

Summary
The human gut microbiome matures toward the adult composition during the first years of life and is implicated in early immune development. Here, we investigate the effects of microbial genomic diversity on gut microbiome development using integrated early childhood datasets collected in the DIABIMMUNE study in Finland, Estonia and Russian Karelia. We show that gut microbial diversity is associated with household location and linear growth of children. Single nucleotide polymorphism (SNP)- and metagenomic assembly-based strain tracking revealed large and highly dynamic microbial pangenomes, especially in the genus Bacteroides, in which we identified evidence of variability deriving from Bacteroides-targeting bacteriophages. Our analyses revealed functional consequences of strain diversity; only 10% of Finnish infants harbored Bifidobacterium longum subsp. infantis, a subspecies specialized in human milk metabolism, whereas Russian infants commonly maintained a probiotic Bifidobacterium bifidum strain in infancy. Groups of bacteria contributing to diverse, characterized metabolic pathways converged to highly subject-specific configurations over the first two years of life. This longitudinal study extends the current view of early gut microbial community assembly based on strain-level genomic variation.
Introduction
Mounting evidence shows the developing gut microbiome, particularly immediately after birth, plays an important role in human health1–3. Immune system maturation is orchestrated by early microbial exposures4,5, and early childhood immune-mediated disorders including type 1 diabetes (T1D)6, asthma7, juvenile rheumatoid arthritis8, allergic disease9, and inflammatory bowel disease 10 are linked to aberrations in gut microbiota. Several human T1D cohort studies reported gut microbiota alterations11 and increased intestinal permeability12 prior to diagnosis, but mechanisms connecting gut health to autoimmune destruction of pancreatic beta cells remain relatively unknown. Complex relationships between the microbiome and the immune system13,14 during the first years of life appear critical to later life health outcomes but have not been explored at the population scale.
Increasingly, microbiome-linked health outcomes appear to be consequences of individual strains of specific microbes15–17. These outcomes can result from structural variants in gene products of individual strains18, the presence or absence of gene cassettes19,20 or currently unexplained mechanisms. Until recently, most culture-independent methods for investigating microbiomes in large-scale human populations (e.g., 16S rRNA gene amplicon sequencing) were limited in their resolution of distinct microbial strains. Now, efficient metagenomic sequencing and culture-independent strain-level analysis methods enable more detailed investigation of the early life microbiome.
Single-nucleotide polymorphisms (SNPs) accruing in microbial genomes over time can be used as a molecular clock to evaluate evolutionary distance between strains of the same species21–24. Such approaches have been instrumental in investigating maternal seeding of infant gut microbiomes23,25–28. Complementing SNP-based methodologies, metagenomic assembly enables detecting and profiling known and previously unknown metagenomic species and genes, as well as gene content-based strain tracking16,29–31. Gene-centric strain profiling also evaluates functional implications of strain-level variability more directly19,20. While SNP- and assembly-based approaches successfully improved the resolution and clinical relevance of many population-level microbiome studies, comparisons of these complementary approaches on a single large microbiome dataset to identify subpopulations and functional adaptations has not been published.
For instance, bifidobacteria are widely-characterized beneficial commensals, commonly dominating the gut during breastfeeding and dissipating throughout life, that possess immunomodulatory functions, produce beneficial metabolites and metabolize a range of diet-derived, nondigestible carbohydrates32. Subspecies found specifically in the infant gut typically harbor a wide variety of genes enabling the sole use of human milk oligosaccharides (HMOs) for energy33. B. longum subsp. infantis (B. infantis)34 and some B. longum subsp. longum strains35 are capable of membrane transport and intracellular degradation of intact HMOs, whereas other B. longum subspecies rely partially on extracellular enzymes for HMO utilization36. Culture-independent detection of B. infantis, or any other Bifidobacterium strains, in metagenomic data is not well-established and requires the above-mentioned strain characterization methods.
Here, we characterize strain-specific genomic variation and its contribution to the early gut microbiome using an integrated and extended dataset from DIABIMMUNE, which includes nearly 300 children with human leukocyte antigen (HLA) haplotypes conferring increased risk to autoimmune disorders (roughly four-fold over the background population) in three neighboring countries: Finland, Estonia, and Russian Karelia. These children were observed for three years from birth by monthly stool sampling, frequent questionnaires about common life events and circumstances, and periodic blood sampling to track different immune parameters. The integrated DIABIMMUNE dataset consists of 16S rRNA gene sequencing of 3,204 samples and metagenomic sequencing of 1,154 samples, together spanning 289 subjects at an average of 11.4 (range 1-36) timepoints per subject. We briefly report association analyses of 16S data followed by SNP- and metagenomic assembly-based strain and pangenome analyses of common early gut species. The integrated DIABIMMUNE microbiome data provide detailed, strain-level characterizations of the developing gut microbiome.
Results
DIABIMMUNE followed Finnish, Estonian, and Russian children for three years from birth by collecting monthly stool samples, periodic serum samples, and frequent questionnaires on early life events. Here, we integrate all published DIABIMMUNE microbiome data generated in multiple studies using 16S rRNA amplicon, metagenomic and virome sequencing techniques37–40. After a unified quality control process, the data consisted of 3,204 16S amplicon and 1,154 metagenomic sequencing profiles from 289 and 269 subjects, respectively (Table 1, Supplementary Figure 1, Supplementary Table 1).
Table 1. DIABIMMUNE microbiome cohort statistics.
Distribution of study subjects, stool samples with sequencing data and several other external variables across the study sites. The table shows the number of study subjects (N) per category unless otherwise specified. T1D autoantibody and diagnosis information as of Nov. 2016.
| Finland | Estonia | Russia | |
|---|---|---|---|
| Study subjects | 140 | 80 | 73 |
| Samples profiled by 16S rRNA gene sequencing (median per subject) | 2,080 (9) | 501 (6) | 623 (7) |
| Samples profiled by metagenomic sequencing (median per subject) | 616 (4) | 221 (3) | 317 (3) |
| Males/Females | 78/62 | 39/41 | 40/33 |
| Caesarean sections | 9 | 6 | 12 |
| Mean maternal age at birth (sd) | 31.1 (4.9) | 29.1 (5.1) | 27.8 (4.7) |
| Born in rural household | 10 (7.7%) | 19 (23.8%) | 0 |
| T1D AAB seropositive subjects | 11 | 4 | 1 |
| Subjects with T1D diagnosis | 5 | 1 | 1 |
Early gut microbiome explorations are complicated by natural dynamics and numerous interactions with intrinsic and extrinsic factors. To further understand early microbial development relative to these factors, we analyzed 16S data using both omnibus and individual association tests. Early growth, household location and antibiotic courses during pregnancy, among other variables, were associated with early gut microbial composition (Supplementary Note 1, Supplementary Figure 1, Supplementary Tables 2–4). Height at age three (Supplementary Figure 1) and growth rate during the first three years of life were positively associated with microbial diversity.
