

Abstract
Background
The anaemia seen in chronic kidney disease (CKD) may be exacerbated by iron deficiency. Iron can be provided through different routes, with advantages and drawbacks of each route. It remains unclear whether the potential harms and additional costs of intravenous (IV) compared with oral iron are justified. This is an update of a review first published in 2012.
Objectives
To determine the benefits and harms of IV iron supplementation compared with oral iron for anaemia in adults and children with CKD, including participants on dialysis, with kidney transplants and CKD not requiring dialysis.
Search methods
We searched the Cochrane Kidney and Transplant Register of Studies up to 7 December 2018 through contact with the Information Specialist using search terms relevant to this review. Studies in the Register are identified through searches of CENTRAL, MEDLINE, and EMBASE, conference proceedings, the International Clinical Trials Register (ICTRP) Search Portal, and ClinicalTrials.gov.
Selection criteria
We included randomised controlled trials (RCTs) and quasi‐RCTs in which IV and oral routes of iron administration were compared in adults and children with CKD.
Data collection and analysis
Two authors independently assessed study eligibility, risk of bias, and extracted data. Results were reported as risk ratios (RR) with 95% confidence intervals (CI) for dichotomous outcomes. For continuous outcomes the mean difference (MD) was used or standardised mean difference (SMD) if different scales had been used. Statistical analyses were performed using the random‐effects model. Subgroup analysis and univariate meta‐regression were performed to investigate between study differences. The certainty of the evidence was assessed using GRADE.
Main results
We included 39 studies (3852 participants), 11 of which were added in this update. A low risk of bias was attributed to 20 (51%) studies for sequence generation, 14 (36%) studies for allocation concealment, 22 (56%) studies for attrition bias and 20 (51%) for selective outcome reporting. All studies were at a high risk of performance bias. However, all studies were considered at low risk of detection bias because the primary outcome in all studies was laboratory‐based and unlikely to be influenced by lack of blinding.
There is insufficient evidence to suggest that IV iron compared with oral iron makes any difference to death (all causes) (11 studies, 1952 participants: RR 1.12, 95% CI 0.64, 1.94) (absolute effect: 33 participants per 1000 with IV iron versus 31 per 1000 with oral iron), the number of participants needing to start dialysis (4 studies, 743 participants: RR 0.81, 95% CI 0.41, 1.61) or the number needing blood transfusions (5 studies, 774 participants: RR 0.86, 95% CI 0.55, 1.34) (absolute effect: 87 per 1,000 with IV iron versus 101 per 1,000 with oral iron). These analyses were assessed as having low certainty evidence. It is uncertain whether IV iron compared with oral iron reduces cardiovascular death because the certainty of this evidence was very low (3 studies, 206 participants: RR 1.71, 95% CI 0.41 to 7.18). Quality of life was reported in five studies with four reporting no difference between treatment groups and one reporting improvement in participants treated with IV iron.
IV iron compared with oral iron may increase the numbers of participants, who experience allergic reactions or hypotension (15 studies, 2607 participants: RR 3.56, 95% CI 1.88 to 6.74) (absolute harm: 24 per 1000 with IV iron versus 7 per 1000) but may reduce the number of participants with all gastrointestinal adverse effects (14 studies, 1986 participants: RR 0.47, 95% CI 0.33 to 0.66) (absolute benefit: 150 per 1000 with IV iron versus 319 per 1000). These analyses were assessed as having low certainty evidence.
IV iron compared with oral iron may increase the number of participants who achieve target haemoglobin (13 studies, 2206 participants: RR 1.71, 95% CI 1.43 to 2.04) (absolute benefit: 542 participants per 1,000 with IV iron versus 317 per 1000 with oral iron), increased haemoglobin (31 studies, 3373 participants: MD 0.72 g/dL, 95% CI 0.39 to 1.05); ferritin (33 studies, 3389 participants: MD 224.84 µg/L, 95% CI 165.85 to 283.83) and transferrin saturation (27 studies, 3089 participants: MD 7.69%, 95% CI 5.10 to 10.28), and may reduce the dose required of erythropoietin‐stimulating agents (ESAs) (11 studies, 522 participants: SMD ‐0.72, 95% CI ‐1.12 to ‐0.31) while making little or no difference to glomerular filtration rate (8 studies, 1052 participants: 0.83 mL/min, 95% CI ‐0.79 to 2.44). All analyses were assessed as having low certainty evidence. There were moderate to high degrees of heterogeneity in these analyses but in meta‐regression, definite reasons for this could not be determined.
Authors' conclusions
The included studies provide low certainty evidence that IV iron compared with oral iron increases haemoglobin, ferritin and transferrin levels in CKD participants, increases the number of participants who achieve target haemoglobin and reduces ESA requirements. However, there is insufficient evidence to determine whether IV iron compared with oral iron influences death (all causes), cardiovascular death and quality of life though most studies reported only short periods of follow‐up. Adverse effects were reported in only 50% of included studies. We therefore suggest that further studies that focus on patient‐centred outcomes with longer follow‐up periods are needed to determine if the use of IV iron is justified on the basis of reductions in ESA dose and cost, improvements in patient quality of life, and with few serious adverse effects.
Keywords: Adult; Child; Humans; Administration, Oral; ; /blood; /therapy; Blood Transfusion; Blood Transfusion/statistics & numerical data; Cause of Death; Ferritins; Ferritins/blood; Hemoglobin A; Hemoglobin A/metabolism; Injections, Intravenous; Iron Compounds; Iron Compounds/administration & dosage; Iron Compounds/adverse effects; Kidney Failure, Chronic; Kidney Failure, Chronic/blood; Kidney Failure, Chronic/complications; Randomized Controlled Trials as Topic; Transferrin; Transferrin/metabolism
Plain language summary
Iron treatment for adults and children with reduced kidney function
What is the issue?
Anaemia (reduction in the number of circulating red blood cells) often occurs in people who have kidney damage, especially those who need dialysis treatment. Anaemia can cause tiredness, reduce exercise tolerance and increase heart size. A common cause of anaemia is reduced production of a hormone, erythropoietin. Iron deficiency can make anaemia worse, and reduce the response to medications that stimulate erythropoietin production. Iron can be taken orally (by mouth) or injected intravenously (via a vein). Intravenous (IV) iron is given under supervision in hospitals. There is uncertainty about whether IV iron should be used rather than oral iron.
What did we do?
We reviewed 39 studies (3852 participants) which compared IV iron supplements with oral iron in participants with chronic kidney disease.
What did we find?
We found that IV iron may increase blood levels of haemoglobin and iron compared with oral iron. However, IV iron may increase the number of allergic reactions though it may reduce side effects such as constipation, diarrhoea, nausea and vomiting seen with oral iron. We did not find sufficient evidence to determine whether IV iron compared with oral iron improved quality of life, altered overall death rate or death due to heart disease.
Conclusions
Although the results suggest that IV iron compared with oral iron may be more effective in raising iron and haemoglobin levels, we found insufficient data to determine if the benefits of IV iron are justified by improved quality of life or mortality despite the small risk of potentially serious allergic effects in some patients given IV iron.
Summary of findings
Summary of findings for the main comparison. Patient‐centred outcomes for oral versus IV iron in adults and children with chronic kidney disease.
| Patient‐centred outcomes for oral versus IV iron in adults and children with CKD | ||||||
| Patient or population: adults and children with CKD Setting: Nephrology departments Intervention: IV iron Comparison: oral iron | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with oral iron | Risk with IV iron | |||||
| Death (all causes) | 30 per 1,000 | 33 per 1,000 (19 to 58) | RR 1.12 (0.64 to 1.94) | 1952 (11) | ⊕⊕⊝⊝ LOW 1 2 | Only 11/38 studies represented with only about 1/3 of patients. |
| Cardiovascular death | 20 per 1,000 | 34 per 1,000 (8 to 142) | RR 1.71 (0.41 to 7.18) | 206 (3) | ⊕⊝⊝⊝ VERY LOW 1 2 4 | ‐ |
| Type of adverse event: allergic reactions/hypotension | 7 per 1,000 | 24 per 1,000 (13 to 46) | RR 3.56 (1.88 to 6.74) | 2607 (15) | ⊕⊕⊝⊝ LOW 1 2 | ‐ |
| Type of adverse event: all gastrointestinal adverse effects | 319 per 1,000 | 150 per 1,000 (105 to 211) | RR 0.47 (0.33 to 0.66) | 1986 (14) | ⊕⊕⊝⊝ LOW 2 3 | ‐ |
| Type of adverse event: infection | 80 per 1,000 | 106 per 1,000 (72 to 157) | RR 1.32 (0.90 to 1.95) | 954 (4) | ⊕⊕⊝⊝ LOW 1 2 | ‐ |
| Numbers of non‐dialysis patients needing to commence dialysis | 46 per 1,000 | 38 per 1,000 (19 to 75) | RR 0.81 (0.41 to 1.61) | 743 (4) | ⊕⊕⊝⊝ LOW 1 2 | ‐ |
| Number requiring transfusion | 101 per 1,000 | 87 per 1,000 (56 to 136) | RR 0.86 (0.55 to 1.34) | 774 (5) | ⊕⊕⊝⊝ LOW 1 2 | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; CKD: chronic kidney disease; IV: intravenous; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level for imprecision
2 Downgraded one level for likely publication bias
3 Downgraded one level for high heterogeneity
4 Downgraded one level for publication bias
Summary of findings 2. Laboratory and pharmaceutical outcomes for adults and children with chronic kidney disease.
| Laboratory and pharmaceutical outcomes for adults and children with CKD | ||||||
| Patient or population: adults and children with CKD Setting: Nephrology departments Intervention: IV iron Comparison: oral iron | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with oral iron | Risk with IV iron | |||||
| Number achieving target Hb or increase ≥1 g/dL | 317 per 1,000 | 542 per 1,000 (453 to 646) | RR 1.71 (1.43 to 2.04) | 2206 (13) | ⊕⊕⊝⊝ LOW 1 2 | Risk of bias (ROB) downgraded as little info on random sequence generation (RSG) and allocation concealment. Heterogeneity 60% |
| Hb: final or change (g/dL) | The mean Hb level was 0.72 g/dL higher with IV iron compared to oral iron (0.39 to 1.05 higher) | ‐ | 3373 (31) | ⊕⊕⊝⊝ LOW 1 2 | 21/31 are at ROB for RSG &/or allocation concealment. Heterogeneity 94% | |
| Ferritin: final or change (µg/L) | The mean ferritin level was 224.84 µg/L higher with IV iron compared to oral iron (165.85 to 283.83 higher) | ‐ | 3389 (33) | ⊕⊕⊝⊝ LOW 1 2 | 8/13 are at ROB for RSG or allocation concealment. Heterogeneity 60%. | |
| TSAT: final or change (%) | The mean TSAT was 7.69% higher with IV iron compared to oral iron (5.1 to 10.28 higher) | ‐ | 3089 (27) | ⊕⊕⊝⊝ LOW 1 2 | 11/27 only are at low risk of bias and heterogeneity is 97%. | |
| HCT (%) | The mean HCT was 1.18% higher with IV iron compared to oral iron (2.17 lower to 4.52 higher) | ‐ | 152 (4) | ⊕⊝⊝⊝ VERY LOW 1 2 3 4 | Only 4 studies in this analysis, all with unknown risk of selection bias. Heterogeneity 96%. | |
| ESA dose: final or change | The SMD for ESA dose was 0.72 lower with IV iron compared to oral iron (0.31 to 1.12 lower) | ‐ | 522 (11) | ⊕⊕⊝⊝ LOW 1 2 | ROB downgraded as little information on RSG and Allocation concealment. Heterogeneity 77% | |
| eGFR: final or change (mL/min) | the mean eGFR was 0.83 mL/min higher with IV iron compared to oral iron (0.79 lower to 2.44 higher) | ‐ | 1052 (8) | ⊕⊕⊝⊝ LOW 1 3 | Half of the studies are at ROB for RSG & allocation concealment. | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; CKD: chronic kidney disease; eGFR: estimated glomerular filtration rate; ESA: erythrocyte‐stimulating agent; Hb: haemoglobin; HCT: haematocrit; IV: intravenous; RR: Risk ratio; TSAT: transferrin saturation | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level for risk of bias
2 Downgraded one level for inconsistency
3 Downgraded one level for imprecision
4 Downgraded one level for likely publication bias
Background
Description of the condition
A reduction in the number of circulating red blood cells is termed anaemia. The prevalence of anaemia in patients with chronic kidney disease (CKD) is twice that in the general population. As kidney function deteriorates, the prevalence of anaemia increases from 8.4% at CKD stage 1 to 53.4% at CKD stage 5 (Stauffer 2014). The cause of anaemia in CKD is multifactorial though largely driven by decreased kidney production of erythropoietin. Iron deficiency can exacerbate the degree of anaemia and reduce the response to erythropoietin‐stimulating agents (ESAs). Anaemia has been found to contribute to a number of pathological processes. Observational studies have shown anaemia to be associated with increased mortality (at an haemoglobin level (Hb) < 11.0 g/dL) (Kovesdy 2006; Levin 2006), increased hospital stay (Li 2004), increased cardiovascular events (Li 2004; Vlagopoulos 2005; Weiner 2005) and decreased quality of life (Fukuhara 2007). Limited data have also shown that an increase in Hb can improve a number of these indices (Levin 2006; Moreno 2000). However, a systematic review of studies assessing the effects of targeting higher Hb concentrations in patients with CKD by using higher doses of ESA showed a significantly higher risk of death (all causes) (risk ratio (RR) 1.17) and arteriovenous access thrombosis (RR 1.34) in the higher Hb target group compared with the lower Hb group (Phrommintikul 2007). National (CARI 2008; Moist 2008; NICE 2015) and international guidelines (KDIGO 2008; Locatelli 2010) recommend target Hb levels of between 10 g/dL to 12 g/dL for patients with CKD. The most recent guidelines (KDIGO 2012) suggest that the Hb in adult CKD patients should not exceed 11.5 g/dL.
Iron is an essential mineral to maintain health. It is required in many intracellular processes including DNA synthesis, mitochondrial energy generation and enzymatic reactions. It is used in the production of myoglobin in muscles and Hb, the oxygen carrying component of the red blood cells. Determining iron deficiency in CKD can be challenging as it is often a functional deficiency caused by insufficient iron availability despite adequate body iron stores. The aetiology of iron deficiency in CKD is complex but includes reduced dietary intake and blood loss, particularly from the gastrointestinal tract, due to uraemia induced platelet dysfunction (Hedges 2007). These losses are compounded in patients on haemodialysis (HD) by the use of heparin, losses from clotted dialysis lines and blood sampling, which can lead to losses of 2 litres to 5 litres of blood per year (Sargent 2004). Lastly, chronic inflammation and uraemia result in an upregulation and reduced clearance of hepcidin, inhibiting the release of iron from macrophages and decreasing gastrointestinal iron absorption (Lopez 2015).
Description of the intervention
Therapeutic iron can be given orally. Four iron preparations are commonly used: ferrous sulphate, ferrous sulphate exsiccated, ferrous gluconate, and ferrous fumarate. It can be given intramuscularly in the form of iron dextran or it can be given intravenously. Six main forms of intravenous (IV) iron are currently available: iron sucrose, ferric gluconate, ferric carboxymaltose, iron isomaltoside‐1000, ferumoxytol, and iron dextran (low‐molecular‐weight forms) (Lopez 2015).
Oral iron frequently causes gastrointestinal side effects including heartburn, nausea, vomiting, diarrhoea, and constipation (Lopez 2015). These events affect patient compliance and can limit total intake. Although serious adverse events related to IV forms of iron are rare, the effects can be life threatening and include pulmonary embolism, anaphylactic reaction, loss of consciousness, circulatory collapse, hypotension, dyspnoea, pruritus, hypersensitivity and urticaria (Bailie 2012; Lopez 2015). The cumulative rate for all adverse events, for all IV iron preparations, is 14.1 adverse events per million units sold though it appears to be higher in products such as ferumoxytol compared with iron sucrose (Bailie 2012). IV iron preparations require administration under supervision and this need increases the cost of administration and is inconvenient for patients who are not receiving in‐centre HD. IV iron has also been linked to an increased risk of infection and cardiovascular disease; iron can act as a growth factor for some bacteria and free iron has been shown to impair neutrophil and T cell function as well as increase reactive oxygen species (Fishbane 2014; Ishida 2014). The majority of the literature to date supports these associations although the most recent cohort study of nearly 23,000 HD patients suggested no difference in length of stay, death or readmission for infection in those who received IV iron during admission and those who did not (Ishida 2015). Furthermore there is increasing evidence that free iron plays a role in direct injury to kidney tissue, which could result in more rapid deterioration in kidney function (Shah 2011).
Controversies remain about the most effective and safe way to provide iron supplementation in patients with CKD (Fishbane 2007; Macdougall 2016). Current parameters used to monitor iron status include serum ferritin levels, serum iron, transferrin saturation (TSAT), per cent of hypochromic red blood cells, and reticulocyte Hb content. There is debate about the most valuable measures to assess iron status, and the setting of optimum levels of these measures in patients with CKD to increase Hb and optimise ESA response. Novel markers being developed but not yet in routine use include hepcidin, soluble transferrin receptor one and non‐transferrin bound iron (Gaweda 2015).
How the intervention might work
Iron deficiency is the most common cause of anaemia in CKD and of hypo‐responsiveness to ESAs (Kwack 2006). ESAs accelerate erythropoiesis by increasing iron utilisation and depleting iron stores. Optimal efficacy of ESAs depends on the availability of iron to achieve and maintain target Hb levels. Patients with CKD stage 5D require higher targets for ferritin and TSAT levels to achieve increased Hb levels compared with patients whose kidney function is normal. Two studies targeting ferritin levels of 400 ng/mL or 30% to 50% TSAT resulted in significant reductions in the ESA dose required to maintain Hb levels compared with targeting a ferritin level of 200 ng/mL or TSAT levels of 20% to 30% (Besarab 2000; DeVita 2003). However, such high ferritin and TSAT levels increase the risk of iron overload and its associated complications. The Kidney Disease Outcomes Quality Initiative (KDOQI) guidelines (KDOQI 2007), the Canadian (Madore 2008) and the European guidelines (Locatelli 2009) recommend serum ferritin of > 200 ng/mL and TSAT > 20% in patients receiving HD. KDIGO 2012 recommend that iron can be given until TSAT > 30% or serum ferritin > 500 ng/mL. In patients with less severe degrees of CKD, serum ferritin levels > 100 ng/mL and TSAT > 20% are recommended.
Why it is important to do this review
The original study published in 2012 found strong evidence for increased ferritin and TSAT levels and a small increase in Hb with IV iron compared with oral iron. There was limited evidence that this came with a reduction in ESA use. Only half of the studies reported on adverse events. There have been several studies done over the last six years which have looked at the adverse event rate of the many preparations of IV iron and also included hard end points including all cause and cardiovascular death. At present the majority of HD patients receive IV iron and the use of IV iron in the peritoneal dialysis (PD) and CKD populations is increasing. We felt it was important to update this review to ensure that patient focused adverse events were analysed as well as providing up to date evidence on the efficacy and safety of IV iron. In this review, we aimed to explore all possible causes of heterogeneity of study results in detail by subgroup analysis and to further investigate the effects of IV iron in patients with CKD who were not on dialysis.
Objectives
Our objective was to determine the benefits and harms of IV iron supplementation compared with oral iron for anaemia in patients with CKD, treated with HD, PD, not receiving dialysis and post transplant. The review aimed to examine the effects of these interventions on patient centred outcomes including death, requirements for transfusion, hospitalisation, cardiac function, quality of life and change in eGFR as well as iron parameters, achieving target levels of Hb, reducing doses of ESA required, and to determine adverse effects of the therapies.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) and quasi‐RCTs (studies in which allocation to treatment was obtained by alternation, use of alternate medical records, date of birth or other predictable methods) in which oral and IV routes of administration of iron were compared in patients with CKD.
Types of participants
Inclusion criteria
We included adult and paediatric patients with CKD (stages 3 to 5D; glomerular filtration rate (GFR) < 60 mL/min/1.73 m2). Studies in patients receiving HD, PD, or those not requiring dialysis, were included. Studies of kidney transplant patients were also included.
Exclusion criteria
Studies of iron administration in patients comparing different IV or oral iron preparations and different doses of the same IV or oral preparation were excluded. Studies in patients with acute kidney injury were excluded.
Types of interventions
We examined different IV iron supplements (including iron sucrose, dextran, ferric gluconate, ferric carboxymaltose, ferumoxytol) and oral iron preparations (including oral iron preparations which contain folic acid, vitamin C or both).
We included studies using different doses and durations of IV iron compared with oral iron preparations provided that the control group received oral iron supplements only.
Types of outcome measures
Primary outcomes
Death (all causes)
Cardiovascular death
Quality of life
Secondary outcomes
-
Hb
Number achieving target Hb level
Time to achieve target Hb
Final or change in Hb at end of study
Increase in Hb > 10 g/L or other target during study
-
Iron
Number achieving target levels of iron (ferritin, TSAT, per cent of hypochromic red blood cells)
Final or change in ferritin levels at the end of study
Final or change in TSAT at end of study
Per cent of hypochromic red blood cells
-
Erythrocyte stimulating agents (ESAs)
Reduction in required dose of ESA
Number needing to increase ESA dose
Number needing to decrease ESA dose or cease ESA
Infection
Change in GFR in non‐dialysis patients
Number needing transfusions
-
Any adverse events of treatment
Adverse effects of oral iron
Adverse effects of IV iron supplements including hypersensitivity reactions
Number of patients needing to cease oral or IV supplements because of adverse effects
Other outcomes
Haematocrit (HCT)
Reticulocyte Hb concentration
Numbers of non‐dialysis patients needing to commence dialysis
Hospitalisation (other than for iron infusions and dialysis)
Exercise tolerance
Left ventricular function
Sexual function
Nutritional status
Adherence to therapy
Numbers and costs of hospitalisations/professional supervision required for IV iron supplements
Iron overload (as defined by the triallists)
Search methods for identification of studies
Electronic searches
We searched the Cochrane Kidney and Transplant Register of Studies up to 7 December 2018 through contact with the Information Specialist using search terms relevant to this review. The Register contains studies identified from the following sources.
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL)
Weekly searches of MEDLINE OVID SP
Handsearching of kidney‐related journals and the proceedings of major kidney conferences
Searching of the current year of EMBASE OVID SP
Weekly current awareness alerts for selected kidney and transplant journals
Searches of the International Clinical Trials Register (ICTRP) Search Portal and ClinicalTrials.gov.
Studies contained in the Register are identified through searches of CENTRAL, MEDLINE, and EMBASE based on the scope of Cochrane Kidney and Transplant. Details of search strategies, as well as a list of handsearched journals, conference proceedings and current awareness alerts, are available in the Specialised Register section of information about Cochrane Kidney and Transplant.
See Appendix 1 for search terms used in strategies for this review.
Searching other resources
Reference lists of review articles, relevant studies and clinical practice guidelines.
Letters seeking information about unpublished or incomplete trials to investigators known to be involved in previous studies.
Data collection and analysis
Selection of studies
The search strategy described was used to obtain titles and abstracts of studies that were potentially relevant to the review. The titles and abstracts were screened independently by two authors, who discarded studies that were not applicable. However, studies and reviews that might include relevant data or information on studies were retained initially. Two authors independently assessed retrieved abstracts, and where necessary the full text, of these studies to determine which satisfied the inclusion criteria.
Data extraction and management
Data extraction and assessment of the risk of bias were performed independently by the same authors using standardised data extraction forms. Studies reported in non‐English language journals were translated before assessment. Where more than one publication of one study existed, the publication with the most complete data was reviewed initially. Where relevant outcomes were only published in earlier versions, these data were used. Any discrepancy between published versions was highlighted. Any further information required from the original author was requested by written correspondence and any relevant information obtained in this manner was included in the review. Disagreements were resolved in consultation with a third author.
Assessment of risk of bias in included studies
The following items were assessed independently by two authors using the risk of bias assessment tool (Higgins 2011) (seeAppendix 2).
Was there adequate sequence generation (selection bias)?
Was allocation adequately concealed (selection bias)?
-
Was knowledge of the allocated interventions adequately prevented during the study?
Participants and personnel (performance bias)
Outcome assessors (detection bias)
Were incomplete outcome data adequately addressed (attrition bias)?
Are reports of the study free of suggestion of selective outcome reporting (reporting bias)?
Was the study apparently free of other problems that could put it at a risk of bias?
Measures of treatment effect
For dichotomous outcomes (number reaching target Hb, death) results were expressed as RR with 95% confidence intervals (CI). RR with 95% CI were calculated for adverse effects. Where continuous scales of measurement were used to assess the effects of treatment (Hb level, iron parameters) the mean difference (MD) was used, or the standardised mean difference (SMD) if different scales had been used (end of study ESA dose). Either final levels or change in levels were included in meta‐analyses of continuous scales of measurement. When both measures are provided in a study, final levels were included.
Unit of analysis issues
Cross‐over studies were thought likely to be inappropriate means of examining IV and oral iron because of carry over effects related to achieved Hb levels and iron parameters. Therefore, only data from the first period of cross‐over studies were included where these were reported separately, and included all or most patients who completed the first period, rather than only those who completed both treatment periods.
Dealing with missing data
Where necessary, we contacted triallists to request missing patient data due to loss to follow‐up and exclusion from study analyses in an effort to conduct intention‐to‐treat analyses. Eight authors responded to our requests. Where missing dichotomous or continuous data were few, and unlikely to affect the overall results, we analysed available data. Where possible we imputed missing standard deviations and standard errors if data was presented alternatively, using methods stated in the Cochrane handbook (Higgins 2011a).
Assessment of heterogeneity
Heterogeneity was analysed using a Chi2 test on N‐1 degrees of freedom, with an alpha of 0.05 used for statistical significance and with the I2 test (Higgins 2003). I2 values of 25%, 50% and 75% correspond to low, medium and high levels of heterogeneity.
Assessment of reporting biases
Cochrane Kidney and Transplant's Specialised Register includes studies obtained from searching major databases, conference proceedings and prospective trial registers without language restriction in an attempt to reduce publication bias related to failure of authors to publish negative results or their inability to publish negative results in journals indexed in major databases. When sufficient studies were available, we created funnel plots and calculated Eggers' test to assess publication bias. Where multiple publications of the same study were identified, data were included from the most recent publication, and preferably, the definitive publication. However, all publications were reviewed to identify outcomes not reported in the index publication in an attempt to reduce outcome reporting bias.
Data synthesis
Data were pooled using the random‐effects model for dichotomous and continuous data.
Subgroup analysis and investigation of heterogeneity
To explore clinical differences among studies that could influence the magnitude of the treatment effect for the outcomes of differences in ferritin, TSAT and Hb, subgroup analyses and univariate meta‐regression were performed using STATA software (StataCorp LP, Texas, USA) using restricted maximum‐likelihood to estimate between study variance. The potential sources of variability were defined a priori and were related to study rationale (CKD stage, whether aiming to increase or maintain Hb, concurrent use of erythropoietin co‐intervention, timing of initiation of erythropoietin co‐intervention), dose delivered and duration of IV and oral iron therapy, and study sponsorship. Where subgroup analysis findings suggested that more than one factor could influence the magnitude of observed differences, we planned to conduct multivariate meta‐regression.
Underlying cause of end‐stage kidney disease (ESKD), baseline iron status, and previous iron therapy were not examined in subgroup analyses because most studies did not provide this information. All studies, except two paediatric studies, included adults of similar ages so different age groups could not be examined in subgroup analyses. Only one study (Li 2008 PD) included solely PD patients so it was not possible to examine different types of renal replacement therapy in subgroup analyses.
Sensitivity analysis
Sensitivity analyses were performed to test decisions where inclusion of a study, with a much higher MD in Hb, might have altered meta‐analysis results.
'Summary of findings' tables
We presented the main results of the review in 'Summary of findings' tables. These tables present key information concerning the quality of the evidence, the magnitude of the effects of the interventions examined, and the sum of the available data for the main outcomes (Schünemann 2011a). The 'Summary of findings' tables also include an overall grading of the evidence related to each of the main outcomes using the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) approach (GRADE 2008; GRADE 2011). The GRADE approach defines the quality of a body of evidence as the extent to which one can be confident that an estimate of effect or association is close to the true quantity of specific interest. The quality of a body of evidence involves consideration of within‐trial risk of bias (methodological quality), directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias (Schünemann 2011b). We presented the following outcomes in the 'Summary of findings' tables.
-
Death (all causes)
Cardiovascular death
Allergic reactions/hypotension
All gastrointestinal adverse effects
Infection
Numbers of non‐dialysis patients needing to commence dialysis
Number requiring transfusion
-
Number achieving target Hb or increase ≥1 g/dL
Hb: final or change
Ferritin: final or change
TSAT: final or change
HCT
End of treatment or change in ESA dose
eGFR end or change
Results
Description of studies
Results of the search
The initial study resulted in a total of 522 study reports from the Cochrane Kidney and Transplant Specialised Register to March 2010, CENTRAL (in The Cochrane Library Issue 1, 2010), MEDLINE (to October week 5, 2008) and EMBASE (to week 45, 2008). From these 522 reports, 28 studies (46 reports) were included in the systematic review while 28 studies were excluded; there were three ongoing studies.
For the 2019 update of this review, a search of the Cochrane Kidney and Transplant Specialised Register identified 49 new reports. From these we identified 11 new included studies (31 reports) (Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Kalra 2016 CKD; Lu 2010 CKD; Mudge 2009 TX; Nagaraju 2013 CKD; NCT01155375 HD,PD,CKD; Pisani 2014 CKD; Ragab 2007 HD; Tsuchida 2010 HD; Winney 1977 HD), three new excluded studies (4 reports) and 14 additional reports of previously included studies.. The additional reports included the full publication of Qunibi 2011 CKD. Of the 11 new included studies, four were publications of trials identified as ongoing trials in the 2010 review (Agarwal 2015 CKD; Kalra 2016 CKD; Mudge 2009 TX; NCT01155375 HD,PD,CKD). The paediatric study (NCT01155375 HD,PD,CKD) was terminated because of challenges with enrolment with minimal data reported. Search results are shown in Figure 1. One new report contained further information on two already included studies (Li 2008 HD; Li 2008 PD). Spinowitz 2008 CKD included all nine reports, which included data for one new included study (Lu 2010 CKD). This 2019 update contains 44 studies (101 reports).
1.

