 1999. Reactions of type II restriction endonucleases with 8-base pair recognition sites. J Biol Chem 274:36379–36386. doi: 10.1074/jbc.274.51.36379. [DOI] [PubMed] [Google Scholar]
- 20.Bitinaite J, Schildkraut I. 2002. Self-generated DNA termini relax the specificity of SgrAI restriction endonuclease. Proc Natl Acad Sci U S A 99:1164–1169. doi: 10.1073/pnas.022346799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniels LE, Wood KM, Scott DJ, Halford SE. 2003. Subunit assembly for DNA cleavage by restriction endonuclease SgrAI. J Mol Biol 327:579–591. [DOI] [PubMed] [Google Scholar]
- 22.Hingorani-Varma K, Bitinaite J. 2003. Kinetic analysis of the coordinated interaction of SgrAI restriction endonuclease with different DNA targets. J Biol Chem 278:40392–40399. doi: 10.1074/jbc.M304603200. [DOI] [PubMed] [Google Scholar]
- 23.Wood KM, Daniels LE, Halford SE. 2005. Long-range communications between DNA sites by the dimeric restriction endonuclease SgrAI. J Mol Biol 350:240–253. doi: 10.1016/j.jmb.2005.04.053. [DOI] [PubMed] [Google Scholar]
- 24.Park CK, Sanchez JL, Barahona C, Basantes LE, Sanchez J, Hernandez C, Horton NC. 2018. The run-on oligomer filament enzyme mechanism of SgrAI. Part 1. Assembly kinetics of the run-on oligomer filament. J Biol Chem 293:14585–14598. doi: 10.1074/jbc.RA118.003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park CK, Sanchez JL, Barahona C, Basantes LE, Sanchez J, Hernandez C, Horton NC. 2018. The run-on oligomer filament enzyme mechanism of SgrAI. Part 2. Kinetic modeling of the full DNA cleavage pathway. J Biol Chem 293:14599–14615. doi: 10.1074/jbc.RA118.003682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaisdell BE, Campbell AM, Karlin S. 1996. Similarities and dissimilarities of phage genomes. Proc Natl Acad Sci U S A 93:5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlin S, Mrazek J, Campbell AM. 1997. Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol 179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelfand MS, Koonin EV. 1997. Avoidance of palindromic words in bacterial and archaeal genomes: a close connection with restriction enzymes. Nucleic Acids Res 25:2430–2439. doi: 10.1093/nar/25.12.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharp PM. 1986. Molecular evolution of bacteriophages: evidence of selection against the recognition sites of host restriction enzymes. Mol Biol Evol 3:75–83. doi: 10.1093/oxfordjournals.molbev.a040377. [DOI] [PubMed] [Google Scholar]
- 30.Moineau S, Pandian S, Klaenhammer TR. 1993. Restriction/modification systems and restriction endonucleases are more effective on lactococcal bacteriophages that have emerged recently in the dairy industry. Appl Environ Microbiol 59:197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, Ward TJ, Siletzky RM, Kathariou S. 2012. Two novel type II restriction-modification systems occupying genomically equivalent locations on the chromosomes of Listeria monocytogenes strains. Appl Environ Microbiol 78:2623–2630. doi: 10.1128/AEM.07203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasarjian JK, Iida M, Ryu J. 2003. New restriction enzymes discovered from Escherichia coli clinical strains using a plasmid transformation method. Nucleic Acids Res 31:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michalowski CB, Little JW. 2005. Positive autoregulation of cI is a dispensable feature of the phage lambda gene regulatory circuitry. J Bacteriology 187:6430–6442. doi: 10.1128/JB.187.18.6430-6442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong H, Wenham LS, Dalton M. 2000. Method for cloning and producing the SgrAI restriction endonuclease. New England Biolabs, Inc., Ipswich, MA, USA. [Google Scholar]
- 35.Lin PM, Lee CH, Robert RJ. 1989. Cloning and characterization of the genes encoding the MspI restriction modification system. Nucleic Acids Res 17:3001–3011. doi: 10.1093/nar/17.8.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rippe K, von Hippel PH, Langowski J. 1995. Action at a distance: DNA-looping and initiation of transcription. Trends Biochem Sci 20:500–506. doi: 10.1016/S0968-0004(00)89117-3. [DOI] [PubMed] [Google Scholar]
- 37.Handa N, Ichige A, Kusano K, Kobayashi I. 2000. Cellular responses to postsegregational killing by restriction-modification genes. J Bacteriology 182:2218–2229. doi: 10.1128/JB.182.8.2218-2229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pleska M, Qian L, Okura R, Bergmiller T, Wakamoto Y, Kussell E, Guet CC. 2016. Bacterial autoimmunity due to a restriction-modification system. Curr Biol 26:404–409. doi: 10.1016/j.cub.2015.12.041. [DOI] [PubMed] [Google Scholar]