Strain diversity and ecology in the early gut
To expand the analysis beyond the genus level typical for 16S data, we leveraged shotgun metagenomic data to perform in-depth, strain-level analyses using SNPs and gene content. Strain analysis can delineate microbial sub-populations22,41 and identify potential functional adaptations in the gut microbiome31. Particularly, de novo strain identification is important for species with a limited number of isolates, and the gut microbiome has many such understudied species despite large cultivation efforts42.
We first characterized dominant strains for the most abundant species in the metagenomic data by calling SNPs on conserved and unique species-specific marker genes selected from their core genomes (i.e., genes shared across all strains within the species)21. This resulted in a marker gene-based SNP haplotype of the dominant strain per species, hereby referred to as SNP haplotype. We then compared SNP haplotypes by sequence similarity and stratified them in intra- and inter-subject comparisons (Fig. 1A, Supplementary Figure 2, Supplementary Table 5). Longitudinal, intra-subject comparisons showed more similar strains compared to inter-subject comparisons, as previously observed22,43. We found a wide range of strain diversities among investigated bacterial species (Fig. 1A, Supplementary Table 5): Haemophilus parainfluenzae and Faecalibacterium prausnitzii were among the most diverse species, with strains having less than 95% sequence similarity in SNP haplotype comparisons. Conversely, all investigated members of genus Bacteroides had very low sequence variability, with virtually identical SNP haplotypes in intra-subject comparisons (mean sequence similarity 99.96% over an average of 44.3kb of core genome per species) and over 99.6% sequence similarity on average in inter-subject comparisons. All other species analyzed had an average inter-subject similarity of 98.9%.
Figure 1. Strain diversity across species in early gut metagenomes.
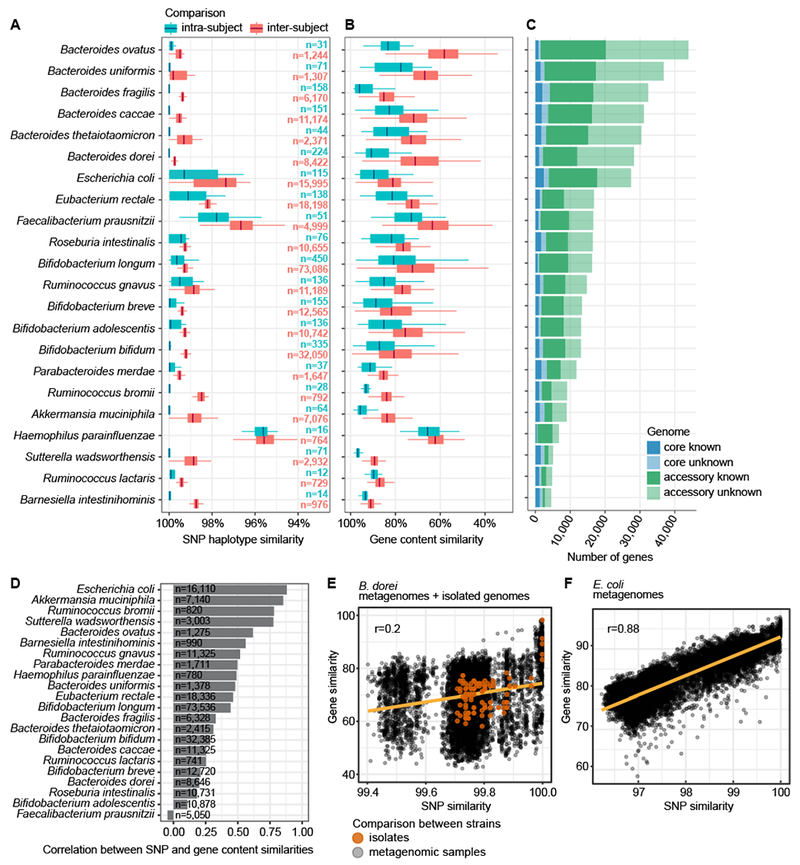
A SNP haplotype similarities per species based on all pairwise comparisons (dominant strain per species per sample) and stratified to intra-subject and inter-subject comparisons. Species containing >10 comparisons in both strata are shown. B Gene content similarities (the percentage of shared genes in the smallest of the two genomes) per species, evaluated on pangenomes generated by metagenomic assembly. Boxplots as in panel A. The box (A, B) shows the interquartile range (IQR), the vertical line shows the median and the whiskers show the range of the data (up to 1.5 times IQR). Sample size (n) per boxplot in panel A gives number of comparisons per panels A and B. C The size of core and accessory genomes per species stratified by the functional annotation of genes using eggNOG. Panels A-C are ordered according to the size of the metagenomic pangenome. D Pearson’s correlation coefficients between SNP- and gene content-based similarity measures between strains. Sample size (n) is indicated. E B. dorei strains’ SNP and gene content similarities show low Pearson’s correlation (r=0.2, n=8646 comparisons from metagenomes, n=136 comparisons of isolate genomes). Comparisons between isolate genomes are shown in orange for reference. F E. coli strains’ SNP and gene content similarities (Pearson’s r=0.88, n=16,110 comparisons).
The observed high level of sequence identity in Bacteroides spp. contradicted existing evidence of diversity in their gene content44. We speculated that the SNP haplotypes did not capture all means of microbial genetic diversification, which include lateral gene transfer (LGT) and niche adaptation. Previously, we identified Bacteroides dorei as a highly abundant species potentially interfering with early immune maturation39. Therefore, we investigated genome diversity further in this species by isolating and sequencing eight B. dorei strains from human stool (Supplementary Note 2, Supplementary Figure 2, Supplementary Tables 6–7). Each isolate genome harbored between 276 and 1,168 (median 750) unique accessory genes representing, on average, 13% of their genomes. Given the numbers of unique accessory genes across these B. dorei isolates, we wondered whether this diversity partially owed to LGT by bacteriophages.
Bacteriophages contribute to genome plasticity in Bacteroides spp.
Bacteroides-targeting bacteriophages (phages) are among the most common members of the highly diverse human gut virome, leading us to evaluate their contribution to the observed genome plasticity in this genus. To investigate bacteria-phage interactions in the gut, we utilized viral contigs (viromes) assembled using data from virus-like particle preparations of 22 DIABIMMUNE subjects40. Using bacterial metagenomes of subjects with viral contig data and a designated computational method45 (Supplementary Note 3), we reconstructed metagenomic CRISPR arrays that harbor spacer sequences targeting phages to which bacteria have adapted a response. We found the number of CRISPR spacers in the metagenomes reflected the microbiomes’ increasing diversity and overall maturation (Supplementary Figure 3). In total, we identified 2,463 CRISPR spacers matching sequences in the gut virome across all samples, of which 223 matched sequences in the gut virome of the same subject (Supplementary Figure 4, Supplementary Table 8). We found 658 of these spacers in CRISPR cassettes on assembled metagenomes, covering 32 bacterial taxa, with B. vulgatus harboring the most (84, 28.5% of the spacers matching contigs annotated on species level), B. dorei harboring 9, and all Bacteroides spp. collectively harboring 138 spacer sequences (Supplementary Table 9). Additionally, when mapped against the virome, 105 (4.2%) spacers matched viral contigs annotated as Bacteroides spp phages. These data suggest Bacteroides spp. were exposed to an extensive phage repertoire in the children’s guts, providing a plausible mechanism for increased genomic plasticity in Bacteroides.