Flow diagram of studies included in the systematic review
Included studies
The 11 new included studies (31 reports) provided an additional 1754 participants bringing the total to 3852 participants. Of the new studies, seven included 1653 participants with CKD, three included 75 participants on HD and one included 102 transplant patients. One study included both dialysis and non‐dialysis patients but did not specify how many patients were in each group.
Of the 39 included studies, 38 (3832 participants) were parallel group studies, and one (20 patients) was a cross‐over study (Strickland 1977 HD). Only three studies involved paediatric patients (NCT01155375 HD,PD,CKD; Ragab 2007 HD; Warady 2002 HD). Nineteen studies included only HD patients. Li 2008 PD included only patients on PD while Macdougall 1999 HD,PD included both HD and PD patients. Two studies (Ahsan 1997 TX; Mudge 2009 TX) evaluated patients who were in the early phase of post‐kidney transplantation. Results from these studies were pooled with studies of dialysis patients. Thirteen studies included non‐dialysis patients (CKD stages 3 to 5) while two studies (Macdougall 1996 HD,PD,CKD; NCT01155375 HD,PD,CKD) included both dialysis and non‐dialysis patients. Twelve studies were available only as abstracts or from ClinicalTrials.Gov (Ahsan 1997 TX; Broumand 1998 HD; Erten 1998 HD; Leehey 2005 CKD; Lu 2010 CKD; Lye 2000 HD; Macdougall 1999 HD,PD; Michael 2007 HD; NCT01155375 HD,PD,CKD; Souza 1997 HD; Wang 2003 HD; Winney 1977 HD). Thirty‐two studies were designed to increase Hb levels and four studies were designed to maintain Hb stability in iron replete patients and decrease ESA dose (Fishbane 1995 HD; Kotaki 1997 HD; Michael 2007 HD; Warady 2002 HD). One study was designed to examine changes in GFR during (Agarwal 2015 CKD) while one study was designed to determine the time to the start of additional anaemia management other than iron (FIND‐CKD 2014 CKD).
The duration of follow‐up ranged from 35 days to 26 months.
Studies compared different oral and IV iron preparations. The oral iron agents investigated were ferrous sulphate (25 studies), ferrous fumarate (7), ferrous succinate (2), iron gluconate (1), liposomal iron (1), heme iron polypeptide (1) and unnamed agents in two studies. The IV iron agents investigated were iron sucrose (15 studies), iron dextran (7), ferumoxytol (4), sodium ferric gluconate complex (5), ferric carboxymaltose (2), iron isomaltoside (1), ferric citrate (1), and ferric hydroxide polymaltose (3). The IV iron agent was not reported in Kotaki 1997 HD. The calculated total dose of elemental iron ranged from 2520 mg to 63,000 mg in the oral iron groups and from 500 to 10,920 mg in the IV iron groups. Three studies (Erten 1998 HD; FIND‐CKD 2014 CKD; Kalra 2016 CKD) included two IV iron treatment groups. For these studies, data from patients who received the higher total dose of IV iron were included in the meta‐analyses.
In twenty‐two studies all participants were treated with an ESA. ESA therapy was started at study commencement in six studies (Aggarwal 2003 CKD; Charytan 2005 CKD; Hussain 1998 HD; Lye 2000 HD; Macdougall 1996 HD,PD,CKD; Stoves 2001 CKD) and before study commencement in 15 studies (Broumand 1998 HD; Erten 1998 HD; Fishbane 1995 HD; Kotaki 1997 HD; Leehey 2005 CKD; Li 2008 HD; Li 2008 PD; Macdougall 1999 HD,PD; Michael 2007 HD; Mudge 2009 TX; Provenzano 2009 HD; Ragab 2007 HD; Svara 1996 HD; Tsuchida 2010 HD; Warady 2002 HD). It was unclear when ESA treatment was commenced in Wang 2003 HD. Seven studies reported that no included patients received ESA treatment (Agarwal 2006 CKD; Ahsan 1997 TX; Fudin 1998 HD; Kalra 2016 CKD; McMahon 2009 CKD; Strickland 1977 HD; Winney 1977 HD), but nine studies indicated that varying proportions of patients received an ESA (Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Nagaraju 2013 CKD; Lu 2010 CKD; Pisani 2014 CKD; Qunibi 2011 CKD; Spinowitz 2008 CKD; Souza 1997 HD; Van Wyck 2005 CKD).
The outcomes reported in 38 studies are presented in Figure 1. One study was terminated and did not provide any outcomes (NCT01155375 HD,PD,CKD). Final and/or change in Hb, serum ferritin and TSAT levels were reported in 31, 33 and 27 studies respectively. Four studies reported final HCT levels but not Hb levels (Ahsan 1997 TX; Fishbane 1995 HD; Kotaki 1997 HD; Svara 1996 HD). Only 11 studies reported death (all causes) (Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Fishbane 1995 HD; Fudin 1998 HD; Kalra 2016 CKD; McMahon 2009 CKD; Lu 2010 CKD; Provenzano 2009 HD; Qunibi 2011 CKD; Stoves 2001 CKD; Tsuchida 2010 HD) while three studies reported on cardiovascular events including death (Agarwal 2015 CKD; Fudin 1998 HD; Stoves 2001 CKD). Five studies reported on quality of life assessment (Agarwal 2006 CKD; Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Kalra 2016 CKD; Van Wyck 2005 CKD). Eighteen studies reported on adverse events (Agarwal 2006 CKD; Agarwal 2015 CKD; Charytan 2005 CKD; FIND‐CKD 2014 CKD; Fishbane 1995 HD; Hussain 1998 HD; Kalra 2016 CKD; Li 2008 HD; Li 2008 PD; Nagaraju 2013 CKD; Lu 2010 CKD; Pisani 2014 CKD; Provenzano 2009 HD; Qunibi 2011 CKD; Spinowitz 2008 CKD; Strickland 1977 HD; Tsuchida 2010 HD; Van Wyck 2005 CKD).
Excluded studies
From the 2012 review, twenty‐three reports were excluded based on titles and abstracts; one study was not an RCT and the remainder involved ineligible interventions. Five more studies (eight reports) were excluded after full text review because participants were not randomised or compared intramuscular with oral iron.
Three studies (four reports) identified in the search for the 2019 update were excluded. One study (Charytan 2013) involved an ineligible comparator (standard medical care which could be oral or IV iron), one study (HEMATOCRIT 2012) compared two oral iron preparations and one study (Adhikary 2011) included non‐randomised patients.
Risk of bias in included studies
The assessment of risk of bias is shown in Figure 2 and Figure 3. Figure 2 shows relative proportional rankings of studies for each risk of bias indicator. Figure 3 shows the risk of bias items for individual studies.
2.