- 39.Darmon E, Eykelenboom JK, Lopez-Vernaza MA, White MA, Leach DR. 2014. Repair on the go: E. coli maintains a high proliferation rate while repairing a chronic DNA double-strand break. PLoS One 9:e110784. doi: 10.1371/journal.pone.0110784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sargentini NJ, Diver WP, Smith KC. 1983. The effect of growth conditions on inducible, recA-dependent resistance to X rays in Escherichia coli. Radiat Res 93:364–380. [PubMed] [Google Scholar]
- 41.Sam MD, Perona JJ. 1999. Mn2+-dependent catalysis by restriction enzymes: pre-steady-state analysis of EcoRV endonuclease reveals burst kinetics and the origins of reduced activity. J Am Chem Soc 121:1444–1447. doi: 10.1021/ja983787q. [DOI] [Google Scholar]
- 42.Lesser DR, Kurpiewski MR, Jen-Jacobson L. 1990. The energetic basis of specificity in the Eco RI endonuclease-DNA interaction. Science 250:776–786. [DOI] [PubMed] [Google Scholar]
- 43.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 44.Jiang F, Doudna JA. 2017. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys 46:505–529. doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 45.Gong S, Yu HH, Johnson KA, Taylor DW. 2018. DNA unwinding is the primary determinant of CRISPR-Cas9 activity. Cell Rep 22:359–371. doi: 10.1016/j.celrep.2017.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tock MR, Dryden DT. 2005. The biology of restriction and anti-restriction. Curr Opin Microbiol 8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg AH, Lade BN, Chui DS, Lin SW, Dunn JJ, Studier FW. 1987. Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56:125–135. doi: 10.1016/0378-1119(87)90165-X. [DOI] [PubMed] [Google Scholar]
- 49.Mkrtumian NM, Lomovskaya ND. 1972. A mutation affecting the ability of temperature actinophage PhiC31 of Streptomyces coelicolor to lyse and lysogenise. Genetika 8:135–141. [Google Scholar]
- 50.Chater KF, Carter AT. 1979. A new, wide host-range, temperate bacteriophage (R4) of Streptomyces and its interaction with some restriction-modification systems. J Gen Microbiol 115:431–442. doi: 10.1099/00221287-115-2-431. [DOI] [Google Scholar]
- 51.Barry RM, Bitbol AF, Lorestani A, Charles EJ, Habrian CH, Hansen JM, Li HJ, Baldwin EP, Wingreen NS, Kollman JM, Gitai Z. 2014. Large-scale filament formation inhibits the activity of CTP synthetase. Elife 3:e03638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch EM, Hicks DR, Shepherd M, Endrizzi JA, Maker A, Hansen JM, Barry RM, Gitai Z, Baldwin EP, Kollman JM. 2017. Human CTP synthase filament structure reveals the active enzyme conformation. Nat Struct Mol Biol 24:507–514. doi: 10.1038/nsmb.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrovska I, Nuske E, Munder MC, Kulasegaran G, Malinovska L, Kroschwald S, Richter D, Fahmy K, Gibson K, Verbavatz JM, Alberti S. 2014. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. Elife 3:e02409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Webb BA, Dosey AM, Wittmann T, Kollman JM, Barber DL. 2017. The glycolytic enzyme phosphofructokinase-1 assembles into filaments. J Cell Biol 216:2305–2313. doi: 10.1083/jcb.201701084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, Maier T. 2018. Structural basis for regulation of human acetyl-CoA carboxylase. Nature 558:470–474. doi: 10.1038/s41586-018-0201-4. [DOI] [PubMed] [Google Scholar]
- 56.Dunten PW, Little EJ, Gregory MT, Manohar VM, Dalton M, Hough D, Bitinaite J, Horton NC. 2008. The structure of SgrAI bound to DNA; recognition of an 8 base pair target. Nucleic Acids Res 36:5405–5416. doi: 10.1093/nar/gkn510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aggarwal AK. 1990. Crystallization of DNA binding proteins with oligodeoxynucleotides. Methods 1:83–90. doi: 10.1016/S1046-2023(05)80150-1. [DOI] [PubMed] [Google Scholar]
- 58.Fasman GD. 1975. CRC handbook of biochemistry and molecular biology, 3rd ed CRC, Cleveland, OH. [Google Scholar]
- 59.Johnson KA, Simpson ZB, Blom T. 2009. Global Kinetic Explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem 387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 60.Johnson KA. 2009. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol 467:601–626. [DOI] [PubMed] [Google Scholar]
- 61.Park CK, Joshi HK, Agrawal A, Ghare MI, Little EJ, Dunten PW, Bitinaite J, Horton NC. 2010. Domain swapping in allosteric modulation of DNA specificity. PLoS Biol 8:e1000554. doi: 10.1371/journal.pbio.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.