Metagenomic assembly highlights strain diversification patterns within gut species
To explore accessory genome variation more broadly and across all taxa, we turned to de novo metagenome assembly of DIABIMMUNE metagenomes. This expanded the gene pool (number of observed non-redundant genes) to 6,328,944 gene families, compared to 1,932,010 gene families found using NCBI isolate genomes. Using a co-abundance technique30, we binned the assembled metagenomes into metagenomic species and constructed pangenomes for 22 species (Fig. 1B, C).
Among all analyzed species, Bacteroides spp. and E. coli harbored the largest assembled pangenomes, each with over 25,000 genes (Fig. 1C), consistent with high genome plasticity reported in these taxa44,46. Consistent with the SNP haplotype analysis, strains recovered from the same individual were more similar to each other than to strains found in different individuals (Fig. 1B, Supplementary Table 5). The magnitude of variability, however, was much higher for gene content than for SNP haplotypes (Fig. 1A, B). These measures were highly correlated in most species but showed low or no correlation in a minor subset, including F. prausnitzii and B. dorei (Fig. 1D). In B. dorei, the results from metagenomic assemblies and isolate genomes showed a similar trend, suggesting the incongruity between SNP haplotypes and assembled genomes was not an artefact of metagenomic assembly (Fig. 1E). Rather, it indicates a more rapid or higher volume diversification in the accessory genome compared to the point mutation rate in the core genome. In contrast, E. coli metagenome assemblies displayed a high correlation between gene content and SNP haplotype similarities (Fig. 1F) as previously reported47.
We again used the longitudinal nature of our study to compare the difference in gene content between strains from the same or different individuals. On average, B. dorei metagenomic assemblies had 88% gene content similarity in intra-subject comparisons and significantly lower (69%) similarity in inter-subject comparisons. Notably, these values are within the observed range comparing B. dorei isolate genomes (Supplementary Figure 2). H. parainfluenzae, an autochthonous oral cavity member, was an outlier with very similar intra- and inter-subject gene similarities (65% and 62%, respectively), conceivably reflecting transient gut colonization events and frequent replacement with new strains transmitted from the oral cavity. A closer investigation of colonization patterns for several common oral species in the guts of these children revealed that many such species bloomed in Finnish and Estonian infants during the first year of life (Supplementary Note 4, Supplementary Figure 5, Supplementary Table 10).
Strain-level variation in Bifidobacterium spp. reflects breastfeeding patterns and geography
Our strain diversity analysis identified relatively high intra-subject variability in B. longum compared to other common Bifidobacterium species B. bifidum or B. breve (Fig. 1A, B). Given implications of bifidobacteria in immune development and early microbial community assembly2,48, we sought to explore in more detail the functional consequences of this strain-level variation during infancy. To first identify a known B. longum subspecies clade, B. infantis, we surveyed the metagenomic data for genes of a well-characterized B. infantis cluster responsible for HMO transport and degradation34. Presence of these genes corresponded with the SNP haplotype-based phylogeny of B. longum strains (Fig. 2A, B) and two B. infantis reference sequences (ATCC 15697) clustered with 70 strains harboring these genes (highlighted red in Fig. 2A). We found evidence of this gene cluster in 14 additional samples, which possibly harbored multiple B. longum strains, of which B. infantis was non-dominant, and resulted in a SNP haplotype profile not based on B. infantis.
Figure 2. Bifidobacterium strains in DIABIMMUNE children.
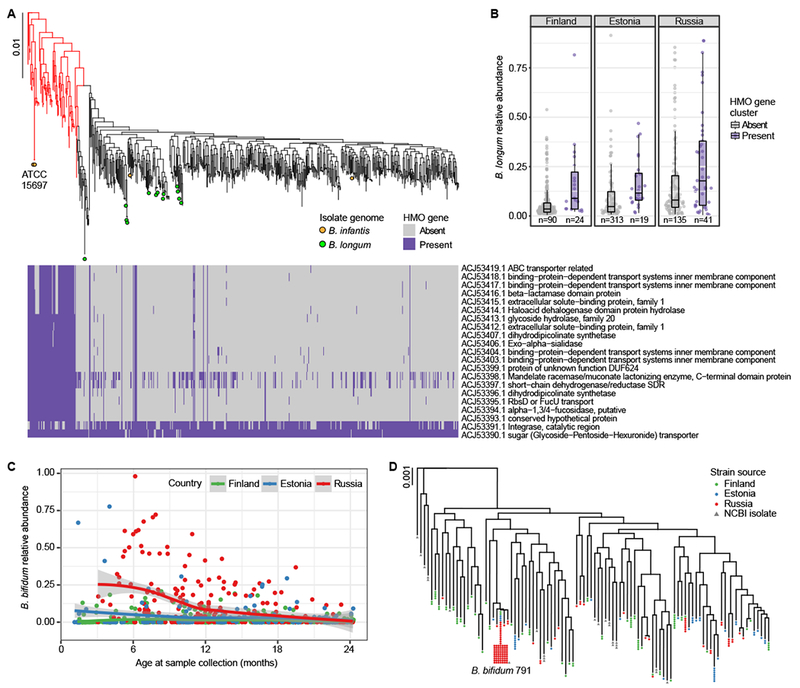
A Phylogenetic tree of B. longum strains in DIABIMMUNE stool samples and 18 NCBI B. longum isolate genomes based on SNP haplotypes. Highlighted B. infantis strains (red) include two reference sequences (ATCC 15697). The heatmap illustrates strain-specific carriage of 21 genes in the B. infantis HMO gene cluster responsible for intracellular HMO degradation, evaluated using the metagenomic data. B B. longum relative abundance stratified by country and B. longum strain; B. infantis (highlighted red in panel A) has, on average, higher relative abundance compared to other B. longum strains (mixed effects logistic regression p=0.00049). The box shows the interquartile range (IQR), the vertical line shows the median and the whiskers show the range of the data (up to 1.5 times IQR). Number of samples (n) are indicated below each box and include samples from subjects with no breastfeeding information. C Relative abundance of B. bifidum longitudinally stratified by the countries up to 24 months of age (n = 864). Russians have more B. bifidum, especially during the first year of life. The curves show locally weighted scatterplot smoothing (LOESS) fits for the relative abundances, and shaded areas show 95% confidence interval for each fit, as implemented in geom_smooth function in ggplot2 R package. D Phylogenetic tree of B. bifidum strains in the DIABIMMUNE stool samples based on SNP haplotypes. Strains with >99.5% sequence similarity have been collapsed into a single tip. A known strain, B. bifidum 791, was found in 79 stool samples. The scale bars on phylogenetic trees denote difference in sequence similarity of SNP haplotypes.