Risk of bias graph: Review authors' judgements about each risk of bias item presented as percentages across all included studies
3.

Risk of bias summary: Review authors' judgements about each risk of bias item for each included study
Allocation
Random sequence generation was at low risk of bias in 20 studies (Agarwal 2006 CKD; Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Fudin 1998 HD; Kalra 2016 CKD; Leehey 2005 CKD; Li 2008 HD; Li 2008 PD; Macdougall 1996 HD,PD,CKD; McMahon 2009 CKD; Mudge 2009 TX; Nagaraju 2013 CKD; Lu 2010 CKD; Pisani 2014 CKD; Qunibi 2011 CKD; Spinowitz 2008 CKD; Stoves 2001 CKD; Strickland 1977 HD; Van Wyck 2005 CKD; Warady 2002 HD). Random sequence generation was not reported in 18 studies.
Allocation concealment was at low risk of bias in 14 studies (Agarwal 2006 CKD; Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Kalra 2016 CKD; Leehey 2005 CKD; Macdougall 1996 HD,PD,CKD; Mudge 2009 TX; Nagaraju 2013 CKD; Lu 2010 CKD; Pisani 2014 CKD; Provenzano 2009 HD; Qunibi 2011 CKD; Spinowitz 2008 CKD; Van Wyck 2005 CKD); at high risk of bias in two studies (Fudin 1998 HD; Lye 2000 HD), and for the remaining 23 studies allocation concealment was unclear.
Blinding
No studies blinded either participants or personnel so were considered to be at high risk of bias. As all studies used laboratory data as primary outcomes, all studies were judged as having a low risk of bias for outcome assessment.
Incomplete outcome data
Outcomes data reporting was considered to be complete with a low risk of bias in 22 studies. Five studies (Charytan 2005 CKD; Fishbane 1995 HD; Fudin 1998 HD; Stoves 2001 CKD; Strickland 1977 HD) reported that from 7% to 36% of patients were excluded from the analyses, so were considered to be at high risk of bias. The risk of bias was unclear in 12 studies because there was insufficient information provided to determine if data from all patients who entered the study were included in the analysis.
Selective reporting
We identified 20 studies that were considered to have reported all outcomes based on the detailed protocols described in the trial methods. Eight studies (Agarwal 2015 CKD; Aggarwal 2003 CKD; Broumand 1998 HD; Charytan 2005 CKD; Leehey 2005 CKD; Ragab 2007 HD; Stoves 2001 CKD; Strickland 1977 HD) reported outcomes incompletely so that either outcomes could not be included in meta‐analyses or included only with imputed standard deviations or as incidence rates. It was unclear if outcomes were selectively reported in 11 studies.
Other potential sources of bias
Sixteen studies reported receiving monetary support from pharmaceutical companies; three studies reported funding from non‐pharmaceutical company sources and the remainder did not report on how their study was funded. In funnel plots, patient centred outcomes showed funnel plot symmetry (example provided in Figure 4), suggesting a low likelihood of publication and other biases. However, for biochemical outcomes, there was some funnel plot asymmetry (example provided in Figure 5) which suggests that the meta‐analyses of these outcomes may be affected by some bias.
4.
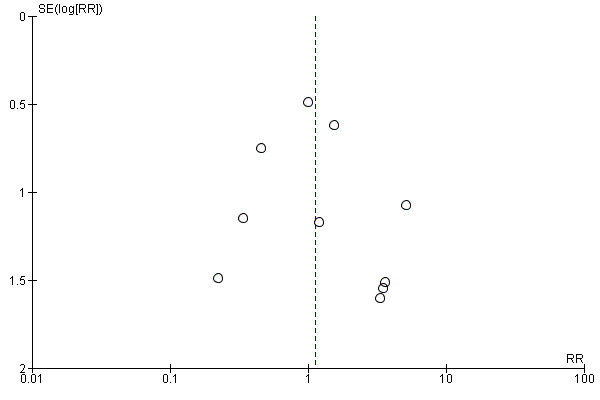
Funnel plot of comparison: 1 Patient‐centred outcomes.Outcome: 1.1 Death (all causes)
Eggers test P = 0.25
5.

Funnel plot of comparison: 2 Laboratory/pharmaceutical outcomes, outcome: 2.4 Transferrin saturation: Final or change [%].
Eggers test P = 0.00
Effects of interventions
Effects of IV iron compared with oral iron on patient‐centred outcomes
Death (all causes) was only reported in 11 studies. There was insufficient evidence to determine whether IV iron compared with oral iron may makes any difference to death (low certainty evidence) (Analysis 1.1 (11 studies, 1952 participants): RR 1.12, 95% CI 0.64 to 1.94; I2 = 0%). The absolute risk was 33 per 1000 with IV iron compared with 30 per 1000 with oral iron.
Cardiovascular death was reported in only three studies. It is uncertain whether IV iron compared with oral iron reduces cardiovascular death because the certainty of this evidence was very low (Analysis 1.2 (3 studies, 206 participants): RR 1.71, 95% CI 0.41 to 7.18; I2 = 0%).
Quality of life was only reported in five studies (Agarwal 2006 CKD; Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Kalra 2016 CKD; Van Wyck 2005 CKD). Agarwal 2006 CKD reported that the SF12 physical composite score improved by 4.8% in patients treated with IV iron, but there was no change in patients treated with oral iron. Kidney disease quality of life score (KDQOL) items ‐ improvement in the ability to do moderate activities and undertake work; and satisfaction with sex life ‐ were reported to be improved among patients treated with IV iron. Scores for a number of factors, including feelings of imposing a burden on family, were lower in patients who received IV iron. In contrast, Van Wyck 2005 CKD found no differences between treatment groups when health concept categories in the SF36 instrument were applied. Agarwal 2015 CKD reported no difference between groups or over time using the KDQOL. FIND‐CKD 2014 CKD reported no difference between groups using the SF‐36 tool. Using the Linear Analogue Scale Assessment score, Kalra 2016 CKD identified an improvement in quality of life from baseline to eight weeks in both treatment groups with no difference between groups (Analysis 1.3 (1 study, 312 participants): MD 1.45, 95% CI ‐5.89 to 8.79).
IV iron compared with oral iron may make little or no difference to the number of participants needing to start dialysis (low certainty evidence) (Analysis 1.4 (4 studies, 743 participants): RR 0.81, 95% CI 0.41 to 1.61; I2 = 0%). The absolute risk for starting dialysis was 38 per 1000 with IV iron and 46 per 1000 with oral iron.
IV iron compared with oral iron may make little or no difference to the need for transfusion (low certainty evidence) (Analysis 1.5 (5 studies, 774 participants): RR 0.86, 95% CI 0.55 to 1.34; I2 = 0%). The absolute risk for needing transfusion was 87 per 1000 with IV iron and 101 with oral iron.
Although nine studies reported that patient adherence to oral iron was assessed, only three provided numerical data (Charytan 2005 CKD; Van Wyck 2005 CKD; Pisani 2014 CKD). Mean adherence rates for IV iron therapy were 95%, 97% and 96% respectively, and adherence to oral iron therapy was 85% and 88% and 95.8%.
The certainty of the evidence was downgraded because of imprecision, heterogeneity between studies and publication bias.
1.1. Analysis.

Comparison 1 Patient centred outcomes, Outcome 1 Death (all causes).
1.2. Analysis.

Comparison 1 Patient centred outcomes, Outcome 2 Cardiovascular death.
1.3. Analysis.

Comparison 1 Patient centred outcomes, Outcome 3 Quality of life.
1.4. Analysis.

Comparison 1 Patient centred outcomes, Outcome 4 Number of non‐dialysis patients needing to commence dialysis.
1.5. Analysis.

Comparison 1 Patient centred outcomes, Outcome 5 Number requiring transfusion.
See Table 1.
Effect of IV iron compared with oral iron on laboratory outcomes
The numbers of patients reaching target Hb or increasing Hb by at least 1 g/dL were reported in 13 studies. Target Hb or an increase in Hb by 1 g/dL may be achieved by more participants receiving IV iron compared with oral iron (low certainty evidence) (Analysis 2.1 (13 studies, 2206 participants): RR 1.71, 95% CI 1.43 to 2.04; I2 = 60%) in all patients (Table 2) and in the subgroups of dialysis participants and CKD participants (Table 3). There was low to moderate heterogeneity. The absolute benefit for reaching the target Hb was 542 per 1000 for IV iron and 317 per 1000 for oral iron.
2.1. Analysis.
Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 1 Number achieving target haemoglobin or increase ≥ 1 g/dL.
1. Laboratory outcomes in dialysis and chronic kidney disease participants.
| Outcome | Population | Studies | Participants | MD | RR | 95% CI |
| Hb (g/dL) | All studies | 31 | 3373 | 0.72 | ‐ | 0.39 to 1.05 |
| Dialysis | 17 | 917 | 1.01 | ‐ | 0.26 to 1.77 | |
| CKD | 14 | 2456 | 0.41 | ‐ | 0.28 to 0.55 | |
| Ferritin (µg/L) | All studies | 33 | 3389 | 224.8 | ‐ | 165.8 to 283.8 |
| Dialysis | 19 | 1027 | 233.7 | ‐ | 163.4 to 303.9 | |
| CKD | 14 | 2362 | 213.1 | ‐ | 123.7 to 302.6 | |
| TSAT (%) | All studies | 27 | 3089 | 7.69 | ‐ | 5.10 to 10.28 |
| Dialysis | 14 | 781 | 10.55 | ‐ | 3.89 to 17.22 | |
| CKD | 13 | 2308 | 5.32 | ‐ | 2.67 to 7.97 | |
| Achieving target Hb | All studies | 13 | 2206 | ‐ | 1.71 | 1.43 to 2.04 |
| Dialysis | 5 | 508 | ‐ | 2.01 | 1.52 to 2.66 | |
| CKD | 8 | 1698 | ‐ | 1.59 | 1.27 to 1.97 |
CKD ‐ chronic kidney disease; Hb ‐ haemoglobin; TSAT ‐ transferrin saturation
End of study or change (g/dL) in Hb were reported in 31 studies. IV iron compared with oral iron may increase Hb (low certainty evidence) (Analysis 2.2 (31 studies, 3373 participants): MD 0.72 g/dL, 95% CI 0.39 to 1.05); I2 = 94%) in all participants and in subgroups of dialysis and CKD participants (Table 3). There was a high levels of heterogeneity, which persisted when a fixed‐effect model was used for analyses. Excluding a study of 26 months treatment and MD 4.92 g/dL (Fudin 1998 HD) did not significantly reduce heterogeneity. Further analyses of heterogeneity are addressed in later sections.
End of study or change (μg/L) in serum ferritin levels were reported in 33 studies. IV iron compared with oral iron may increase ferritin levels (low certainty evidence) in all participants (Analysis 2.3 (33 studies, 3389 participants): MD 224.84 µg/L, 95% CI 165.85 to 283.83, I2 = 99%) and in subgroups of dialysis and CKD participants (Table 3). There was a high level of heterogeneity in these analyses.
End of study or change (%) in TSAT levels were reported in 27 studies. IV iron compared with oral iron may increase TSAT levels (low certainty evidence) in all participants (Analysis 2.4 (27 studies, 3089 participants): MD 7.69 %, 95% CI 5.10 to 10.28, I2 = 97%) and in subgroups of dialysis and CKD participants (Table 3). There was a high level of heterogeneity in these analyses.
Five studies reported results for HCT rather than Hb. It is uncertain whether IV iron improves HCT because the certainty of this evidence was very low (Analysis 2.5.1 (5 studies, 180 participants): MD 1.09%, 95% CI ‐2.19 to 4.37, I2 = 96%). There was a high level of heterogeneity in this analysis.
Eleven studies reported end of study or change in ESA dose. IV iron probably leads to a reduction in ESA dose compared with oral iron (low certainty evidence) (Analysis 2.6 (11 studies, 522 participants): SMD ‐0.72, 95% CI ‐1.12 to ‐0.31) with a high level of heterogeneity (I2 = 77%).
IV iron compared with oral iron may make little or no difference to eGFR at the end of study (low certainty evidence) (Analysis 2.7 (8 studies,1052 participants): MD 0.83 mL/min, 95% CI ‐0.79 to 2.44). There was low to moderate heterogeneity (I2 = 38%).
The certainty of the evidence was downgraded because of high risk of bias, inconsistency, imprecision and possible publication bias.
2.2. Analysis.

Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 2 Haemoglobin: final or change (all patients).
2.3. Analysis.

Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 3 Ferritin: final or change (all patients).
2.4. Analysis.

Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 4 Transferrin saturation: final or change.
2.5. Analysis.

Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 5 Haematocrit.
2.6. Analysis.

Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 6 End of treatment or change in ESA dose.
2.7. Analysis.
Comparison 2 Laboratory/pharmaceutical outcomes, Outcome 7 eGFR end or change.
See Table 2.
Adverse effects
18 studies provided some information on adverse effects of therapy.
IV iron compared with oral iron may increase the numbers of participants, who experience allergic reactions or hypotension (low certainty evidence) (Analysis 1.6.1 (15 studies, 2607 participants): RR 3.56, 95% CI 1.88 to 6.74; I2 = 0%). The absolute risk for allergic reactions/hypotension was 24 per 1000 with IV iron and 7 per 1000 with oral iron.
Only four studies reported data on infection. The most commonly reported infections were respiratory and urinary tract infections. IV iron compared with oral iron may make little or no difference to the risk of infection (low certainty evidence) (Analysis 1.6.2 (4 studies, 954 participants): RR 1.32, 95% CI 0.90 to 1.95; I2 = 2%).
IV iron compared with oral iron may be associated with fewer participants with all gastrointestinal adverse effects (low certainty evidence) (Analysis 1.6.3 (14 studies, 1986 participants): RR 0.47, 95% CI 0.33 to 0.66; I2 = 63%), fewer participants with constipation (Analysis 1.6.4 (10 studies, 1618 participants): RR 0.32, 95% CI 0.18 to 0.57; I2 = 19%) and possibly with diarrhoea (Analysis 1.6.5 (10 studies, 1625 participants): RR 0.70, 95% CI 0.47 to 1.05; I2 = 0%). The absolute risk of all gastrointestinal adverse effects was 150 per 1000 with IV iron and 319 per 1000 with oral iron.
Only three studies reported data on iron overload. Each of these studies defined iron overload as ferritin levels > 800 ng/mL. IV iron compared with oral iron may make little or no difference to the risk of iron overload (low certainty evidence) (Analysis 1.6.8 (3 studies, 158 participants): RR 6.58, 95% CI 0.81 to 53.51; I2 = 0%).
1.6. Analysis.