B. infantis was found in 23.7% (42/177) of stool samples collected during breastfeeding but only 3.2% (11/343) of samples collected after weaning (excluding samples with low relative abundance of B. longum precluding strain identification and samples from subjects with no breastfeeding information), reflecting a clear strain shift relative to breastfeeding cessation. Overall, 10% of Finns, 20% of Estonians and 23% of Russians harbored B. infantis in at least one stool sample (either during breastfeeding or after cessation), suggesting that most subjects in this cohort never obtained B. infantis in their gut ecosystems. Comparing B. infantis with other B. longum strains revealed evidence of a competitive advantage, conferred by the HMO gene cluster or other genomic differences, that allows B. infantis to reach higher relative abundances on average (Fig. 2B), albeit with modest effect sizes.
Probiotics supplements and foods containing commercial strains of Bifidobacterium spp. are also a common source of bifidobacteria in early life. One such species, B. bifidum, showed contrasting relative abundances between the countries: unlike Finnish and Estonian samples, Russian samples commonly contained over 10% of B. bifidum (Fig. 2C). B. bifidum SNP haplotypes revealed that 79 samples from 34 Russians, 3 Estonians, and 2 Finns harbored the same B. bifidum strain with greater than 99.9% sequence similarity (Fig. 2D). This SNP haplotype was identical to the NCBI isolate genome B. bifidum 791, which was isolated from a healthy human gut in Nizhny Novgorod, Russia and has been patented for medical use in Russia. B. bifidum relative abundance was over 10% in 57 of 79 (75%) samples containing this strain. While these observations are not direct evidence for engraftment of this strain, they show that a probiotic strain can obtain high (>50%) relative abundance in the infant gut.
Contributional diversity of microbial functions
Finally, we approached the developing microbiome from a function-centric view49. We binned species based on shared functional pathways and assessed their contributional diversity (i.e., how diverse sets of species encode and have a potential to perform a given function per sample)49 for 365 Gene Ontology (GO) biological process terms present in over 100 metagenomes. Most GO terms displayed increasing within-sample functional diversity (Gini-Simpson index) with increasing age that coincides with microbiome maturation and increasing taxonomic diversity (Fig. 3A, Supplementary Table 11). Many widely distributed pathways such as sporulation (GO:0030435), glycolysis (GO:0006096) and riboflavin biosynthesis (GO:0009231) followed this pattern. Contrastingly, a few specific pathways displayed the opposite trend: aerobic electron transport chain (r=−0.16, q=0.001, GO:0019646), viral release from host cell (r=−0.05, q=1.0, GO:0019076) and siderophore biosynthetic process (r=−0.07, q=1.0, GO:0015891) showed decreasing or stable functional diversity within samples over time (Fig. 3A, Supplementary Table 11).
Figure 3. Contributional diversity of microbial pathways.
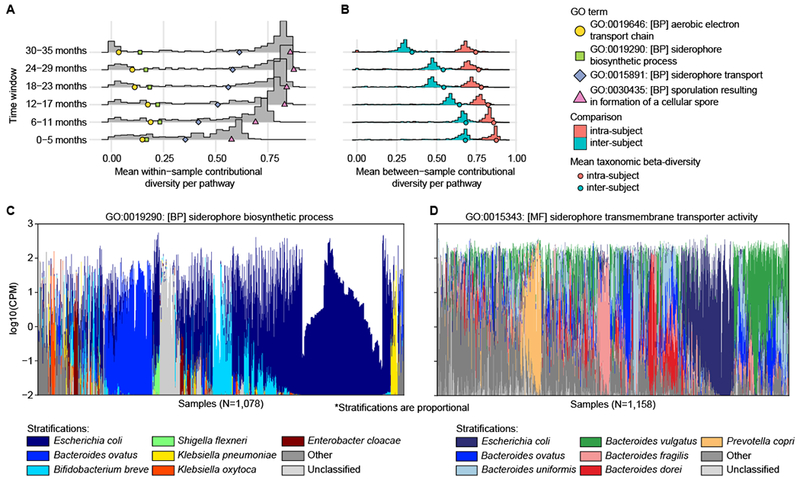
A-B We applied alpha- (A) and beta-diversity (B) to the distribution of species contributing to functional categories (GO biological process terms), measuring their contributional diversities. The histograms show the mean alpha- (A) and beta-diversities (B) per GO term stratified by time windows. Colored shapes show (A) examples of pathways with different trends and (B) mean intra- and inter-subject beta-diversities of taxonomic profiles. C-D Species contributing to (C) siderophore biosynthetic process and (D) siderophore transport. Colors displaying the contributions of individual species are linearly scaled within the log-scaled total bar height depicting the total abundance of the pathway.
Between-sample contributional diversities (beta-diversity, Bray-Curtis dissimilarity) reflect the stability of functional contributions per pathway and can be assessed longitudinally within and across subjects. We observed a decreasing longitudinal trend in contributional beta-diversities with increasing age (Fig. 3B, correlation between age and beta-diversity, Pearson r=−0.28, p<2.2e-16), reflecting a change towards a more stable, adult microbial composition. Microbial contributions to pathways were more stable within individuals (Student’s t-test p<1e-20 in all time windows), as reflected by lower beta-diversities, and the gap between intra- and inter-subject comparisons tended to widen with time, similar to the average beta-diversities of taxonomic profiles (Fig. 3B). This provides another perspective of early stabilization of gut microbial communities: as pathways in some cases reflect ecological niches (e.g., aerobic electron transport), the above trend may mirror convergence to specific ecological attractor states, which in turn results in stabilization after community adaptation and competition over the niche has resolved (Fig. 3B).
Pathways related to bacterial acquisition of iron by siderophores (Fig. 3A) provide an example interpreting contributional diversities49. Bacteria secrete iron-binding siderophores to harvest iron, but extracellular siderophores are exploited by other bacteria. According to the black queen hypothesis, the ability to produce such costly but necessary molecules is under negative selection until the production is minimal but sufficient to support the microbial community50. Indeed, according to our data, a single dominant species per community contributes to siderophore biosynthesis (i.e., low contributional alpha-diversity, Fig. 3A, C), whereas siderophore transport-related genes are more widely distributed across community members (Fig. 3A, D).