Comparison 1 Patient centred outcomes, Outcome 6 Type of adverse event.
See Table 1.
Exploration of heterogeneity using subgroup analyses: effect of different doses of IV or oral iron on haemoglobin, ferritin and TSAT
Subgroup analysis using testing for interaction was applied to investigate the effects of different total doses of IV iron (≤ 1000 mg, 1000 to 2000 mg, > 2000 mg), different doses/month of IV iron (≤ 400 mg/month, > 400 to 700 mg/month, > 700 mg/month), different total doses of oral iron (< 12,000 mg, 12,000 to 30,000 mg, > 30,000 mg), and different doses/month of oral iron (< 4000 mg/month, 4000 to <6000 mg/month, ≥ 6000 mg/month) on levels of Hb, ferritin and TSAT. These values were chosen based on tertiles of doses investigated in the included studies. Results for the outcomes of Hb, ferritin and TSAT are shown as SMD in Table 4; Table 5 and Table 6 respectively.
2. Subgroup analysis and meta‐regression to examine heterogeneity in haemoglobin meta‐analyses.
| Total studies (N) | Studies | SMD (95% CI) | P | |
| Dose IV iron/study month | ||||
| ≥ 400 mg/month | 12 | 8 | 0.17 (‐0.18 to 0.52) | 0.12 |
| > 400 to 700 mg/month | 7 | 6 | 0.76 (0.29 to 1.24) | ‐ |
| > 700 mg/month | 9 | 8 | 0.74 (0.41 to 1.06) | ‐ |
| Dose IV iron (mg total dose) | ||||
| ≤ 1000 mg | 11 | 8 | 0.46 (0.25 to 0.66) | 0.21 |
| 1000 to 1999 mg | 12 | 10 | 0.48 (0.11 to 0.84) | ‐ |
| > 2000 mg | 5 | 4 | 0.89 (0.04 to 1.73) | ‐ |
| Oral dose iron/study month | ||||
| < 4000 mg/month | 12 | 10 | 0.87 (0.37 to 1.38) | 0.15 |
| ≥ 4000 and < 6000 mg/month | 12 | 11 | 0.46 (0.28 to 0.64) | ‐ |
| ≥ 6000 mg/month | 7 | 5 | 0.37 (0.16 to 0.59) | ‐ |
| Dose oral iron (mg total dose) | ||||
| ≥ 12,000 mg | 13 | 12 | 0.60 (0.38 to 0.82) | 0.86 |
| 1200 to 30,000 mg | 10 | 8 | 0.66 (0.29 to 1.03) | ‐ |
| > 30,000 mg | 18 | 11 | 0.45 (‐0.05 to 0.94) | ‐ |
| Any ESA use | ||||
| No EPO | 8 | 6 | 0.57 (0.05 to 1.08) | 0.34 |
| EPO | 27 | 22 | 0.55 (0.32 to 0.78) | ‐ |
| ESA timing of use | ||||
| Start of study | 8 | 7 | 0.40 (0.08 to 0.72) | 0.90 |
| Before study | 19 | 15 | 0.57 (0.28 to 0.85) | ‐ |
| CKD stage | ||||
| 1 to 5 | 15 | 14 | 0.37 (0.26 to 0.50) | 0.10 |
| Dialysis (5D) | 22 | 16 | 0.80 (0.37 to 1.24) | ‐ |
| Study duration | ||||
| ≥ 2 months | 14 | 12 | 0.55 (0.35 to 0.75) | 0.81 |
| > 2 to ≤ 4 months | 9 | 7 | 0.74 (0.28 to 1.19) | ‐ |
| > 4 months | 14 | 11 | 0.46 (0.002 to 0.91) | ‐ |
| Intervention aim | ||||
| Increase Hb | 24 | 20 | 1.00 (0.51 to 1.50) | 0.18 |
| Maintain Hb | 4 | 2 | ‐0.09 (‐0.53 to 0.36) | ‐ |
| Pharmaceutical company sponsorship | ||||
| Unclear | 23 | 18 | 0.81 (0.40 to 1.23) | 0.08 |
| Sponsored | 15 | 13 | 0.38 (0.28 to 0.48) | ‐ |
| Imputed SD | ||||
| Not imputed | ‐ | 5 | 0.42 (0.02 to 0.81) | 0.52 |
| Imputed | ‐ | 26 | 0.55 (0.35 to 0.76) | ‐ |
CKD: chronic kidney disease; EPO ‐ erythropoietin; ESA: erythropoiesis‐stimulating agent; Hb: haemoglobin; SD: standard deviation
3. Subgroup analysis and meta‐regression to examine heterogeneity in ferritin meta‐analyses .
| Total studies (N) | Studies | SMD (95% CI) | P | |
| Dose IV iron/study month | ||||
| ≥ 400 mg/month | 12 | 8 | 1.59 (0.73 to 2.44) | 0.02 |
| > 400 to 700 mg/month | 7 | 6 | 1.62 (1.41 to 1.83) | ‐ |
| >700 mg/month | 9 | 9 | 1.32 (0.85 to 1.78) | ‐ |
| Dose IV iron (mg total dose) | ||||
| ≥ 1000 mg | 11 | 9 | 1.67 (1.03 to 2.30) | 0.08 |
| 1000 to 1999 mg | 12 | 10 | 1.12 (0.83 to 1.42) | ‐ |
| > 2000 mg | 5 | 4 | 2.27 (0.55 to 3.99) | ‐ |
| Oral dose iron/study month | ||||
| <4000 mg/month | 12 | 10 | 1.44 (0.77 to 2.11) | 0.04 |
| ≥ 4000 to < 6000 mg/month | 12 | 11 | 1.43 (1.16 to 1.69) | ‐ |
| ≥ 6000 mg/month | 7 | 7 | 2.16 (1.18 to 3.14) | ‐ |
| Dose oral iron (mg total dose) | ||||
| ≥ 12,000 mg | 13 | 13 | 1.44 (1.05 to 1.83) | 0.40 |
| 12000 to 30,000 mg | 10 | 8 | 1.69 (1.05 to 2.34) | ‐ |
| > 30,000 mg | 18 | 12 | 1.79 (1.15 to 2.43) | ‐ |
| Any ESA use | ||||
| No EPO | 8 | 5 | 1.27 (0.46 to 2.08) | 0.91 |
| EPO | 27 | 25 | 1.62 (1.28 to 1.96) | ‐ |
| ESA timing of use | ||||
| Start of study | 8 | 6 | 1.75 (0.88 to 2.62) | 0.70 |
| Before study | 19 | 18 | 1.64 (1.22 to 2.06) | ‐ |
| CKD stage | ||||
| 1 to 5 | 15 | 14 | 1.70 (1.29 to 2.11) | 0.66 |
| Dialysis (5D) | 22 | 18 | 1.50 (1.07 to 1.92) | ‐ |
| Study duration | ||||
| ≥ 2 months | 10 | 13 | 1.18 (0.86 to 1.49) | 0.54 |
| > 2 to ≤ 4 months | 7 | 8 | 2.64 (1.45 to 3.82) | ‐ |
| > 4 months | 9 | 11 | 1.54 (1.05 to 2.04) | ‐ |
| Intervention aim | ||||
| Increase Hb | 24 | 20 | 336 (84 to 588) | 0.12 |
| Maintain Hb | 4 | 4 | 282 (177 to 261) | ‐ |
| Pharmaceutical company sponsorship | ||||
| Unclear | 23 | 20 | 1.84 (1.31 to 2.37) | 0.63 |
| Sponsored | 15 | 13 | 1.36 (1.02 to 1.71) | ‐ |
| Imputed SD | ||||
| Not imputed | ‐ | 5 | 1.18 (0.51 to 1.86) | 0.62 |
| Imputed | ‐ | 26 | 1.63 (1.31 to 1.94) | ‐ |
CKD: chronic kidney disease; EPO ‐ erythropoietin; ESA: erythropoiesis‐stimulating agent; Hb: haemoglobin; SD: standard deviation
4. Subgroup analysis and meta‐regression to examine heterogeneity in transferrin saturation meta‐analyses.
| Total studies (N) | Studies | SMD (95% CI) | P | |
| Dose IV iron/study month | ||||
| ≥ 400 mg/month | 12 | 7 | 0.69 (0.39 to 1.00) | 0.20 |
| > 400 to 700 mg/month | 7 | 5 | 0.46 (0.14 to 0.78) | ‐ |
| > 700 mg/month | 9 | 7 | 2.00 (0.55 to 3.45) | ‐ |
| Dose IV iron (mg total dose) | ||||
| ≥ 1000 mg | 11 | 8 | 0.62 (0.34 to 0.90) | 0.06 |
| 1000 to 1999 mg | 12 | 9 | 0.41 (0.07 to 0.74) | ‐ |
| > 2000 mg | 5 | 2 | 3.5 (‐1.46 to 8.39) | ‐ |
| Oral dose iron/study month | ||||
| < 4000 mg/month | 12 | 9 | 0.56 (0.25 to 0.86) | 0.21 |
| ≥ 4000 to < 6000 mg/month | 12 | 9 | 0.54 (0.20 to 0.87) | ‐ |
| ≥ 6000 mg/month | 7 | 6 | 1.64 (0.69 to 2.59) | ‐ |
| Dose oral iron (mg total dose) | ||||
| ≥ 12,000 mg | 13 | 11 | 0.56 (0.41 to 0.72) | 0.15 |
| 1200 to 30,000 mg | 10 | 8 | 0.56 (0.00 to 1.13) | ‐ |
| > 30,000 mg | 18 | 8 | 1.59 (0.55 to 2.63) | ‐ |
| Any ESA use | ||||
| No EPO | 8 | 6 | 0.83 (0.36 to 1.31) | 0.83 |
| EPO | 27 | 19 | 0.73 (0.42 to 1.03) | ‐ |
| ESA timing of use | ||||
| Start of study | 8 | 5 | 0.29 (‐0.42 to 1.00) | 0.57 |
| Before study | 19 | 14 | 0.85 (0.51 to 1.20) | ‐ |
| CKD stage | ||||
| 1 to 5 | 15 | 13 | 0.55 (0.35, 0.74) | 0.08 |
| Dialysis (5D) | 22 | 13 | 1.27 (0.75 to 1.80) | ‐ |
| Study duration | ||||
| ≥ 2 months | 14 | 11 | 0.56 (0.41, 0.72) | 0.93 |
| > 2 to ≤ 4 months | 9 | 9 | 1.34 (0.32 to 2.35) | ‐ |
| > 4 months | 14 | 7 | 0.67 (0.16 to 1.18) | ‐ |
| Intervention aim | ||||
| Increase Hb | 24 | 14 | 7.59 (4.07 to 17.11) | 0.18 |
| Maintain Hb | 4 | 4 | 18.28 (‐3.73 to 40.30) | ‐ |
| Pharmaceutical company sponsorship | ||||
| Unclear | 23 | 15 | 1.07 (0.52 to 1.62) | 0.26 |
| Sponsored | 15 | 12 | 0.52 (0.34 to 0.71) | ‐ |
| Imputed SD | ||||
| Not imputed | ‐ | 3 | 0.26 (‐0.24 to 0.77) | 0.45 |
| Imputed | ‐ | 21 | 0.72 (0.47 to 0.97) | ‐ |
CKD: chronic kidney disease; EPO ‐ erythropoietin; ESA: erythropoiesis‐stimulating agent; Hb: haemoglobin; SD: standard deviation
There were no significant differences in total dose administered of IV iron or of IV iron/month between subgroups for Hb or TSAT. There was a significant difference found in the SMD for ferritin in the doses of IV iron per month, though the relationship did not appear to be linear (P = 0.02); there was no difference in ferritin levels with increasing total IV iron dose.
There were no significant differences in total oral iron dose or oral iron/month dose for Hb between subgroups for Hb or TSAT. There was a significant difference found in the SMD for ferritin in the dose of oral iron per month but not with total oral iron dose.
In subgroup analyses no significant differences in results were detected on testing for interaction among studies in which SDs were imputed and other studies (Table 4; Table 5; Table 6).
Exploration of heterogeneity using subgroup analyses: effects of erythrocyte‐stimulating agents (ESAs) on the response to iron therapy
Subgroup analysis was used to investigate the differential response of Hb, ferritin and TSAT levels in patients who did or did not receive an ESA during iron therapy, and in patients who began ESA therapy at study commencement compared with those already on ESA. No significant differences were found among subgroups (Table 4; Table 5; Table 6).
Other subgroup analyses
Subgroup analyses of study duration (≤ 2 months, ≥ 2 to 4 months, ≥ 4 months) showed no significant difference on testing for interaction (Table 4; Table 5; Table 6) for final levels or changes in levels in Hb, ferritin or TSAT. There was significant heterogeneity.
Pharmaceutical company sponsorship previously showed some association with a lower mean reported Hb. With additional data in this updated review, no significant association could be demonstrated (Table 4). There were no significant differences for ferritin or TSAT levels (Table 5; Table 6).
Discussion
Summary of main results
This review included 39 studies which compared IV iron with oral iron therapy in patients with CKD. Eleven studies were added to the original review; one paediatric study was terminated and provided no outcome data. There was considerable variability among studies in the total dose and duration of IV and oral iron therapies prescribed. Durations of studies ranged from 35 days to 26 months with only 14 studies having durations greater than four months. The doses/month of iron ranged from 200 mg to 1000 mg for IV iron and 840 mg to 10,500 mg for oral iron. Use of ESAs also varied. Eight studies reported that ESAs were not administered. Of the studies that reported ESA use, some maintained ESA doses unchanged and others altered the dose to maintain Hb within a target range.
Patient‐centred outcomes such as death (all causes) (11 studies), cardiovascular death (three studies), and quality of life (five studies) were rarely reported with studies concentrating on surrogate laboratory outcomes. While no differences overall in these outcomes were detected between treatment groups, the data available were limited and of low quality (GRADE) so we have low certainty evidence that IV iron compared with oral iron makes little or no difference to these outcomes.
Compared with oral iron, IV iron increased levels of Hb (31 studies), serum ferritin (33 studies) and TSAT (27 studies). The final weighted mean increase with IV iron compared with oral iron was 0.72 g/dL in Hb, 225 µg/L in ferritin and 8% in TSAT. The proportion of patients who reached the targeted Hb or increased their Hb by 1 g/dL was 71% higher among those treated with IV iron compared with oral iron. Increases in these outcomes were seen in dialysis and non‐dialysis participants (Table 3). The required ESA dose was reduced in patients treated with IV iron compared with oral iron, but was reported in only 11 studies involving 522 participants. eGFR did not decline more rapidly with IV iron compared with oral iron. However, the quality of the evidence (GRADE) was considered low for all outcomes indicating that we have low certainty evidence to support the findings above.
Adverse effects were reported in 18 studies. Gastrointestinal adverse effects were more common with oral iron while allergic reactions and/or hypotension were more common with IV iron. However, the quality of the evidence (GRADE) was considered low indicating that we have low certainty evidence to support these findings.
There was considerable heterogeneity between studies so that subgroup analyses using meta‐regression was carried out to investigate possible reasons for this heterogeneity. Subgroup analyses investigated the effect of different monthly and total doses of oral or IV iron, different uses of ESA, CKD stage, and different durations of treatment on Hb, ferritin and transferrin levels. Other than an increase in ferritin levels with increasing IV and oral iron per month, no differences were found in these analyses. Comparing the results of these subgroup analyses with those in the initial version of this systematic review, no increase in Hb SMD with increased oral iron dose/month could now be demonstrated. There was no longer a significant increase in ferritin levels with total IV or oral iron dose. The additional data from newly identified studies showed that studies sponsored by pharmaceutical companies were no longer associated with a significantly lower increase in MD in Hb compared with studies that did not report sponsorship. Heterogeneity among studies therefore remains largely unexplained, but was likely to be related to the significant variation in the relative doses of IV and oral iron used in each study.
Overall completeness and applicability of evidence
Most included studies reported on laboratory assessments of response to IV and oral iron treatment in patients with CKD stages 3 to 5 including those receiving dialysis. Our meta‐analyses identified that there are probably small increases in laboratory parameters of Hb, ferritin and transferrin in both dialysis and non‐dialysis patients though the certainty of the evidence was low. However, key patient‐centred outcomes were reported in only a few studies so we were unable to make definitive conclusions about the influence of IV iron compared with oral iron therapy on death (all causes), cardiovascular death, morbidity, or on quality of life. This review confirmed that gastrointestinal disorders are found to be more common in patients taking oral iron while hypotension and allergic reactions are more common in patients receiving IV iron. Although ESA dose was probably lower in patients treated with IV iron, only 11 studies (522 participants) reported on ESA dosage at the end of the study and all studies providing these data were in dialysis patients.
The observed Hb increase of 1.01 g/dL in dialysis patients, together with a significant reduction in ESA dose, provides some support for the practice of administering IV iron to these patients, particularly among those unable to tolerate oral iron. However, studies have identified that high Hb levels achieved with IV iron and ESA are associated with increased cardiovascular death and morbidity (Phrommintikul 2007).
The Hb increase in non‐dialysis patients was modest (0.41 g/dL), but this was not significantly different from the response in dialysis patients. None of the included studies assessed if the patient‐centred benefits of achieving higher Hb levels outweighed financial costs or disruption to patients not on dialysis as a result of additional or prolonged hospital visits. While three further large studies (Agarwal 2015 CKD; FIND‐CKD 2014 CKD; Kalra 2016 CKD) in CKD patients assessed quality of life, none identified improved quality of life with the higher Hb associated with IV iron therapy so that only one of five studies, which assessed quality of life, identified some improvement in quality of life in non‐dialysis patients receiving IV iron (Agarwal 2006 CKD). A systematic review of RCTs identified no significant benefit on quality of life of higher Hb levels achieved with ESAs and iron supplements in CKD patients (Collister 2016).There were no data relating to non‐dialysis patients to determine if ESA requirements were reduced. We were therefore unable to derive a definitive conclusion on the relative benefits and harms of IV iron for non‐dialysis patients.
The applicability of the conclusions in children, PD patients and kidney transplant patients may be limited since few studies were identified for each of these patient groups. However, the magnitude and direction of results in these studies did not differ from the overall results.
Quality of the evidence
Our review included 39 studies, which involved 3852 participants. Twenty‐one studies enrolled dialysis patients, two involved transplant patients, two enrolled dialysis and non‐dialysis patients and the remainder enrolled CKD patients. There was considerable variation among studies in dose and duration of IV and oral iron administration.
Of the 39 included studies, 12 were available only as abstracts. Twenty studies reported adequate random sequence generation while only 14 studies demonstrated adequate allocation concealment. Studies that lack adequate allocation concealment are considered to be at increased risk of bias (Moyer 1998; Schultz 1995). Blinding of participants and personnel was not reported in any study. No study reported blinding of outcome assessment, but because primary outcomes were laboratory measurements and unlikely to be influenced by lack of blinding, all studies were considered to be at low risk of bias for blinding of outcome assessment. Twenty‐two studies reported complete outcome data while 20 studies were at low risk of selective reporting. The authors of 15 included studies indicated receiving some form of sponsorship from pharmaceutical companies.
Although administration of IV iron consistently resulted in an increase in Hb or HCT, ferritin and TSAT, there was considerable heterogeneity among studies in the results of these laboratory outcomes. This effect could not be explained after examining for interactions related to participants, interventions and risk of bias items as reported.
The certainty of the evidence for patient centred outcomes was considered low or very low because of small patient numbers included in these analyses and high risk or unclear risk of bias for allocation concealment in many studies (Table 1). Similarly the certainty of the evidence for laboratory and pharmaceutical outcomes was considered to be low or very low because of considerable heterogeneity in study results and the high or unclear risk for allocation concealment in many studies (Table 2).
Potential biases in the review process
The literature search has been run several times (up to December 2018) since the publication of the original review in 2012 to reduce the likelihood that additional studies eligible for inclusion were missed. Although the Cochrane Kidney and Transplant Specialised Register also includes references of reports of studies identified by handsearching resources including conference proceedings, it is a possibility that relevant studies may have been missed.
The relatively high proportion of included studies that were available only as abstracts (12/39; 31%) is a potential source of bias as abstracts may not contain complete results or provide detailed information on risk of bias attributes. To address reporting gaps in studies, we contacted authors to seek additional information. Responses from nine study authors were received but information received related principally to risk of bias attributes. In this update, we only identified the full publication of one study (Qunibi 2011 CKD) previously included as an abstract. A large completed RCT (Lu 2010 CKD) comparing IV ferumoxytol with oral iron enrolled 519 participants but has only been published in abstract form in combination with other similar studies.
Some outcomes were reported in only a few studies which increased the risk of selection bias. In particular, the final or change in ESA dose was reported in only 11 studies (522 patients) so that the observed significant decrease in ESA dose with IV iron therapy compared with oral iron may not be generalisable to the dialysis population. Similarly, adverse effects were reported in only 18 (46%) of the included studies.
Agreements and disagreements with other studies or reviews