Discussion
Here, we report a longitudinal, strain-level investigation of the developing gut microbiome utilizing the DIABIMMUNE cohort and its rich metadata of various life events. We integrated all published microbiome data, resulting in 3,204 16S amplicon and 1,154 metagenomic sequencing profiles from 289 and 269 subjects, respectively. This integrated dataset will serve as a reference resource for the microbiome research community. Further, our analyses contribute to taxonomic and functional understandings of early gut communities.
A strain can be defined experimentally — a single clonal isolate — or operationally — a combination of variants detected in a metagenomic assembly, phased haplotype, or collection of reads — as used here. SNP and gene content profiling offer complementary means of tracking microbial strains in metagenomic data. While metagenomes can provide information on many strains simultaneously, depth of resolution on any one strain is limited. SNP-based methods usually operate within a few percentages of each genome that serves as a marker region for evaluating evolutionary distance within a population21–24. Evaluating the gene content of microbial strains offers more direct means for functional interpretation of any observed differences19,20. For most species, these approaches provided highly concordant phylogenetic population structures, as evidenced by high correlation between SNP haplotype and gene content similarities. In some species such as F. prausnitzii and B. dorei, however, these measures did not correlate. F. prausnitzii is a phylogenetically diverse clade, consisting of distinct subspecies clades that blur the distinction between strain tracking and species differentiation and potentially confound the methodologies for tracking strains41. We isolated and sequenced eight high-quality B. dorei genomes that confirmed this observation. For these and similar species, such as B. adolescentis and R. intestinalis, the observed lack of correlation may stem from the difference between the time scales at which the measures operate; rapid genetic adaptation driven by promiscuous LGT and gene loss contrasted by slower, long-term SNP acquisition may confound the correlation51. The consequence of most of these adaptations for strain fitness or symbiosis with the host early in life, especially considering the known immunomodulatory effects of specific strains , remains to be elucidated.
This study contributes several observations on another group of bacteria common in early childhood, Bifidobacterium. We observed virtually identical B. bifidum strains in 79, mostly Russian, samples. This analysis demonstrates that microbial strains may be shared on the population level and such strain-level trends can be detected from metagenomic data. The observed strain, B. bifidum 791, has been patented for medical use in Russia (http://russianpatents.com/patent/216/2165454.html), and local regulation allows adding such bacterial components to infant formulas (GOST 30626-98 “Dry milk products for infant feeding” http://gostexpert.ru/data/files/30626-98/0f82d40248598989307bf0a50b573429.pdf). Our communication with locals confirmed this strain is common in baby formulas and other infant food products. Therefore, these 34 Russian infants potentially obtained this strain, which may achieve stable engraftment, as a probiotic supplement. This observation supports the idea that early gut microbial assembly can be intervened by probiotics48, which can confer beneficial effects such as restoration of healthy growth52 and protection against immune-mediated diseases53 or adverse effects of antibiotic courses54.
There are consequential differences in HMO processing capabilities within Bifidobacterium species that underscore the importance of identifying bacterial strains in this genus. We detected B. infantis in metagenomic data by both its HMO processing genes and SNP haplotype profile. We observed B. infantis in only 10% of Finns in this cohort, suggesting that this keystone species may be less prevalent in Finnish gut ecosystems. Among other effects, this may lead to elevated fecal pH levels, further promoting inflammation-favoring bacteria and gut dysbiosis55. A probiotic trial adding B. infantis to breast milk during the first weeks of life demonstrated persistent B. infantis engraftment and beneficial alterations in intestinal fermentation48. Our data corroborates the notion that intracellular HMO utilization provides B. infantis a competitive advantage over other HMO-consuming species, allowing B. infantis to dominate the infant gut during breastfeeding. Based on these findings and literature (reviewed by Insel and Knip56), we hypothesize that natively resident or supplemented B. infantis during breastfeeding drives a shift in gut microbial community structure, shaping subsequent ecology and potentially immune development and/or protection against immune disorders in genetically predisposed populations. This could be tested by further characterization of the associated bifidobacterial functional diversity and by randomized, placebo-controlled clinical trials in humans.
Methods
DIABIMMUNE cohort
The DIABIMMUNE cohort recruitment took place between September 2008 and July 2011 in Finland, Estonia and Russia. Families with a newborn infant with HLA DR-DQ alleles conferring increased risk for autoimmunity, determined by a cord blood test, were invited to join the study. The parents gave their written informed consent prior to sample collection. The study participants were monitored for infections, use of antibiotics, breastfeeding, introduction of complementary foods, and other life events on study visits at months 3, 6, 12, 18, 24, and 36 from birth. Maternal information and events during the pregnancy were collected using a questionnaire on these visits. Serum samples were collected from all subjects during visits to the clinic at the following time points: 0 (cord blood), 3, 6, 12, 18, 24, and 36 months. Diabetes-associated autoantibodies were analyzed as previously described37. The DIABIMMUNE study was conducted according to the guidelines in the Declaration of Helsinki, and all procedures involving human subjects were approved by Ethical Committee for Psychiatric Diseases and Diseases in Children and Adolescents, Helsinki and Uusimaa Hospital District (Finland), Ethics Review Committee on Human Research of the University of Tartu (Estonia) and Ethical Committee, Ministry of Health and Social Development, Karelian Republic of the Russian Federation (Russia). More information about the cohort and data collection can be found in other DIABIMMUNE publications37–39 and online at http://www.diabimmune.org/ and https://pubs.broadinstitute.org/diabimmune/.
For statistical association testing described below, the additional information (external variables) of subjects was preprocessed as follows. The external variables were categorized into two categories: generic and complex variables. Here, generic variable (maternal age at delivery, gestational diabetes, gestational age in days, mode of delivery, gender, country of birth, cohort, and HLA risk class) information was available for all subjects and contained no missing values. Complex variables, on the other hand, contained missing values and in many cases required pre-processing and exact defining beforehand (e.g., antibiotics courses, maternal illnesses during pregnancy, urban or rural family location when the child was born, daycare attendance, elder siblings, etc.). As breastfeeding information was not available for all the subjects and reduced the sample sizes significantly in cross-sectional analyses, it was not considered a generic variable. While the associations between the generic variables and the gut microbial communities were modeled together in one analysis, the associations of complex variables were determined by modeling them one-by-one with all generic variables.
16S sequencing analysis
16S rRNA gene sequencing was conducted essentially as previously described57. Paired-end sequencing reads were demultiplexed using ea-utils command line tools (https://code.google.com/p/ea-utils/) and clustered into operational taxonomic units (OTUs) using the UPARSE pipeline58. Reads were quality-filtered using the UPARSE quality-filtering threshold of Emax=1, at which the most probable number of base errors per read is zero for filtered reads59. Filtered reads were trimmed to a fixed length, singletons removed, and clustered de novo into OTUs, with simultaneous chimera filtering. Taxonomic classification of OTUs was performed against the Greengenes version 13.8 16S rDNA database60. The full OTU table was filtered by removing samples with less than 3,000 OTU counts and by removing OTUs appearing in less than 5% of samples (178 samples). This resulted in an OTU table consisting of 3,204 samples from 289 subjects and 920 OTUs.
PERMANOVA analysis between the external variables and gut microbiomes were performed on 16S rRNA amplicon sequencing data of samples collected roughly at 2 (between 0 and 90 days), 6 (170 and 260 days) and 18 months (510 to 600 days) of age using adonis function in vegan R package (default parameters). Per each subject, the sample closest to the exact cross-section time under analysis was chosen, resulting in 140, 184 and 202 samples per time window, respectively. The order of external variables in the multivariable PERMANOVA model formula was determined by first analyzing each variable individually using univariable PERMANOVA model and then ordering the variables based on the significance of their association (i.e., permuted p-value) from the most to the least significant in the multivariable PERMANOVA model. Statistical significance of PERMANOVA results was evaluated by permutation test with 10,000 permutations.
Individual associations between bacterial genera and external variables were tested using MaAsLin, which conducts outlier removal, feature selection and linear modeling61. Association analyses were performed in both cross-sectional and longitudinal manners. The cross-sectional analyses were conducted on the same samples from the time windows chosen for the PERMANOVA analyses, where all variables of the analyses (only generic variables or generic and one added complex variable) were used as fixed effects. In the longitudinal analyses, subject IDs were used as a random effect, and all the generic variables and breastfeeding information were used as fixed effects. In the case a complex variable was added to the analysis, it was also used as a fixed effect. With these effect settings in longitudinal analyses, a total of 2,586 samples from 237 subjects were available, where the numbers varied according to the complex variable added to the analysis and the amount of missing values it introduced. For both the cross-sectional and longitudinal analyses, genus-level 16S rRNA microbiome data was used for identifying taxonomic level associations of the external variables.
Metagenomic sequencing
The metagenomic shotgun sequencing was conducted as previously described37–39. Additional sequencing data was generated for 45 samples which were excluded from a previous investigation39 due to low read count and are indicated in Supplementary Table 1. The quality control for the metagenomic shotgun sequencing data was conducted using kneadData v0.4.6.1 with additional automatic adapter detection and trimming at a minimum overlap of 5bp by Trim Galore!. Taxonomic profiles were generated using MetaPhlAn v2.662 and functional profiling was done by HUMAnN2 v0.10.049, which provides gene family level (here, 90% similarity) quantifications of microbial genes that are further stratified by contributing organisms. The gene families were further mapped to Gene Ontology (GO)63 terms as previously described39. Strain SNP haplotypes were generated using StrainPhlAn21 by requiring minimum coverage of 10 bases for SNP calling (“--min_read_depth 10” command line parameter for sample2markers.py).
Metagenomic assembly
Metagenomic reads were assembled into contigs using MegaHIT64 individually for each sample, followed by an open reading frame prediction using Prodigal65. A non-redundant gene catalogue was constructed in a fashion similar to earlier approaches29 by clustering genes based on sequence similarity at 95% identity and 90% coverage of the shorter sequence using CD-HIT66. Subsequently, the gene catalogue was merged with the IGC gene catalogue67 using the same criteria to create a more comprehensive reference gene catalogue for the gut microbiome. Only genes detected in DIABIMMUNE samples (~6M) were used in the downstream analysis. Gene abundance was estimated by mapping quality trimmed reads from each sample to the gene catalogue with BWA68. This served as an input for binning genes into metagenomic species using canopy clustering that finds core genes for each metagenomic species30. Metagenomic species with at least 400 genes were retained for further analysis. To extend the analysis beyond core genes, we detected accessory genes in each sample in which the metagenomic species was present in the following manner. We recruited genes co-assembled on the same contigs as core genes as long as the abundances of these genes was between the 10th and 90th percentiles of the abundances of core genes in a sample. Core genes and accessory genes grouped together in this manner across all samples defined the metagenomic pangenome of a species. To define a metagenomic strain, we used the same 10th and 90th percentiles of the abundances of core genes criteria to determine the specific accessory genes from the pangenome associated with the core genes of a metagenomic species in a sample. Similarity between pairs of metagenomic strains within species was measured using the percentage of shared genes in the smallest of the two genomes, as established previously47. Assembled genes were annotated with COG, KEGG and GO terms using eggnog mapper69 and at species, genus and phylum levels with NCBI RefSeq (version July 2017) as described previously67.
Phylogenetic trees
Phylogenetic trees (Fig. 2A, D, Supplementary Figure 5) were generated based on StrainPhlAn SNP haplotypes using the phangorn R package70. Briefly, similarities between strain haplotypes were computed using Jukes and Cantor (JC69) model, and an initial tree was constructed using UPGMA hierarchical clustering. The tree was optimized using maximum likelihood method, by iterative optimization of edge lengths, base frequencies and topology. Visualizations were generated using ggtree R package. For Bifidobacterium bifidum (Fig. 2D), strains with >99.5% sequence similarity were collapsed to a single tip and represented by the strain with the lowest average distance to other strains prior to optimizing the phylogenetic tree.
B. dorei isolate genomes
Bacteroides dorei colonies were isolated from serial dilutions of DIABIMMUNE and PRISM (Prospective Registry in IBD Study at Massachusetts General Hospital) stool samples plated on selective and non-selective media after being incubated anaerobically at 37 °C for 72 hours. To isolate high molecular weight DNA for PacBio sequencing (Pacific Biosciences, Menlo Park, USA), the isolates were grown on brain heart infusion agar supplemented (sBHI) with 10% fetal bovine serum (Hyclone), 1% hemin/vitamin K solution (BD), 1% trace vitamins (ATCC), 1% trace minerals (ATCC), 0.5 g/L cysteine hydrochloride (Sigma), 1 g/L maltose, 1 g/L fructose (VWR), and 1 g/L cellubiose (Sigma) anaerobically at 37 °C for 72 hours. Colonies were transferred to 30 mL sBHI broth and grown anaerobically for 48 hours. Cells were centrifuged at 4,450 rpm for 10 minutes and supernatant was discarded. DNA was extracted using the Genomic-tip 500/G kit (Qiagen) according to the manufacturer’s instruction. After isopropanol treatment, precipitated DNA was spooled and transferred to 70% ethanol 1.2 mL tube and left to dry in a clean PCR hood for 4 hours. Dried DNA was resuspended in elution buffer (Qiagen). DNA fragment size was measured with 4200 TapeStation (Agilent) using a Genomic DNA ScreenTape (Agilent). PacBio sequencing libraries were constructed by the blunt-ended ligation of SMRT bell adapter sequences to needle-sheared genomic DNA as per the manufacturer’s instructions (Pacific Biosciences). The libraries were damage-repaired using the SMRTbell Damage Repair Kit (Pacific Biosciences) following the manufacturer’s instructions and subsequently size-selected on a Blue Pippin with an 8 kb cut-off and then loaded on a Sequel sequencing instrument with MagBeads as per the manufacturer’s instructions (Pacific Biosciences). The genomes were assembled using the internal PacBio assembler HGAP4. Reads less than 6 kb in length were excluded from the assembly process.