A systematic review published in 2008 that included 13 studies applied a comprehensive literature review strategy that included searching some conference proceedings (American Society of Nephrology, European Renal Association ‐ European Dialysis and Transplant Association) (Rozen‐Zvi 2008). This systematic review was updated to October 2015 and included 24 RCTs (Shepshelovich 2016). Our updated review includes 22 of the 24 studies included by Shepshelovich 2016. We excluded one study included in the 2008 systematic review and one included in the 2015 review because they included both randomised and non‐randomised data (Allegra 1991; Adhikary 2011). Shepshelovich 2016 identified 13 studies involving CKD participants and 11 studies involving dialysis patients. Both reviews reported similar increases in mean Hb, ferritin, TSAT and the proportion of participants achieving an increase in Hb in patients treated with IV iron compared with oral iron. In their initial systematic review, Rozen‐Zvi 2008 demonstrated a significant correlation between Hb and IV iron dose/month in dialysis patients but not non‐dialysis patients. We were unable to demonstrate an overall correlation or a correlation in dialysis patients alone in our 2012 review or in this update. Both reviews reported considerable heterogeneity for the outcomes of Hb, ferritin and TSAT concentrations which could not be explained. A systematic review of 103 RCTs has evaluated the safety of IV iron compared with oral iron, no iron, placebo or intramuscular iron (Avni 2015). The review found no increase in overall serious adverse effects with IV iron. As in this review, the authors found that serious infusion reactions were more common and gastrointestinal adverse reactions were less common with IV iron, while there was no increase in infections with IV iron. Another systematic review (Susantitaphong 2014) included 34 single‐arm and RCTs evaluating IV iron in HD patients with relative or functional iron deficiency as defined by ferritin levels > 200 ± TSAT < 30%; studies of patients with absolute iron deficiency were excluded. Therefore only one study (Provenzano 2009 HD) included in our review was also included in that review. The review also concluded that IV iron resulted in increases in Hb, ferritin and TSAT and reductions in ESA dose though the changes were less evident in RCTs compared with single arm studies.
Authors' conclusions
Implications for practice.
This systematic review identified evidence to indicate that compared with oral iron therapy, IV iron therapy contributed increased ferritin and TSAT levels, reduced ESA dose required, and provided a small but significant increase in Hb. Limited patient‐centred outcomes data (death, cardiovascular disease, quality of life) were reported in the included studies. These data support the current practice of administering IV iron to in‐centre HD patients to increase iron stores, and probably, reduce both the ESA dose required, and its cost.
While this update identified a few more studies, which addressed patient‐centred outcomes, including adverse effects, to determine if benefits exceed harms for all patients with CKD, only 11 studies reported on death (all causes), three reported on cardiovascular death and five reported on quality of life. However, because of small number of studies reporting these outcomes and low quality of evidence, the relative effects of different iron regimens on these patient‐centred outcomes remain uncertain. More studies reported on allergic reactions/hypotension (14 studies) and gastrointestinal adverse effects (13 studies), While gastrointestinal adverse effects with oral iron are common with oral iron, these effects must be balanced against the rare, but potentially life threatening adverse effects seen with IV iron.
There are now additional large studies examining IV and oral iron in CKD participants with longer follow up periods. These confirm that IV iron compared with oral iron in CKD participants increases laboratory indices compared with oral iron, increases the number achieving target Hb without changing the rate of decline in kidney function. However, there are still no studies, which have assessed whether these benefits outweigh the disadvantages of increased numbers and durations of hospital visits for treatment.
Implications for research.
Further large RCTs with longer durations of treatment and follow‐up periods are still required. These need to assess patient‐centred outcomes including death (all causes), cardiovascular death, cardiac morbidity using cardiac function tests, hospitalisations, quality of life and patient inconvenience created by hospital or clinic visits for IV iron in non‐dialysis or PD patients as well as common haematological parameters. The costs of all aspects of IV therapy must also be determined to assess overall value of IV iron, especially in non‐dialysis and PD patients. The doses of oral and IV iron should be standardised across studies in an effort to reduce the heterogeneity seen in this systematic review.
What's new
| Date | Event | Description |
|---|---|---|
| 20 February 2019 | New search has been performed | New studies added (11) |
| 20 February 2019 | New citation required and conclusions have changed | GRADE used to assess the evidence |
History
Protocol first published: Issue 3, 2009 Review first published: Issue 1, 2012
| Date | Event | Description |
|---|---|---|
| 12 March 2014 | Amended | Search strategies updated |
Acknowledgements
We would like to thank the referees for their comments and feedback during the preparation of this review update.
We would like to thank Drs Broumand, Fudin, Macdougall, Provenzano, Richardson, Spinowitz, Van Wyck and Warady, and Ms Dahl, for their responses to our queries about their studies.
We would like to thank Dr Jumana Albaramki, who carried out all the steps in the original version of this review
We would like to thank Dr Melani Mahendran for her help in obtaining the data for Figure 1
Appendices
Appendix 1. Electronic search strategies
| Database | Search terms |
| CENTRAL |
|
| MEDLINE |
|
| EMBASE |
|
Appendix 2. Risk of bias assessment tool
| Potential source of bias | Assessment criteria |
|
Random sequence generation Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence |
Low risk of bias: Random number table; computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots; minimization (minimization may be implemented without a random element, and this is considered to be equivalent to being random). |
| High risk of bias: Sequence generated by odd or even date of birth; date (or day) of admission; sequence generated by hospital or clinic record number; allocation by judgement of the clinician; by preference of the participant; based on the results of a laboratory test or a series of tests; by availability of the intervention. | |
| Unclear: Insufficient information about the sequence generation process to permit judgement. | |
|
Allocation concealment Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment |
Low risk of bias: Randomisation method described that would not allow investigator/participant to know or influence intervention group before eligible participant entered in the study (e.g. central allocation, including telephone, web‐based, and pharmacy‐controlled, randomisation; sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes). |
| High risk of bias: Using an open random allocation schedule (e.g. a list of random numbers); assignment envelopes were used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered); alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. | |
| Unclear: Randomisation stated but no information on method used is available. | |
|
Blinding of participants and personnel Performance bias due to knowledge of the allocated interventions by participants and personnel during the study |
Low risk of bias: No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding; blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding; blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Blinding of outcome assessment Detection bias due to knowledge of the allocated interventions by outcome assessors. |
Low risk of bias: No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding; blinding of outcome assessment ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Incomplete outcome data Attrition bias due to amount, nature or handling of incomplete outcome data. |
Low risk of bias: No missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes not enough to have a clinically relevant impact on observed effect size; missing data have been imputed using appropriate methods. |
| High risk of bias: Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size; ‘as‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation. | |
| Unclear: Insufficient information to permit judgement | |
|
Selective reporting Reporting bias due to selective outcome reporting |
Low risk of bias: The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way; the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon). |
| High risk of bias: Not all of the study’s pre‐specified primary outcomes have been reported; one or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified; one or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis; the study report fails to include results for a key outcome that would be expected to have been reported for such a study. | |
| Unclear: Insufficient information to permit judgement | |
|
Other bias Bias due to problems not covered elsewhere in the table |
Low risk of bias: The study appears to be free of other sources of bias. |
| High risk of bias: Had a potential source of bias related to the specific study design used; stopped early due to some data‐dependent process (including a formal‐stopping rule); had extreme baseline imbalance; has been claimed to have been fraudulent; had some other problem. | |
| Unclear: Insufficient information to assess whether an important risk of bias exists; insufficient rationale or evidence that an identified problem will introduce bias. |
Data and analyses
Comparison 1. Patient centred outcomes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death (all causes) | 11 | 1952 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.64, 1.94] |
| 2 Cardiovascular death | 3 | 206 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.41, 7.18] |
| 3 Quality of life | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Number of non‐dialysis patients needing to commence dialysis | 4 | 743 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.41, 1.61] |
| 5 Number requiring transfusion | 5 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.55, 1.34] |
| 6 Type of adverse event | 21 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Allergic reactions/hypotension | 15 | 2607 | Risk Ratio (M‐H, Random, 95% CI) | 3.56 [1.88, 6.74] |
| 6.2 Infection | 4 | 954 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.90, 1.95] |
| 6.3 All gastrointestinal adverse effects | 14 | 1986 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.33, 0.66] |
| 6.4 Constipation | 10 | 1618 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.18, 0.57] |
| 6.5 Diarrhoea | 10 | 1625 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.47, 1.05] |
| 6.6 Nausea and vomiting | 9 | 1573 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.45, 1.29] |
| 6.7 Taste disturbances | 4 | 851 | Risk Ratio (M‐H, Random, 95% CI) | 3.78 [0.84, 16.97] |
| 6.8 Iron overload | 3 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 6.58 [0.81, 53.51] |
Comparison 2. Laboratory/pharmaceutical outcomes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number achieving target haemoglobin or increase ≥ 1 g/dL | 13 | 2206 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.43, 2.04] |
| 2 Haemoglobin: final or change (all patients) | 31 | 3373 | Mean Difference (IV, Random, 95% CI) | 0.72 [0.39, 1.05] |
| 3 Ferritin: final or change (all patients) | 33 | 3389 | Mean Difference (IV, Random, 95% CI) | 224.84 [165.85, 283.83] |
| 4 Transferrin saturation: final or change | 27 | 3089 | Mean Difference (IV, Random, 95% CI) | 7.69 [5.10, 10.28] |
| 5 Haematocrit | 4 | 152 | Mean Difference (IV, Random, 95% CI) | 1.18 [‐2.17, 4.52] |
| 6 End of treatment or change in ESA dose | 11 | 522 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.72 [‐1.12, ‐0.31] |
| 7 eGFR end or change | 8 | 1052 | Mean Difference (IV, Random, 95% CI) | 0.83 [‐0.79, 2.44] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Agarwal 2006 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomisation of blocks of 4 |
| Allocation concealment (selection bias) | Low risk | Central randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data balanced in both groups, reason for missing data unlikely to be related to true outcome |
| Selective reporting (reporting bias) | Low risk | Study protocol available in paper and all of the pre‐specified outcomes reported |
| Other bias | High risk | Funded by Watson Laboratories Inc |
Agarwal 2015 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised in a 1:1 ratio using permuted blocks. The randomisation sequence was computer generated by a statistician |
| Allocation concealment (selection bias) | Low risk | Opaque and concealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded and lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome is a laboratory outcome and unlikely to be influenced by lack of blinding. Adverse events adjudicated by blinded personnel. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | At 3 months < 20% lost to follow‐up (13/136). Trial stopped early |
| Selective reporting (reporting bias) | High risk | All prespecified outcomes reported but no standard deviations reported |
| Other bias | Low risk | Supported in part by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (U01‐DK71633) and Indiana Institute for Medical Research |
Aggarwal 2003 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All reported patients included in follow up, but unclear whether any patients included were initially excluded from analysis |
| Selective reporting (reporting bias) | High risk | Some outcomes, such as symptoms of fatigue and shortness of breath, were reported incompletely and could not be included in the meta‐analysis |
| Other bias | Unclear risk | Funding source not reported |
Ahsan 1997 TX.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scant data available from abstract |
| Selective reporting (reporting bias) | Unclear risk | Limited information to judge |
| Other bias | Unclear risk | Funding source not reported |
Broumand 1998 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐intervention
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were included in analysis |
| Selective reporting (reporting bias) | High risk | Limited information on methods. SDs imputed |
| Other bias | Unclear risk | Funding source not reported |
Charytan 2005 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for patients not completing the trial were not provided, patients with missing data were excluded from analysis (19% missing in IV, 8% missing in oral group). Data were provided as percentages with unclear denominators |
| Selective reporting (reporting bias) | High risk | Data were not provided with SD. SD imputed to enable entry in meta‐analyses |
| Other bias | High risk | Funded by American Regent Inc |
Erten 1998 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron group 1
IV iron group 2
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes not affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one patient excluded from analysis |
| Selective reporting (reporting bias) | Unclear risk | Limited information on methods |
| Other bias | Unclear risk | Funding source not reported |
FIND‐CKD 2014 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron low‐ferritin arm
IV iron high‐ferritin arm
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Via a central interactive voice‐response system in a 1:1:2 ratio, with randomisation blocks distributed by country |
| Allocation concealment (selection bias) | Low risk | Central interactive voice‐response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were accounted for |
| Selective reporting (reporting bias) | Low risk | Review's prespecified primary outcomes reported in either full publication or abstract |
| Other bias | High risk | Funding source bias: "This work was supported by Vifor Pharma" |
Fishbane 1995 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Large number excluded from analysis, 20% in oral group, 36% in IV group |
| Selective reporting (reporting bias) | Low risk | Study protocol available in paper and all of the pre‐specified outcomes reported |
| Other bias | Unclear risk | Funding source not reported |
Fudin 1998 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Table of 1000 random digits generated by multiplicative congruent method |
| Allocation concealment (selection bias) | High risk | Open random allocation schedule |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 16% in both groups did not complete and were excluded from the analysis |
| Selective reporting (reporting bias) | Unclear risk | Limited information on methods |
| Other bias | Unclear risk | Funding source not reported |
Hussain 1998 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear whether results of all patients were included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Limited information on methods |
| Other bias | Unclear risk | Funding source not reported |
Kalra 2016 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV Iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system (IWRS) was used to randomise the patients |
| Allocation concealment (selection bias) | Low risk | Unique identifier number via IWRS |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded and lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not blinded but primary outcome is laboratory measurement and unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 7 (2%) lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes reported |
| Other bias | High risk | Funded by Pharmacosmos A/S |
Kotaki 1997 HD.
| Methods |
|
|
| Participants |
Exclusion criteria
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data balanced across groups |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes were reported |
| Other bias | Unclear risk | Funding source not reported |
Leehey 2005 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomisation schedule |
| Allocation concealment (selection bias) | Low risk | Central randomisation in blocks of 4 at a 1:1 ratio. Investigators had no prior knowledge of allocation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory based outcome unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only |
| Selective reporting (reporting bias) | High risk | Abstract only. Reported change in Hb and ferritin. No SDs provided. SD imputed for inclusion in meta‐analyses |
| Other bias | High risk | Funded by Watson Laboratories Inc |
Li 2008 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised random number list |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing data. All participants completed the study and were included in the analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funding source not reported |
Li 2008 PD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer random number list |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing data |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funding source not reported |
Lu 2010 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 3:1 automated pre‐programmed interactive voice response system |
| Allocation concealment (selection bias) | Low risk | Telephone based |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could have influenced management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open label but outcomes were laboratory based and unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data of primary endpoint balanced between groups,5.3% IV,12% oral |
| Selective reporting (reporting bias) | Low risk | Study protocol not available but identical to Spinowitz 2008. Information on outcomes provided by AMAG Pharmaceuticals |
| Other bias | High risk | Funded by AMAG Pharmaceuticals whose employees identified study sites, monitored the study and performed data analyses according to a predefined statistical analysis plan |
Lye 2000 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract only. No information provided |
| Allocation concealment (selection bias) | High risk | Inadequate allocation. Author reported that patients were allocated alternately to each group |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Abstract only. No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Abstract only. Outcome was laboratory based and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only. No information provided |
| Selective reporting (reporting bias) | Unclear risk | Abstract only. Reported end of study Hb and ferritin levels. Patient numbers provided by the author |
| Other bias | Unclear risk | Abstract only |
Macdougall 1996 HD,PD,CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sealed envelopes containing random numbers |
| Allocation concealment (selection bias) | Low risk | Central, by pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were accounted for |
| Selective reporting (reporting bias) | Low risk | Primary outcomes reported in either full publication or abstract |
| Other bias | Unclear risk | Funding source not reported |
Macdougall 1999 HD,PD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract only; not reported |
| Allocation concealment (selection bias) | Unclear risk | Abstract only; not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Abstract only. No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Abstract only. Outcome was laboratory based and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only. Unclear if all patients completed study |
| Selective reporting (reporting bias) | Unclear risk | Abstract only. Reported end of study Hb, ferritin and per cent of participants who had a rise in Hb |
| Other bias | Unclear risk | Abstract only |
McMahon 2009 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple block randomisation from block randomisation lists generated with Graphpad Statmate |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open label study but outcomes based on laboratory results unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were accounted for. Patients not completing 6 months were excluded a priori |
| Selective reporting (reporting bias) | Low risk | Primary outcomes (end Hb, ferritin, TSAT) reported in either full publication or abstract |
| Other bias | High risk | Grant/research support from Vifor (NCT 000202345) |
Michael 2007 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear if all patients completed study |
| Selective reporting (reporting bias) | Unclear risk | No clear protocol |
| Other bias | High risk | Grant/Research support: Watson Laboratories |
Mudge 2009 TX.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated sequence with blocks of 10 |
| Allocation concealment (selection bias) | Low risk | Was performed by the use of sequentially numbered, sealed, opaque envelopes with stratification for calcineurin inhibitor type (cyclosporine or tacrolimus), in a one‐to‐one ratio |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients completed the study |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | Funded by Princess Alexandra Hospital Research Foundation |
Nagaraju 2013 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence |
| Allocation concealment (selection bias) | Low risk | Group allocation was stored in sealed opaque sequentially numbered envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single blind (investigator) was blinded but no blinding of care givers or patients. Lack of blinding could have influenced management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were laboratory based and unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 7/40 (17.5%) dropped out but were included in analyses (last result carried forward). All randomised patients were followed until the end of the study, reasons for dropout provided |
| Selective reporting (reporting bias) | Low risk | Outcomes were reported as median and IQR and could not be entered in meta‐analyses; we have changed this to low risk as we have used the median as the mean and imputed standard deviations |
| Other bias | Low risk | Baseline age imbalance between the two groups but group ages were not significantly different. Funded by Ottawa Hospital Research Institute |
NCT01155375 HD,PD,CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV ferumoxytol
Oral ferrous fumarate
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome laboratory based |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | High risk | Study set up by AMAG Pharmaceuticals, Inc. |
Pisani 2014 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral Iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list generated by computer |
| Allocation concealment (selection bias) | Low risk | numbered, sealed envelopes opened in sequence by staff personnel not involved in patient care |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded and lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | outcomes were laboratory based and unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 99/109 patients analysed, only 6 lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes were all reported |
| Other bias | Unclear risk | Funding not stated |
Provenzano 2009 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐intervention
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Low risk | Telephone‐based system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients who were randomised were included in the analysis |
| Selective reporting (reporting bias) | Low risk | All outcomes defined in study registration reported |
| Other bias | High risk | Funded by AMAG Pharmaceuticals whose employees identified study sites, monitored the study and performed data analyses according to a predefined statistical analysis plan |
Qunibi 2011 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised interactive voice‐response system. Stratified by severity of CKD |
| Allocation concealment (selection bias) | Low risk | Centralised interactive voice‐response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label study. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10/255 (4%) not included in analyses |
| Selective reporting (reporting bias) | Low risk | Data provided on expected outcomes including adverse effects |
| Other bias | High risk | Funding support from American Reagent/Luipold Pharmaceuticals |
Ragab 2007 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV Iron
Oral Iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were "randomly subdivided" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded, lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory based outcome, unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No patients lost to follow‐up |
| Selective reporting (reporting bias) | High risk | No prespecified outcomes and only medians reported with no IQR |
| Other bias | Unclear risk | Funding not reported |
Souza 1997 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐intervention
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear if all patients completed study |
| Selective reporting (reporting bias) | Unclear risk | No clear protocol |
| Other bias | Unclear risk | Funding source not reported |
Spinowitz 2008 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 3:1 automated pre‐programmed interactive voice response system |
| Allocation concealment (selection bias) | Low risk | Telephone based |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data of primary endpoint balanced between groups,10% IV,13% oral |
| Selective reporting (reporting bias) | Low risk | Study protocol available in paper and all of the prespecified outcomes reported |
| Other bias | High risk | Funded by AMAG Pharmaceuticals whose employees identified study sites, monitored the study and performed data analyses according to a predefined statistical analysis plan |
Stoves 2001 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐based |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 29% did not complete the study; this large number could induce bias the results |
| Selective reporting (reporting bias) | High risk | Outcomes reported as median and IQR and could not be entered in meta‐analyses |
| Other bias | High risk | Imbalance between ACEi treatment in each group Sponsored by Janssen Cilag and Syner‐Med |
Strickland 1977 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced allocation within strata using a method similar to the minimisation procedure |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 25% of participants were excluded from analysis |
| Selective reporting (reporting bias) | High risk | Data combined in crossover study and could not be incorporated in meta‐analyses |
| Other bias | High risk | Funding support from Abbott Laboratories Ltd and Fisons Pharmaceuticals Ltd |
Svara 1996 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Three patients were excluded from oral, one from IV group |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in methods were reported |
| Other bias | Unclear risk | Funding source not reported |
Tsuchida 2010 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No flow chart provided, only data about included patients is provided |
| Selective reporting (reporting bias) | Low risk | Expected outcomes of haematological outcomes and adverse effects reported |
| Other bias | Unclear risk | Funding source not reported |
Van Wyck 2005 CKD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sequential random numbers |
| Allocation concealment (selection bias) | Low risk | Central randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data balanced between groups |
| Selective reporting (reporting bias) | Low risk | All of outcomes have been reported |
| Other bias | High risk | Supported by American Regent, Inc |
Wang 2003 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome is laboratory based and unlike to be altered by blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Selective reporting (reporting bias) | Unclear risk | Reported end Hb and ferritin |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Warady 2002 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding. Lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were included in the analysis (information from authors) |
| Selective reporting (reporting bias) | Low risk | All of outcomes have been reported |
| Other bias | High risk | Supported by a grant from Watson Laboratories |
Winney 1977 HD.
| Methods |
|
|
| Participants |
|
|
| Interventions | IV iron
Oral iron
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly allocated" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding and lack of blinding could influence results |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcomes unlikely to be affected by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Selective reporting (reporting bias) | Unclear risk | Abstract only. Only information provided is for Hb/HCT |
| Other bias | Unclear risk | No information provided on funding |
ACE ‐ angiotensin‐converting enzyme; ACEi ‐ angiotensin‐converting enzyme inhibitor; AKI ‐ acute kidney injury; CAD ‐ coronary artery disease; CAPD ‐ continuous ambulatory peritoneal dialysis; CHF ‐ congestive heart failure; CHr ‐ reticulocyte haemoglobin content; CKD ‐ chronic kidney disease; Cr ‐ creatinine; CrCl ‐ creatinine clearance; CRP ‐ C‐reactive protein; DGF ‐ delayed graft function; eGFR ‐ estimated glomerular filtration rate; (rHu)EPO ‐ (recombinant human) erythropoietin; ESA ‐ erythrocyte‐stimulating agent/s; FOBT ‐ faecal occult blood test; GFR ‐ glomerular filtration rate; Hb ‐ haemoglobin; HCT ‐ haematocrit; HCV ‐ hepatitis C virus; HD ‐ haemodialysis; HIV ‐ human immunosuppressive virus; HTN ‐ hypertension; IHD ‐ ischaemic heart disease; IQR ‐ interquartile range; IV ‐ intravenous; Kt/V ‐ dialyser urea clearance adequacy; M/F ‐ male/female; MDRD ‐ Modified Diet in Renal Disease; PCKD ‐ polycystic kidney disease; PCV ‐ packed cell volume; PD ‐ peritoneal dialysis; PTH ‐ parathyroid hormone; RBC ‐ red blood cell/s; SD ‐ standard deviation; SE ‐ standard error; SFGC ‐ sodium ferric gluconate complex; TIBC ‐ total iron binding capacity; TSAT ‐ transferrin saturation; URR ‐ urea reduction ratio
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adhikary 2011 | Said to be RCT but included some non‐randomised participants |
| Allegra 1991 | Said to be RCT but results included some non‐randomised patients |
| Charytan 2013 | Wrong comparator: IV iron is compared to standard medical therapy, which could be oral or IV iron. No separate data available for patients receiving oral iron |
| HEMATOCRIT 2012 | Wrong intervention: Compares two oral iron preparations |
| Lye 1997 | Wrong intervention: compares intramuscular and oral routes |
RCT ‐ randomised controlled trial
Contributions of authors
Draft the protocol: JA, EH, JC
Study selection: JA, EH, EO'L
Extract data from studies: JA, EH, EO'L
Enter data into RevMan: JA, EH, EO'L
Carry out the analysis: JA, EH, AW, EO'L
Interpret the analysis: JA, EH, AW, JC, EO'L
Draft the final review: JA, EH, AW, JC, EO'L
Disagreement resolution: AW, JC
Update the review: EO'L, EH, AW, DB, IN, JC
Declarations of interest
Emma L O'Lone: none known
Elisabeth M Hodson: none known
Ionut Nistor: none known
Davide Bolignano: none known
Angela C Webster: none known
Jonathan C Craig: none known
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Agarwal 2006 CKD {published and unpublished data}
- Agarwal R. Sodium ferric gluconate complex in sucrose (SFGC) injection improves energy level in chronic kidney disease (CKD) patients independent of increase in hemoglobin levels [abstract no: F‐PO337]. Journal of the American Society of Nephrology 2004;15(Oct):140A. [CENTRAL: CN‐01657325] [Google Scholar]
- Agarwal R, Rizkala AR, Bastani B, Kaskas MO, Leehey DJ, Besarab A. A randomized controlled trial of oral versus intravenous iron in chronic kidney disease. American Journal of Nephrology 2006;26(5):445‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Agarwal 2015 CKD {published data only}
- Agarwal R, Kusek JW, Pappas MK. A randomized trial of intravenous and oral iron in chronic kidney disease. Kidney International 2015;88(4):905‐14. [DOI: 10.1038/ki.2015.163; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Aggarwal 2003 CKD {published data only}
- Aggarwal HK, Kumar H, Singh S, Nand N. Iron therapy in patients of CRF receiving recombinant human EPO which route of iron? [abstract]. Indian Journal of Nephrology 2002;12(4):205. [CENTRAL: CN‐00460253] [Google Scholar]
- Aggarwal HK, Nand N, Singh S, Singh M, Hemant, Kaushik G. Comparison of oral versus intravenous iron therapy in predialysis patients of chronic renal failure receiving recombinant human erythropoietin. Journal of the Association of Physicians of India 2003;51:170‐4. [MEDLINE: ] [PubMed] [Google Scholar]
- Aggarwal SH, Hemant K, Singh S, Nand N, Sharma BD. Comparison of oral versus intravenous iron therapy in patients of chronic renal failure receiving recombined human erythropoietin (R‐HUEPO) [abstract no: 56]. Journal of the Association of Physicians of India 2001;50(Oct). [CENTRAL: CN‐00663209] [PubMed] [Google Scholar]
Ahsan 1997 TX {published data only}
- Ahsan N, Holman MJ, O'Brien B, Langhoff EG, Yang HC. Intravenous infusion of total is superior to oral iron in the treatment of post‐kidney transplant functional anemia [abstract no: A3131]. Journal of the American Society of Nephrology 1997;8(Program & Abstracts):672A. [CENTRAL: CN‐00676050] [Google Scholar]
Broumand 1998 HD {published and unpublished data}
- Broumand B, Ghods A, Taheri FM, Hanjani MR. Intravenous versus oral iron supplementation in the management of anemia in end stage renal disease [abstract]. 35th Congress. European Renal Association. European Dialysis and Transplantation Association; 1998 Jun 6‐9; Rimini, Italy. 1998:330. [CENTRAL: CN‐00483339]
Charytan 2005 CKD {published data only}
- Charytan C, Qunibi W, Bailie GR, Venofer Clinical Studies Group. Comparison of intravenous iron sucrose to oral iron in the treatment of anemic patients with chronic kidney disease not on dialysis. Nephron 2005;100(3):c55‐62. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Charytan C, Qunibi W, Singh H, Schwenk M, Aronoff G, Besarab A. Safety of Venofer®, [iron sucrose injection] administered by rapid IV push (IVP) in predialysis CKD patients [abstract no: SU‐PO260]. Journal of the American Society of Nephrology 2002;13(September, Program & Abstracts):521a. [CENTRAL: CN‐00444759] [Google Scholar]
Erten 1998 HD {published data only}
- Erten Y, Ozdemir FN, Guz G, Sezer S, Haberal A, Kaya S, et al. Comparison of the effect of intravenous and oral iron therapies on hemodialysis patients [abstract no: M384]. Journal of the American Society of Nephrology 1998;9(Program & Abstracts):248A. [CENTRAL: CN‐01657553] [Google Scholar]
- Erten Y, Ozdemir FN, Guz G, Sezer S, Haberal A, Kaya S, et al. Comparison of the effect of intravenous and oral iron therapies on hemodialysis patients [abstract]. 35th Congress. European Renal Association. European Dialysis and Transplantation Association; 1998 Jun 6‐9; Rimini, Italy. 1998:337. [CENTRAL: CN‐00445251]
FIND‐CKD 2014 CKD {published data only}
- Gaillard CA, Bock AH, Carrera F, Eckardt K, Wyck DB, Bansal SS, et al. Hepcidin response to intravenous (IV) or oral iron in the randomized FIND‐CKD trial of patients with non‐dialysis CKD (ND‐CKD) [abstract no: TH‐OR040]. Journal of the American Society of Nephrology 2015;26(Abstract Suppl):11A. [Google Scholar]
- Gaillard CA, Bock AH, Carrera F, Eckardt KU, Wyck DB, Bansal SS, et al. Hepcidin response to iron therapy in patients with non‐dialysis dependent CKD: an analysis of the FIND‐CKD trial. PLoS ONE [Electronic Resource] 2016;11(6):e0157063. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall I, Bock AH, Carrera F, Eckardt KU, Gaillard CA, Wyck DB, et al. A model to predict haematopoietic non‐response to oral iron in patients with non‐dialysis dependent CKD (ND‐CKD): an analysis from the FIND‐CKD trial [abstract]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i193‐4. [EMBASE: 72326413] [Google Scholar]
- Macdougall IC, Bock A, Carrera F, Cushway T, Mori C, Roger S. Rationale and design of a new RCT of iron therapy in anemic non‐dialysis CKD patients: FIND‐CKD trial [abstract no: SA‐PO2402]. Journal of the American Society of Nephrology 2009;20(Abstract Suppl):660A. [Google Scholar]