The assembled B. dorei genomes were analyzed using PanPhlAn71 (default settings) together with five existing isolate genomes in NCBI. The resulting non-redundant gene catalogue was annotated by translated DIAMOND search72 against the UniRef90 and UniRef50 databases and by enforcing UniRef’s clustering criteria. We primarily used UniRef90 annotations, if available, but applied UniRef50 annotation in absence of UniRef90 annotation.
B. longum gene analysis
B. longum HMO gene presence in the metagenomic samples (Fig. 2A) was determined as follows. We identified UniRef90 gene families corresponding to the protein sequences in B. infantis HMO gene cluster34 (protein sequences Blon_2331-Blon_2361 in NCBI protein sequence database) by translated BLAST search against B. longum pangenome in ChocoPhlAn pangenome collection73 utilized by HUMAnN2. Specifically, we required ≥90% alignment identify and ≥80% mutual coverage (corresponding to the definition of UniRef90 gene families) and accepted only the best hit per protein sequence. Combining this information with HUMAnN2 species-stratified UniRef90 gene family quantification enabled calling these genes present given that they had sufficient read coverage, here defined as log10(counts-per-million / B. longum relative abundance) > 1.
Contributional diversities of metagenomic functions
Contributional diversities of the metagenomic functions were analyzed as previously described49. Briefly, stratified abundances of metagenomic functions were first renormalized after excluding any “unclassified” relative abundance. Contributional diversity for a given metagenomic function was then calculated by applying ecological similarity measures to the stratified abundance of that function. Gini-Simpson index was used for alpha-diversity and Bray-Curtis dissimilarity was used for beta-diversity.
CRISPR array detection and mapping
CRISPR spacers and repeat sequences were searched using Crass version 0.3.845. We mapped all identified 42,412 CRISPR spacers sequences to viral contigs of 112 samples with viromic data using bowtie2 version 2.3.4.174 using the parameters `-N 1 --local --no-unal` and exported results in bam format using `samtools view -bS -` with an overall alignment rate of 5.81% (2,463 aligned spacers). Alignment of the 42,412 spacers and 3,272 repeats against the full set of metagenomic assembled contigs (n=5,368,547) were performed with the same bowtie2 setting, resulting in an overall alignment rate of 73% and 95% for spacers and repeats, respectively. We determined the number of spacer and repeat matching the contigs of the DIABIMMUNE assembly and marked 658 spacers matching a virome contig in which also repeat matches were found. The majority of the repeats (93%) had multiple matches in the assembly, as expected for CRISPR repeats.
Data availability
All 16S rRNA and metagenomic sequencing data are available in NCBI Sequence Read Archive under BioProject PRJNA497734 and through the DIABIMMUNE microbiome website at https://pubs.broadinstitute.org/diabimmune/.
Code availability
The analysis software used for quality control and taxonomic and functional profiling is publicly available in bioBakery at https://bitbucket.org/biobakery/biobakery/ and referenced as appropriate. More detailed analysis scripts will be made available upon request.
Supplementary Material
Acknowledgements
We thank Tiffany Poon and Scott Steelman (Broad Institute) for help in sequence production and sample management, Ali Rahnavard for help in HMP SNP haplotype analysis, Dmitry Shungin for discussions and connections regarding the use of infant milk products in Russia, Katriina Koski and Matti Koski (University of Helsinki) for the coordination and database work in the DIABIMMUNE study and Theresa Reimels for editorial help in writing and figure generation. T.V. was supported by funding from Juvenile Diabetes Research Foundation (JDRF). A.B.H. is a Merck Fellow of the Helen Hay Whitney Foundation. P.C.M. received funding from the German Research Foundation (Grant number 315980449). C.H. was supported by funding from the JDRF (3-SRA-2016-141-Q-R) and the National Institutes of Health (R24DK110499). M.K. was supported by the European Union Seventh Framework Programme FP7/2007-2013 (202063) and the Academy of Finland Centre of Excellence in Molecular Systems Immunology and Physiology Research (250114). R.J.X. was supported by funding from JDRF (2-SRA-2016-247-S-B and 2-SRA-2018-548-S-B), the National Institutes of Health (DK43351 and AI110498), and the Center for Microbiome Informatics and Therapeutics.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Kundu P, Blacher E, Elinav E & Pettersson S Our Gut Microbiome: The Evolving Inner Self. Cell 171, 1481–1493, doi: 10.1016/j.cell.2017.11.024 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Backhed F et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 690–703, doi: 10.1016/j.chom.2015.04.004 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Chu DM et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med 23, 314–326, doi: 10.1038/nm.4272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bach JF The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol 18, 105–120, doi: 10.1038/nri.2017.111 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Haahtela T et al. The biodiversity hypothesis and allergic disease: world allergy organization position statement. World Allergy Organ J 6, 3, doi: 10.1186/1939-4551-6-3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rewers M & Ludvigsson J Environmental risk factors for type 1 diabetes. Lancet 387, 2340–2348, doi: 10.1016/S0140-6736(16)30507-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrieta MC et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med 7, 307ra152, doi: 10.1126/scitranslmed.aab2271 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Arvonen M et al. Gut microbiota-host interactions and juvenile idiopathic arthritis. Pediatr Rheumatol Online J 14, 44, doi: 10.1186/s12969-016-0104-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simonyte Sjodin K, Vidman L, Ryden P & West CE Emerging evidence of the role of gut microbiota in the development of allergic diseases. Curr Opin Allergy Clin Immunol 16, 390–395, doi: 10.1097/ACI.0000000000000277 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Lewis JD et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500, doi: 10.1016/j.chom.2015.09.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knip M & Siljander H The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol 12, 154–167, doi: 10.1038/nrendo.2015.218 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Maffeis C et al. Association between intestinal permeability and faecal microbiota composition in Italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab Res Rev 32, 700–709, doi: 10.1002/dmrr.2790 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Thaiss CA, Zmora N, Levy M & Elinav E The microbiome and innate immunity. Nature 535, 65–74, doi: 10.1038/nature18847 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Honda K & Littman DR The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84, doi: 10.1038/nature18848 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Lebreton F et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. MBio 4, doi: 10.1128/mBio.00534-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall AB et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med 9, 103, doi: 10.1186/s13073-017-0490-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schonherr-Hellec S et al. Clostridial strain-specific characteristics associated with necrotizing enterocolitis. Appl Environ Microbiol, doi: 10.1128/AEM.02428-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bron PA, van Baarlen P & Kleerebezem M Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nat Rev Microbiol 10, 66–78, doi: 10.1038/nrmicro2690 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Ward DV et al. Metagenomic Sequencing with Strain-Level Resolution Implicates Uropathogenic E. coli in Necrotizing Enterocolitis and Mortality in Preterm Infants. Cell Rep 14, 2912–2924, doi: 10.1016/j.celrep.2016.03.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazen TH et al. Genomic diversity of EPEC associated with clinical presentations of differing severity. Nat Microbiol 1, 15014, doi: 10.1038/nmicrobiol.2015.14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truong DT, Tett A, Pasolli E, Huttenhower C & Segata N Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27, 626–638, doi: 10.1101/gr.216242.