- Macdougall IC, Bock A, Carrera F, Eckardt KU, Gaillard C, Wyck D, et al. The FIND‐CKD study‐‐a randomized controlled trial of intravenous iron versus oral iron in non‐dialysis chronic kidney disease patients: background and rationale. Nephrology Dialysis Transplantation 2014;29(4):843‐50. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Bock A, Carrera F, Eckardt KU, Gaillard C, Wyck DB, et al. FIND‐CKD: a 56‐week randomized trial of intravenous ferric carboxymaltose versus oral iron in anemic patients with chronic kidney disease and iron deficiency [abstract no: SA‐PO1079]. Journal of the American Society of Nephrology 2013;24(Abstracts):3B. [Google Scholar]
- Macdougall IC, Bock A, Carrera F, Eckardt KU, Gaillard C, Wyck DB, et al. Intravenous ferric carboxymaltose vs oral iron in anaemic patients with non‐dialysis dependent chronic kidney disease and iron deficiency: results of the randomised FIND‐CKD study [abstract no: SO049]. Nephrology Dialysis Transplantation 2014;27(Suppl 3):ii21. [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt K, Gaillard C, Wyck D, et al. Efficacy of oral iron for treating iron deficiency in anaemic patients with non‐dialysis dependent CKD (ND‐CKD) [abstract]. Nephrology Dialysis Transplantation 2017;32(Suppl 3):iii581. [EMBASE: 617290898] [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt K, Gaillard CA, Wyck DB, et al. Iron administration to non‐dialysis CKD patients for one year does not cause worsening of renal function: results from the FIND‐CKD trial [abstract no: FR‐PO809]. Journal of the American Society of Nephrology 2014;25(Abstract Suppl):555A. [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard C, Wyck D, et al. Erythropoietic response to oral iron in patients with nondialysis‐dependent chronic kidney disease in the FIND‐CKD trial. Clinical Nephrology 2017;88(12):301‐10. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard C, Wyck D, et al. FIND‐CKD: a randomized trial of intravenous ferric carboxymaltose versus oral iron in patients with chronic kidney disease and iron deficiency anaemia. Nephrology Dialysis Transplantation 2014;29(11):2075‐84. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard C, Wyck D, et al. Renal function in patients with non‐dialysis chronic kidney disease receiving intravenous ferric carboxymaltose: an analysis of the randomized FIND‐CKD trial. BMC Nephrology 2017;18(1):24. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard CA, Wyck DB, et al. Hepcidin response to iron therapy in patients with non‐dialysis CKD: an analysis of the FIND‐CKD trial [abstract]. Nephrology Dialysis Transplantation 2015;30(Suppl 3):iii8‐9. [EMBASE: 72206297] [Google Scholar]
- Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard CA, Wyck DB, et al. Low rate of early response to oral iron in patients with non‐dialysis dependent CKD (ND‐CKD) [abstract no: TH‐PO906]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):304A. [Google Scholar]
- Roger S, Bock AH, Carrera F, Eckardt K, Gaillard CA, Wyck DB, et al. The safety of intravenous (IV) ferric carboxymaltose (FCM) in patients with non‐dialysis dependent CKD (ND‐CKD): 1‐year results from the FIND‐CKD trial [abstract]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i15‐6. [EMBASE: 72325953] [Google Scholar]
- Roger SD, Bock A, Carrera F, Eckardt KU, Gaillard C, Wyck DB, et al. IV ferric carboxymaltose (FCM) is a convenient and well‐tolerated first‐line therapy for the treatment of anaemia in patients with iron deficiency and non‐dialysis dependent chronic kidney disease (ND‐CKD): results of the randomised FIND‐CKD study [abstract no: SO048]. Nephrology Dialysis Transplantation 2014;27(Suppl 3):ii21. [Google Scholar]
- Roger SD, Bock AH, Carrera F, Eckardt KU, Gaillard CA, Wyck DB, et al. Intravenous (IV) ferric carboxymaltose (FCM) versus oral iron in the randomized FIND‐CKD trial of patients with non‐dialysis dependant CKD (ND‐CKD): a safety analysis [abstract no: TH‐PO652]. Journal of the American Society of Nephrology 2015;26(Abstract Suppl):238A. [Google Scholar]
Fishbane 1995 HD {published data only}
- Fishbane S, Frei GL, Maesaka J. Reduction in recombinant human erythropoietin doses by the use of chronic intravenous iron supplementation. American Journal of Kidney Diseases 1995;26(1):41‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fudin 1998 HD {published and unpublished data}
- Fudin R, Jaichenko J, Shostak A, Bennett M, Gotloib L. Correction of uremic iron deficiency anemia in hemodialyzed patients: a prospective study. Nephron 1998;79(3):299‐305. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hussain 1998 HD {published data only}
- Hussain R, Chishti SH, Naqvi S. Experience of iron saccharate supplementation in haemodialysis patients treated with erythropoietin. Nephrology 1998;4(1‐2):105‐8. [EMBASE: 1998205059] [Google Scholar]
- Hussain R, Chishti SH, Naqvi SA. Experience of iron sucrose supplementation in haemodialysis patients treated with erythropoietin [abstract]. 3rd International Congress of Nephrology, Urology & Transplantation Society of SAARC Countries; 1999 Feb 18‐21; Colombo, Sri Lanka. 1999:152. [CENTRAL: CN‐00460975]
Kalra 2016 CKD {published data only}
- Kalra PA, Bhandari S, Saxena S, Agarwal D, Wirtz G, Kletzmayr J, et al. A randomized trial of iron isomaltoside 1000 versus oral iron in non‐dialysis‐dependent chronic kidney disease patients with anaemia. Nephrology Dialysis Transplantation 2016;31(4):646‐55. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra PA, Bhandari S, Thomsen LL. Iron isomaltoside 1000 (monofer) compared to oral iron sulphate in European patients with non‐dialysis dependent chronic kidney disease (NDD‐CKD) [abstract]. Nephrology Dialysis Transplantation 2015;30(Suppl 3):iii200. [EMBASE: 72206812] [Google Scholar]
- Kalra PA, Bhandari S, Thomsen LL. Iron isomaltoside1000 (monofer) compared to oral iron sulphate in patients with non‐dialysis dependent chronic kidney disease (NDD‐CKD) [abstract no: SA‐PO1106]. Journal of the American Society of Nephrology 2014;25(Abstract Suppl):B7. [Google Scholar]
Kotaki 1997 HD {published data only}
- Kotaki M, Uday K, Henriquez M, Blum S, Dave M. Intravenous iron therapy in hemodialysis [abstract no: A1021]. Journal of the American Society of Nephrology 1996;7(9):1452. [CENTRAL: CN‐01657494] [Google Scholar]
- Kotaki M, Uday K, Henriquez M, Blum S, Dave M. Maintenance therapy with intravenous iron in hemodialysis patients receiving erythropoietin. Clinical Nephrology 1997;48(1):63‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Leehey 2005 CKD {published and unpublished data}
- Leehey DJ, Kaskas MO, Bastani B, Ferrlecit CKD Study Group. Sodium ferric gluconate complex (SFGC) in the treatment of chronic kidney disease (CKD) patients on stable erythropoietic therapy [abstract no: F‐PO966]. Journal of the American Society of Nephrology 2005;16:547A. [CENTRAL: CN‐00747316] [Google Scholar]
Li 2008 HD {published data only}
- Li H, Wang SX. Intravenous iron sucrose in Chinese hemodialysis patients with renal anemia. Blood Purification 2008;26(2):151‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Li H, Wang SX. Intravenous iron sucrose in maintenance dialysis patients with renal anemia: a clinical study. Chung‐Hua i Hsueh Tsa Chih [Chinese Medical Journal] 2009;89(7):457‐62. [MEDLINE: ] [PubMed] [Google Scholar]
Li 2008 PD {published data only}
- Li H, Wang SX. Intravenous iron sucrose in maintenance dialysis patients with renal anemia: a clinical study. Chung‐Hua i Hsueh Tsa Chih [Chinese Medical Journal] 2009;89(7):457‐62. [MEDLINE: ] [PubMed] [Google Scholar]
- Li H, Wang SX. Intravenous iron sucrose in peritoneal dialysis patients with renal anemia. Peritoneal Dialysis International 2008;28(2):149‐54. [MEDLINE: ] [PubMed] [Google Scholar]
Lu 2010 CKD {published and unpublished data}
- Besarab A, Coyne D, Bolton WK, Sharma A, Foti A, Brenner L. Ferumoxytol as an intravenous iron replacement therapy: safety results from two phase III studies in subjects with chronic kidney disease (CKD) [abstract no: SU‐PO0805]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):762A. [CENTRAL: CN‐00747286] [Google Scholar]
- Bolton WK, Besarab A, Germain M, Kovesdy CP, Hutchinson J, Krausz A. Ferumoxytol as an IV iron replacement therapy: efficacy results from two Phase III studies in subjects with chronic kidney disease (CKD) [abstract no: SU‐PO798]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):761A. [CENTRAL: CN‐00747287] [Google Scholar]
- Bolton WK, Fishbane S, Li J, Milich L, Brenner R. Increases in hemoglobin and the effect of ESA use in CKD patients treated with IV ferumoxytol [abstract]. American Journal of Kidney Diseases 2009;53(4):A30. [EMBASE: 70141518] [Google Scholar]
- Fishbane S, Bolton WK, Winkelmayer WC, Strauss W, Li Z, Pereira BJ. Factors affecting response and tolerability to ferumoxytol in nondialysis chronic kidney disease patients. Clinical Nephrology 2012;78(3):181‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Germain M, Brenner L, Dioguardi J, Gilbertson D, Besarab A. Comparison of safety and efficacy of ferumoxytol in CKD subtypes: kidney transplant recipients, hemodialysis patients and non‐hemodialysis patients [abstract no: SA‐PO2530]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):678A. [CENTRAL: CN‐00747289] [Google Scholar]
- Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. American Journal of Hematology 2010;85(5):315‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Spinowitz B, Besarab A, Bolton WK, Pereira BJ, Provenzano R, Rao M, et al. Ferumoxytol as intravenous iron replacement therapy in chronic kidney disease (CKD) patients not on dialysis ‐ evaluation of safety and efficacy in two phase III studies [abstract no: F‐PO026]. Journal of the American Society of Nephrology 2006;17(Abstracts):342A. [CENTRAL: CN‐00644267] [Google Scholar]
- Spinowitz B, Kausz A, Miller P, Brenner L, Singh A. Achievement of target hemoglobin with ferumoxytol versus oral iron in anemic CKD patients [abstract no: SU‐PO1031]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):814A. [CENTRAL: CN‐00688845] [Google Scholar]
- Spinowitz B, Krauz A, Dioguardi J, Kovesdy C. Evaluation of ferumoxytol safety and efficacy in all stages of chronic kidney disease (CKD) [abstract no: 249]. American Journal of Kidney Diseases 2008;51(4):A90. [CENTRAL: CN‐00747323] [Google Scholar]
Lye 2000 HD {published data only}
- Lye WC. Ferric gluconate polymaltose complex (Ferrum) is safe and effective for intravenous use in hemodialysis (HD) patients [abstract no: A1489]. Journal of the American Society of Nephrology 2000;11(Sept):284A. [Google Scholar]
Macdougall 1996 HD,PD,CKD {published and unpublished data}
- Macdougall IC, Tucker B, Thompson J, Baker IR, Raine AE. A randomised controlled study of iron supplementation in patients treated with erythropoietin [abstract]. Journal of the American Society of Nephrology 1993;4(Program & Abstracts):428. [CENTRAL: CN‐00484929] [Google Scholar]
- Macdougall IC, Tucker B, Thompson J, Tomson CR, Baker LR, Raine AE. A randomized controlled study of iron supplementation in patients treated with erythropoietin. Kidney International 1996;50(5):1694‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Macdougall IC, Tucker B, Thompson J, Tomson CR, Baker LR, Raine AE. Randomised controlled study of iron supplementation in patients treated with erythropoietin [abstract]. Nephrology Dialysis Transplantation 1993;8(12):1424. [CENTRAL: CN‐00260914] [DOI] [PubMed] [Google Scholar]
- Macdougall IC, Tucker B, Thompson J, Tomson CRV, Baker LR, Raine AE. Randomised controlled study of iron supplementation in patients treated with erythropoietin [abstract]. Nephrology Dialysis Transplantation 1993;8(9):960. [CENTRAL: CN‐01657602] [Google Scholar]
Macdougall 1999 HD,PD {published data only}
- Macdougall IC, UK Multicentre IV Iron Study Group. UK multicentre randomized controlled study of IV vs oral iron supplementation in dialysis patients receiving epoetin [abstract no: A1472]. Journal of the American Society of Nephrology 1999;10(Program & Abstracts):291A. [CENTRAL: CN‐00747320] [Google Scholar]
McMahon 2009 CKD {published data only}
- McMahon L, Kent AB, Roger S, Kerr P, Healy H, Irish A, et al. IV iron sucrose versus oral iron for the anaemia of chronic kidney disease (CKD) ‐ a randomized controlled trial [abstract no: O82]. Nephrology 2008;13(Suppl 3):A120. [CENTRAL: CN‐00747281] [Google Scholar]
- McMahon LP, Kent AB, Kerr PG, Healy H, Irish AB, Cooper B, et al. Maintenance of elevated versus physiological iron indices in non‐anaemic patients with chronic kidney disease: a randomized controlled trial. Nephrology Dialysis Transplantation 2010;25(3):920‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- McMahon LP, Kent AB, Roger SD, Kerr PG, Healy H, Irish AB, et al. IV iron sucrose versus oral iron for the anemia of chronic kidney disease (CKD) ‐ a randomized controlled trial [abstract no: SU‐PO1028]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):813A. [CENTRAL: CN‐00690668] [Google Scholar]
Michael 2007 HD {published data only}
- Michael B, Trout JR, Hoel G, Volinn W, Jorgensen N, Dahl NV, et al. Effectiveness of continuous low‐dose intravenous ferric gluconate therapy for maintaining Hgb and decreasing epoetin requirements in hemodialysis patients [abstract no: F‐PO849]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):289A. [CENTRAL: CN‐00689934] [Google Scholar]
Mudge 2009 TX {published data only}
- Mudge DW, Tan K, Miles R, Johnson DW, Campbell SB, Hawley CM, et al. Intravenous versus oral iron for post‐transplant anaemia: a randomised controlled trial [abstract no: 149]. Transplantation 2010;90(Suppl 1):679. [EMBASE: 71532404] [Google Scholar]
- Mudge DW, Tan KS, Miles R, Johnson DW, Badve SV, Campbell SB, et al. A randomized controlled trial of intravenous or oral iron for posttransplant anemia in kidney transplantation. Transplantation 2012;93(8):822‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Mudge DW, Tan KS, Miles R, Johnson DW, Campbell SB, Hawley CM, et al. Intravenous versus oral iron supplementation for correction of post‐transplant anaemia in renal transplant patients. BMC Nephrology 2009;10:14. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nagaraju 2013 CKD {published data only}
- Nagaraju SP, Cohn A, Akbari A, Davis JL, Zimmerman DL. Heme iron polypeptide for the treatment of iron deficiency anemia in non‐dialysis chronic kidney disease patients: a randomized controlled trial. BMC Nephrology 2013;14:64. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01155375 HD,PD,CKD {published data only}
- Strauss MD. A randomized, open‐label, active‐controlled study of the safety, efficacy, and pharmacokinetics of ferumoxytol compared with oral iron for the treatment of iron deficiency anemia in pediatric subjects with chronic kidney disease. www.clinicaltrials.gov/ct2/show/NCT01155375 (first received 1 July 2010).
Pisani 2014 CKD {published data only}
- Pisani A, Riccio E, Sabbatini M, Andreucci M, Rio A, Visciano B. Effect of oral liposomal iron versus intravenous iron for treatment of iron deficiency anaemia in CKD patients: a randomized trial. Nephrology Dialysis Transplantation 2014;30(4):645‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Visciano B, Nazzaro P, Riccio E, Rio A, Mozzillo GR, Pisani A. Liposomial iron for the treatment of iron deficiency anemia in non dialysis chronic kidney disease patients: a randomized controlled trial [abstract]. Nephrology Dialysis Transplantation 2014;29(5 Suppl 3):iii141‐2. [EMBASE: 71491842] [Google Scholar]
- Visciano B, Nazzaro P, Tarantino G, Taddei A, Rio A, Mozzillo GR, et al. Liposomial iron: a new proposal for the treatment of anaemia in chronic kidney disease [Il ferro liposomiale: una nuova proposta per il trattamento dell’anemia nei pazienti affetti da insufficenza renale cronica]. Giornale Italiano di Nefrologia 2013;30(5). [MEDLINE: ] [PubMed] [Google Scholar]
Provenzano 2009 HD {published and unpublished data}
- Germain M, Brenner L, Dioguardi J, Gilbertson D, Besarab A. Comparison of safety and efficacy of ferumoxytol in CKD subtypes: kidney transplant recipients, hemodialysis patients and non‐hemodialysis patients [abstract no: SA‐PO2530]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):678A. [CENTRAL: CN‐00747289] [Google Scholar]
- Lu M, Cohen MH, Rieves D, Pazdur R. FDA report. Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. American Journal of Hematology 2010;85(5):315‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Provenzano R, Schiller B, Rao M, Coyne D, Brenner L, Pereira BJ. Ferumoxytol as an intravenous iron replacement therapy in hemodialysis patients. Clinical Journal of the American Society of Nephrology: CJASN 2009;4(2):386‐93. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinowitz B, Krauz A, Dioguardi J, Kovesdy C. Evaluation of ferumoxytol safety and efficacy in all stages of chronic kidney disease (CKD) [abstract no: 249]. American Journal of Kidney Diseases 2008;51(4):A90. [CENTRAL: CN‐00688845] [Google Scholar]
Qunibi 2011 CKD {published data only}
- Benjamin J. A randomized controlled trial comparing IV ferric carboxymaltose (FCM) to oral iron in anemic nondialysis dependent CKD patients (with or without erythropoiesis stimulating agent use) [abstract no: M574]. World Congress of Nephrology; 2009 May 22‐26; Milan, Italy. 2009. [CENTRAL: CN‐01658222]
- Benjamin J, Qunibi W. Comparison of intravenous (IV) ferric carboxymaltose (FCM) to oral iron in anemic non‐dialysis dependent (NDD)‐CKD patients with or without ESA therapy [abstract no: SA‐PO2422]. Journal of the American Society of Nephrology 2009;20(Abstract Suppl):666A. [Google Scholar]
- Qunibi W, Martinez C, Smith M, Benjamin J, Dinh Q. A randomized controlled trial comparing IV ferric carboxymaltose (FCM) to oral iron in anemic patients with non‐dialysis‐dependent CKD [abstract no: SU‐PO1030]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):814A. [CENTRAL: CN‐00689191] [Google Scholar]
- Qunibi WY, Martinez C, Smith M, Benjamin J, Mangione A, Roger SD. A randomized controlled trial comparing intravenous ferric carboxymaltose with oral iron for treatment of iron deficiency anaemia of non‐dialysis‐dependent chronic kidney disease patients. Nephrology Dialysis Transplantation 2011;26(5):1599‐607. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ragab 2007 HD {published data only}
- Ragab M, Mahmoud K, Ragab A. Maintenance intravenous iron sucrose therapy in children under regular hemodialysis. Journal of Medical Sciences 2007;7(7):1112‐6. [EMBASE: 2007615596] [Google Scholar]
Souza 1997 HD {published data only}
- Souza RM, Defferrari R, Karohl C, Barros E, Thome F. Iron status evaluation and iron supplementation therapy in hemodialysis (HD) chronic renal failure (CRF) patients: a randomized clinical trial [abstract]. Nephrology 1997;3(Suppl 1):S307. [CENTRAL: CN‐00461775] [Google Scholar]
Spinowitz 2008 CKD {published and unpublished data}
- Besarab A, Coyne D, Bolton WK, Sharma A, Foti A, Brenner L. Ferumoxytol as an intravenous iron replacement therapy: safety results from two phase III studies in subjects with chronic kidney disease (CKD) [abstract no: SU‐PO0805]. Journal of the American Society of Nephrology 2007;18(Abstracts issue):762A. [CENTRAL: CN‐00747286] [Google Scholar]
- Bolton WK, Besarab A, Germain M, Kovesdy CP, Hutchinson J, Krausz A. Ferumoxytol as an IV iron replacement therapy: efficacy results from two Phase III studies in subjects with chronic kidney disease (CKD) [abstract no: SU‐PO798]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):761A. [CENTRAL: CN‐00747287] [Google Scholar]
- Bolton WK, Fishbane S, Li J, Milich L, Brenner R. Increases in hemoglobin and the effect of ESA use in CKD patients treated with IV ferumoxytol [abstract]. American Journal of Kidney Diseases 2009;53(4):A30. [EMBASE: 70141518] [Google Scholar]
- Brenner L, Miller P, Rodriguez S, Parikh N, Coyne DW. Treatment of iron‐deficiency anemia with IV ferumoxytol in CKD patients: efficacy compared with oral iron across different age groups [abstract]. Blood 2007;110(11). [CENTRAL: CN‐00646734] [Google Scholar]
- Fishbane S, Bolton WK, Winkelmayer WC, Strauss W, Li Z, Pereira BJ. Factors affecting response and tolerability to ferumoxytol in nondialysis chronic kidney disease patients. Clinical Nephrology 2012;78(3):181‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Germain M, Brenner L, Dioguardi J, Gilbertson D, Besarab A. Comparison of safety and efficacy of ferumoxytol in CKD subtypes: kidney transplant recipients, hemodialysis patients and non‐hemodialysis patients [abstract no: SA‐PO2530]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):678A. [CENTRAL: CN‐00747289] [Google Scholar]
- Horl WH. Comparing the efficacy of intravenous iron and oral iron in nondialysis patients with chronic kidney disease. Nature Clinical Practice Nephrology 2008;4(10):530‐1. [EMBASE: 2008457134] [DOI] [PubMed] [Google Scholar]
- Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. American Journal of Hematology 2010;85(5):315‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Spinowitz B, Bernardo M, Noble S, Baptista J, Pereira B, Brenner L. Safety and efficacy of ferumoxytol as an intravenous iron replacement therapy: results from a phase III study of chronic kidney disease (CKD) patients not on dialysis [abstract no: 30]. American Journal of Kidney Diseases 2007;49(4):A32. [CENTRAL: CN‐00644268] [Google Scholar]
- Spinowitz B, Besarab A, Bolton WK, Pereira BJ, Provenzano R, Rao M, et al. Ferumoxytol as intravenous iron replacement therapy in chronic kidney disease (CKD) patients not on dialysis ‐ evaluation of safety and efficacy in two phase III studies [abstract no: F‐PO026]. Journal of the American Society of Nephrology 2006;17(Abstracts):342A. [CENTRAL: CN‐00644267] [Google Scholar]
- Spinowitz B, Kausz A, Miller P, Brenner L, Singh A. Achievement of target hemoglobin with ferumoxytol versus oral iron in anemic CKD patients [abstract no: SU‐PO1031]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):814A. [CENTRAL: CN‐00688845] [Google Scholar]
- Spinowitz B, Krauz A, Dioguardi J, Kovesdy C. Evaluation of ferumoxytol safety and efficacy in all stages of chronic kidney disease (CKD) [abstract no: 249]. American Journal of Kidney Diseases 2008;51(4):A90. [CENTRAL: CN‐00747323] [Google Scholar]
- Spinowitz BS, Kausz AT, Baptista J, Noble SD, Sothinathan R, Bernardo MV, et al. Ferumoxytol for treating iron deficiency anemia in CKD. Journal of the American Society of Nephrology 2008;19(8):1599‐605. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stoves 2001 CKD {published data only}
- Stoves J, Inglis H, Newstead CG. A randomized study of oral vs intravenous iron supplementation in patients with progressive renal insufficiency treated with erythropoietin. Nephrology Dialysis Transplantation 2001;16(5):967‐74. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Strickland 1977 HD {published data only}
- Strickland ID, Chaput de Saintonge DM, Boulton FE, Francis B, Roubikova J, Waters JI. The therapeutic equivalence of oral and intravenous iron in renal dialysis patients. Clinical Nephrology 1977;7(2):55‐7. [MEDLINE: ] [PubMed] [Google Scholar]
Svara 1996 HD {published data only}
- Svara F, Sulkova S, Kvasnicka J, Polakovic V. Iron supplementation during erythropoietin therapy in patients on hemodialysis [Doplnovani zeleza pri lecbe erytropoetinem u hemodialyzovanych pacientu]. Vnitrni Lekarstvi 1996;42(12):849‐52. [MEDLINE: ] [PubMed] [Google Scholar]
Tsuchida 2010 HD {published data only}
- Tsuchida A, Paudyal B, Paudyal P, Ishii Y, Hiromura K, Nojima Y, et al. Effectiveness of oral iron to manage anemia in long‐term hemodialysis patients with the use of ultrapure dialysate. Experimental & Therapeutic Medicine 2010;1(5):777‐81. [EMBASE: 2010419867] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Wyck 2005 CKD {published and unpublished data}
- Wyck DB, Roppolo M, Martinez CO, Mazey RM, McMurray S, United States Iron Sucrose (Venofer) Clinical Trials Group. A randomized, controlled trial comparing IV iron sucrose to oral iron in anemic patients with nondialysis‐dependent CKD. Kidney International 2005;68(6):2846‐56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Wyck DB, Roppolo M, Martinez CO, McMurray SD, Mazey R. A randomized controlled trial comparing IV iron sucrose to oral iron in anemic patients with non‐dialysis‐dependent CKD [abstract no: F‐FC051]. Journal of the American Society of Nephrology 2005;16:49A. [CENTRAL: CN‐01657327] [Google Scholar]
Wang 2003 HD {published data only}
- Wang L, Li G, Liao C, Wang F. The effects of oral vs venous iron supplement in treatment of iron deficiency of maintained patients with anemia [abstract no: PUB324]. Journal of the American Society of Nephrology 2003;14(Nov):842A. [CENTRAL: CN‐00747318] [Google Scholar]
Warady 2002 HD {published and unpublished data}
- Warady BA, Kausz A, Lerner G, Brewer ED, Chadha V, Brugnara C, et al. Iron therapy in the pediatric hemodialysis population. Pediatric Nephrology 2004;19(6):655‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Warady BA, Kausz AT, Lerner G, Brewer ED, Chadha V, Brugnara C, et al. A comparison of intravenous and oral iron therapy in children receiving hemodialysis [abstract no: F‐PO789]. Journal of the American Society of Nephrology 2002;13(Program & Abstracts):221A. [CENTRAL: CN‐01657600] [Google Scholar]
Winney 1977 HD {published data only}
- Winney RJ, Swainson CP, Parker A, Bone JM, Robson JS. Iron therapy in hemodialysis patients: Oral or parenteral? [abstract]. Kidney International 1977;12(1):88. [CENTRAL: CN‐00747346] [PubMed] [Google Scholar]
References to studies excluded from this review
Adhikary 2011 {published data only}
- Adhikary L, Acharya S. Efficacy of IV iron compared to oral iron for increment of haemoglobin level in anemic chronic kidney disease patients on erythropoietin therapy. Jnma, Journal of the Nepal Medical Association 2011;51(183):133‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Allegra 1991 {published data only}
- Allegra V, Mengozzi G, Vasile A. Iron deficiency in maintenance hemodialysis patients: Assessment of diagnosis criteria and of three different iron treatments. Nephron 1991;57(2):175‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Charytan 2013 {published data only}
- Charytan C, Bernardo MV, Koch TA, Butcher A, Morris D, Bregman DB. Intravenous ferric carboxymaltose versus standard medical care in the treatment of iron deficiency anemia in patients with chronic kidney disease: a randomized, active‐controlled, multi‐center study. Nephrology Dialysis Transplantation 2013;28(4):953‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
HEMATOCRIT 2012 {published data only}
- Barraclough KA, Brown F, Hawley CM, Leary D, Noble E, Campbell SB, et al. A randomized controlled trial of oral heme iron polypeptide versus oral iron supplementation for the treatment of anaemia in peritoneal dialysis patients: HEMATOCRIT trial. Nephrology Dialysis Transplantation 2012;27(11):4146‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Barraclough KA, Noble E, Leary D, Brown B, Hawley CM, Campbell SB, et al. Rationale and design of the oral HEMe iron polypeptide Against Treatment with Oral Controlled Release Iron Tablets trial for the correction of anaemia in peritoneal dialysis patients (HEMATOCRIT trial). BMC Nephrology 2009;10:20. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lye 1997 {published data only}
- Lye WC, Chin S, Lee WT, Fan KS, Tan SY. Prospective randomised comparison of three routes of iron supplementation during erythropoietin (rHuEPO) therapy in hemodialysis patients [abstract no: A1334]. Journal of the American Society of Nephrology 1999;10(Program & Abstracts):264A. [CENTRAL: CN‐00677754] [Google Scholar]
- Lye WC, Chin S, Wong KC, Fan KS. A prospective randomised comparison of three routes of iron administration during erythropoietin (RHEUPO) therapy in dialysis patients [abstract no: P949]. Nephrology 1997;3(Suppl 1):S311. [CENTRAL: CN‐00520359] [Google Scholar]
- Lye WC, Chin S, Wong KC, Fan KS. Prospective randomised comparison of three routes of iron administration during erythropoietin (rHuEPO) therapy in hemodialysis patients [abstract no: A1018]. Journal of the American Society of Nephrology 1997;8(Program & Abstracts):220A. [CENTRAL: CN‐00446509] [Google Scholar]
Additional references
Avni 2015
- Avni T, Bieber A, Grossman A, Green H, Leibovici L, Gafter‐Gvili A. The safety of intravenous iron preparations: systematic review and meta‐analysis. Mayo Clinic Proceedings 2015;90(1):12‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bailie 2012
- Bailie GR. Adverse events associated with intravenous iron preparations: a comparison of reported rates. Clinical Advances in Hematology & Oncology 2012;10(9):600‐2. [MEDLINE: ] [PubMed] [Google Scholar]
Besarab 2000
- Besarab A, Amin N, Ahsan M, Vogel SE, Zazuwa G, Frinak S, et al. Optimization of epoetin therapy with intravenous iron therapy in hemodialysis patients. Journal of the American Society of Nephrology 2000;11(3):530‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
CARI 2008
- McMahon L. Haemoglobin. Nephrology 2008;13(s2):S44‐56. [DOI: 10.1111/j.1440-1797.2008.00997.x] [DOI] [PubMed] [Google Scholar]
Collister 2016
- Collister D, Komenda P, Hiebert B, Gunasekara R, Xu Y, Eng F, et al. The effect of erythropoietin‐stimulating agents on health‐related quality of life in anemia of chronic kidney disease: a systematic review and meta‐analysis. Annals of Internal Medicine 2016;164(7):472‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
DeVita 2003
- DeVita MV, Frumkin D, Mittal S, Kamran A, Fishbane S, Michelis MF. Targeting higher ferritin concentrations with intravenous iron dextran lowers erythropoietin requirement in hemodialysis patients. Clinical Nephrology 2003;60(5):335‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fishbane 2007
- Fishbane S. Iron management in nondialysis‐dependent CKD. American Journal of Kidney Diseases 2007;49(6):736‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fishbane 2014
- Fishbane S, Mathew A, Vaziri ND. Iron toxicity: relevance for dialysis patients. Nephrology Dialysis Transplantation 2014;29(2):255‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fukuhara 2007
- Fukuhara S, Yamazaki S, Marumo F, Akiba T, Akizawa T, Fujimi S, et al. Health‐related quality of life of predialysis patients with chronic renal failure. Nephron 2007;105(1):c1‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gaweda 2015
- Gaweda AE, Ginzburg YZ, Chait Y, Germain MJ, Aronoff GR, Rachmilewitz E. Iron dosing in kidney disease: inconsistency of evidence and clinical practice. Nephrology Dialysis Transplantation 2015;30(2):187‐96. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2011
- Guyatt G, Oxman A D, Akl E A, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction‐GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64:383‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hedges 2007
- Hedges SJ, Dehoney SB, Hooper JS, Amanzadeh J, Busti AJ. Evidence‐based treatment recommendations for uremic bleeding. Nature Clinical Practice Nephrology 2007;3(3):138‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Ishida 2014
- Ishida JH, Johansen KL. Iron and infection in hemodialysis patients. Seminars in Dialysis 2014;27(1):26‐36. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ishida 2015
- Ishida JH, Marafino BJ, McCulloch CE, Dalrymple LS, Dudley RA, Grimes BA, et al. Receipt of intravenous iron and clinical outcomes among hemodialysis patients hospitalized for infection. Clinical Journal of The American Society of Nephrology: CJASN 2015;10(10):1799‐805. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
KDIGO 2008
- Locatelli F, Nissenson AR, Barrett BJ, Walker RG, Wheeler DC, Eckardt KU, et al. Clinical practice guidelines for anemia in chronic kidney disease: problems and solutions. A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney International 2008;74(10):1237‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
KDIGO 2012
- Kidney Disease: Improving Global Outcomes. Summary of recommendation statements. Kidney International Supplements 2012;2(4):283‐7. [DOI: 10.1038/kisup.2012.41] [DOI] [PMC free article] [PubMed] [Google Scholar]
KDOQI 2007
- Eckardt KU, Wyck D. KDOQI 2007 update on hemoglobin target: recommendation and evidence base. www.kdigo.org/wp‐content/uploads/2017/01/Eckardt‐anemia‐in‐ckd‐2007.pdf (accessed 11 February 2019).
Kovesdy 2006
- Kovesdy CP, Trivedi BK, Kalantar‐Zadeh K, Anderson JE. Association of anemia with outcomes in men with moderate and severe chronic kidney disease.. Kidney International 2006;69(3):560‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kwack 2006
- Kwack C, Balakrishnan VS. Managing erythropoietin hyporesponsiveness. Seminars in Dialysis 2006;19(2):146‐51. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Levin 2006
- Levin A, Djurdjev O, Duncan J, Rosenbaum D, Werb R. Haemoglobin at time of referral prior to dialysis predicts survival: an association of haemoglobin with long‐term outcomes. Nephrology Dialysis Transplantation 2006;21(2):370‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Li 2004
- Li S, Collins AJ. Association of hematocrit value with cardiovascular morbidity and mortality in incident hemodialysis patients. Kidney International 2004;65(2):626‐33. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Locatelli 2009
- Locatelli F, Covic A, Eckardt K‐U, Wiecek A, Vanholder R, ERA‐EDTA ERBP Advisory Board. Anaemia management in patients with chronic kidney disease: a position statement by the Anaemia Working Group of European Renal Best Practice (ERBP). Nephrology Dialysis Transplantation 2009;24(2):348‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Locatelli 2010
- Locatelli F, Aljama P, Canaud B, Covic A, Francisco A, Macdougall IA, et al. Target haemoglobin to aim for with erythropoiesis‐stimulating agents: a position statement by ERBP following publication of the Trial to Reduce Cardiovascular Events with Aranesp® Therapy (TREAT) Study. Nephrology Dialysis Transplantation 2010;25(9):2846‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lopez 2015
- Lopez A, Cacoub P, Macdougall IC, Peyrin‐Biroulet L. Iron deficiency anaemia. Lancet 2016;387(10021):907‐16. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Macdougall 2016
- Macdougall IC, Bircher AJ, Eckardt KU, Obrador GT, Pollock CA, Stenvinkel P, et al. Iron management in chronic kidney disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney International 2016;89(1):28‐39. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Madore 2008
- Madore F, White CT, Foley RN, Barrett BJ, Moist LM, Klarenbach SW, et al. Clinical Practice Guidelines for assessment and management of iron deficiency. Kidney International ‐ Supplement 2008, (110):S7‐11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moist 2008
- Moist LM, Foley RN, Barrett BJ, Madore F, White CT, Klarenbach SW, et al. Clinical practice guidelines for evidence‐based use of erythropoietic‐stimulating agents. Kidney International ‐ Supplement 2008, (110):S12‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moreno 2000
- Moreno F, Sanz‐Guajardo D, Lopez‐Gomez JM, Jofre R, Valderrabano F. Increasing the hematocrit has a beneficial effect on quality of life and is safe in selected hemodialysis patients. Spanish Cooperative Renal Patients Quality of Life Study Group of the Spanish Society of Nephrology. Journal of the American Society of Nephrology 2000;11(2):335‐42. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moyer 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
NICE 2015
- National Institute of Health and Care Excellence. Chronic kidney disease: managing anaemia. www.nice.org.uk/guidance/ng8 June 2015. [Google Scholar]
Phrommintikul 2007
- Phrommintikul A, Haas SJ, Elsik M, Krum H. Mortality and target haemoglobin concentrations in anaemic patients with chronic kidney disease treated with erythropoietin: a meta‐analysis. Lancet 2007;369(9559):381‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rozen‐Zvi 2008
- Rozen‐Zvi B, Gafter‐Gvilli A, Paul M, Leibovici L, Shpilberg O, Gafter U. Intravenous versus oral iron supplementation for the treatment of anemia in CKD: systematic review and meta‐analysis. American Journal of Kidney Diseases 2008;52(5):897‐906. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sargent 2004
- Sargent JA, Acchiardo SR. Iron requirements in hemodialysis. Blood purification 2004;22(1):112‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schultz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JP, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Higgins JP, Deeks JJ, Glasziou P, Guyatt GH. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Shah 2011
- Shah SV, Rajapurkar MM, Baliga R. The role of catalytic iron in acute kidney injury. Clinical Journal of The American Society of Nephrology: CJASN 2011;6(10):2329‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shepshelovich 2016
- Shepshelovich D, Rozen‐Zvi B, Avni T, Gafter U, Gafter‐Gvili A. Intravenous versus oral iron supplementation for the treatment of anemia in CKD: an updated systematic review and meta‐analysis. American Journal of Kidney Diseases 2016;68(5):677‐90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stauffer 2014
- Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS ONE [Electronic Resource] 2014;9(1):e84943. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Susantitaphong 2014
- Susantitaphong P, Alqahtani F, Jaber BL. Efficacy and safety of intravenous iron therapy for functional iron deficiency anemia in hemodialysis patients: a meta‐analysis. American Journal of Kidney Diseases 2014;39(2):130‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vlagopoulos 2005
- Vlagopoulos PT, Tighiouart H, Weiner DE, Griffith J, Pettitt D, Salem DN, et al. Anemia as a risk factor for cardiovascular disease and all‐cause mortality in diabetes: the impact of chronic kidney disease. Journal of the American Society of Nephrology 2005;16(11):3403‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Weiner 2005
- Weiner DE, Tighiouart H, Vlagopoulos PT, Griffith JL, Salem DN, Levey AS, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. Journal of the American Society of Nephrology 2005;16(6):1803‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Albaramki 2009
- Albaramki J, Hodson EM, Craig JC. Parenteral versus oral iron therapy for adults and children with chronic kidney disease. Cochrane Database of Systematic Reviews 2009, Issue 7. [DOI: 10.1002/14651858.CD007857] [DOI] [PubMed] [Google Scholar]
Albaramki 2012
- Albaramki J, Hodson EM, Craig JC, Webster AC. Parenteral versus oral iron therapy for adults and children with chronic kidney disease. Cochrane Database of Systematic Reviews 2012, Issue 1. [DOI: 10.1002/14651858.CD007857.pub2] [DOI] [PubMed] [Google Scholar]