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd-Price J et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66, doi: 10.1038/nature23889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korpela K et al. Selective maternal seeding and environment shape the human gut microbiome. Genome Res 28, 561–568, doi: 10.1101/gr.233940.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mende DR, Sunagawa S, Zeller G & Bork P Accurate and universal delineation of prokaryotic species. Nat Methods 10, 881–884, doi: 10.1038/nmeth.2575 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Asnicar F et al. Studying Vertical Microbiome Transmission from Mothers to Infants by Strain-Level Metagenomic Profiling. mSystems 2, doi: 10.1128/mSystems.00164-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nayfach S, Rodriguez-Mueller B, Garud N & Pollard KS An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res 26, 1612–1625, doi: 10.1101/gr.201863.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yassour M et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 24, 146–154 e144, doi: 10.1016/j.chom.2018.06.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferretti P et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145 e135, doi: 10.1016/j.chom.2018.06.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin J et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65, doi: 10.1038/nature08821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen HB et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol 32, 822–828, doi: 10.1038/nbt.2939 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Scher JU et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202, doi: 10.7554/eLife.01202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bottacini F, van Sinderen D & Ventura M Omics of bifidobacteria: research and insights into their health-promoting activities. Biochem J 474, 4137–4152, doi: 10.1042/BCJ20160756 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Sela DA & Mills DA Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol 18, 298–307, doi: 10.1016/j.tim.2010.03.008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sela DA et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A 105, 18964–18969, doi: 10.1073/pnas.0809584105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrido D et al. A novel gene cluster allows preferential utilization of fucosylated milk oligosaccharides in Bifidobacterium longum subsp. longum SC596. Sci Rep 6, 35045, doi: 10.1038/srep35045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sela DA Bifidobacterial utilization of human milk oligosaccharides. Int J Food Microbiol 149, 58–64, doi: 10.1016/j.ijfoodmicro.2011.01.025 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Kostic AD et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17, 260–273, doi: 10.1016/j.chom.2015.01.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yassour M et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med 8, 343ra381, doi: 10.1126/scitranslmed.aad0917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vatanen T et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 165, 842–853, doi: 10.1016/j.cell.2016.04.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao G et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A 114, E6166–E6175, doi: 10.1073/pnas.1706359114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He Q et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. Gigascience 6, 1–11, doi: 10.1093/gigascience/gix050 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browne HP et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546, doi: 10.1038/nature17645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schloissnig S et al. Genomic variation landscape of the human gut microbiome. Nature 493, 45–50, doi: 10.1038/nature11711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange A et al. Extensive Mobilome-Driven Genome Diversification in Mouse Gut-Associated Bacteroides vulgatus mpk. Genome Biol Evol 8, 1197–1207, doi: 10.1093/gbe/evw070 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skennerton CT, Imelfort M & Tyson GW Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res 41, e105, doi: 10.1093/nar/gkt183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Land M et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15, 141–161, doi: 10.1007/s10142-015-0433-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snel B, Bork P & Huynen MA Genome phylogeny based on gene content. Nat Genet 21, 108–110, doi: 10.1038/5052 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Frese SA et al. Persistence of Supplemented Bifidobacterium longum subsp. infantis EVC001 in Breastfed Infants. mSphere 2, doi: 10.1128/mSphere.00501-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franzosa EA et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods, doi: 10.1038/s41592-018-0176-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris JJ, Lenski RE & Zinser ER The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 3, doi: 10.1128/mBio.00036-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andreani NA, Hesse E & Vos M Prokaryote genome fluidity is dependent on effective population size. ISME J 11, 1719–1721, doi: 10.1038/ismej.2017.36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subramanian S et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417–421, doi: 10.1038/nature13421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uusitalo U et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr 170, 20–28, doi: 10.1001/jamapediatrics.2015.2757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fox MJ, Ahuja KD, Robertson IK, Ball MJ & Eri RD Can probiotic yogurt prevent diarrhoea in children on antibiotics? A double-blind, randomised, placebo-controlled study. BMJ Open 5, e006474, doi: 10.1136/bmjopen-2014-006474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henrick BM et al. Elevated Fecal pH Indicates a Profound Change in the Breastfed Infant Gut Microbiome Due to Reduction of Bifidobacterium over the Past Century. mSphere 3, doi: 10.1128/mSphere.00041-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Insel R & Knip M Prospects for Primary Prevention of Type 1 Diabetes by Restoring a Disappearing Microbe. Pediatr Diabetes, doi: 10.1111/pedi.12756 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392, doi: 10.1016/j.chom.2014.02.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996–998, doi: 10.1038/nmeth.2604 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Edgar RC & Flyvbjerg H Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482, doi: 10.1093/bioinformatics/btv401 (2015). [DOI] [PubMed] [Google Scholar]
- 60.McDonald D et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618, doi: 10.1038/ismej.2011.139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79, doi: 10.1186/gb-2012-13-9-r79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Segata N et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9, 811–814, doi: 10.1038/nmeth.2066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, doi: 10.1038/75556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li D, Liu CM, Luo R, Sadakane K & Lam TW MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676, doi: 10.1093/bioinformatics/btv033 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119, doi: 10.1186/1471-2105-11-119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu L, Niu B, Zhu Z, Wu S & Li W CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, doi: 10.1093/bioinformatics/bts565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32, 834–841, doi: 10.1038/nbt.2942 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huerta-Cepas J et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol Biol Evol 34, 2115–2122, doi: 10.1093/molbev/msx148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schliep KP phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593, doi: 10.1093/bioinformatics/btq706 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scholz M et al. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 13, 435–438, doi: 10.1038/nmeth.3802 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60, doi: 10.1038/nmeth.3176 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Huang K et al. MetaRef: a pan-genomic database for comparative and community microbial genomics. Nucleic Acids Res 42, D617–624, doi: 10.1093/nar/gkt1078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All 16S rRNA and metagenomic sequencing data are available in NCBI Sequence Read Archive under BioProject PRJNA497734 and through the DIABIMMUNE microbiome website at https://pubs.broadinstitute.org/diabimmune/.